Co-reporter:Marius Koch, Mykhaylo Myahkostupov, Daniel G. Oblinsky, Siwei Wang, Sofia Garakyaraghi, Felix N. Castellano, and Gregory D. Scholes
Journal of the American Chemical Society April 19, 2017 Volume 139(Issue 15) pp:5530-5530
Publication Date(Web):March 29, 2017
DOI:10.1021/jacs.7b01630
The intramolecular charge-transfer (CT) dynamics of a rigid and strongly conjugated perylenediimide-bridge-perylene dyad (PDIPe) has been investigated in dichloromethane using ultrafast transient electronic absorption spectroscopy and quantum chemical calculations. The strong electronic coupling between the dyad units gives rise to a CT band. Its photoexcitation forms a delocalized CT state with well-preserved ion bands despite the strong coupling. In the dyad, the electronic transition dipole moment of the electron donor perylene is aligned along the axis of the electric field vector with respect to the CT species. This alignment makes the donor sensitive to the Stark effect and thus charge density fluctuations in the CT state. Charge localization on the picosecond time scale manifests as a time-dependent Stark shift in the visible region. Quantum chemical calculations reveal a twist around the acetylene bridging unit to be the responsible mechanism generating a partial to an almost complete CT state. An estimate of the electric field strength in the CT state yields approximately 25 MV/cm, which increases to around 31 MV/cm during charge localization. Furthermore, the calculations illustrate the complexity of electronic structure in this strongly delocalized superchromophore and reflect the complications in the interpretation of transient absorption results when compared to steady-state approaches such as spectroelectrochemistry and model chromophore experiments such as photoinduced bimolecular charge transfer.
Co-reporter:Sofia Garakyaraghi;Petr Koutnik
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 25) pp:16662-16668
Publication Date(Web):2017/06/28
DOI:10.1039/C7CP03343E
Copper(I) phenanthroline complexes represent viable earth-abundant alternatives to the ubiquitous Ru(II) tris-bipyridine photosensitizers owing to their similar metal-to-ligand charge transfer (MLCT) properties. A well-established complication of Cu(I) phenanthroline complexes is that they can undergo significant photo-induced structural rearrangements, leading to excited states that are highly susceptible to exciplex formation and short-lived. In this work, a comprehensive analysis of the photo-induced structural distortions and singlet–triplet intersystem crossing dynamics of a series of four sterically encumbered Cu(I) phenanthroline chromophores has been conducted, namely, [Cu(dsbp)2]+ (dsbp = 2,9-di-sec-butyl-1,10-phenanthroline), [Cu(dsbtmp)2]+ (dsbtmp = 2,9-di-sec-butyl-3,4,7,8-tetramethyl-1,10-phenanthroline), [Cu(dipp)2]+ (dipp = 2,9-di-isopropyl-1,10-phenanthroline), and [Cu(diptmp)2]+ (diptmp = 2,9-di-isopropyl-3,4,7,8-tetramethyl-1,10-phenanthroline). Upconverted fluorescence decay kinetics were measured at wavelengths along the blue side of the photoluminescence spectrum. The experimental results displayed strong wavelength dependence of the singlet emission, with rapid sub-picosecond decay dominating at higher energies. At lower emission energies, increasing contribution of a longer decay component was revealed. This wavelength dependence is a signature of the excited state structural rearrangement of the phenanthroline ligands which concomitantly lower the excited state energy. The obtained time constants were in excellent agreement with those measured in the complementary ultrafast transient absorption experiments. The sub-picosecond component (prompt fluorescence) is associated with the photo-induced structural rearrangement that lowers the energy of the singlet excited state. The longer decay component represents the lifetime of the S1 excited state, and thus the time-scale of singlet–triplet intersystem crossing. Lastly, the observed dual emission was further characterized by constructing picosecond time-resolved emission spectra from the measured kinetic data. These qualitative luminescence spectra capture the resulting emission from both the S1 initial state and the S1 flattened state, providing further insight into the energy-lowering excited state distortion across the series.
Co-reporter:Karim A. El Roz
Chemical Communications 2017 vol. 53(Issue 85) pp:11705-11708
Publication Date(Web):2017/10/24
DOI:10.1039/C7CC07188D
A major challenge remaining in photochemical upconversion lies in identifying appropriate chromophore combinations that function in pure water in the absence of hydrophobic hosts or surfactant additives. The current investigation achieves this goal using combinations of water soluble Ru(II) metal-to-ligand charge transfer (MLCT) sensitizers in concert with 9-anthracenecarboxylate (AnCO2−) and 1-pyrenecarboxylate (PyCO2−) in neat H2O.
Co-reporter:Cédric Mongin;Sofia Garakyaraghi;Natalia Razgoniaeva;Mikhail Zamkov
Science 2016 Vol 351(6271) pp:369-372
Publication Date(Web):22 Jan 2016
DOI:10.1126/science.aad6378
A different way to put triplets in play
Most molecules adopt a singlet spin configuration: All their electrons are arranged in pairs. Unpaired triplet states engage in a variety of useful reactions but are hard to produce. Quantum mechanics dictates that photo-excitation from singlet to triplet states is inefficient. Instead, chemists rely on sensitizers, which populate the triplet states of their neighbors through energy transfer after absorbing light themselves. Mongin et al. now show that certain nanoparticles can act as triplet sensitizers.
