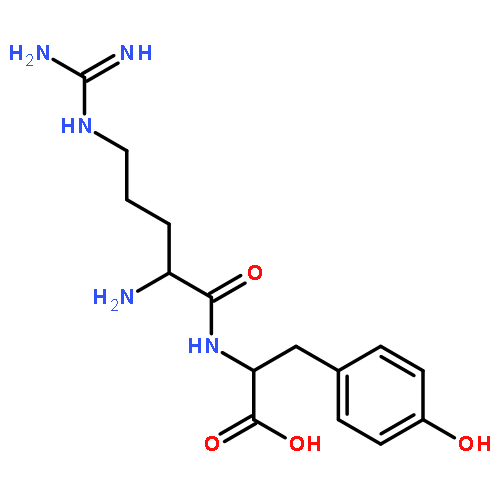
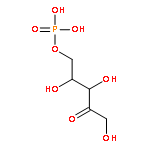
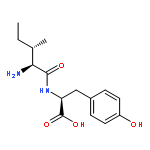
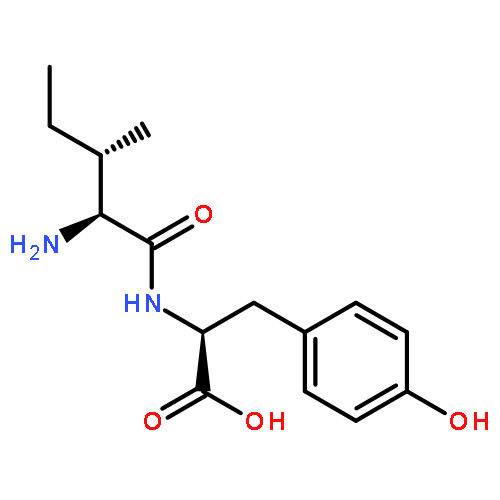
Co-reporter:Xinshuai Zhang;Michael S. Carter;Matthew W. Vetting;Suwen Zhao;Nawar F. Al-Obaidi;Brian San Francisco;Jose O. Solbiati;Jennifer J. Thiaville;Valérie de Crécy-Lagard;Matthew P. Jacobson;Steven C. Almo
PNAS 2016 Volume 113 (Issue 29 ) pp:E4161-E4169
Publication Date(Web):2016-07-19
DOI:10.1073/pnas.1605546113
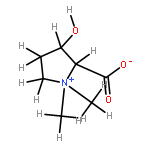
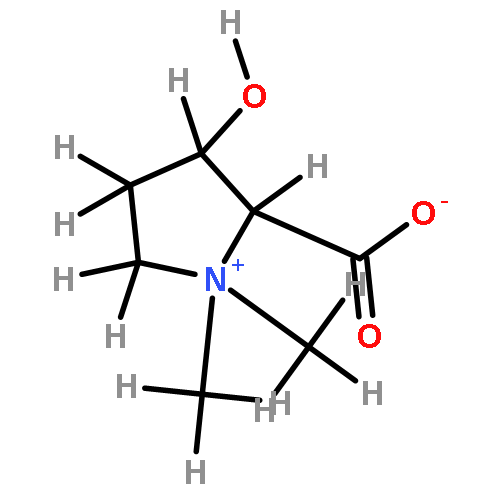
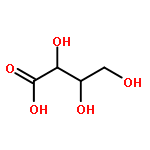
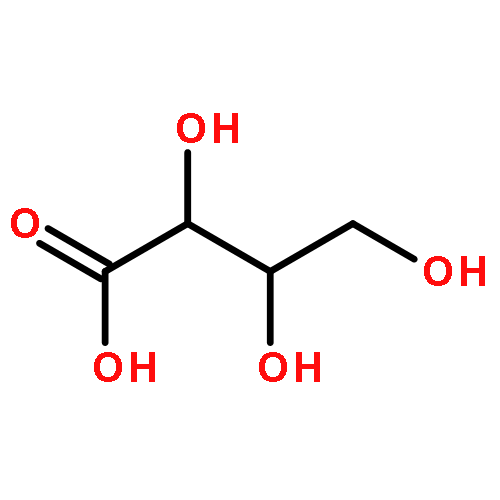
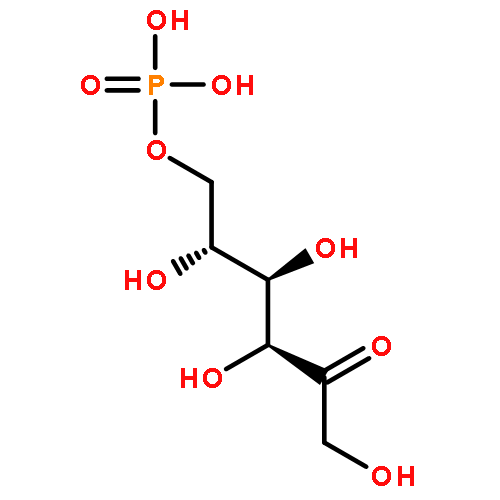
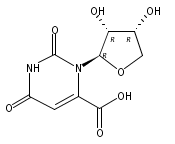
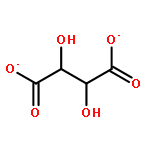
Using a large-scale “genomic enzymology” approach, we (i) assigned novel ATP-dependent four-carbon acid sugar kinase functions to members of the DUF1537 protein family (domain of
unknown function; Pfam families PF07005 and PF17042) and (ii) discovered novel catabolic pathways for d-threonate, l-threonate, and d-erythronate. The experimentally determined ligand specificities of several solute binding proteins (SBPs) for TRAP (tripartite
