Co-reporter:Huixian Wu;Chong Wang;Karen J. Gregory;Gye Won Han;Hyekyung P. Cho;Yan Xia;Colleen M. Niswender;Vsevolod Katritch;Jens Meiler;Vadim Cherezov;P. Jeffrey Conn
Science 2014 Volume 344(Issue 6179) pp:58-64
Publication Date(Web):04 Apr 2014
DOI:10.1126/science.1249489
Completing the Set
G protein–coupled receptors (GPCRs) are membrane proteins that transduce extracellular signals to activate diverse signaling pathways. Significant insight into GPCR function has come from structures of three of four classes of GPCRs—A, B, and Frizzled. Wu et al. (p. 58, published online 6 March) complete the picture by reporting the structure of metabotropic glutamate receptor 1, a class C GPCR. The structure shows differences in the seven-transmembrane (7TM) domain between class C and other classes; however, the overall fold is preserved. Class C GPCRs are known to form dimers through their extracellular domains; however, the structure suggests additional interactions between the 7TM domains mediated by cholesterol.
Co-reporter:Raymond C. Stevens,
Vadim Cherezov,
Vsevolod Katritch,
Ruben Abagyan,
Peter Kuhn,
Hugh Rosen
&
Kurt Wüthrich
Nature Reviews Drug Discovery 2013 12(1) pp:25
Publication Date(Web):2012-12-14
DOI:10.1038/nrd3859
G protein-coupled receptors (GPCRs) are targeted by ~30–40% of marketed drugs, and their key roles in normal physiology and in disease demonstrate that an understanding of their structure and function is valuable to researchers in both basic science and drug discovery. However, until recently, detailed structural information on this protein family was limited by challenges in X-ray crystallographic analysis of such membrane proteins. The GPCR Network was created in 2010 with the goal of structurally characterizing 15–25 representative human GPCRs within 5 years, based on an active outreach programme addressing an interdisciplinary community of scientists interested in GPCR structure, chemistry and biology. Here, we provide an overview of how this collaborative effort has enabled the structural determination and characterization of eight human GPCRs so far, and discuss some of the challenges that remain in gaining more detailed insights into structure–function relationships in this receptor superfamily.
Co-reporter:Chong Wang;Yi Jiang;Jinming Ma;Huixian Wu;Daniel Wacker;Vsevolod Katritch;Gye Won Han;Wei Liu;Eyal Vardy;Xi-Ping Huang;John D. McCorvy;Xiang Gao;X. Edward Zhou;Karsten Melcher;Chenghai Zhang;Fang Bai;Huaiyu Yang;Linlin Yang;Hualiang Jiang;Bryan L. Roth;Vadim Cherezov;H. Eric Xu
Science 2013 Volume 340(Issue 6132) pp:610-614
Publication Date(Web):03 May 2013
DOI:10.1126/science.1232807
Dissecting Serotonin Receptors
Serotonin receptors are the targets for many widely used drugs prescribed to treat ailments from depression to obesity and migraine headaches (see the Perspective by Palczewski and Kiser). C. Wang et al. (p. 610, published online 21 March) and Wacker et al. (p. 615, published online 21 March) describe crystal structures of two members of the serotonin family of receptors bound to antimigraine medications or to a precursor of the hallucinogenic drug LSD. Subtle differences in the way particular ligands bind to the receptors cause substantial differences in the signals generated by the receptor and the consequent biological responses. The structures reveal how the same ligand can activate one or both of the two main serotonin receptor signaling mechanisms, depending on which particular receptor it binds.
Co-reporter:Daniel Wacker;Chong Wang;Vsevolod Katritch;Gye Won Han;Xi-Ping Huang;Eyal Vardy;John D. McCorvy;Yi Jiang;Meihua Chu;Fai Yiu Siu;Wei Liu;H. Eric Xu;Vadim Cherezov;Bryan L. Roth
Science 2013 Volume 340(Issue 6132) pp:615-619
Publication Date(Web):03 May 2013
DOI:10.1126/science.1232808
Dissecting Serotonin Receptors
Serotonin receptors are the targets for many widely used drugs prescribed to treat ailments from depression to obesity and migraine headaches (see the Perspective by Palczewski and Kiser). C. Wang et al. (p. 610, published online 21 March) and Wacker et al. (p. 615, published online 21 March) describe crystal structures of two members of the serotonin family of receptors bound to antimigraine medications or to a precursor of the hallucinogenic drug LSD. Subtle differences in the way particular ligands bind to the receptors cause substantial differences in the signals generated by the receptor and the consequent biological responses. The structures reveal how the same ligand can activate one or both of the two main serotonin receptor signaling mechanisms, depending on which particular receptor it binds.
Co-reporter:Michael A. Hanson;Christopher B. Roth;Euijung Jo;Mark T. Griffith;Fiona L. Scott;Greg Reinhart;Hans Desale;Bryan Clemons;Stuart M. Cahalan;Stephan C. Schuerer;M. Germana Sanna;Gye Won Han;Peter Kuhn;Hugh Rosen
Science 2012 Volume 335(Issue 6070) pp:851-855
Publication Date(Web):17 Feb 2012
DOI:10.1126/science.1215904
Co-reporter:Jeffrey J. Liu;Reto Horst;Vsevolod Katritch;Kurt Wüthrich
Science 2012 Vol 335(6072) pp:1106-1110
Publication Date(Web):02 Mar 2012
DOI:10.1126/science.1215802
Co-reporter:Eugene Chun;Wei Liu;Aaron A. Thompson;Pavel Chubukov;Fei Xu;Vsevolod Katritch;Gye Won Han;Christopher B. Roth;Laura H. Heitman;Adriaan P. IJzerman;Vadim Cherezov
Science 2012 Volume 337(Issue 6091) pp:232-236
Publication Date(Web):13 Jul 2012
DOI:10.1126/science.1219218
GPCR Close-Up
Structures of G protein–coupled receptors (GPCRs) determined in the past few years, have provided insight into the function of this important family of membrane proteins. Liu et al. (p. 232) used a protein-engineering strategy to produce a stabilized version of the human A2Aadenosine receptor (A2AAR). The high-resolution structure reveals the position of about 60 internal waters, which suggests an almost continuous channel in the GPCR and can explain the allosteric effects of Na+ on ligand binding and how cholesterol may contribute to GPCR stabilization.
Co-reporter:Fei Xu;Huixian Wu;Vsevolod Katritch;Gye Won Han;Kenneth A. Jacobson;Zhan-Guo Gao;Vadim Cherezov
Science 2011 Vol 332(6027) pp:322-327
Publication Date(Web):15 Apr 2011
DOI:10.1126/science.1202793
Changes associated with conformationally selective agonist binding shed light on G protein–coupled receptor activation.
Co-reporter:Mauro Mileni ; Joie Garfunkle ; Cyrine Ezzili ; Benjamin F. Cravatt ; Raymond C. Stevens ;Dale L. Boger
Journal of the American Chemical Society 2011 Volume 133(Issue 11) pp:4092-4100
Publication Date(Web):February 28, 2011
DOI:10.1021/ja110877y
Two cocrystal X-ray structures of the exceptionally potent α-ketoheterocycle inhibitor 1 (Ki = 290 pM) bound to a humanized variant of rat fatty acid amide hydrolase (FAAH) are disclosed, representing noncovalently and covalently bound states of the same inhibitor with the enzyme. Key to securing the structure of the noncovalently bound state of the inhibitor was the inclusion of fluoride ion in the crystallization conditions that is proposed to bind the oxyanion hole precluding inhibitor covalent adduct formation with stabilization of the tetrahedral hemiketal. This permitted the opportunity to detect important noncovalent interactions stabilizing the binding of the inhibitor within the FAAH active site independent of the covalent reaction. Remarkably, noncovalently bound 1 in the presence of fluoride appears to capture the active site in the same “in action” state with the three catalytic residues Ser241−Ser217−Lys142 occupying essentially identical positions observed in the covalently bound structure of 1, suggesting that this technique of introducing fluoride may have important applications in structural studies beyond inhibiting substrate or inhibitor oxyanion hole binding. Key insights to emerge from the studies include the observations that noncovalently bound 1 binds in its ketone (not gem diol) form, that the terminal phenyl group in the acyl side chain of the inhibitor serves as the key anchoring interaction overriding the intricate polar interactions in the cytosolic port, and that the role of the central activating heterocycle is dominated by its intrinsic electron-withdrawing properties. These two structures are also briefly compared with five X-ray structures of α-ketoheterocycle-based inhibitors bound to FAAH recently disclosed.
Co-reporter:Aaron A. Thompson, Guan-Sheng Jiao, Seongjin Kim, April Thai, Lynne Cregar-Hernandez, Stephen A. Margosiak, Alan T. Johnson, Gye Won Han, Sean O’Malley, and Raymond C. Stevens
Biochemistry 2011 Volume 50(Issue 19) pp:
Publication Date(Web):March 24, 2011
DOI:10.1021/bi2001483
Neurotoxins synthesized by Clostridium botulinum bacteria (BoNT), the etiological agent of human botulism, are extremely toxic proteins making them high-risk agents for bioterrorism. Small molecule inhibitor development has been focused on the light chain zinc-dependent metalloprotease domain of the neurotoxin, an effort that has been hampered by its relatively flexible active site. Developed in concert with structure−activity relationship studies, the X-ray crystal structures of the complex of BoNT serotype A light chain (BoNT/A LC) with three different micromolar-potency hydroxamate-based inhibitors are reported here. Comparison with an unliganded BoNT/A LC structure reveals significant changes in the active site as a result of binding by the unique inhibitor scaffolds. The 60/70 loop at the opening of the active site pocket undergoes the largest conformational change, presumably through an induced-fit mechanism, resulting in the most compact catalytic pocket observed in all known BoNT/A LC structures.
Co-reporter:Ellen Y. T. Chien;Wei Liu;Qiang Zhao;Vsevolod Katritch;Gye Won Han;Michael A. Hanson;Lei Shi;Amy Hauck Newman;Jonathan A. Javitch;Vadim Cherezov
Science 2010 Vol 330(6007) pp:1091-1095
Publication Date(Web):19 Nov 2010
DOI:10.1126/science.1197410
Co-reporter:Beili Wu;Ellen Y. T. Chien;Clifford D. Mol;Gustavo Fenalti;Wei Liu;Vsevolod Katritch;Ruben Abagyan;Alexei Brooun;Peter Wells;F. Christopher Bi;Damon J. Hamel;Peter Kuhn;Tracy M. Handel;Vadim Cherezov
Science 2010 Vol 330(6007) pp:1066-1071
Publication Date(Web):19 Nov 2010
DOI:10.1126/science.1194396
Co-reporter:Daniel Wacker ; Gustavo Fenalti ; Monica A. Brown ; Vsevolod Katritch ; Ruben Abagyan ; Vadim Cherezov
Journal of the American Chemical Society 2010 Volume 132(Issue 33) pp:11443-11445
Publication Date(Web):July 29, 2010
DOI:10.1021/ja105108q
G protein-coupled receptors (GPCRs) represent a large fraction of current pharmaceutical targets, and of the GPCRs, the β2 adrenergic receptor (β2AR) is one of the most extensively studied. Previously, the X-ray crystal structure of β2AR has been determined in complex with two partial inverse agonists, but the global impact of additional ligands on the structure or local impacts on the binding site are not well-understood. To assess the extent of such ligand-induced conformational differences, we determined the crystal structures of a previously described engineered β2AR construct in complex with two inverse agonists: ICI 118,551 (2.8 Å), a recently described compound (2.8 Å) (Kolb et al, 2009), and the antagonist alprenolol (3.1 Å). The structures show the same overall fold observed for the previous β2AR structures and demonstrate that the ligand binding site can accommodate compounds of different chemical and pharmacological properties with only minor local structural rearrangements. All three compounds contain a hydroxy-amine motif that establishes a conserved hydrogen bond network with the receptor and chemically diverse aromatic moieties that form distinct interactions with β2AR. Furthermore, receptor ligand cross-docking experiments revealed that a single β2AR complex can be suitable for docking of a range of antagonists and inverse agonists but also indicate that additional ligand−receptor structures may be useful to further improve performance for in-silico docking or lead-optimization in drug design.
Co-reporter:Vsevolod Katritch ; Veli-Pekka Jaakola ; J. Robert Lane ; Judy Lin ; Adriaan P. IJzerman ; Mark Yeager ; Irina Kufareva ; Raymond C. Stevens ;Ruben Abagyan
Journal of Medicinal Chemistry 2010 Volume 53(Issue 4) pp:1799-1809
Publication Date(Web):January 22, 2010
DOI:10.1021/jm901647p
The recent progress in crystallography of G-protein coupled receptors opens an unprecedented venue for structure-based GPCR drug discovery. To test efficiency of the structure-based approach, we performed molecular docking and virtual ligand screening (VLS) of more than 4 million commercially available “drug-like” and ‘‘lead-like’’ compounds against the A2AAR 2.6 Å resolution crystal structure. Out of 56 high ranking compounds tested in A2AAR binding assays, 23 showed affinities under 10 μM, 11 of those had sub-μM affinities and two compounds had affinities under 60 nM. The identified hits represent at least 9 different chemical scaffolds and are characterized by very high ligand efficiency (0.3−0.5 kcal/mol per heavy atom). Significant A2AAR antagonist activities were confirmed for 10 out of 13 ligands tested in functional assays. High success rate, novelty, and diversity of the chemical scaffolds and strong ligand efficiency of the A2AAR antagonists identified in this study suggest practical applicability of receptor-based VLS in GPCR drug discovery.
Co-reporter:Mayako Michino,
Enrique Abola,
GPCR Dock 2008 participants,
Charles L. Brooks, III,
J. Scott Dixon,
John Moult
&
Raymond C. Stevens
Nature Reviews Drug Discovery 2009 8(6) pp:455
Publication Date(Web):2009-05-22
DOI:10.1038/nrd2877
Recent breakthroughs in the determination of the crystal structures of G protein-coupled receptors (GPCRs) have provided new opportunities for structure-based drug design strategies targeting this protein family. With the aim of evaluating the current status of GPCR structure prediction and ligand docking, a community-wide, blind prediction assessment — GPCR Dock 2008 — was conducted in coordination with the publication of the crystal structure of the human adenosine A2A receptor bound to the ligand ZM241385. Twenty-nine groups submitted 206 structural models before the release of the experimental structure, which were evaluated for the accuracy of the ligand binding mode and the overall receptor model compared with the crystal structure. This analysis highlights important aspects for success and future development, such as accurate modelling of structurally divergent regions and use of additional biochemical insight such as disulphide bridges in the extracellular loops.
Co-reporter:Kay Ahn, Douglas S. Johnson, Mauro Mileni, David Beidler, Jonathan Z. Long, Michele K. McKinney, Eranthie Weerapana, Nalini Sadagopan, Marya Liimatta, Sarah E. Smith, Scott Lazerwith, Cory Stiff, Satwik Kamtekar, Keshab Bhattacharya, Yanhua Zhang, Stephen Swaney, Keri Van Becelaere, Raymond C. Stevens, Benjamin F. Cravatt
Chemistry & Biology 2009 Volume 16(Issue 4) pp:411-420
Publication Date(Web):24 April 2009
DOI:10.1016/j.chembiol.2009.02.013
Endocannabinoids are lipid signaling molecules that regulate a wide range of mammalian behaviors, including pain, inflammation, and cognitive/emotional state. The endocannabinoid anandamide is principally degraded by the integral membrane enzyme fatty acid amide hydrolase (FAAH), and there is currently much interest in developing FAAH inhibitors to augment endocannabinoid signaling in vivo. Here, we report the discovery and detailed characterization of a highly efficacious and selective FAAH inhibitor, PF-3845. Mechanistic and structural studies confirm that PF-3845 is a covalent inhibitor that carbamylates FAAH's serine nucleophile. PF-3845 selectively inhibits FAAH in vivo, as determined by activity-based protein profiling; raises brain anandamide levels for up to 24 hr; and produces significant cannabinoid receptor-dependent reductions in inflammatory pain. These data thus designate PF-3845 as a valuable pharmacological tool for in vivo characterization of the endocannabinoid system.
Co-reporter:Vadim Cherezov, Jeffrey Liu, Mark Griffith, Michael A. Hanson and Raymond C. Stevens
Crystal Growth & Design 2008 Volume 8(Issue 12) pp:4307-4315
Publication Date(Web):November 7, 2008
DOI:10.1021/cg800778j
Fluorescence recovery after photobleaching was used to study the diffusion of two integral membrane proteins, bacteriorhodopsin and beta2-adrenergic receptor, in lipidic cubic phase (LCP). We found that the diffusion properties within the LCP matrix strongly depend on the protein construct and applied screening conditions. Common precipitants often induce restriction on diffusion of proteins in LCP and thereby impede their chances for crystallization. A high protein mobile fraction and a fast diffusion rate correlate well with known crystallization conditions. Using this knowledge, one can now prescreen precipitant conditions with microgram quantities of material to rule out conditions that are not conducive to diffusion, nucleation, and crystal growth. The results of this assay will narrow membrane protein crystallization space by identifying suitable protein constructs, stabilizing compounds and precipitant conditions amenable to in meso crystallization. Crystallization prescreening will significantly increase the chances of obtaining initial crystal hits, expediting efforts in generating high-resolution structures of challenging membrane protein targets.
Co-reporter:Ian M. Slaymaker, Michael Bracey, Mauro Mileni, Joie Garfunkle, Benjamin F. Cravatt, Dale L. Boger, Raymond C. Stevens
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 22) pp:5847-5850
Publication Date(Web):15 November 2008
DOI:10.1016/j.bmcl.2008.06.086
The melting curves of fatty acid amide hydrolase (FAAH) in the presence of 29 reversible inhibitors were measured using a thiol-reactive fluorophore. The thermal stability (Tm) of the FAAH/inhibitor complex varied significantly depending on the chemical characteristics of the inhibitors, notably variations in the head group. Two separate distributions were observed when Tm was plotted against Ki. The majority of the inhibitors showed a positive correlation between binding affinity and Tm, however inhibitors with a pyridine carboxylic acid moiety in the head group fell in a distinct and uncorrelated distribution when tail groups were varied.
Co-reporter:Mauro Mileni;Douglas S. Johnson;Zhigang Wang;Daniel S. Everdeen;Marya Liimatta;Brandon Pabst;Keshab Bhattacharya;Richard A. Nugent;Satwik Kamtekar;Benjamin F. Cravatt;Kay Ahn;
Proceedings of the National Academy of Sciences 2008 105(35) pp:12820-12824
Publication Date(Web):August 27, 2008
DOI:10.1073/pnas.0806121105
The integral membrane enzyme fatty acid amide hydrolase (FAAH) hydrolyzes the endocannabinoid anandamide and related amidated
signaling lipids. Genetic or pharmacological inactivation of FAAH produces analgesic, anxiolytic, and antiinflammatory phenotypes
but not the undesirable side effects of direct cannabinoid receptor agonists, indicating that FAAH may be a promising therapeutic
target. Structure-based inhibitor design has, however, been hampered by difficulties in expressing the human FAAH enzyme.
Here, we address this problem by interconverting the active sites of rat and human FAAH using site-directed mutagenesis. The
resulting humanized rat (h/r) FAAH protein exhibits the inhibitor sensitivity profiles of human FAAH but maintains the high-expression
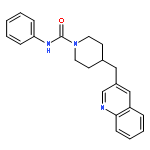
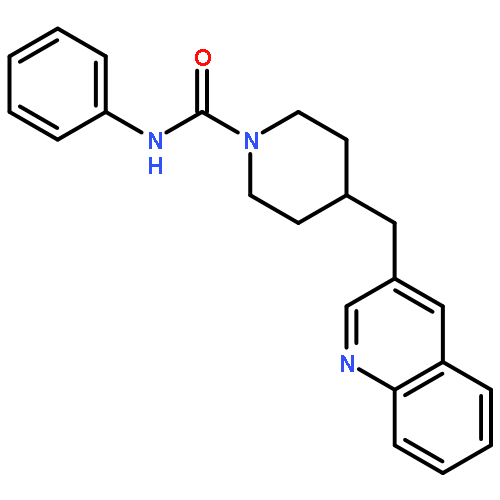
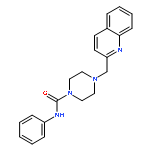
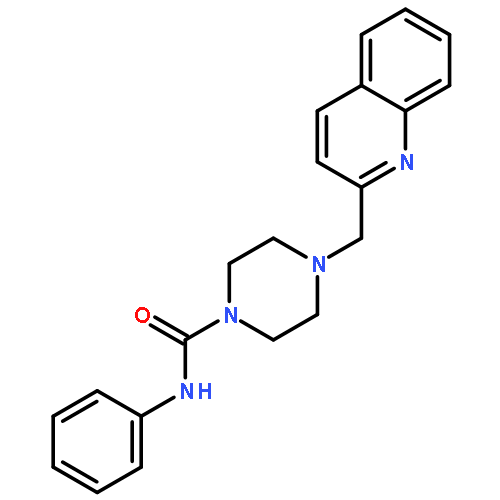
yield of the rat enzyme. We report a 2.75-Å crystal structure of h/rFAAH complexed with an inhibitor, N-phenyl-4-(quinolin-3-ylmethyl)piperidine-1-carboxamide (PF-750), that shows strong preference for human FAAH. This structure
offers compelling insights to explain the species selectivity of FAAH inhibitors, which should guide future drug design programs.
Co-reporter:Mark T. Griffith;Veli-Pekka Jaakola;Michael A. Hanson;Vadim Cherezov;Ellen Y. T. Chien;J. Robert Lane;Adriaan P. IJzerman
Science 2008 Volume 322(Issue 5905) pp:1211-1217
Publication Date(Web):21 Nov 2008
DOI:10.1126/science.1164772
Abstract
The adenosine class of heterotrimeric guanine nucleotide–binding protein (G protein)–coupled receptors (GPCRs) mediates the important role of extracellular adenosine in many physiological processes and is antagonized by caffeine. We have determined the crystal structure of the human A2A adenosine receptor, in complex with a high-affinity subtype-selective antagonist, ZM241385, to 2.6 angstrom resolution. Four disulfide bridges in the extracellular domain, combined with a subtle repacking of the transmembrane helices relative to the adrenergic and rhodopsin receptor structures, define a pocket distinct from that of other structurally determined GPCRs. The arrangement allows for the binding of the antagonist in an extended conformation, perpendicular to the membrane plane. The binding site highlights an integral role for the extracellular loops, together with the helical core, in ligand recognition by this class of GPCRs and suggests a role for ZM241385 in restricting the movement of a tryptophan residue important in the activation mechanism of the class A receptors.
Co-reporter:Guillermo A. Asmar-Rovira;Aloysha M. Asseo-García
The Journal of Membrane Biology 2008 Volume 223( Issue 1) pp:13-26
Publication Date(Web):2008 May
DOI:10.1007/s00232-008-9107-7