Science, this issue p. 369
Co-reporter:Cédric Mongin, Jessica H. Golden, and Felix N. Castellano
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 36) pp:24038
Publication Date(Web):August 1, 2016
DOI:10.1021/acsami.6b05697
This article proposes the exploitation of widely available, inexpensive, innocuous “green” liquid polyethylene glycol (PEG) polymers containing the oxygen scavenger oleic acid (OA) as promising media for studying oxygen-sensitive photochemical processes. Here we report the successful application of this media to detailed investigations of triplet-sensitized photochemical upconversion, previously established as being readily poisoned by dissolved oxygen. Three different PEG materials were investigated with increasing molecular weight from 200 to 600 g/mol, coded as PEG-200, PEG-400, and PEG-600. These fluidic polymers facilitate an oxygen-depleted environment in comparison to commonly employed organic solvents while providing high solubility and diffusion for the dissolved chromophores. Moreover, the low oxygen permeation afforded by these PEG solvents allows them to remain deoxygenated in open containers under ambient conditions for extended time periods. OA, 9,10-dimethylanthracene (DMA), and 2,5-dimethylfuran (DMF) are shown to efficiently and quantitatively consume dissolved oxygen in the PEG environment in the presence of the photoactivated triplet sensitizer platinum(II) tetraphenyltetrabenzoporphyrin (PtTPBP). Oxygen consumption was directly correlated with systematically increasing sensitizer excited-state lifetimes that eventually reach the same plateau as achieved through extensive N2 sparging. Diffusion-controlled bimolecular triplet–triplet energy transfer quenching between PtTPBP and the acceptor/annihilator 9,10-bisphenylethynylanthracene (BPEA) was observed in all three PEG formulations investigated. Subsequent triplet–triplet annihilation, between triplet excited BPEA acceptors, achieves bright and stable upconverted singlet fluorescence from BPEA with no decrease in intensity over 20 h under ambient conditions. In the champion composition (PEG 200), the upconversion quantum efficiency reached 31% under conditions where triplet–triplet annihilation was maximized. This is in stark contrast for the same upconverting pair measured in toluene under ambient conditions, which rapidly decomposes upon exposure to visible light. To illustrate that these PEG compositions could be translated into a suitable solid-state format, these viscous solutions were embedded in a transparent polyurethane polymer shell yielding a flexible and long-term stable upconverting cell that can be manipulated for possible real-world applications. Although the current investigation focused on photochemical upconversion, the oxygen-depleted environments developed here can be utilized to study a plethora of oxygen-intolerant photochemical reactions.Keywords: liquid polymers; oxygen scavenging; oxygen-depleted materials; photochemical upconversion; singlet oxygen; triplet energy transfer
Co-reporter:Michelle M. McGoorty, Rony S. Khnayzer and Felix N. Castellano
Chemical Communications 2016 vol. 52(Issue 50) pp:7846-7849
Publication Date(Web):24 May 2016
DOI:10.1039/C6CC03932D
Two water-soluble anionic cyclometalated Ir(III) complexes, Ir(ppy)2BPS [1] and Ir(F-mppy)2BPS [2] have been synthesized and display clear evidence of self-assembly in water. Concentration-induced aggregation enhances the excited-state properties of both complexes, blue-shifting the photoluminescence emission energies as well as increasing the corresponding excited state lifetimes and quantum yields up to a factor of 5.
Co-reporter:Felix N. Castellano
Inorganic Chemistry 2016 Volume 55(Issue 24) pp:12483-12487
Publication Date(Web):December 19, 2016
DOI:10.1021/acs.inorgchem.6b02830
Co-reporter:Sofia Garakyaraghi, Peter D. Crapps, Catherine E. McCusker, and Felix N. Castellano
Inorganic Chemistry 2016 Volume 55(Issue 20) pp:10628-10636
Publication Date(Web):September 28, 2016
DOI:10.1021/acs.inorgchem.6b01880
In the interest of expanding the inventory of available long lifetime, photochemically robust, and strongly reducing Cu(I) MLCT sensitizers, we present detailed structural, photophysical, and electrochemical characterization of [Cu(dipp)2]+, dipp = 2,9-diisopropyl-1,10-phenanthroline, and its sterically encumbered tetramethyl analogue [Cu(diptmp)2]+, diptmp = 2,9-diisopropyl-3,4,7,8-tetramethyl-1,10-phenanthroline. The achiral isopropyl substituents enable similar steric bulk effects to the previously investigated sec-butyl substituents while eliminating the complex NMR structural analyses associated with the presence of two chiral centers in the latter. The photophysical properties of [Cu(diptmp)2]+ are impressive, possessing a 2.3 μs lifetime in deaerated CH2Cl2 and a photoluminescence quantum yield of 4.7%, which were slightly attenuated in coordinating tetrahydrofuran (THF) solutions. Nanosecond transient absorption spectroscopy results matched the transient photoluminescence kinetics enabling complete characterization of MLCT excited-state decay in these molecules. The calculated excited-state potential for the Cu2+/Cu+* couple (E = −1.74 V vs Fc+/0) indicated that [Cu(diptmp)2]+* is a strong photoreductant potentially useful for myriad applications. Ultrafast transient absorption measurements performed in THF solutions are also reported, yielding the relative time scales for both the pseudo-Jahn–Teller distortion (0.4–0.8 ps in [Cu(dipp)2]+ and 0.12–0.5 ps in [Cu(diptmp)2]+) and singlet–triplet intersystem crossing (6.4–10.1 ps for [Cu(dipp)2]+ and 3.5–5.4 ps for [Cu(diptmp)2]+) within these molecules. The disparity in the time scales of pseudo-Jahn–Teller distortion and intersystem crossing between two complexes with different anticipated excited-state geometries suggests that strongly impeded structural distortion in the MLCT excited state (i.e., [Cu(diptmp)2]+) enables more rapid surface crossings in the initial deactivation dynamics.
Co-reporter:James E. Yarnell;Catherine E. McCusker;Alexer J. Leeds;Josué M. Breaux
European Journal of Inorganic Chemistry 2016 Volume 2016( Issue 12) pp:1808-1818
Publication Date(Web):
DOI:10.1002/ejic.201600194
Abstract
The electronic structure and photophysical properties of a luminescent IrIII bis-cyclometalated complex covalently attached to one 4-piperidinyl-1,8-naphthalimide (PNI) chromophore through a coordinated 1,10-phenanthroline, [Ir(ppy)2(phen-PNI)](PF6), is presented. This bichromophore represents a new class of visible light-harvesting IrIII complexes that exhibit markedly enhanced room-temperature excited-state lifetimes (τ = 8.8 ms) as a result of intervening ligand-centered triplet states present on the pendant naphthalimide chromophore. In this IrIII complex, the intense singlet fluorescence of the pendant PNI chromophore is nearly quantitatively quenched and was found to sensitize the IrIII metal/ligand-to-ligand charge-transfer (MLLCT) excited state. The excited state ultimately returns to the PNI chromophore as a long-lived excited triplet that disposes of its energy by equilibrating with the photoluminescent IrIII MLLCT excited state. Evidence of the excited-state equilibrium is provided through static and dynamic photoluminescence spectroscopy, transient absorption spectroscopy, and time-dependent density functional theory calculations.
Co-reporter:Masato Hirai, Mykhaylo Myahkostupov, Felix N. Castellano, and François P. Gabbaï
Organometallics 2016 Volume 35(Issue 11) pp:1854-1860
Publication Date(Web):May 12, 2016
DOI:10.1021/acs.organomet.6b00233
In the context of our work on main group-based anion sensors, we have synthesized the bromide salts of a series of tetraarylstibonium cations of general formula [ArSbPh3]+ with Ar = 9-phenanthryl ([1]+), 1-pyrenyl- ([2]+), and 3-perylenyl ([3]+). While [1]+ is not stable in water, we found that [2]+ and [3]+ can be used as sensors for the sub-ppm detection of fluoride anions in aqueous solutions consisting of 9/1 (v/v) H2O/DMSO (pH 4.8). Fluoride sensing, which rests on the formation of the fluorostiboranes 2–F and 3–F, is accompanied by a distinct turn-on fluorescence response. This response is especially marked upon conversion of [3]+ into 3–F, with a fluorescence intensity enhancement by ∼8 fold and a quantum yield of 59.2% for 3–F. The relevance of this study is established by demonstrating that [3]+ can be used as a selective fluoride sensor for bottle or tap water.