ATP-independent permease) transporters for four-carbon acids, including d-erythronate and l-erythronate, were used to constrain the substrates for the catabolic pathways that degrade the SBP ligands to intermediates
in central carbon metabolism. Sequence similarity networks and genome neighborhood networks were used to identify the enzyme
components of the pathways. Conserved genome neighborhoods encoded SBPs as well as permease components of the TRAP transporters,
members of the DUF1537 family, and a member of the 4-hydroxy-l-threonine 4-phosphate dehydrogenase (PdxA) oxidative decarboxylase, class II aldolase, or ribulose 1,5-bisphosphate carboxylase/oxygenase,
large subunit (RuBisCO) superfamily. Because the characterized substrates of members of the PdxA, class II aldolase, and RuBisCO
superfamilies are phosphorylated, we postulated that the members of the DUF1537 family are novel ATP-dependent kinases that
participate in catabolic pathways for four-carbon acid sugars. We determined that (i) the DUF1537/PdxA pair participates in a pathway for the conversion of d-threonate to dihydroxyacetone phosphate and CO2 and (ii) the DUF1537/class II aldolase pair participates in pathways for the conversion of d-erythronate and l-threonate (epimers at carbon-3) to dihydroxyacetone phosphate and CO2. The physiological importance of these pathways was demonstrated in vivo by phenotypic and genetic analyses.