The nicotinic acetylcholine receptor (nAChR) of Torpedo electric rays has been extensively characterized over the last three decades. However, high-resolution structural studies have been hampered by the lack of mechanistic molecular models that describe how detergents influence membrane protein stability and function. Furthermore, elucidation of the dynamic detergent–lipid–protein interactions of solubilized membrane proteins is a largely unexplored research field. This study examines the effects of nine detergents on: (1) nAChR-lipid composition (gas chromatography with flame ionization; GC-FID and/or mass selective detectors; GC-MSD), (2) stability and aggregation state (analytical size exclusion chromatography; A-SEC and electron microscopy; EM) and (3) ion channel function (planar lipid bilayers). Detergent solubilization of nAChR-enriched membranes did not result in significant native lipid depletion or destabilization. Upon purification, native lipid depletion occurred in all detergents, with lipid-analogue detergents CHAPS {(3-[(3-cholamidopropyl)-dimethylammonio]-1-propane sulfonate}, FC-12 (n-dodecylphosphocholine) and sodium cholate (3α,7α,12α-trihydroxy-5β-cholan-24-oic acid) maintaining stability and supporting ion channel function, and non-lipid-analogue detergents Cymal-6 (6-cyclohexyl-1-hexyl-β-D-maltoside), DDM (n-dodecyl-β-D-maltopyranoside), LDAO (lauryldimethylamine-N-oxide) and OG (n-octyl-β-d-glucopyranoside) decreasing stability and significantly reducing or completely suppressing ion channel function. Anapoe-C12E9 (polyoxyethylene-[9]-dodecyl ether) and BigCHAP (N,N’-bis-[3-d-gluconamidopropyl] cholamide) retained residual amounts of native lipid, maintaining moderate stability and ion channel function compared to lipid-analogue detergents. Therefore, the nAChR can be stable and functional in lipid-analogue detergents or in detergents that retain moderate amounts of residual native lipids, but not in non-lipid-analogue detergents.
Co-reporter:Christineh N. Sarkissian;Alejandra Gámez;Paul Fitzpatrick;Marilyse Charbonneau;Laurie Tsuruda;Michael Vellard;Amy Lambert;Carroll Henschell;Sean M. Bell;Lin Wang;Charles R. Scriver;Jeffrey F. Lemontt;Bin Zhao
PNAS 2008 Volume 105 (Issue 52 ) pp:20894-20899
Publication Date(Web):2008-12-30
DOI:10.1073/pnas.0808421105
Phenylketonuria (PKU) is a metabolic disorder, in which loss of phenylalanine hydroxylase activity results in neurotoxic levels
of phenylalanine. We used the Pahenu2/enu2 PKU mouse model in short- and long-term studies of enzyme substitution therapy with PEGylated phenylalanine ammonia lyase
(PEG-PAL conjugates) from 4 different species. The most therapeutically effective PAL (Av, Anabaena variabilis) species was one without the highest specific activity, but with the highest stability; indicating the importance of protein
stability in the development of effective protein therapeutics. A PEG-Av-p.C503S/p.C565S-PAL effectively lowered phenylalanine levels in both vascular space and brain tissue over a >90 day trial
period, resulting in reduced manifestations associated with PKU, including reversal of PKU-associated hypopigmentation and
enhanced animal health. Phenylalanine reduction occurred in a dose- and loading-dependent manner, and PEGylation reduced the
neutralizing immune response to the enzyme. Human clinical trials with PEG-Av-p.C503S/p.C565S-PAL as a treatment for PKU are underway.
Co-reporter:Daniel M. Rosenbaum;Vadim Cherezov;Michael A. Hanson;Tong Sun Kobilka;Hee-Jung Choi;Foon Sun Thian;Søren G. F. Rasmussen;Xiao-Jie Yao;William I. Weis;Brian K. Kobilka
Science 2007 Volume 318(Issue 5854) pp:1266-1273
Publication Date(Web):23 Nov 2007
DOI:10.1126/science.1150609
Abstract
The β2-adrenergic receptor (β2AR) is a well-studied prototype for heterotrimeric guanine nucleotide–binding protein (G protein)–coupled receptors (GPCRs) that respond to diffusible hormones and neurotransmitters. To overcome the structural flexibility of the β2AR and to facilitate its crystallization, we engineered a β2AR fusion protein in which T4 lysozyme (T4L) replaces most of the third intracellular loop of the GPCR (“β2AR-T4L”) and showed that this protein retains near-native pharmacologic properties. Analysis of adrenergic receptor ligand-binding mutants within the context of the reported high-resolution structure of β2AR-T4L provides insights into inverse-agonist binding and the structural changes required to accommodate catecholamine agonists. Amino acids known to regulate receptor function are linked through packing interactions and a network of hydrogen bonds, suggesting a conformational pathway from the ligand-binding pocket to regions that interact with G proteins.
Co-reporter:Daniel M. Rosenbaum;Vadim Cherezov;Søren G. F. Rasmussen;Foon Sun Thian;Michael A. Hanson;Tong Sun Kobilka;Hee-Jung Choi;Peter Kuhn;Brian K. Kobilka;William I. Weis
Science 2007 Volume 318(Issue 5854) pp:1258-1265
Publication Date(Web):23 Nov 2007
DOI:10.1126/science.1150577
Abstract
Heterotrimeric guanine nucleotide–binding protein (G protein)–coupled receptors constitute the largest family of eukaryotic signal transduction proteins that communicate across the membrane. We report the crystal structure of a human β2-adrenergic receptor–T4 lysozyme fusion protein bound to the partial inverse agonist carazolol at 2.4 angstrom resolution. The structure provides a high-resolution view of a human G protein–coupled receptor bound to a diffusible ligand. Ligand-binding site accessibility is enabled by the second extracellular loop, which is held out of the binding cavity by a pair of closely spaced disulfide bridges and a short helical segment within the loop. Cholesterol, a necessary component for crystallization, mediates an intriguing parallel association of receptor molecules in the crystal lattice. Although the location of carazolol in the β2-adrenergic receptor is very similar to that of retinal in rhodopsin, structural differences in the ligand-binding site and other regions highlight the challenges in using rhodopsin as a template model for this large receptor family.
Co-reporter:Qing Chai,
Joseph W. Arndt,
Min Dong,
William H. Tepp,
Eric A. Johnson,
Edwin R. Chapman
&
Raymond C. Stevens
Nature 2006 444(7122) pp:1096
Publication Date(Web):
DOI:10.1038/nature05411
Co-reporter:Angel L. Pey;Heidi Erlandsen;Belén Pérez;Alejandra Gámez;Lourdes R. Desviat;Cristina Aguado;Richard Koch;Sankar Surendran;Stephen Tyring;Reuben Matalon;Charles R. Scriver;Magdalena Ugarte;Aurora Martínez
PNAS 2004 Volume 101 (Issue 48 ) pp:16903-16908
Publication Date(Web):2004-11-30
DOI:10.1073/pnas.0407256101
Phenylketonuria patients harboring a subset of phenylalanine hydroxylase (PAH) mutations have recently shown normalization
of blood phenylalanine levels upon oral administration of the PAH cofactor tetrahydrobiopterin [(6R)-l-erythro-5,6,7,8-tetrahydrobiopterin (BH4)]. Several hypotheses have been put forward to explain BH4 responsiveness, but the molecular basis for the corrective effect(s) of BH4 has not been understood. We have investigated the biochemical, kinetic, and structural changes associated with BH4-responsive mutations (F39L, I65T, R68S, H170D, E178G, V190A, R261Q, A300S, L308F, A313T, A373T, V388M, E390G, P407S, and
Y414C). The biochemical and kinetic characterization of the 15 mutants studied points toward a multifactorial basis for the
BH4 responsiveness; the mutants show residual activity (>30% of WT) and display various kinetic defects, including increased
K
m (BH4) and reduced cooperativity of substrate binding, but no decoupling of cofactor (BH4) oxidation. For some, BH4 seems to function through stabilization and protection of the enzyme from inactivation and proteolytic degradation. In the
crystal structures of a phenylketonuria mutant, A313T, minor changes were seen when compared with the WT PAH structures, consistent
with the mild effects the mutant has upon activity of the enzyme both in vitro and in vivo. Truncations made in the A313T mutant PAH form revealed that the N and C termini of the enzyme influence active site binding.
Of fundamental importance is the observation that BH4 appears to increase Phe catabolism if at least one of the two heterozygous mutations has any residual activity remaining.
Co-reporter:
Nature Structural and Molecular Biology 2004 11(4) pp:293-295
Publication Date(Web):
DOI:10.1038/nsmb0404-293
Two camps continue to evolve in the field of structural biology—a 'systems-oriented' camp, which studies proteins or complexes carefully one system at a time, and a 'discovery-oriented' one, which studies proteins of entire families, pathways or genomes. The end goals of both camps are the same: to decipher the atomic-resolution structures and mechanisms of biological macromolecules and understand them in the context of the living cell.
Co-reporter:Jun Yin;Scott E. Andryski;Albert E. Beuscher IV;Peter G. Schultz
PNAS 2003 Volume 100 (Issue 3 ) pp:856-861
Publication Date(Web):2003-02-04
DOI:10.1073/pnas.0235873100
The crystal structure of the Michaelis complex between the Fab fragment of ferrochelatase antibody 7G12 and its substrate
mesoporphyrin has been solved to 2.6-Å resolution. The antibody-bound mesoporphyrin clearly adopts a nonplanar conformation
and reveals that the antibody catalyzes the porphyrin metallation reaction by straining/distorting the bound substrate toward
the transition-state configuration. The crystal structures of the Fab fragment of the germ-line precursor antibody to 7G12
and its complex with the hapten N-methylmesoporphyrin have also been solved. A comparison of these structures with the corresponding structures of the affinity-matured
antibody 7G12 reveals the molecular mechanism by which the immune system evolves binding energy to catalyze this reaction.
Co-reporter:Jie Song, Quan Cheng, Raymond C. Stevens
Chemistry and Physics of Lipids 2002 Volume 114(Issue 2) pp:203-214
Publication Date(Web):February 2002
DOI:10.1016/S0009-3084(02)00007-5
Morphological transformations of bolaamphiphilic polydiacetylene (L-Glu-Bis-3) lipid assemblies from helical ribbons to vesicles and flat sheets through controlled doping are described, and the role of specific lipid dopants in these processes is discussed. Upon doping with cell surface receptor GM1 ganglioside, fluid vesicular structures start to emerge, coexisting with the micro-crystalline helical ribbons. The vesicle formation is further facilitated and stabilized by the introduction of cholesterol into the system, presumably through surface curvature variation induced by inhomogeneous distribution and dynamic clustering of GM1 and cholesterol within the doped assemblies. Extended helical ribbons are ‘truncated’ into patches of flat sheets when a sufficient amount of Bis-1, a structurally compatible symmetric bolaamphiphilic diacetylene lipid, is doped. The results reaffirm the important roles of packing geometry and headgroup chirality in the formation of extended helical ribbon structures. The doped assemblies of bolaamphiphiles allow for capture of intermediate structures of morphological transformation using transmission electron microscopy (TEM). A vesicle-to-ribbon transformation mechanism via lateral reorganization within relatively fluid vesicular microstructures has been suggested. Understanding of the doping-induced transformation process provides useful information for the design of advanced materials where the microscopic morphology of material is crucial to its function.
Co-reporter:
Nature Structural and Molecular Biology 2001 8(3) pp:238-242
Publication Date(Web):
DOI:10.1038/84981
Directed evolution can be a powerful tool to predict antibiotic resistance. Resistance involves the accumulation of mutations beneficial to the pathogen while maintaining residue interactions and core packing that are critical for preserving function. The constraint of maintaining stability, while increasing activity, drastically reduces the number of possible mutational combination pathways. To test this theory, TEM-1 -lactamase was evolved using a hypermutator E. coli-based directed evolution technique with cefotaxime selection. The selected mutants were compared to two previous directed evolution studies and a database of clinical isolates. In all cases, evolution resulted in the generation of the E104K/M182T/G238S combination of mutations (500-fold increased resistance), which is equivalent to clinical isolate TEM-52. The structure of TEM-52 was determined to 2.4 Å. G238S widens access to the active site by 2.8 Å whereas E104K stabilizes the reorganized topology. The M182T mutation is located 17 Å from the active site and appears to be a global suppressor mutation that acts to stabilize the new enzyme structure. Our results demonstrate that directed evolution coupled with structural analysis can be used to predict future mutations that lead to increased antibiotic resistance.
Co-reporter:
Nature Structural and Molecular Biology 2001 8(8) pp:664
Publication Date(Web):
DOI:10.1038/90364
In response to the Comment by Rupp and Segelke concerning our paper published in Nature Structural Biology last year, we apologize for omitting that the occupancy of the peptide is estimated to be 30−40%. Our published study was a rapid soak freeze-trap experiment, and in order for us to observe the product bound form of the peptide, it is not unexpected that the peptide is of lower occupancy in the crystal than the protein molecule. However, we should have noted this in our publication. The stereochemical quality of the peptide is low, and we believe this to be due to the occupancy of the peptide. However, since the peptide is disordered in solution, we did not want to constrain the peptide to an artificially high quality stereochemical model. Attempts to obtain higher occupancy binding of the peptide were not successful. The peptide is most clearly observed when one compares the active site of both the apo (1f82.pdb, r1f82sf.ent) and peptide bound (1f83.pdb, r1f83sf.ent) forms. This latter comparison helps to resolve any potential phase bias that might have occurred from the molecular replacement solution using the holotoxin model.More germane to the science is the discovery that the synaptobrevin peptide binds in the same location as the translocation domain of the holo-botulinum neurotoxin after translocation domain separation. This observation is consistent with published biochemical studies as cited in our publication and it provides a potential explanation for previously unexplained observations in the field of botulinum neurotoxin research. Additional biochemical data are accumulating that support our observation and I suspect we will read more articles in the near future on this topic. Based on our 2.2 Å apo and 2.0 Å substrate-bound structures and additional structural studies that we have conducted since publication, we continue to support our published proposal on how the neurotoxin cleaves synaptic vesicle proteins. However, our goal is to accurately understand how the neurotoxin works and if our proposal is not correct, we would like to know as soon as possible to advance the field in a forward direction. We look forward to seeing this matter resolved in the peer-reviewed literature in the near future.In summary, we thank Rupp and Segelke for pointing out that we did not state the occupancy of the peptide in our published article, and we apologize to the community for this omission.
Co-reporter:
Nature Structural and Molecular Biology 2000 7(8) pp:687-692
Publication Date(Web):
DOI:10.1038/77997
Botulinum neurotoxin serotype B is a zinc protease that disrupts neurotransmitter release by cleaving synaptobrevin-II (Sb2), one of three SNARE proteins involved in neuronal synaptic vesicle fusion. The three-dimensional crystal structure of the apo botulinum neurotoxin serotype B catalytic domain (BoNT/B-LC) has been determined to 2.2 Å resolution, and the complex of cleaved Sb2 with the catalytic domain (Sb2−BoNT/B-LC) has been determined to 2.0 Å resolution. A comparison of the holotoxin catalytic domain and the isolated BoNT/B-LC structure shows a rearrangement of three active site loops. This rearrangement exposes the BoNT/B active site. The Sb2−BoNT/B-LC structure illustrates two distinct binding regions, which explains the specificity of each botulinum neurotoxin for its synaptic vesicle protein. This observation provides an explanation for the proposed cooperativity between binding of full-length substrate and catalysis and suggest a mechanism of synaptobrevin proteolysis employed by the clostridial neurotoxins.
Co-reporter:
Nature Structural and Molecular Biology 2000 7(11s) pp:973-977
Publication Date(Web):
DOI:10.1038/80754
Structure-based biological discovery is entering a new era with the development
of industrialized macromolecular structure determination pipelines. Intense,
highly focused X-rays from integrated synchrotron radiation beam lines combined
with significant advances in protein expression, purification, and micro-crystallization
automation allow for the full streamlining of the traditionally tedious and
time consuming process of determining the three dimensional structures of
macromolecules.
Co-reporter:Michael A. Hanson, Alexei Brooun, Kent A. Baker, Veli-Pekka Jaakola, Chris Roth, Ellen Y.T. Chien, Alexander Alexandrov, Jeffrey Velasquez, Leila Davis, Mark Griffith, Kin Moy, Barbie K. Ganser-Pornillos, Yuanzi Hua, Peter Kuhn, Sam Ellis, Mark Yeager, Raymond C. Stevens
Protein Expression and Purification (November 2007) Volume 56(Issue 1) pp:85-92
Publication Date(Web):1 November 2007
DOI:10.1016/j.pep.2007.06.003
Production of structure-grade mammalian membrane proteins in substantial quantities has been hindered by a lack of methods for effectively profiling multiple constructs expression in higher eukaryotic systems such as insect or mammalian cells. To address this problem, a specialized small-scale eukaryotic expression platform by Thomson Instrument Company (Vertiga-IM) was developed and used in tandem with a Guava EasyCyte microcapillary 96-well cytometer to monitor cell density and health and evaluate membrane protein expression. Two proof of concept experiments were conducted using the human β2-adrenergic receptor (β2AR) and the gap junction protein connexin26 (Cx26) in a baculovirus expression system. First, cell surface expression was used to assess the expression levels of 14 β2AR truncation variants expressed using the Vertiga-IM shaker. Three of these variants were then compared to wild-type β2AR using three metrics: cell surface expression, saturation ligand binding and protein immunoblot analysis of dodecylmaltoside extracted material. Second, a series of systematic Cx26 truncation variants were evaluated for expression by protein immunoblot analysis. The cumulative results for these two systems show that the Vertiga-IM instrument can be used effectively in the parallel insect cell microexpression of membrane protein variants, and that the expression of cell surface molecules as monitored with the Guava EasyCyte instrument can be used to rapidly assess the production of properly folded proteins in the baculovirus expression system. This approach expedites the in vitro evaluation of a large number of mammalian membrane protein variants.
Co-reporter:Kaspar Hollenstein, Chris de Graaf, Andrea Bortolato, Ming-Wei Wang, Fiona H. Marshall, Raymond C. Stevens
Trends in Pharmacological Sciences (January 2014) Volume 35(Issue 1) pp:12-22
Publication Date(Web):1 January 2014
DOI:10.1016/j.tips.2013.11.001
•GCGR and CRF1 structures show different features compared to class A GPCRs.•Class B structures and structure-based drug discovery for peptide hormone GPCRs.•Glucagon, GLP1, and GIP peptide molecular recognition and diabetes.•Corticotropin-releasing factor and stress.The secretin-like (class B) family of G protein-coupled receptors (GPCRs) are key players in hormonal homeostasis and are interesting drug targets for the treatment of several metabolic disorders (such as type 2 diabetes, osteoporosis, and obesity) and nervous system diseases (such as migraine, anxiety, and depression). The recently solved crystal structures of the transmembrane domains of the human glucagon receptor and human corticotropin-releasing factor receptor 1 have opened up new opportunities to study the structure and function of class B GPCRs. The current review shows how these structures offer more detailed explanations to previous biochemical and pharmacological studies of class B GPCRs, and provides new insights into their interactions with ligands.
Co-reporter:Vsevolod Katritch, Vadim Cherezov, Raymond C. Stevens
Trends in Pharmacological Sciences (January 2012) Volume 33(Issue 1) pp:17-27
Publication Date(Web):1 January 2012
DOI:10.1016/j.tips.2011.09.003
G protein-coupled receptors (GPCRs) comprise the most ‘prolific’ family of cell membrane proteins, which share a general mechanism of signal transduction, but greatly vary in ligand recognition and function. Crystal structures are now available for rhodopsin, adrenergic, and adenosine receptors in both inactive and activated forms, as well as for chemokine, dopamine, and histamine receptors in inactive conformations. Here we review common structural features, outline the scope of structural diversity of GPCRs at different levels of homology, and briefly discuss the impact of the structures on drug discovery. Given the current set of GPCR crystal structures, a distinct modularity is now being observed between the extracellular (ligand-binding) and intracellular (signaling) regions. The rapidly expanding repertoire of GPCR structures provides a solid framework for experimental and molecular modeling studies, and helps to chart a roadmap for comprehensive structural coverage of the whole superfamily and an understanding of GPCR biological and therapeutic mechanisms.
Co-reporter:Tse Siang Kang, Lin Wang, Christineh N. Sarkissian, Alejandra Gámez, ... Raymond C. Stevens
Molecular Genetics and Metabolism (January 2010) Volume 99(Issue 1) pp:4-9
Publication Date(Web):1 January 2010
DOI:10.1016/j.ymgme.2009.09.002
Phenylalanine ammonia lyase (PAL) has long been recognized as a potential enzyme replacement therapeutic for treatment of phenylketonuria. However, various strategies for the oral delivery of PAL have been complicated by the low intestinal pH, aggressive proteolytic digestion and circulation time in the GI tract. In this work, we report 3 strategies to address these challenges. First, we used site-directed mutagenesis of a chymotrypsin cleavage site to modestly improve protease resistance; second, we used silica sol–gel material as a matrix to demonstrate that a silica matrix can provide protection to entrapped PAL proteins against intestinal proteases, as well as a low pH of 3.5; finally, we demonstrated that PEGylation of AvPAL surface lysines can reduce the inactivation of the enzyme by trypsin.
Co-reporter:Alejandra Gámez, Lin Wang, Christineh N. Sarkissian, Dan Wendt, ... Raymond C. Stevens
Molecular Genetics and Metabolism (August 2007) Volume 91(Issue 4) pp:325-334
Publication Date(Web):1 August 2007
DOI:10.1016/j.ymgme.2007.04.015