Co-reporter:Samantha E. Brown-Xu, Matthew S. J. Kelley, Kelly A. Fransted, Arnab Chakraborty, George C. Schatz, Felix N. Castellano, and Lin X. Chen
The Journal of Physical Chemistry A 2016 Volume 120(Issue 4) pp:543-550
Publication Date(Web):January 13, 2016
DOI:10.1021/acs.jpca.5b11233
The influence of molecular structure on excited-state properties and dynamics of a series of cyclometalated platinum dimers was investigated through a combined experimental and theoretical approach using femtosecond transient absorption (fs TA) spectroscopy and density functional theory (DFT) calculations. The molecules have the general formula [Pt(ppy)(μ-R2pz)]2, where ppy = 2-phenylpyridine, pz = pyrazolate, and R = H, Me, Ph, or tBu, and are strongly photoluminescent at room temperature. The distance between the platinum centers in this A-frame geometry can be varied depending on the steric bulk of the bridging pyrazolate ligands that exert structural constraints and compress the Pt–Pt distance. At large Pt–Pt distances there is little interaction between the subunits, and the chromophore behaves similar to a monomer with excited states described as mixtures of ligand-centered and metal-to-ligand charge transfer (LC/MLCT) transitions. When the Pt(II) centers are brought closer together with bulky bridging ligands, they interact through their dz2 orbitals and the S1 and T1 states are best characterized as metal–metal-to-ligand charge transfer (MMLCT) in character. The results of the femtoseconds TA experiments reveal that intersystem crossing (ISC) occurs on ultrafast time scales (τS1 < 200 fs), while there are two relaxation processes occurring within the triplet manifold, τ1 = 0.5–3.2 ps and τ2 = 20–70 ps; the longer time constants correspond to the presence of bulkier bridging ligands. DFT calculations illustrate that the Pt–Pt distances further contract in the T1 3MMLCT states; therefore, slower relaxation may be related to a larger structural reorganization. Subsequent investigations using faster time resolution are planned to measure the ISC process as well as to identify any potential coherent interaction(s) between the platinum centers that may occur.
Co-reporter:Jonah W. Jurss, Rony S. Khnayzer, Julien A. Panetier, Karim A. El Roz, Eva M. Nichols, Martin Head-Gordon, Jeffrey R. Long, Felix N. Castellano and Christopher J. Chang
Chemical Science 2015 vol. 6(Issue 8) pp:4954-4972
Publication Date(Web):09 Jun 2015
DOI:10.1039/C5SC01414J
Mononuclear metalloenzymes in nature can function in cooperation with precisely positioned redox-active organic cofactors in order to carry out multielectron catalysis. Inspired by the finely tuned redox management of these bioinorganic systems, we present the design, synthesis, and experimental and theoretical characterization of a homologous series of cobalt complexes bearing redox-active pyrazines. These donor moieties are locked into key positions within a pentadentate ligand scaffold in order to evaluate the effects of positioning redox non-innocent ligands on hydrogen evolution catalysis. Both metal- and ligand-centered redox features are observed in organic as well as aqueous solutions over a range of pH values, and comparison with analogs bearing redox-inactive zinc(II) allows for assignments of ligand-based redox events. Varying the geometric placement of redox non-innocent pyrazine donors on isostructural pentadentate ligand platforms results in marked effects on observed cobalt-catalyzed proton reduction activity. Electrocatalytic hydrogen evolution from weak acids in acetonitrile solution, under diffusion-limited conditions, reveals that the pyrazine donor of axial isomer 1-Co behaves as an unproductive electron sink, resulting in high overpotentials for proton reduction, whereas the equatorial pyrazine isomer complex 2-Co is significantly more active for hydrogen generation at lower voltages. Addition of a second equatorial pyrazine in complex 3-Co further minimizes overpotentials required for catalysis. The equatorial derivative 2-Co is also superior to its axial 1-Co congener for electrocatalytic and visible-light photocatalytic hydrogen generation in biologically relevant, neutral pH aqueous media. Density functional theory calculations (B3LYP-D2) indicate that the first reduction of catalyst isomers 1-Co, 2-Co, and 3-Co is largely metal-centered while the second reduction occurs at pyrazine. Taken together, the data establish that proper positioning of non-innocent pyrazine ligands on a single cobalt center is indeed critical for promoting efficient hydrogen catalysis in aqueous media, akin to optimally positioned redox-active cofactors in metalloenzymes. In a broader sense, these findings highlight the significance of electronic structure considerations in the design of effective electron–hole reservoirs for multielectron transformations.
Co-reporter:Catherine E. McCusker and Felix N. Castellano
Inorganic Chemistry 2015 Volume 54(Issue 12) pp:6035-6042
Publication Date(Web):June 2, 2015
DOI:10.1021/acs.inorgchem.5b00907
The current investigation compares the photochemical upconversion sensitization properties of two long lifetime Cu(I) metal-to-ligand charge transfer (MLCT) chromophores to 3 distinct anthryl-based triplet acceptors. The sensitizers [Cu(dsbtmp)2](PF6) (1, dsbtmp = 2,9-di(sec-butyl)-3,4,7,8-tetramethyl-1,10-phenanthroline) and [Cu(dsbp)2](PF6) (2, dsbp = 2,9-di(sec-butyl-1,10-phenanthroline) were selectively excited in the presence of anthracene, 9,10-diphenylanthracene (DPA), and 9,10-dimethylanthracene (DMA) in degassed dichloromethane solutions. In all instances, triplet energy transfer was observed from selective excitation of the Cu(I) MLCT chromophore to each respective anthryl species. The bimolecular triplet–triplet energy transfer quenching rate constants were extracted from dynamic Stern–Volmer analyses in each case, yielding values below the diffusion limit in dichloromethane. However, the Stern–Volmer quenching constants (KSV’s) were sizable enough (up to ∼2300 M–1 with 1 as a sensitizer) to support efficient photochemical upconversion. As such, visible to near-UV photochemical upconversion was observed in every instance, along with the anticipated quadratic-to-linear incident light power dependence when pumping at 488 nm. The latter verified that it is indeed sensitized triplet–triplet annihilation responsible for the generation of the anthryl-based singlet fluorescence. Photochemical upconversion quantum efficiencies were evaluated using a relative actinometric method as both a function of incident light power density as well as anthryl acceptor/annihilator concentration. When 1 was used as the sensitizer, upconversion quantum yields as large as 9.2% and 17.8% were observed for DMA and DPA, respectively. Finally, the combination of 1 with DMA was shown to be quite robust, showing no obvious signs of decomposition during 12 h of continuous 488 nm photolysis.