Co-reporter:Xinshuai Zhang; Ritesh Kumar; Matthew W. Vetting; Suwen Zhao; Matthew P. Jacobson; Steven C. Almo
Journal of the American Chemical Society 2015 Volume 137(Issue 4) pp:1388-1391
Publication Date(Web):January 21, 2015
DOI:10.1021/ja5103986
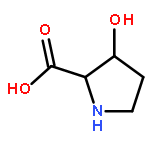
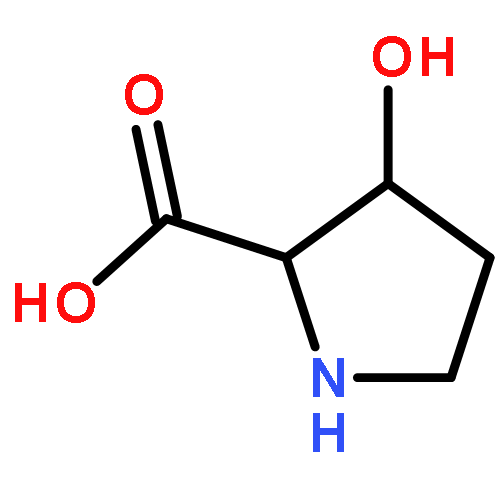
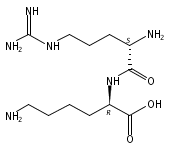
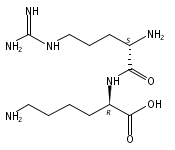
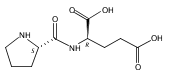
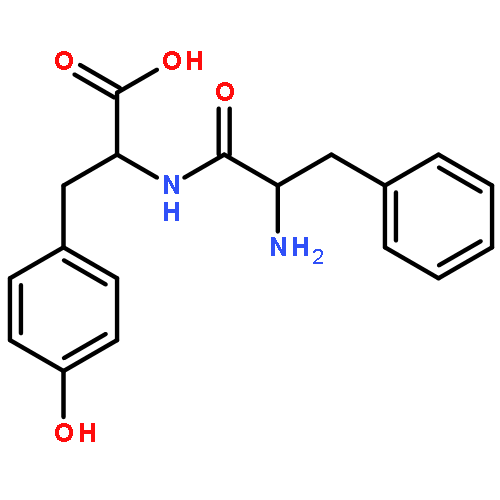
The genome of Labrenzia aggregata IAM 12614 encodes an uncharacterized member of the muconate lactonizing enzyme (MLE) subgroup of the enolase superfamily (UniProt ID A0NXQ8). The gene encoding A0NXQ8 is located between genes that encode members of the proline racemase superfamily, 4R-hydroxyproline 2-epimerase (UniProt ID A0NXQ7; 4HypE) and trans-3-hydroxy-l-proline dehydratase (UniProt ID A0NXQ9; t3LHypD). A0NXQ8 was screened with a library of proline analogues; two reactions were observed with cis-3-hydroxy-l-proline (c3LHyp), competing 2-epimerization to trans-3-hydroxy-d-proline (1,1-proton transfer) and dehydration to Δ1-pyrroline-2-carboxylate (β-elimination; c3LHyp dehydratase), with eventual total dehydration. The genome context encoding A0NXQ8 both (1) confirms its novel c3LHyp dehydratase function and (2) provides evidence for metabolic pathways that allow L. aggregata to utilize several isomeric 3- and 4-hydroxyprolines as sole carbon sources.
Co-reporter:Matthew W. Vetting, Nawar Al-Obaidi, Suwen Zhao, Brian San Francisco, Jungwook Kim, Daniel J. Wichelecki, Jason T. Bouvier, Jose O. Solbiati, Hoan Vu, Xinshuai Zhang, Dmitry A. Rodionov, James D. Love, Brandan S. Hillerich, Ronald D. Seidel, Ronald J. Quinn, Andrei L. Osterman, John E. Cronan, Matthew P. Jacobson, John A. Gerlt, and Steven C. Almo
Biochemistry 2015 Volume 54(Issue 3) pp:909-931
Publication Date(Web):December 25, 2014
DOI:10.1021/bi501388y
The rate at which genome sequencing data is accruing demands enhanced methods for functional annotation and metabolism discovery. Solute binding proteins (SBPs) facilitate the transport of the first reactant in a metabolic pathway, thereby constraining the regions of chemical space and the chemistries that must be considered for pathway reconstruction. We describe high-throughput protein production and differential scanning fluorimetry platforms, which enabled the screening of 158 SBPs against a 189 component library specifically tailored for this class of proteins. Like all screening efforts, this approach is limited by the practical constraints imposed by construction of the library, i.e., we can study only those metabolites that are known to exist and which can be made in sufficient quantities for experimentation. To move beyond these inherent limitations, we illustrate the promise of crystallographic- and mass spectrometric-based approaches for the unbiased use of entire metabolomes as screening libraries. Together, our approaches identified 40 new SBP ligands, generated experiment-based annotations for 2084 SBPs in 71 isofunctional clusters, and defined numerous metabolic pathways, including novel catabolic pathways for the utilization of ethanolamine as sole nitrogen source and the use of d-Ala-d-Ala as sole carbon source. These efforts begin to define an integrated strategy for realizing the full value of amassing genome sequence data.