Protein and peptide therapeutics are of growing importance as medical treatments but can frequently induce an immune response. This work describes the combination of complementary approaches to map the potential immunogenic regions of the yeast Rhodosporidium toruloides phenylalanine ammonia-lyase (PAL, EC 4.3.1.5) and to engineer the protein as a human therapeutic agent for the treatment of phenylketonuria (PKU), an inherited metabolic disorder. The identification of B and T cell epitopes on the PAL protein was performed by computational predictions based on the antigenicity and hydrophilicity of proteins, as well as by experimental epitope mapping using a PepSpots™ peptide array (Jerini AG). Human T cell epitope mapping was performed by applying the computational EpiMatrix™ algorithm (EpiVax, Inc.) for MHC Class I and Class II associated T cell epitopes on PAL, which predicts which sequences are associated with binding to several different HLA alleles, a requirement for antigen presentation and subsequent primary immune response. By chemical modification through PEGylation of surface lysine residues, it is possible to cover the immunogenic regions of a protein. To evaluate this strategy, we used mass spectrometry to determine which of the immunogenic epitopes are covered by the covalent PEGylation modification strategy. This approach has allowed us to determine whether additional lysines are needed in specific residue locations, or whether certain lysine residues can be removed in order to accomplish complete molecular coverage of the therapeutic enzyme.
Co-reporter:Fai Y. Siu, Raymond C. Stevens
Structure (8 September 2010) Volume 18(Issue 9) pp:1067-1068
Publication Date(Web):8 September 2010
DOI:10.1016/j.str.2010.08.004
The cocrystal structure of the heterodimer GPCR extracellular domain complex of the CGRP receptor (CLR and RAMP1) has been determined in an apo form and with two different antagonists olcegepant and telcagepant (ter Haar et al., 2010).
Co-reporter:Fei Xu, Raymond C. Stevens
Structure (7 September 2011) Volume 19(Issue 9) pp:1204-1207
Publication Date(Web):7 September 2011
DOI:10.1016/j.str.2011.08.007
Three new X-ray structures of a thermostabilized A2A adenosine receptor in complex with different antagonists reported by Doré et al. in this issue have advanced the understanding of molecular recognition for this receptor.
Co-reporter:Fei Xu, Raymond C. Stevens
Structure (7 September 2011) Volume 19(Issue 9) pp:1204-1207
Publication Date(Web):7 September 2011
DOI:10.1016/j.str.2011.08.007
Three new X-ray structures of a thermostabilized A2A adenosine receptor in complex with different antagonists reported by Doré et al. in this issue have advanced the understanding of molecular recognition for this receptor.
Co-reporter:Christopher B. Roth, Michael A. Hanson, Raymond C. Stevens
Journal of Molecular Biology (7 March 2008) Volume 376(Issue 5) pp:1305-1319
Publication Date(Web):7 March 2008
DOI:10.1016/j.jmb.2007.12.028
G protein-coupled receptor (GPCR) instability represents one of the most profound obstacles in the structural study of GPCRs that bind diffusible ligands. The introduction of targeted mutations at nonconserved residues that lie proximal to helix interfaces has the potential to enhance the fold stability of the receptor helix bundle while maintaining wild-type receptor function. To test this hypothesis, we studied the effect of amino acid substitutions at Glu1223.41 in the well-studied β2-adrenergic receptor (β2AR), which was predicted from sequence conservation to lie at a position equivalent to a tryptophan residue in rhodopsin at the 3,4,5 helix interface among transmembrane (TM) domains 3, 4, and 5. Replacement of Glu1223.41 with bulky hydrophobic residues, such as tryptophan, tyrosine, and phenylalanine, increases the yield of functionally folded β2AR by as much as 5-fold. Receptor stability in detergent solution was studied by isothermal denaturation, and it was found that the E122W and E122Y mutations enhanced the β2AR thermal half-life by 9.3- and 6.7-fold, respectively, at 37 °C. The β1AR was also stabilized by the introduction of tryptophan at Glu1473.41, and the effect on protein behavior was similar to the rescue of the unstable wild-type receptor by the antagonist propranolol. Molecular modeling of the E122W and E122Y mutants revealed that the tryptophan ring edge and tyrosine hydroxyl are positioned proximal to the helical break in TM5 introduced by the conserved Pro2115.50 and may stabilize the helix by interacting favorably with the unpaired carbonyl oxygen of Val2065.45. Conformational flexibility of TM5 is likely to be a general property of class A GPCRs; therefore, engineering of the TM4–TM3–TM5 interface at the 3.41 position may provide a general strategy for the stabilization of other receptors.
Co-reporter:Eugene Chun, Aaron A. Thompson, Wei Liu, Christopher B. Roth, ... Raymond C. Stevens
Structure (6 June 2012) Volume 20(Issue 6) pp:967-976
Publication Date(Web):6 June 2012
DOI:10.1016/j.str.2012.04.010
Structural studies of human G protein-coupled receptors (GPCRs) have recently been accelerated through the use of a fusion partner that was inserted into the third intracellular loop. Using chimeras of the human β2-adrenergic and human A2A adenosine receptors, we present the methodology and data for the initial selection of an expanded set of fusion partners for crystallizing GPCRs. In particular, use of the thermostabilized apocytochrome b562RIL as a fusion partner displays certain advantages over previously utilized fusion proteins, resulting in a significant improvement in stability and structure of GPCR-fusion constructs.Highlights► A method was developed for the selection of fusion domains for GPCR crystallization ► Apocytochrome b562RIL has advantages over previously utilized T4 lysozyme ► Diffraction quality crystals of two engineered GPCRs were successfully grown ► The method led to the crystal structure of the A2A adenosine receptor at 1.8 Å
Co-reporter:Irina Kufareva, Vsevolod Katritch, Participants of GPCR Dock 2013, Raymond C. Stevens, Ruben Abagyan
Structure (5 August 2014) Volume 22(Issue 8) pp:1120-1139
Publication Date(Web):5 August 2014
DOI:10.1016/j.str.2014.06.012
•The four targets in the 2013 assessment presented diverse challenges for modelers•Highly accurate predictions for close homology models and rigid orthosteric ligands•Alignment inaccuracies are the major hurdle in modeling by distant homology•Predictions of loops and receptor activation states remain unsolved problemsDespite tremendous successes of GPCR crystallography, the receptors with available structures represent only a small fraction of human GPCRs. An important role of the modeling community is to maximize structural insights for the remaining receptors and complexes. The community-wide GPCR Dock assessment was established to stimulate and monitor the progress in molecular modeling and ligand docking for GPCRs. The four targets in the present third assessment round presented new and diverse challenges for modelers, including prediction of allosteric ligand interaction and activation states in 5-hydroxytryptamine receptors 1B and 2B, and modeling by extremely distant homology for smoothened receptor. Forty-four modeling groups participated in the assessment. State-of-the-art modeling approaches achieved close-to-experimental accuracy for small rigid orthosteric ligands and models built by close homology, and they correctly predicted protein fold for distant homology targets. Predictions of long loops and GPCR activation states remain unsolved problems.Download high-res image (559KB)Download full-size image
Co-reporter:Hugo Gutiérrez-de-Terán, Arnault Massink, David Rodríguez, Wei Liu, ... Raymond C. Stevens
Structure (3 December 2013) Volume 21(Issue 12) pp:2175-2185
Publication Date(Web):3 December 2013
DOI:10.1016/j.str.2013.09.020
•A multidisciplinary study explores the role of the sodium binding site on A2AAR•The allosteric modulation by sodium ions and amilorides on A2AAR is rationalized•Sodium ions selectively bind and stabilize the inactive conformation of the A2AAR•The binding of sodium ions and agonists is mutually exclusiveThe function of G protein-coupled receptors (GPCRs) can be modulated by a number of endogenous allosteric molecules. In this study, we used molecular dynamics, radioligand binding, and thermostability experiments to elucidate the role of the recently discovered sodium ion binding site in the allosteric modulation of the human A2A adenosine receptor, conserved among class A GPCRs. While the binding of antagonists and sodium ions to the receptor was noncompetitive in nature, the binding of agonists and sodium ions appears to require mutually exclusive conformational states of the receptor. Amiloride analogs can also bind to the sodium binding pocket, showing distinct patterns of agonist and antagonist modulation. These findings suggest that physiological concentrations of sodium ions affect functionally relevant conformational states of GPCRs and can help to design novel synthetic allosteric modulators or bitopic ligands exploiting the sodium ion binding pocket.Download high-res image (187KB)Download full-size image
Co-reporter:Mauro Mileni, Satwik Kamtekar, David C. Wood, Timothy E. Benson, ... Raymond C. Stevens
Journal of Molecular Biology (23 July 2010) Volume 400(Issue 4) pp:743-754
Publication Date(Web):23 July 2010
DOI:10.1016/j.jmb.2010.05.034
The endocannabinoid system regulates a wide range of physiological processes including pain, inflammation, and cognitive/emotional states. URB597 is one of the best characterized covalent inhibitors of the endocannabinoid-degrading enzyme fatty acid amide hydrolase (FAAH). Here, we report the structure of the FAAH–URB597 complex at 2.3 Å resolution. The structure provides insights into mechanistic details of enzyme inactivation and experimental evidence of a previously uncharacterized active site water molecule that likely is involved in substrate deacylation. This water molecule is part of an extensive hydrogen-bonding network and is coordinated indirectly to residues lining the cytosolic port of the enzyme. In order to corroborate our hypothesis concerning the role of this water molecule in FAAH's catalytic mechanism, we determined the structure of FAAH conjugated to a urea-based inhibitor, PF-3845, to a higher resolution (2.4 Å) than previously reported. The higher-resolution structure confirms the presence of the water molecule in a virtually identical location in the active site. Examination of the structures of serine hydrolases that are non-homologous to FAAH, such as elastase, trypsin, or chymotrypsin, shows a similarly positioned hydrolytic water molecule and suggests a functional convergence between the amidase signature enzymes and serine proteases.
Co-reporter:Lin Wang, Alejandra Gamez, Holly Archer, Enrique E. Abola, ... Raymond C. Stevens
Journal of Molecular Biology (18 July 2008) Volume 380(Issue 4) pp:623-635
Publication Date(Web):18 July 2008
DOI:10.1016/j.jmb.2008.05.025
We have recently observed promising success in a mouse model for treating the metabolic disorder phenylketonuria with phenylalanine ammonia lyase (PAL) from Rhodosporidium toruloides and Anabaena variabilis. Both molecules, however, required further optimization in order to overcome problems with protease susceptibility, thermal stability, and aggregation. Previously, we optimized PAL from R. toruloides, and in this case we reduced aggregation of the A. variabilis PAL by mutating two surface cysteine residues (C503 and C565) to serines. Additionally, we report the structural and biochemical characterization of the A. variabilis PAL C503S/C565S double mutant and carefully compare this molecule with the R. toruloides engineered PAL molecule. Unlike previously published PAL structures, significant electron density is observed for the two active-site loops in the A. variabilis C503S/C565S double mutant, yielding a complete view of the active site. Docking studies and N-hydroxysuccinimide–biotin binding studies support a proposed mechanism in which the amino group of the phenylalanine substrate is attacked directly by the 4-methylidene-imidazole-5-one prosthetic group. We propose a helix-to-loop conformational switch in the helices flanking the inner active-site loop that regulates accessibility of the active site. Differences in loop stability among PAL homologs may explain the observed variation in enzyme efficiency, despite the highly conserved structure of the active site. A. variabilis C503S/C565S PAL is shown to be both more thermally stable and more resistant to proteolytic cleavage than R. toruloides PAL. Additional increases in thermal stability and protease resistance upon ligand binding may be due to enhanced interactions among the residues of the active site, possibly locking the active-site structure in place and stabilizing the tetramer. Examination of the A. variabilis C503S/C565S PAL structure, combined with analysis of its physical properties, provides a structural basis for further engineering of residues that could result in a better therapeutic molecule.
Co-reporter:Pål Stenmark, Min Dong, Jérôme Dupuy, Edwin R. Chapman, Raymond C. Stevens
Journal of Molecular Biology (16 April 2010) Volume 397(Issue 5) pp:1287-1297
Publication Date(Web):16 April 2010
DOI:10.1016/j.jmb.2010.02.041
Botulinum neurotoxins (BoNTs) typically bind the neuronal cell surface via dual interactions with both protein receptors and gangliosides. We present here the 1.9-Å X-ray structure of the BoNT serotype G (BoNT/G) receptor binding domain (residues 868–1297) and a detailed view of protein receptor and ganglioside binding regions. The ganglioside binding motif (SxWY) has a conserved structure compared to the corresponding regions in BoNT serotype A and BoNT serotype B (BoNT/B), but several features of interactions with the hydrophilic face of the ganglioside are absent at the opposite side of the motif in the BoNT/G ganglioside binding cleft. This may significantly reduce the affinity between BoNT/G and gangliosides. BoNT/G and BoNT/B share the protein receptor synaptotagmin (Syt) I/II. The Syt binding site has a conserved hydrophobic plateau located centrally in the proposed protein receptor binding interface (Tyr1189, Phe1202, Ala1204, Pro1205, and Phe1212). Interestingly, only 5 of 14 residues that are important for binding between Syt-II and BoNT/B are conserved in BoNT/G, suggesting that the means by which BoNT/G and BoNT/B bind Syt diverges more than previously appreciated. Indeed, substitution of Syt-II Phe47 and Phe55 with alanine residues had little effect on the binding of BoNT/G, but strongly reduced the binding of BoNT/B. Furthermore, an extended solvent-exposed hydrophobic loop, located between the Syt binding site and the ganglioside binding cleft, may serve as a third membrane association and binding element to contribute to high-affinity binding to the neuronal membrane. While BoNT/G and BoNT/B are homologous to each other and both utilize Syt-I/Syt-II as their protein receptor, the precise means by which these two toxin serotypes bind to Syt appears surprisingly divergent.
Co-reporter:Michael A. Hanson, Raymond C. Stevens
Structure (14 January 2009) Volume 17(Issue 1) pp:8-14
Publication Date(Web):14 January 2009
DOI:10.1016/j.str.2008.12.003
G-protein-coupled receptors (GPCRs) are the largest family of proteins in the human genome. Within the last year, we have witnessed a relative explosion in the amount of structural information available for the GPCR family with two new structures of opsin in the presence and absence of transducin peptide, four new structures of β-adrenergic receptors, and a recent structure of the human adenosine A2A receptor. The new biological insight being gained, such as the highly divergent extracellular loops and areas of structural convergence within the transmembrane helices, allows us to chart a course for further investigation into this important class of membrane proteins.
Co-reporter:Raymond C. Stevens
Structure (13 December 2007) Volume 15(Issue 12) pp:1517-1519
Publication Date(Web):13 December 2007
DOI:10.1016/j.str.2007.11.003
Co-reporter:Graham M. West, Ellen Y.T. Chien, Vsevolod Katritch, Jovylyn Gatchalian, ... Patrick R. Griffin
Structure (12 October 2011) Volume 19(Issue 10) pp:1424-1432
Publication Date(Web):12 October 2011
DOI:10.1016/j.str.2011.08.001
Mechanism of G protein-coupled receptor (GPCR) activation and their modulation by functionally distinct ligands remains elusive. Using the technique of amide hydrogen/deuterium exchange coupled with mass spectrometry, we examined the ligand-induced changes in conformational states and stability within the beta-2-adrenergic receptor (β2AR). Differential HDX reveals ligand-specific alterations in the energy landscape of the receptor's conformational ensemble. The inverse agonists timolol and carazolol were found to be most stabilizing even compared with the antagonist alprenolol, notably in intracellular regions where G proteins are proposed to bind, while the agonist isoproterenol induced the largest degree of conformational mobility. The partial agonist clenbuterol displayed conformational effects found in both the inverse agonists and the agonist. This study highlights the regional plasticity of the receptor and characterizes unique conformations spanning the entire receptor sequence stabilized by functionally selective ligands, all of which differ from the profile for the apo receptor.Graphical AbstractDownload high-res image (192KB)Download full-size imageHighlights► Comprehensive HDX study comparing apo β2AR to β2AR in complex with five ligands ► Each complex affords a unique HDX fingerprint ► Inverse agonists and an antagonist show varying degrees of increased stability ► Full agonists increase conformational mobility in helix VIII on long timescales
Co-reporter:Alexander I. Alexandrov, Mauro Mileni, Ellen Y.T. Chien, Michael A. Hanson, Raymond C. Stevens
Structure (11 March 2008) Volume 16(Issue 3) pp:351-359
Publication Date(Web):11 March 2008
DOI:10.1016/j.str.2008.02.004
Systematic efforts to understand membrane protein stability under a variety of different solution conditions are not widely available for membrane proteins, mainly due to technical problems stemming from the presence of detergents necessary to keep the proteins in the solubilized state and the background that such detergents usually generate during biophysical characterization. In this report, we introduce an efficient microscale fluorescent stability screen using the thiol-specific fluorochrome N-[4-(7-diethylamino-4-methyl-3-coumarinyl)phenyl]maleimide (CPM) for stability profiling of membrane proteins under different solution and ligand conditions. The screen uses the chemical reactivity of the native cysteines embedded in the protein interior as a sensor for the overall integrity of the folded state. The thermal information gained by thorough investigation of the protein stability landscape can be effectively used to guide purification and biophysical characterization efforts including crystallization. To evaluate the method, three different protein families were analyzed, including the Apelin G protein-coupled receptor (APJ).
Co-reporter:Michael A. Hanson, Vadim Cherezov, Mark T. Griffith, Christopher B. Roth, ... Raymond C. Stevens
Structure (11 June 2008) Volume 16(Issue 6) pp:897-905
Publication Date(Web):11 June 2008
DOI:10.1016/j.str.2008.05.001
The role of cholesterol in eukaryotic membrane protein function has been attributed primarily to an influence on membrane fluidity and curvature. We present the 2.8 Å resolution crystal structure of a thermally stabilized human β2-adrenergic receptor bound to cholesterol and the partial inverse agonist timolol. The receptors pack as monomers in an antiparallel association with two distinct cholesterol molecules bound per receptor, but not in the packing interface, thereby indicating a structurally relevant cholesterol-binding site between helices I, II, III, and IV. Thermal stability analysis using isothermal denaturation confirms that a cholesterol analog significantly enhances the stability of the receptor. A consensus motif is defined that predicts cholesterol binding for 44% of human class A receptors, suggesting that specific sterol binding is important to the structure and stability of other G protein-coupled receptors, and that this site may provide a target for therapeutic discovery.
Co-reporter:Irina Kufareva, Manuel Rueda, Vsevolod Katritch, GPCR Dock 2010 participants, ... Ruben Abagyan
Structure (10 August 2011) Volume 19(Issue 8) pp:1108-1126
Publication Date(Web):10 August 2011
DOI:10.1016/j.str.2011.05.012
The community-wide GPCR Dock assessment is conducted to evaluate the status of molecular modeling and ligand docking for human G protein-coupled receptors. The present round of the assessment was based on the recent structures of dopamine D3 and CXCR4 chemokine receptors bound to small molecule antagonists and CXCR4 with a synthetic cyclopeptide. Thirty-five groups submitted their receptor-ligand complex structure predictions prior to the release of the crystallographic coordinates. With closely related homology modeling templates, as for dopamine D3 receptor, and with incorporation of biochemical and QSAR data, modern computational techniques predicted complex details with accuracy approaching experimental. In contrast, CXCR4 complexes that had less-characterized interactions and only distant homology to the known GPCR structures still remained very challenging. The assessment results provide guidance for modeling and crystallographic communities in method development and target selection for further expansion of the structural coverage of the GPCR universe.Graphical AbstractDownload high-res image (473KB)Download full-size imageHighlights► The GPCR Dock 2010 assessment featured three targets of varying modeling difficulty ► Thirty-five groups submitted 275 GPCR complex models prior to release of X-ray coordinates ► Best predictions capture GPCR-ligand interaction details at atomic resolution level ► Reliable homology modeling requires 35%–40% sequence identity between target and template
.jpg)
![Oxazole,2-[1-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-7-phenylheptyl]-5-(tributylstannyl)-](http://img.cochemist.com/ccimg/808200/808134-98-7.png)
![Oxazole,2-[1-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-7-phenylheptyl]-5-(tributylstannyl)-](http://img.cochemist.com/ccimg/808200/808134-98-7_b.png)