Co-reporter:Felix N. Castellano and Catherine E. McCusker
Dalton Transactions 2015 vol. 44(Issue 41) pp:17906-17910
Publication Date(Web):22 Sep 2015
DOI:10.1039/C5DT03212A
This treatment highlights the historical development of MLCT sensitizers in photochemical upconversion while indentifying current state-of-the-art and exciting opportunities in this arena moving towards the future.
Co-reporter:F. Deng, A. J. Francis, W. W. Weare and F. N. Castellano
Photochemical & Photobiological Sciences 2015 vol. 14(Issue 7) pp:1265-1270
Publication Date(Web):03 Jun 2015
DOI:10.1039/C5PP00106D
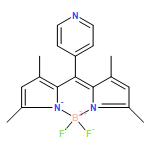
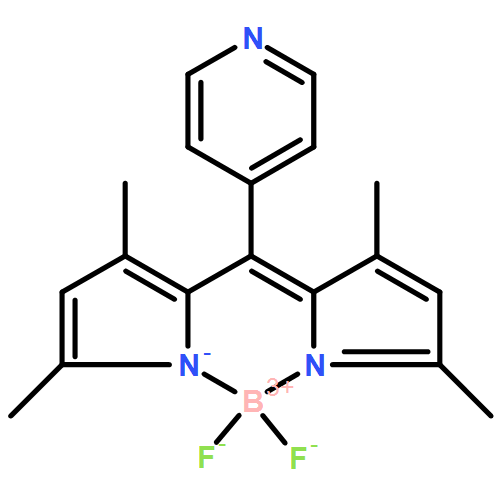
Non-coherent sensitized red-to-green upconversion has been achieved utilizing platinum(II) tetraphenyltetrabenzoporphyrin (PtTPTBP) as the triplet sensitizer and a nearly quantitatively fluorescent meso-(2,6-dichloropyridyl)-substituted boron dipyrromethene (Cl2PyBODIPY) chromophore (Φ = 0.99 in toluene) as the energy acceptor/annihilator in deoxygenated toluene. Dynamic Stern–Volmer analysis revealed that PtTPTBP phosphorescence as quenched by Cl2PyBODIPY occurs with a KSV of 108000 M−1, yielding a triplet–triplet energy transfer rate constant of 2.3 × 109 M−1 s−1. Using a non-coherent red light-emitting diode excitation source centered at 626 nm, the incident power dependence responsible for generating singlet BODIPY fluorescence in the green was shown to traverse quadratic to linear regimes, the latter being achieved near 60 mW cm−2. These data were consistent with a photochemical upconversion mechanism being responsible for generating singlet fluorescence from the Cl2PyBODIPY chromophores through sensitized triplet–triplet annihilation (TTA). Integrated delayed fluorescence transients were utilized to reveal the TTA efficiency for the Cl2PyBODIPY chromophore and saturated near 46%, representing the lower limit for the TTA process. Kinetic modelling of the delayed fluorescence transient produced from 1.5 mJ laser pulses (λex = 615 nm) revealed a maximum limiting TTA efficiency of 64% for this upconverting composition, implying that this is indeed an extremely relevant acceptor/annihilator composition for photochemical upconversion.
Co-reporter:Mykhaylo Myahkostupov, Felix N. Castellano
Tetrahedron 2015 Volume 71(Issue 51) pp:9519-9527
Publication Date(Web):23 December 2015
DOI:10.1016/j.tet.2015.10.083
Two new representative methane- and adamantane-centered ‘antenna’ tetramers bearing bay-substituted π-conjugated phenylethynyl-perylenediimides (PDICCPh) as chromophoric subunits, tetrakis-[1-(4-ethynylphenyl)-N,N′-bis(1-hexylheptyl)-perylene-3,4:9,10-tetracarboxylic diimide]-methane (1) and tetrakis-1,3,5,7-[1-(4-ethynylphenyl)-N,N′-bis(1-hexylheptyl)-perylene-3,4:9,10-tetracarboxylic diimide]-adamantane (2), have been synthesized and their structural aspects have been thoroughly investigated by NMR spectroscopy. These PDI tetramers (1 and 2) represent the first successful example of incorporating the bay-substituted phenylethynyl-perylenediimides into the large rigid core tetrahedral frameworks. In these PDI tetramers, dynamic NMR experiments revealed the existence of perylene-centered conformational dynamic equilibrium (ΔG≠=15−17 kcal/mol), the primary cause of the observed spectral broadening in conventional 1H NMR spectra (295 K). In addition, PDI tetramers 1 and 2 were found to possess exceptional (photo)chemical stability, and their corresponding photophysical properties (ɛmax∼180,000; τFL=6.9 ns; ΦFL∼60%) make them viable candidates for various photonic applications and are in good agreement with other related multichromophoric PDI-based systems.
Co-reporter:Sofia Garakyaraghi, Evgeny O. Danilov, Catherine E. McCusker, and Felix N. Castellano
The Journal of Physical Chemistry A 2015 Volume 119(Issue 13) pp:3181-3193
Publication Date(Web):March 9, 2015
DOI:10.1021/acs.jpca.5b00901
Subpicosecond through supra-nanosecond transient absorption dynamics of the homoleptic Cu(I) metal-to-ligand charge transfer (MLCT) photosensitizers including the benchmark [Cu(dmp)2]+ (dmp =2,9-dimethyl-1,10-phenanthroline) chromophore, as well as [Cu(dsbp)2]+ (dsbp =2,9-di(sec-butyl)-1,10-phenanthroline and [Cu(dsbtmp)2]+ (dsbtmp =2,9-di(sec-butyl)-3,4,7,8-tetramethyl-1,10-phenanthroline) were investigated in dichloromethane and tetrahydrofuran solutions. Visible and near-IR spectroelectrochemical measurements of the singly reduced [Cu(dsbp)2]+ and [Cu(dsbtmp)2]+ species were determined in tetrahydrofuran, allowing for the identification of redox-specific phenanthroline-based radical anion spectroscopic signatures prevalent in the respective transient absorption experiments. This study utilized four different excitation wavelengths (418, 470, 500, and 530 nm) to elucidate dynamics on ultrafast times scales spanning probe wavelengths ranging from the UV to the near-IR (350 to 1450 nm). With the current time resolution of ∼150 fs, initial excited state decay in all three compounds was found to be independent of excitation wavelength. Not surprisingly, there was little to no observed influence of solvent in the initial stages of excited state decay in any of these molecules including [Cu(dmp)2]+, consistent with results from previous investigators. The combined experimental data revealed two ranges of time constants observed on short time scales in all three MLCT chromophores and both components lengthen as a function of structure in the following manner: [Cu(dsbtmp)2]+ < [Cu(dsbp)2]+ < [Cu(dmp)2]+. The molecule with the most inhibited potential for distortion, [Cu(dsbtmp)2]+, possessed the fastest ultrafast dynamics as well as the longest excited state lifetimes in both solvents. These results are consistent with a small degree of excited state distortion, rapid intersystem crossing, and weak vibronic coupling to the ground state. The concomitant systematic variation in both initial time constants, assigned to pseudo-Jahn–Teller distortion and intersystem crossing, suggest that both processes are intimately coupled in all molecules in the series. The variability in these time scales illustrate that strongly impeded structural distortion in Cu(I) MLCT excited state enables more rapid surface crossings in the initial deactivation dynamics.