Co-reporter:John A. Gerlt, Jason T. Bouvier, Daniel B. Davidson, Heidi J. Imker, Boris Sadkhin, David R. Slater, Katie L. Whalen
Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2015 Volume 1854(Issue 8) pp:1019-1037
Publication Date(Web):August 2015
DOI:10.1016/j.bbapap.2015.04.015
•Sequence–function space can be visualized using protein sequence similarity networks.•The EFI-EST webtool is available for generating sequence similarity networks.•A tutorial is provided that describes the use of EFI-EST.•The community is encouraged to use EFI-EST without cost.The Enzyme Function Initiative, an NIH/NIGMS-supported Large-Scale Collaborative Project (EFI; U54GM093342; http://enzymefunction.org/), is focused on devising and disseminating bioinformatics and computational tools as well as experimental strategies for the prediction and assignment of functions (in vitro activities and in vivo physiological/metabolic roles) to uncharacterized enzymes discovered in genome projects. Protein sequence similarity networks (SSNs) are visually powerful tools for analyzing sequence relationships in protein families (H.J. Atkinson, J.H. Morris, T.E. Ferrin, and P.C. Babbitt, PLoS One 2009, 4, e4345). However, the members of the biological/biomedical community have not had access to the capability to generate SSNs for their “favorite” protein families. In this article we announce the EFI-EST (Enzyme Function Initiative-Enzyme Similarity Tool) web tool (http://efi.igb.illinois.edu/efi-est/) that is available without cost for the automated generation of SSNs by the community. The tool can create SSNs for the “closest neighbors” of a user-supplied protein sequence from the UniProt database (Option A) or of members of any user-supplied Pfam and/or InterPro family (Option B). We provide an introduction to SSNs, a description of EFI-EST, and a demonstration of the use of EFI-EST to explore sequence–function space in the OMP decarboxylase superfamily (PF00215). This article is designed as a tutorial that will allow members of the community to use the EFI-EST web tool for exploring sequence/function space in protein families.
Co-reporter:Fiona P. Groninger-Poe, Jason T. Bouvier, Matthew W. Vetting, Chakrapani Kalyanaraman, Ritesh Kumar, Steven C. Almo, Matthew P. Jacobson, and John A. Gerlt
Biochemistry 2014 Volume 53(Issue 25) pp:4192-4203
Publication Date(Web):June 13, 2014
DOI:10.1021/bi5005377
The genome of Agrobacterium tumefaciens C58 encodes 12 members of the enolase superfamily (ENS), eight of which are members of the mandelate racemase (MR) subgroup and, therefore, likely to be acid sugar dehydratases. Using a library of 77 acid sugars for high-throughput screening, one protein (UniProt entry A9CG74; locus tag Atu4196) showed activity with both m-galactarate and d-galacturonate. Two families of galactarate dehydratases had been discovered previously in the ENS, GalrD/TalrD [Yew, W. S., et al. (2007) Biochemistry 46, 9564–9577] and GalrD-II [Rakus, J. F., et al. (2009) Biochemistry 48, 11546–11558]; these have different active site acid/base catalysis and have no activity with d-galacturonate. A9CG74 dehydrates m-galactarate to form 2-keto-3-deoxy-galactarate but does not dehydrate d-galacturonate as expected. Instead, when A9CG74 is incubated with d-galacturonate, 3-deoxy-d-xylo-hexarate or 3-deoxy-d-lyxo-hexarate is formed. In this reaction, instead of abstracting the C5 proton α to the carboxylate group, the expected reaction for a member of the ENS, the enzyme apparently abstracts the proton α to the aldehyde group to form 3-deoxy-d-threo-hexulosuronate that undergoes a 1,2-hydride shift similar to the benzylic acid rearrangement to form the observed product. A. tumefaciens C58 does not utilize m-galactarate as a carbon source under the conditions tested in this study, although it does utilize d-galacturonate, which is a likely precursor to m-galactarate. The gene encoding A9CG74 and several genome proximal genes were upregulated with d-galacturonate as the carbon source. One of these, a member of the dihydrodipicolinate synthase superfamily, catalyzes the dehydration and subsequent decarboxylation of 2-keto-3-deoxy-d-galactarate to α-ketoglutarate semialdehyde, thereby providing a pathway for the conversion of m-galactarate to α-ketoglutarate semialdehyde.