METHANOL](http://img.cochemist.com/ccimg/785900/785835-79-2.png)
METHANOL](http://img.cochemist.com/ccimg/785900/785835-79-2_b.png)

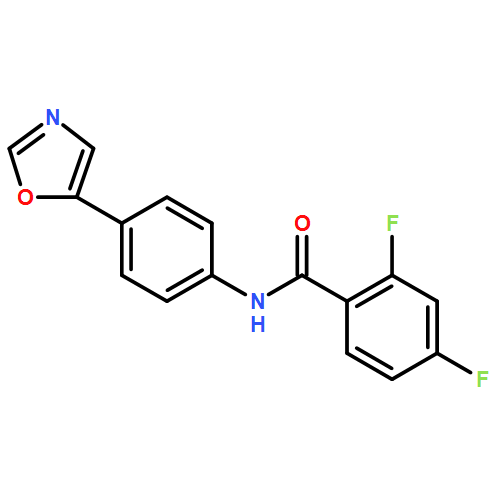
![(2R)-1-(Phenylmethyl)-N-[3-(spiro[isobenzofuran-1(3H),4'-piperidin]-1-yl)propyl-2-pyrrolidinecarboxamide](http://img.cochemist.com/ccimg/475200/475150-69-7.png)
![(2R)-1-(Phenylmethyl)-N-[3-(spiro[isobenzofuran-1(3H),4'-piperidin]-1-yl)propyl-2-pyrrolidinecarboxamide](http://img.cochemist.com/ccimg/475200/475150-69-7_b.png)