Co-reporter:Jean-Hubert Olivier, Yusong Bai, Hyounsoo Uh, Hyejin Yoo, Michael J. Therien, and Felix N. Castellano
The Journal of Physical Chemistry A 2015 Volume 119(Issue 22) pp:5642-5649
Publication Date(Web):May 11, 2015
DOI:10.1021/acs.jpca.5b03199
We report four supermolecular chromophores based on (porphinato)zinc(II) (PZn) and (polypyridyl)metal units bridged via ethyne connectivity (Pyr1RuPZn2, Pyr1RuPZnRuPyr1, Pyr1RuPZn2RuPyr1, and OsPZn2Os) that fulfill critical sensitizer requirements for NIR-to-vis triplet–triplet annihilation upconversion (TTA-UC) photochemistry. These NIR sensitizers feature: (i) broad, high oscillator strength NIR absorptivity (700 nm < λmax(NIR) < 770 nm; 6 × 104 M–1 cm–1 < extinction coefficient (λmax(NIR)) < 1.6 × 105 M–1 cm–1; 820 cm–1 < fwhm < 1700 cm–1); (ii) substantial intersystem crossing quantum yields; (iii) long, microsecond time scale T1 state lifetimes; and (iv) triplet states that are energetically poised for exergonic energy transfer to the molecular annihilator (rubrene). Using low-power noncoherent illumination at power densities (1–10 mW cm–2) similar to that of terrestrial solar photon illumination conditions, we demonstrate that Pyr1RuPZn2, Pyr1RuPZn2RuPyr1, and Pyr1RuPZnRuPyr1 sensitizers can be used in combination with the rubrene acceptor/annihilator to achieve TTA-UC: these studies represent the first examples whereby a low-power noncoherent NIR light source drives NIR-to-visible upconverted fluorescence centered in a spectral window within the bandgap of amorphous silicon.
Co-reporter:Dr. Lorenzo Mosca;Dr. Rony S. Khnayzer;Dr. Megan S. Lazorski;Dr. Evgeny O. Danilov; Felix N. Castellano; Pavel Anzenbacher Jr.
Chemistry - A European Journal 2015 Volume 21( Issue 10) pp:4056-4064
Publication Date(Web):
DOI:10.1002/chem.201405717
Abstract
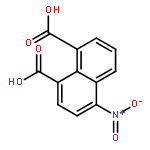
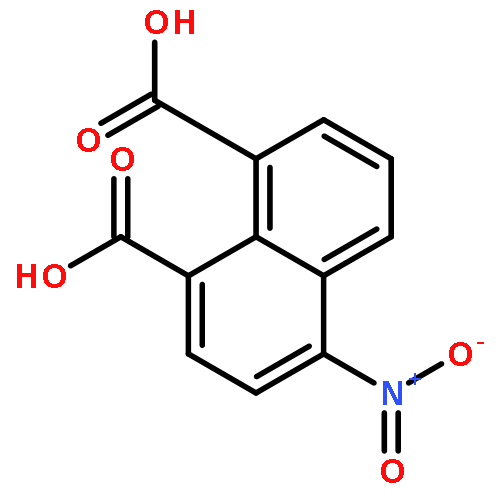
A series of metal–organic chromophores containing RuII or IrIII were studied for the luminometric detection of nitroaromatic compounds, including trinitrotoluene (TNT). These complexes display long-lived, intense photoluminescence in the visible region and are demonstrated to serve as luminescent sensors for nitroaromatics. The solution-based behavior of these photoluminescent molecules has been studied in detail in order to identify the mechanism responsible for metal-to-ligand charge-transfer (MLCT) excited state quenching upon addition of TNT and 2,4-dinitrotoluene (2,4-DNT). A combination of static and dynamic spectroscopic measurements unequivocally confirmed that the quenching was due to a photoinduced electron transfer (PET) process. Ultrafast transient absorption experiments confirmed the formation of the TNT radical anion product following excited state electron transfer from these metal complexes. Reported for the first time, photoluminescence quenching realized through ink-jet printing and solid-state titrations was used for the solid-state detection of TNT; achieving a limit-of-quantitation (LOQ) as low as 5.6 ng cm−2. The combined effect of a long-lived excited state and an energetically favorable driving force for the PET process makes the RuII and IrIII MLCT complexes discussed here particularly appealing for the detection of nitroaromatic volatiles and related high-energy compounds.