Co-reporter:Daniel J. Wichelecki, D. Sean Froese, Jolanta Kopec, Joao R.C. Muniz, Wyatt W. Yue, and John A. Gerlt
Biochemistry 2014 Volume 53(Issue 16) pp:
Publication Date(Web):April 3, 2014
DOI:10.1021/bi500349e
In humans, the gene encoding a reverse thymidylate synthase (rTS) is transcribed in the reverse direction of the gene encoding thymidylate synthase (TS) that is involved in DNA biosynthesis. Three isoforms are found: α, β, and γ, with the transcript of the α-isoform overlapping with that of TS. rTSβ has been of interest since the discovery of its overexpression in methotrexate and 5-fluorouracil resistant cell lines. Despite more than 20 years of study, none of the rTS isoforms have been biochemically or structurally characterized. In this study, we identified rTSγ as an l-fuconate dehydratase and determined its high-resolution crystal structure. Our data provide an explanation for the observed difference in enzymatic activities between rTSβ and rTSγ, enabling more informed proposals for the possible function of rTSβ in chemotherapeutic resistance.
Co-reporter:Daniel J. Wichelecki, Bryan M. Balthazor, Anthony C. Chau, Matthew W. Vetting, Alexander A. Fedorov, Elena V. Fedorov, Tiit Lukk, Yury V. Patskovsky, Mark B. Stead, Brandan S. Hillerich, Ronald D. Seidel, Steven C. Almo, and John A. Gerlt
Biochemistry 2014 Volume 53(Issue 16) pp:
Publication Date(Web):April 4, 2014
DOI:10.1021/bi500264p
The continued increase in the size of the protein sequence databases as a result of advances in genome sequencing technology is overwhelming the ability to perform experimental characterization of function. Consequently, functions are assigned to the vast majority of proteins via automated, homology-based methods, with the result that as many as 50% are incorrectly annotated or unannotated (Schnoes et al. PLoS Comput. Biol. 2009, 5 (12), e1000605). This manuscript describes a study of the d-mannonate dehydratase (ManD) subgroup of the enolase superfamily (ENS) to investigate how function diverges as sequence diverges. Previously, one member of the subgroup had been experimentally characterized as ManD [dehydration of d-mannonate to 2-keto-3-deoxy-d-mannonate (equivalently, 2-keto-3-deoxy-d-gluconate)]. In this study, 42 additional members were characterized to sample sequence–function space in the ManD subgroup. These were found to differ in both catalytic efficiency and substrate specificity: (1) high efficiency (kcat/KM = 103 to 104 M–1 s–1) for dehydration of d-mannonate, (2) low efficiency (kcat/KM = 101 to 102 M–1 s–1) for dehydration of d-mannonate and/or D-gluconate, and 3) no-activity with either d-mannonate or d-gluconate (or any other acid sugar tested). Thus, the ManD subgroup is not isofunctional and includes d-gluconate dehydratases (GlcDs) that are divergent from the GlcDs that have been characterized in the mandelate racemase subgroup of the ENS (Lamble et al. FEBS Lett. 2004, 576, 133–136) (Ahmed et al. Biochem. J. 2005, 390, 529–540). These observations signal caution for functional assignment based on sequence homology and lay the foundation for the studies of the physiological functions of the GlcDs and the promiscuous ManDs/GlcDs.