![<br>3-Amino-N-(4-nitrophenyl)-5,6,7,8-tetrahydrothieno[2,3-b]quinoline-2-carbox amide](http://img.cochemist.com/ccimg/361200/361198-72-3.png)
![<br>3-Amino-N-(4-nitrophenyl)-5,6,7,8-tetrahydrothieno[2,3-b]quinoline-2-carbox amide](http://img.cochemist.com/ccimg/361200/361198-72-3_b.png)
![(2r,3r,4s,5s,6r)-2-[(2r,3s,4r,5r,6r)-6-(7-cyclohexylheptoxy)-4,5-dihydroxy-2-(hydroxymethyl)oxan-3-yl]oxy-6-(hydroxymethyl)oxane-3,4,5-triol](http://img.cochemist.com/ccimg/349500/349477-49-2.png)
![(2r,3r,4s,5s,6r)-2-[(2r,3s,4r,5r,6r)-6-(7-cyclohexylheptoxy)-4,5-dihydroxy-2-(hydroxymethyl)oxan-3-yl]oxy-6-(hydroxymethyl)oxane-3,4,5-triol](http://img.cochemist.com/ccimg/349500/349477-49-2_b.png)




![2,4,7,10-Tetraoxaundecane, 11-[4-[(1E)-2-phenylethenyl]phenyl]-](http://img.cochemist.com/ccimg/329800/329767-72-8.png)
![2,4,7,10-Tetraoxaundecane, 11-[4-[(1E)-2-phenylethenyl]phenyl]-](http://img.cochemist.com/ccimg/329800/329767-72-8_b.png)