Co-reporter:R. S. Khnayzer, V. S. Thoi, M. Nippe, A. E. King, J. W. Jurss, K. A. El Roz, J. R. Long, C. J. Chang and F. N. Castellano
Energy & Environmental Science 2014 vol. 7(Issue 4) pp:1477-1488
Publication Date(Web):19 Feb 2014
DOI:10.1039/C3EE43982H
Homogeneous aqueous solutions of photocatalytic ensembles, consisting of [Ru(bpy)3]2+ as a photosensitizer, ascorbic acid/ascorbate as the electron source, and 10 distinct Co2+-based molecular catalysts, were evaluated for visible-light induced hydrogen evolution using high-throughput screening. The combined results demonstrate that Co2+ complexes bearing tetradentate ligands yield more active photocatalytic compositions than their congeners with pentadentate ligands while operating with high catalyst stability. Additionally, molecular Co2+ catalysts with cis open coordination sites appear to be significantly more active for hydrogen evolution than those with trans open sites. As evidenced by mass spectrometric analysis of the reactor headspace and associated deuteration experiments, the H2 gas generated in all instances was derived from aqueous protons. One of the most promising cis-disposed Co2+ species, [Co(bpyPY2Me)(CH3CN)(CF3SO3)](CF3SO3) (1), engages in highly efficient hydrogen evolving photocatalysis, achieving a turnover number of 4200 (H2/Co) and a turnover frequency of 3200 (H2/Co per h) at pH 4 under simulated sunlight (AM 1.5G, 100 mW cm−2) at room temperature. At equimolar concentrations of photosensitizer and 1, the total hydrogen produced appears to be exclusively limited by the photostability of [Ru(bpy)3]2+, which was observed to decompose into an Ru(bpy)2–ascorbate adduct, as evidenced by HPLC and ESI-MS experiments. Lowering the operating temperature from 27 to 5 °C significantly attenuates bpy dissociation from the sensitizer, resulting in a net ∼two-fold increase in hydrogen production from this composition. The primary electron transfer steps of this photocatalytic ensemble were investigated by nanosecond transient absorption spectroscopy. Photoexcited [Ru(bpy)3]2+ undergoes reductive quenching by ascorbic acid/ascorbate (kq = 2.6 × 107 M−1 s−1), releasing [Ru(bpy)3]+ from the encounter solvent cage with an efficiency of 55 ± 5%. In the presence of catalyst 1, [Ru(bpy)3]+ generated in the initial flash-quench experiment transfers an electron (ket = 2 × 109 M−1 s−1) at an efficiency of 85 ± 10% to the catalyst, which is believed to enter the hydrogen evolution cycle subsequently. Using a combinatorial approach, all ten Co2+ catalysts were evaluated for their potential to operate under neutral pH 7.0 conditions. Catalyst 7, [Co(PY4MeH2)(CH3CN)(CF3SO3)](CF3SO3), was revealed to be most promising, as its performance metrics were only marginally affected by pH and turnover numbers greater than 1000 were easily obtained in photocatalytic hydrogen generation. These comprehensive findings provide guidelines for the development of molecular compositions capable of evolving hydrogen from purely aqueous media.
Co-reporter:Catherine E. McCusker, Delphine Hablot, Raymond Ziessel, and Felix N. Castellano
Inorganic Chemistry 2014 Volume 53(Issue 23) pp:12564-12571
Publication Date(Web):November 13, 2014
DOI:10.1021/ic502169a
The synthesis, structural characterization, and excited-state dynamics of series of diketopyrrolopyrrole (DPP) bridged homodinuclear Ir(III) and heterodinuclear Ir(III)/Pt(II) complexes is described. Steady-state and time-resolved photoluminescence along with transient absorption measurements were used to probe the nature of the emissive and long-lived excited states. Upon excitation into the 1DPP ligand-localized excited state in the presence of coordinated Ir(III) or Pt(II) metal centers, the intersystem crossing is enhanced, leading to a quenching of the 1DPP fluorescence and the formation of the long-lived (τ ≈ 30–40 μs) 3DPP excited state in all instances.
Co-reporter:Fan Deng, Wenfang Sun and Felix N. Castellano
Photochemical & Photobiological Sciences 2014 vol. 13(Issue 5) pp:813-819
Publication Date(Web):20 Mar 2014
DOI:10.1039/C4PP00037D
Near-IR (NIR) absorption from a Cd(II) texaphyrin (TXP) has been successfully coupled with rubrene triplet acceptors/annihilators in vacuum degassed dichloromethane to upconvert NIR (670–800 nm) incident photons into yellow fluorescence through sensitized triplet–triplet annihilation. Stern–Volmer analysis of dynamic energy transfer quenching of TXP by rubrene using transient absorption spectroscopy revealed Stern–Volmer and bimolecular quenching constants of 21000 M−1 and 5.7 × 108 M−1 s−1 respectively, for the triplet–triplet energy transfer process. The upconverted emission intensity with respect to the incident excitation power density at 750 nm was shown to vary between quadratic and linear, illustrating the expected kinetic limits for the light producing photochemistry under continuous wave illumination. Furthermore, with increasing TXP sensitizer concentration, the characteristic quadratic-to-linear crossover point shifted to lower incident photon power density. This is consistent with the notion that stronger photon capture in the sensitizer leads to experimental conditions promoting upconversion under milder excitation conditions. The maximum quantum yield of the TXP-sensitized rubrene upconverted fluorescence was 1.54 ± 0.04% under dilute conditions determined relative to [Os(phen)3](PF6)2 under continuous wave excitation conditions. This saturating quantum efficiency was realized when the incident light power dependence reached the quadratic-to-linear crossover point and was constant over the region where the composition displayed linear response to incident light power density. In pulsed laser experiments at higher sensitizer concentrations, the triplet–triplet annihilation quantum yield was determined to saturate at approximately 13%, corresponding to an upconversion yield of ∼10%, suggesting that the dichloromethane solvent either lowers the T2 state of the rubrene acceptor or is somehow attenuating the annihilation reaction between excited rubrene triplets.
Co-reporter:Timothy W. Schmidt and Felix N. Castellano
The Journal of Physical Chemistry Letters 2014 Volume 5(Issue 22) pp:4062-4072
Publication Date(Web):October 30, 2014
DOI:10.1021/jz501799m
Incoherent photochemical upconversion is a process by which low-energy light can be converted into a higher-energy form with promising applications in solar energy conversion and storage, photocatalysis, biological imaging, and photochemical drug activation. Despite intensive research in recent years, there remains an underappreciation of the chemical kinetics that controls the efficiency of the upconversion process. Here, we provide a brief overview of research into photochemical upconversion and provide a tutorial to guide the design of efficient upconversion compositions. We further provide our perspective on where this area of research is heading and how very efficient systems will be developed.
Co-reporter:Megan S. Lazorski, Felix N. Castellano
Polyhedron 2014 Volume 82() pp:57-70
Publication Date(Web):4 November 2014
DOI:10.1016/j.poly.2014.04.060
The need to develop low-cost, sustainable, earth abundant fuel sources is becoming paramount as the rate of global energy consumption continues to increase. Toward this goal, solar energy conversion is an obvious choice, yet the current molecular based technologies still rely heavily on expensive, non-earth abundant photosensitizers, which limits the net benefits of these systems. Complexes of copper(I) have been recognized for decades as viable low-cost, earth abundant alternative photosensitizers in solar energy conversion technologies; however, when used in solution based applications, issues such as geometrical distortions associated with photoexcitation and ligand lability has frustrated numerous research efforts. Fortunately, these investigations have not been in vain, and many investigations have successfully circumvented the aforementioned issues. Recent reports on Cu(I) based photosensitizers demonstrate that they are beginning to rival the performance metrics of the more costly, less earth abundant species typically used in solution-based solar energy conversion schemes. Therefore, this minireview focuses on the most recent and influential advances made in the field of Cu(I) based photosensitizers.This mini-review focuses on recently conceived Cu(I)-based photosensitizers and the exploitation of their excited state properties in solar energy conversion schemes and related phenomena.