![1-Piperazinamine,N-[(3,5-dimethyl-1-phenyl-1H-pyrazol-4-yl)methylene]-4-(phenylmethyl)-](http://img.cochemist.com/ccimg/305000/304909-07-7.png)
![1-Piperazinamine,N-[(3,5-dimethyl-1-phenyl-1H-pyrazol-4-yl)methylene]-4-(phenylmethyl)-](http://img.cochemist.com/ccimg/305000/304909-07-7_b.png)
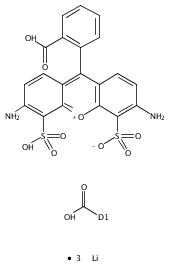
![17-(Cyclopropylmethyl)-4,5alpha-epoxy-3,14-dihydroxy-5'-guanidino-6,7-didehydroindolo[2',3':6,7]morphinan](http://img.cochemist.com/ccimg/219700/219655-56-8.png)
![17-(Cyclopropylmethyl)-4,5alpha-epoxy-3,14-dihydroxy-5'-guanidino-6,7-didehydroindolo[2',3':6,7]morphinan](http://img.cochemist.com/ccimg/219700/219655-56-8_b.png)




![N-[2-[5-Amino-1(S)-[4-(4-pyridinyl)piperazin-1-ylcarbonyl]pentylamino]-1(R)-(3,5-dibromo-4-hydroxybenzyl)-2-oxoethyl]-4-(2-oxo-1,2,3,4-tetrahydroquinazolin-3-yl)piperidine-1-carboxamide](http://img.cochemist.com/ccimg/204700/204697-65-4.png)
![N-[2-[5-Amino-1(S)-[4-(4-pyridinyl)piperazin-1-ylcarbonyl]pentylamino]-1(R)-(3,5-dibromo-4-hydroxybenzyl)-2-oxoethyl]-4-(2-oxo-1,2,3,4-tetrahydroquinazolin-3-yl)piperidine-1-carboxamide](http://img.cochemist.com/ccimg/204700/204697-65-4_b.png)
![1-BROMO-4-[2-(METHOXYMETHOXY)ETHYL]BENZENE](/data/chemimg/442400/203664-53-3.png)
![1-BROMO-4-[2-(METHOXYMETHOXY)ETHYL]BENZENE](/data/chemimg/442400/203664-53-3_b.png)
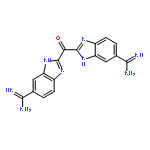
![(2r,3r,4s,5s,6r)-2-[(2r,3s,4r,5r,6r)-6-(4-cyclohexylbutoxy)-4,5-dihydroxy-2-(hydroxymethyl)oxan-3-yl]oxy-6-(hydroxymethyl)oxane-3,4,5-triol](http://img.cochemist.com/ccimg/181200/181135-57-9.png)
![(2r,3r,4s,5s,6r)-2-[(2r,3s,4r,5r,6r)-6-(4-cyclohexylbutoxy)-4,5-dihydroxy-2-(hydroxymethyl)oxan-3-yl]oxy-6-(hydroxymethyl)oxane-3,4,5-triol](http://img.cochemist.com/ccimg/181200/181135-57-9_b.png)


![Benzenemethanol, 4-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/168300/168291-15-4.png)
![Benzenemethanol, 4-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/168300/168291-15-4_b.png)









![Benzene, 1-bromo-4-[(methoxymethoxy)methyl]-](http://img.cochemist.com/ccimg/130600/130534-91-7.png)
![Benzene, 1-bromo-4-[(methoxymethoxy)methyl]-](http://img.cochemist.com/ccimg/130600/130534-91-7_b.png)




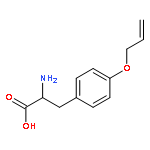
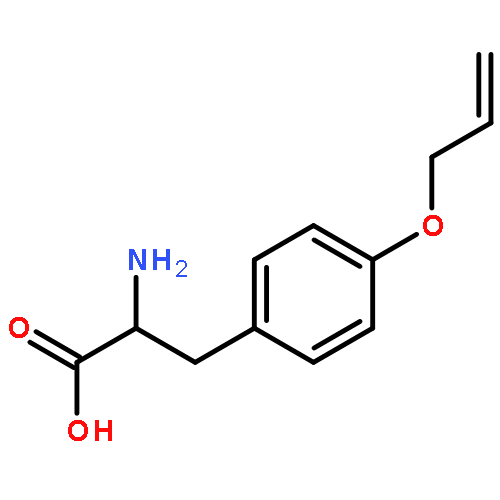

![(2r,3r,4s,5s,6r)-2-[(2r,3s,4r,5r,6r)-4,5-dihydroxy-2-(hydroxymethyl)-6-tridecoxyoxan-3-yl]oxy-6-(hydroxymethyl)oxane-3,4,5-triol](http://img.cochemist.com/ccimg/94000/93911-12-7.png)
![(2r,3r,4s,5s,6r)-2-[(2r,3s,4r,5r,6r)-4,5-dihydroxy-2-(hydroxymethyl)-6-tridecoxyoxan-3-yl]oxy-6-(hydroxymethyl)oxane-3,4,5-triol](http://img.cochemist.com/ccimg/94000/93911-12-7_b.png)
![Benzoic acid, 2-[(3-amino-2-oxo-2H-1-benzopyran-4-yl)amino]-](http://img.cochemist.com/ccimg/87100/87024-59-7.png)
![Benzoic acid, 2-[(3-amino-2-oxo-2H-1-benzopyran-4-yl)amino]-](http://img.cochemist.com/ccimg/87100/87024-59-7_b.png)
![Ethanaminium, 2-[[hydroxy(tridecyloxy)phosphinyl]oxy]-N,N,N-trimethyl-, inner salt](/data/chemimg/1997700/85775-42-4.png)
![Ethanaminium, 2-[[hydroxy(tridecyloxy)phosphinyl]oxy]-N,N,N-trimethyl-, inner salt](/data/chemimg/1997700/85775-42-4_b.png)
![2H-Naphtho[2,1-c]pyran-7-carboxylicacid, 9-(acetyloxy)-2-(3-furanyl)dodecahydro-6a,10b-dimethyl-4,10-dioxo-,methyl ester, (2S,4aR,6aR,7R,9S,10aS,10bR)-](http://img.cochemist.com/ccimg/83800/83729-01-5.png)
![2H-Naphtho[2,1-c]pyran-7-carboxylicacid, 9-(acetyloxy)-2-(3-furanyl)dodecahydro-6a,10b-dimethyl-4,10-dioxo-,methyl ester, (2S,4aR,6aR,7R,9S,10aS,10bR)-](http://img.cochemist.com/ccimg/83800/83729-01-5_b.png)
![1H-Indole-2-carbonitrile,4-[2-hydroxy-3-[(1-methylethyl)amino]propoxy]-](http://img.cochemist.com/ccimg/81700/81608-27-7.png)
![1H-Indole-2-carbonitrile,4-[2-hydroxy-3-[(1-methylethyl)amino]propoxy]-](http://img.cochemist.com/ccimg/81700/81608-27-7_b.png)
![(6AR,9R,10AR)-N-[3-(DIMETHYLAMINO)PROPYL]-N-(ETHYLCARBAMOYL)-7-PROP-2-ENYL-6,6A,8,9,10,10A-HEXAHYDRO-4H-INDOLO[4,3-FG]QUINOLINE-9-CARBOXAMIDE](http://img.cochemist.com/ccimg/81500/81409-90-7.png)
![(6AR,9R,10AR)-N-[3-(DIMETHYLAMINO)PROPYL]-N-(ETHYLCARBAMOYL)-7-PROP-2-ENYL-6,6A,8,9,10,10A-HEXAHYDRO-4H-INDOLO[4,3-FG]QUINOLINE-9-CARBOXAMIDE](http://img.cochemist.com/ccimg/81500/81409-90-7_b.png)
















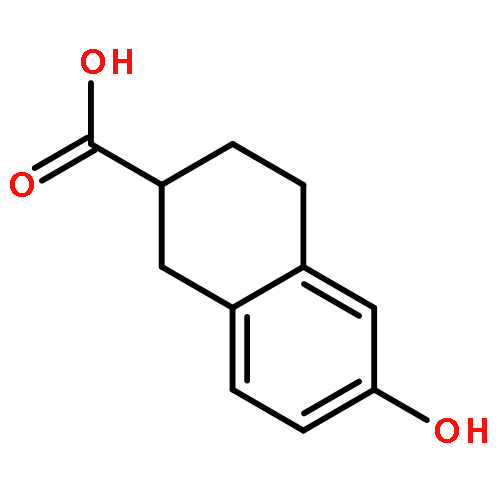
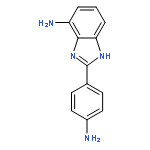


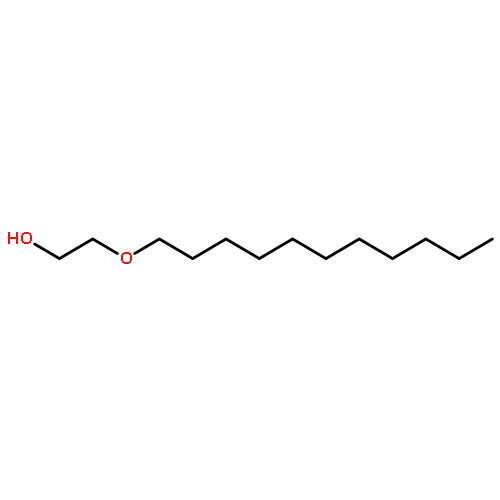









![Ethanaminium,2-[[(dodecyloxy)hydroxyphosphinyl]oxy]-N,N,N-trimethyl-, inner salt](http://img.cochemist.com/ccimg/29600/29557-51-5.png)
![Ethanaminium,2-[[(dodecyloxy)hydroxyphosphinyl]oxy]-N,N,N-trimethyl-, inner salt](http://img.cochemist.com/ccimg/29600/29557-51-5_b.png)
![2-Propanol,1-[(1-methylethyl)amino]-3-[2-(2-propen-1-yl)phenoxy]-, (2S)-](http://img.cochemist.com/ccimg/23900/23846-71-1.png)
![2-Propanol,1-[(1-methylethyl)amino]-3-[2-(2-propen-1-yl)phenoxy]-, (2S)-](http://img.cochemist.com/ccimg/23900/23846-71-1_b.png)
![1-amino-2-[(phenylimino)methyl]anthraquinone](http://img.cochemist.com/ccimg/21900/21810-19-5.png)
![1-amino-2-[(phenylimino)methyl]anthraquinone](http://img.cochemist.com/ccimg/21900/21810-19-5_b.png)
![[5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl [hydroxy-[(3,4,5-trihydroxyoxolan-2-yl)methoxy]phosphoryl] Hydrogen Phosphate](http://img.cochemist.com/ccimg/20800/20762-30-5.png)
![[5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl [hydroxy-[(3,4,5-trihydroxyoxolan-2-yl)methoxy]phosphoryl] Hydrogen Phosphate](http://img.cochemist.com/ccimg/20800/20762-30-5_b.png)


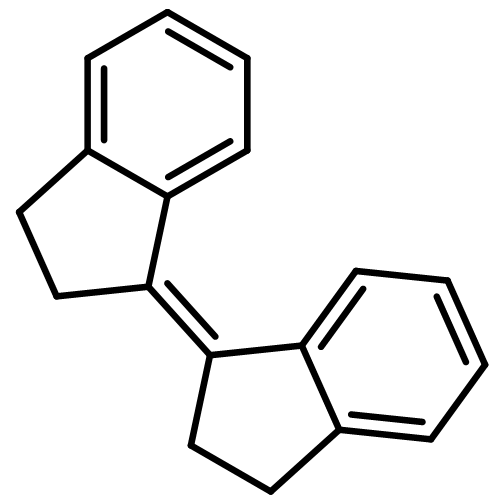


![6-amino-3-methyl-3H-naphtho[1,2,3-de]quinoline-2,7-dione](http://img.cochemist.com/ccimg/14700/14642-72-9.png)
![6-amino-3-methyl-3H-naphtho[1,2,3-de]quinoline-2,7-dione](http://img.cochemist.com/ccimg/14700/14642-72-9_b.png)





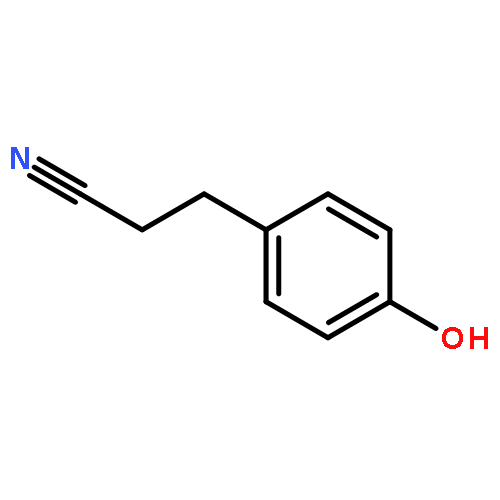





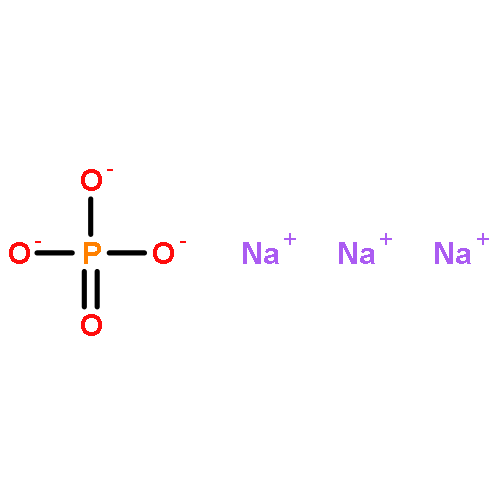
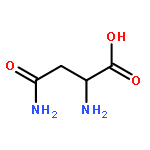