Co-reporter:Catherine E. McCusker, Arnab Chakraborty, and Felix N. Castellano
The Journal of Physical Chemistry A 2014 Volume 118(Issue 45) pp:10391-10399
Publication Date(Web):June 9, 2014
DOI:10.1021/jp503827e
Covalently linking two square planar platinum(II) centers using two pyrazolate bridging ligands allows the filled dz2 orbitals on each Pt center to overlap, producing a Pt–Pt σ interaction and new low energy dσ* → π* metal—metal-to-ligand charge transfer (MMLCT) transitions terminating on an appropriate π-acceptor ligand such as 2-phenylpyridine (ppy). In an effort to extend the lifetime of the associated MMLCT excited state, we decided to append piperidinyl naphthalimide (PNI) chromophores to the 2-phenylpyridine charge transfer ligands. This structural modification introduces low-lying PNI-based triplet states serving as long-lived triplet population reservoirs, thermally capable of repopulating the charge transfer state at room temperature (RT), thereby extending its excited state lifetime. Specifically, [Pt(PNI-ppy)(μ-Ph2pz)]2 (1), where PNI-ppy is N-(2-phenylpyridine)-4-(1-piperidinyl)naphthalene-1,8-dicarboximide and Ph2pz is 3,5-diphenylpyrazolate, was synthesized and structurally characterized. The static and dynamic photophysical behavior of 1 was directly compared to the MMLCT complex [Pt(ppy)(μ-Ph2pz)]2 (2), lacking the PNI substituents, as well as the naked PNI-ppy ligand 3, intended to independently model the MMLCT and NI excited state properties, respectively. Ultimately, experimental evidence for the presence of both the 3PNI and 3MMLCT excited states in 1 were revealed at RT in nanosecond transient absorbance and time-resolved photoluminescence spectroscopy, respectively. Temperature-dependent transient absorption spectroscopy permitted the extraction of an energy gap of 1740 cm–1 between the MMLCT and PNI triplet states in 1 along with the time constants associated with the interconversions between the various excited states resident on this complex chromophore, ultimately decaying back to the ground state with a time constant of 65 μs at RT.
Co-reporter:Jae-Hyuk Kim, Fan Deng, Felix N. Castellano, and Jae-Hong Kim
ACS Photonics 2014 Volume 1(Issue 4) pp:382
Publication Date(Web):March 5, 2014
DOI:10.1021/ph500036m
Microcapsules that achieve multicolor triplet–triplet annihilation (TTA)-based upconversion (UC) in both aqueous and dry phases without deoxygenation are presented for the first time. Platinum(II) tetraphenyltetrabenzoporphyrin (PtTPBP) was used as a sensitizer and perylene, 9,10-bis(phenylethynyl)anthracene (BPEA), and a boron dipyrromethene derivative (BD-2) were employed as acceptors for red to blue, cyan, and green UC, respectively. Additional color tuning was introduced into microcapsules by embedding rose bengal onto the microcapsule shell, resulting in UC-mediated excitation of a distal fluorophore through a trivial process. Microcapsules were further modified to host superparamagnetic nanoparticles for magnetic-induced collection, which permitted sorting and color separation potentially instrumental for various photonics-based applications.Keywords: anti-Stokes emission; energy transfer; microcapsule; microfluidics; upconversion
Co-reporter:Rony S. Khnayzer ; Catherine E. McCusker ; Babatunde S. Olaiya
Journal of the American Chemical Society 2013 Volume 135(Issue 38) pp:14068-14070
Publication Date(Web):September 12, 2013
DOI:10.1021/ja407816f
The Cu(I) metal-to-ligand charge-transfer complex, [Cu(dsbtmp)2]+ (dsbtmp = 2,9-di(sec-butyl)-3,4,7,8-tetramethyl-1,10-phenanthroline), exhibits outstanding stability as a visible-light-absorbing photosensitizer in hydrogen-evolving homogeneous photocatalysis. In concert with the Co(dmgH)2(py)Cl water reduction catalyst and N,N-dimethyl-p-toluidine sacrificial donor in 1:1 H2O:CH3CN, this Cu(I) sensitizer remains active even after 5 days of visible-light-pumped (λex = 452 ± 10 nm) hydrogen evolution catalysis. Deuteration studies illustrate that the hydrogen produced from this composition does indeed originate from aqueous protons.
Co-reporter:Jonah W. Jurss, Rony S. Khnayzer, Julien A. Panetier, Karim A. El Roz, Eva M. Nichols, Martin Head-Gordon, Jeffrey R. Long, Felix N. Castellano and Christopher J. Chang
Chemical Science (2010-Present) 2015 - vol. 6(Issue 8) pp:NaN4972-4972
Publication Date(Web):2015/06/09
DOI:10.1039/C5SC01414J
Mononuclear metalloenzymes in nature can function in cooperation with precisely positioned redox-active organic cofactors in order to carry out multielectron catalysis. Inspired by the finely tuned redox management of these bioinorganic systems, we present the design, synthesis, and experimental and theoretical characterization of a homologous series of cobalt complexes bearing redox-active pyrazines. These donor moieties are locked into key positions within a pentadentate ligand scaffold in order to evaluate the effects of positioning redox non-innocent ligands on hydrogen evolution catalysis. Both metal- and ligand-centered redox features are observed in organic as well as aqueous solutions over a range of pH values, and comparison with analogs bearing redox-inactive zinc(II) allows for assignments of ligand-based redox events. Varying the geometric placement of redox non-innocent pyrazine donors on isostructural pentadentate ligand platforms results in marked effects on observed cobalt-catalyzed proton reduction activity. Electrocatalytic hydrogen evolution from weak acids in acetonitrile solution, under diffusion-limited conditions, reveals that the pyrazine donor of axial isomer 1-Co behaves as an unproductive electron sink, resulting in high overpotentials for proton reduction, whereas the equatorial pyrazine isomer complex 2-Co is significantly more active for hydrogen generation at lower voltages. Addition of a second equatorial pyrazine in complex 3-Co further minimizes overpotentials required for catalysis. The equatorial derivative 2-Co is also superior to its axial 1-Co congener for electrocatalytic and visible-light photocatalytic hydrogen generation in biologically relevant, neutral pH aqueous media. Density functional theory calculations (B3LYP-D2) indicate that the first reduction of catalyst isomers 1-Co, 2-Co, and 3-Co is largely metal-centered while the second reduction occurs at pyrazine. Taken together, the data establish that proper positioning of non-innocent pyrazine ligands on a single cobalt center is indeed critical for promoting efficient hydrogen catalysis in aqueous media, akin to optimally positioned redox-active cofactors in metalloenzymes. In a broader sense, these findings highlight the significance of electronic structure considerations in the design of effective electron–hole reservoirs for multielectron transformations.