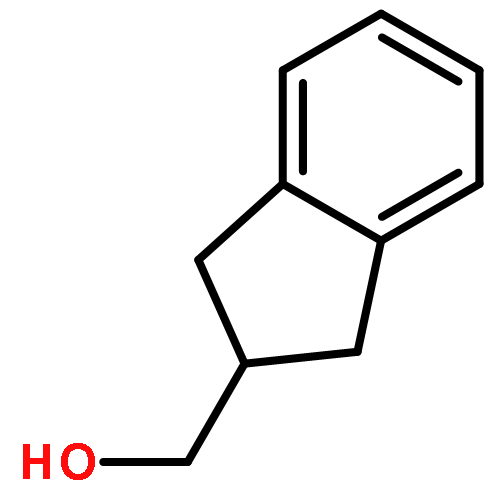
![5,10-Dihydroindeno[2,1-a]indene](http://img.cochemist.com/ccimg/6600/6543-29-9.png)
![5,10-Dihydroindeno[2,1-a]indene](http://img.cochemist.com/ccimg/6600/6543-29-9_b.png)







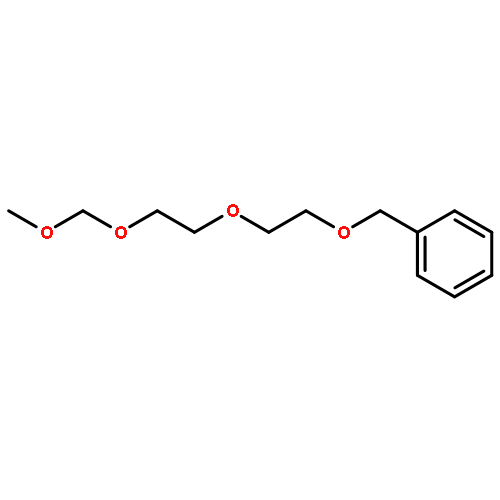
![Benzenamine,4-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/4400/4309-66-4.png)
![Benzenamine,4-[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/4400/4309-66-4_b.png)
![9-Octadecenoic acid(9Z)-,1,1'-[(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-1,2-ethanediyl]ester](http://img.cochemist.com/ccimg/4100/4004-05-1.png)
![9-Octadecenoic acid(9Z)-,1,1'-[(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-1,2-ethanediyl]ester](http://img.cochemist.com/ccimg/4100/4004-05-1_b.png)




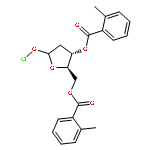
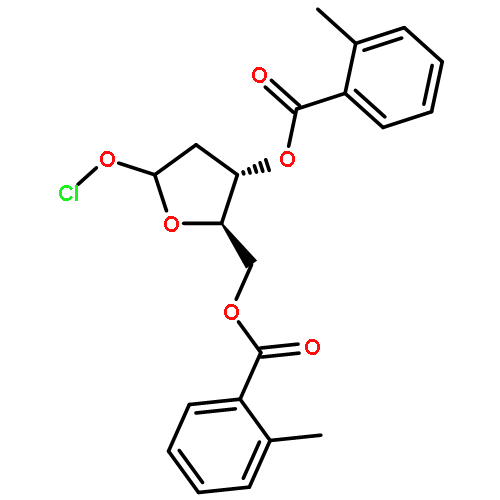
![Ethanol,2-[2-[2-(dodecyloxy)ethoxy]ethoxy]-](http://img.cochemist.com/ccimg/3100/3055-94-5.png)
![Ethanol,2-[2-[2-(dodecyloxy)ethoxy]ethoxy]-](http://img.cochemist.com/ccimg/3100/3055-94-5_b.png)

![9-Octadecenoic acid(9Z)-, 1,1'-[1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-1,2-ethanediyl]ester](http://img.cochemist.com/ccimg/2500/2462-63-7.png)
![9-Octadecenoic acid(9Z)-, 1,1'-[1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-1,2-ethanediyl]ester](http://img.cochemist.com/ccimg/2500/2462-63-7_b.png)






![1,2-Benzenediol,4-[1-hydroxy-2-[(1-methylethyl)amino]butyl]-](http://img.cochemist.com/ccimg/600/530-08-5.png)
![1,2-Benzenediol,4-[1-hydroxy-2-[(1-methylethyl)amino]butyl]-](http://img.cochemist.com/ccimg/600/530-08-5_b.png)
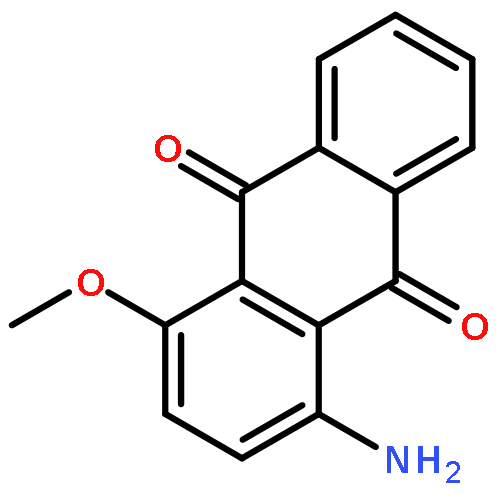





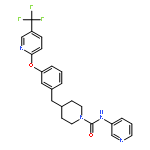
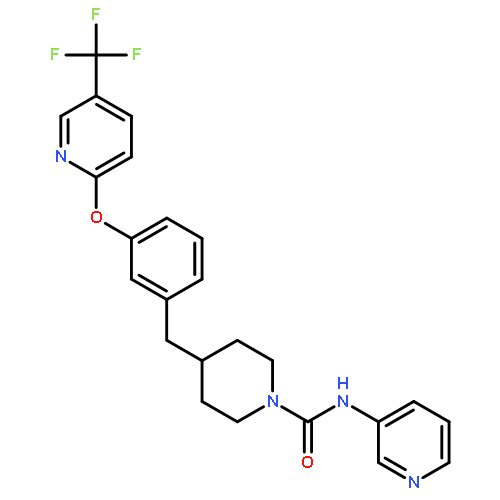








![Benzonitrile,4-[4-[2-[[3-(2-aminoethyl)-1H-indol-5-yl]oxy]acetyl]-1-piperazinyl]-](http://img.cochemist.com/ccimg/171000/170912-52-4.png)
![Benzonitrile,4-[4-[2-[[3-(2-aminoethyl)-1H-indol-5-yl]oxy]acetyl]-1-piperazinyl]-](http://img.cochemist.com/ccimg/171000/170912-52-4_b.png)




![4,8:11,15-Dimethano-20H-bisbenzofuro[2,3-a:3',2'-i]dipyrido[4,3-b:3',4'-h]carbazole-1,8a,10a,18-tetrol,7,12-bis(cyclopropylmethyl)-5,6,7,8,9,10,11,12,13,14,19a,20b-dodecahydro-,(4bS,8R,8aS,10aS,11R,14aS,19aR,20bR)-](http://img.cochemist.com/ccimg/105700/105618-26-6.png)
![4,8:11,15-Dimethano-20H-bisbenzofuro[2,3-a:3',2'-i]dipyrido[4,3-b:3',4'-h]carbazole-1,8a,10a,18-tetrol,7,12-bis(cyclopropylmethyl)-5,6,7,8,9,10,11,12,13,14,19a,20b-dodecahydro-,(4bS,8R,8aS,10aS,11R,14aS,19aR,20bR)-](http://img.cochemist.com/ccimg/105700/105618-26-6_b.png)



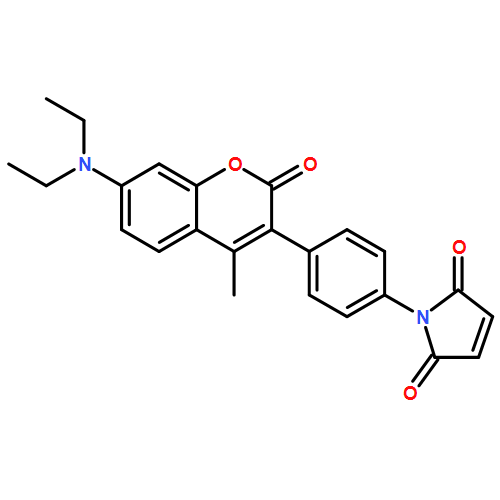


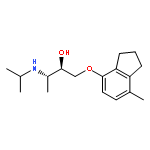






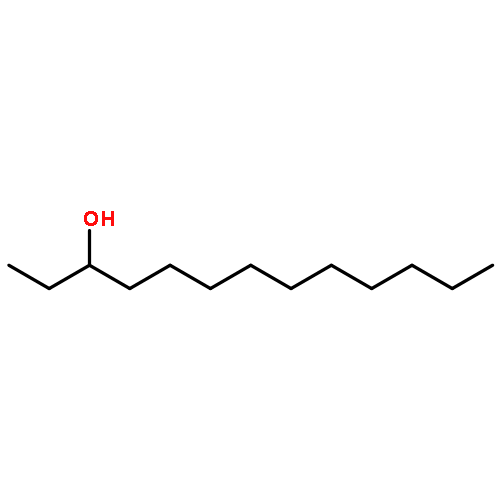




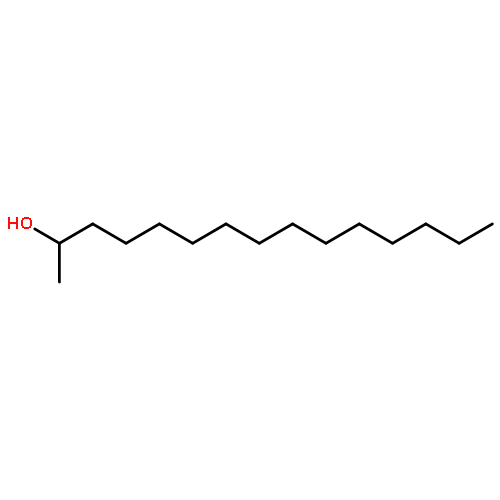





![2-Pyridinecarboxylic acid, 6-[2-(1-oxo-7-phenylheptyl)-5-oxazolyl]-](/data/chemimg/126800/935264-47-4.png)
![2-Pyridinecarboxylic acid, 6-[2-(1-oxo-7-phenylheptyl)-5-oxazolyl]-](/data/chemimg/126800/935264-47-4_b.png)


![Benzamide,3-chloro-5-ethyl-N-[[(2S)-1-ethyl-2-pyrrolidinyl]methyl]-6-hydroxy-2-methoxy-](http://img.cochemist.com/ccimg/84300/84226-12-0.png)
![Benzamide,3-chloro-5-ethyl-N-[[(2S)-1-ethyl-2-pyrrolidinyl]methyl]-6-hydroxy-2-methoxy-](http://img.cochemist.com/ccimg/84300/84226-12-0_b.png)
![Benzenemethanol, 4-amino-3,5-dichloro-α-[[(1,1-dimethylethyl)amino]methyl]-](/data/chemimg/568900/37148-27-9.png)
![Benzenemethanol, 4-amino-3,5-dichloro-α-[[(1,1-dimethylethyl)amino]methyl]-](/data/chemimg/568900/37148-27-9_b.png)



![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-](/data/chemimg/935900/525-66-6.png)
![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-](/data/chemimg/935900/525-66-6_b.png)




![4(3H)-Pteridinone,2-amino-6-[(1R,2S)-1,2-dihydroxypropyl]-7,8-dihydro-](http://img.cochemist.com/ccimg/6800/6779-87-9.png)
![4(3H)-Pteridinone,2-amino-6-[(1R,2S)-1,2-dihydroxypropyl]-7,8-dihydro-](http://img.cochemist.com/ccimg/6800/6779-87-9_b.png)














![2-Propanol,1-(9H-carbazol-4-yloxy)-3-[(1-methylethyl)amino]-](http://img.cochemist.com/ccimg/57800/57775-29-8.png)
![2-Propanol,1-(9H-carbazol-4-yloxy)-3-[(1-methylethyl)amino]-](http://img.cochemist.com/ccimg/57800/57775-29-8_b.png)