Co-reporter:Felix N. Castellano and Catherine E. McCusker
Dalton Transactions 2015 - vol. 44(Issue 41) pp:NaN17910-17910
Publication Date(Web):2015/09/22
DOI:10.1039/C5DT03212A
This treatment highlights the historical development of MLCT sensitizers in photochemical upconversion while indentifying current state-of-the-art and exciting opportunities in this arena moving towards the future.
Co-reporter:Michelle M. McGoorty, Rony S. Khnayzer and Felix N. Castellano
Chemical Communications 2016 - vol. 52(Issue 50) pp:NaN7849-7849
Publication Date(Web):2016/05/24
DOI:10.1039/C6CC03932D
Two water-soluble anionic cyclometalated Ir(III) complexes, Ir(ppy)2BPS [1] and Ir(F-mppy)2BPS [2] have been synthesized and display clear evidence of self-assembly in water. Concentration-induced aggregation enhances the excited-state properties of both complexes, blue-shifting the photoluminescence emission energies as well as increasing the corresponding excited state lifetimes and quantum yields up to a factor of 5.
Co-reporter:Sofia Garakyaraghi, Petr Koutnik and Felix N. Castellano
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 25) pp:NaN16668-16668
Publication Date(Web):2017/06/12
DOI:10.1039/C7CP03343E
Copper(I) phenanthroline complexes represent viable earth-abundant alternatives to the ubiquitous Ru(II) tris-bipyridine photosensitizers owing to their similar metal-to-ligand charge transfer (MLCT) properties. A well-established complication of Cu(I) phenanthroline complexes is that they can undergo significant photo-induced structural rearrangements, leading to excited states that are highly susceptible to exciplex formation and short-lived. In this work, a comprehensive analysis of the photo-induced structural distortions and singlet–triplet intersystem crossing dynamics of a series of four sterically encumbered Cu(I) phenanthroline chromophores has been conducted, namely, [Cu(dsbp)2]+ (dsbp = 2,9-di-sec-butyl-1,10-phenanthroline), [Cu(dsbtmp)2]+ (dsbtmp = 2,9-di-sec-butyl-3,4,7,8-tetramethyl-1,10-phenanthroline), [Cu(dipp)2]+ (dipp = 2,9-di-isopropyl-1,10-phenanthroline), and [Cu(diptmp)2]+ (diptmp = 2,9-di-isopropyl-3,4,7,8-tetramethyl-1,10-phenanthroline). Upconverted fluorescence decay kinetics were measured at wavelengths along the blue side of the photoluminescence spectrum. The experimental results displayed strong wavelength dependence of the singlet emission, with rapid sub-picosecond decay dominating at higher energies. At lower emission energies, increasing contribution of a longer decay component was revealed. This wavelength dependence is a signature of the excited state structural rearrangement of the phenanthroline ligands which concomitantly lower the excited state energy. The obtained time constants were in excellent agreement with those measured in the complementary ultrafast transient absorption experiments. The sub-picosecond component (prompt fluorescence) is associated with the photo-induced structural rearrangement that lowers the energy of the singlet excited state. The longer decay component represents the lifetime of the S1 excited state, and thus the time-scale of singlet–triplet intersystem crossing. Lastly, the observed dual emission was further characterized by constructing picosecond time-resolved emission spectra from the measured kinetic data. These qualitative luminescence spectra capture the resulting emission from both the S1 initial state and the S1 flattened state, providing further insight into the energy-lowering excited state distortion across the series.
.jpg)
![1H-Benz[de]isoquinoline-1,3(2H)-dione, 2-(1,10-phenanthrolin-5-yl)-6-(1-piperidinyl)-](/data/chemimg/133600/357264-48-3.png)
![1H-Benz[de]isoquinoline-1,3(2H)-dione, 2-(1,10-phenanthrolin-5-yl)-6-(1-piperidinyl)-](/data/chemimg/133600/357264-48-3_b.png)
![Platinum, [6,13,20,27-tetraphenyl-29H,31H-tetrabenzo[b,g,l,q]porphinato(2-)-κN29,κN30,κN31,κN32]-, (SP-4-1)-](/data/chemimg/107100/166174-05-6.png)
![Platinum, [6,13,20,27-tetraphenyl-29H,31H-tetrabenzo[b,g,l,q]porphinato(2-)-κN29,κN30,κN31,κN32]-, (SP-4-1)-](/data/chemimg/107100/166174-05-6_b.png)






![Benzene, 1,1',1'',1'''-methanetetrayltetrakis[4-[2-(trimethylsilyl)ethynyl]-](/data/chemimg/137100/177991-00-3.png)
![Benzene, 1,1',1'',1'''-methanetetrayltetrakis[4-[2-(trimethylsilyl)ethynyl]-](/data/chemimg/137100/177991-00-3_b.png)
![Tricyclo[3.3.1.13,7]decane, 1,3,5,7-tetrakis(4-ethynylphenyl)-](/data/chemimg/2941300/144970-32-1.png)
![Tricyclo[3.3.1.13,7]decane, 1,3,5,7-tetrakis(4-ethynylphenyl)-](/data/chemimg/2941300/144970-32-1_b.png)
![Tricyclo[3.3.1.13,7]decane, 1,3,5,7-tetrakis(4-iodophenyl)-](/data/chemimg/102800/144970-30-9.png)
![Tricyclo[3.3.1.13,7]decane, 1,3,5,7-tetrakis(4-iodophenyl)-](/data/chemimg/102800/144970-30-9_b.png)
![2,9-Di(tridecan-7-yl)anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetraone](/data/chemimg/339200/110590-84-6.png)
![2,9-Di(tridecan-7-yl)anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetraone](/data/chemimg/339200/110590-84-6_b.png)



![Tricyclo[3.3.1.13,7]decane, 1,3,5,7-tetraphenyl-](/data/chemimg/530700/16004-75-4.png)
![Tricyclo[3.3.1.13,7]decane, 1,3,5,7-tetraphenyl-](/data/chemimg/530700/16004-75-4_b.png)