Co-reporter:Jason T. Bouvier, Fiona P. Groninger-Poe, Matthew Vetting, Steven C. Almo, and John A. Gerlt
Biochemistry 2014 Volume 53(Issue 4) pp:614-616
Publication Date(Web):January 23, 2014
DOI:10.1021/bi5000492
Agrobacterium tumefaciens strain C58 can utilize d-galacturonate as a sole source of carbon via a pathway in which the first step is oxidation of d-galacturonate to d-galactaro-1,5-lactone. We have identified a novel enzyme, d-galactarolactone isomerase (GLI), that catalyzes the isomerizaton of d-galactaro-1,5-lactone to d-galactaro-1,4-lactone. GLI, a member of the functionally diverse amidohydrolase superfamily, is a homologue of LigI that catalyzes the hydrolysis of 2-pyrone-4,6-dicarboxylate in lignin degradation. The ability of GLI to catalyze lactone isomerization instead of hydrolysis can be explained by the absence of the general basic catalysis used by 2-pyrone-4,6-dicarboxylate lactonase.
Co-reporter:Daniel J. Wichelecki, Dylan C. Graff, Nawar Al-Obaidi, Steven C. Almo, and John A. Gerlt
Biochemistry 2014 Volume 53(Issue 25) pp:4087-4089
Publication Date(Web):June 20, 2014
DOI:10.1021/bi500683x
The d-mannonate dehydratase (ManD) subgroup of the enolase superfamily contains members with varying catalytic activities (high-efficiency, low-efficiency, or no activity) that dehydrate d-mannonate and/or d-gluconate to 2-keto-3-deoxy-d-gluconate [Wichelecki, D. J., et al. (2014) Biochemistry 53, 2722–2731]. Despite extensive in vitro characterization, the in vivo physiological role of a ManD has yet to be established. In this study, we report the in vivo functional characterization of a high-efficiency ManD from Caulobacter crescentus NA1000 (UniProt entry B8GZZ7) by in vivo discovery of its essential role in d-glucuronate metabolism. This in vivo functional annotation may be extended to ∼50 additional proteins.
Co-reporter:Daniel J. Wichelecki, Jean Alyxa Ferolin Vendiola, Amy M. Jones, Nawar Al-Obaidi, Steven C. Almo, and John A. Gerlt
Biochemistry 2014 Volume 53(Issue 35) pp:
Publication Date(Web):August 22, 2014
DOI:10.1021/bi500837w
The sequence/function space in the d-mannonate dehydratase subgroup (ManD) of the enolase superfamily was investigated to determine how enzymatic function diverges as sequence identity decreases [Wichelecki, D. J., et al. (2014) Biochemistry 53, 2722–2731]. That study revealed that members of the ManD subgroup vary in substrate specificity and catalytic efficiency: high-efficiency (kcat/KM = 103–104 M–1 s–1) for dehydration of d-mannonate, low-efficiency (kcat/KM = 10–102 M–1 s–1) for dehydration of d-mannonate and/or d-gluconate, and no activity. Characterization of high-efficiency members revealed that these are ManDs in the d-glucuronate catabolic pathway {analogues of UxuA [Wichelecki, D. J., et al. (2014) Biochemistry 53, 4087–4089]}. However, the genomes of organisms that encode low-efficiency members of the ManDs subgroup encode UxuAs; therefore, these must have divergent physiological functions. In this study, we investigated the physiological functions of three low-efficiency members of the ManD subgroup and identified a novel physiologically relevant pathway for l-gulonate catabolism in Chromohalobacter salexigens DSM3043 as well as cryptic pathways for l-gulonate catabolism in Escherichia coli CFT073 and l-idonate catabolism in Salmonella enterica subsp. enterica serovar Enteritidis str. P125109. However, we could not identify physiological roles for the low-efficiency members of the ManD subgroup, allowing the suggestion that these pathways may be either evolutionary relics or the starting points for new metabolic potential.
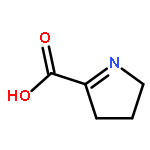
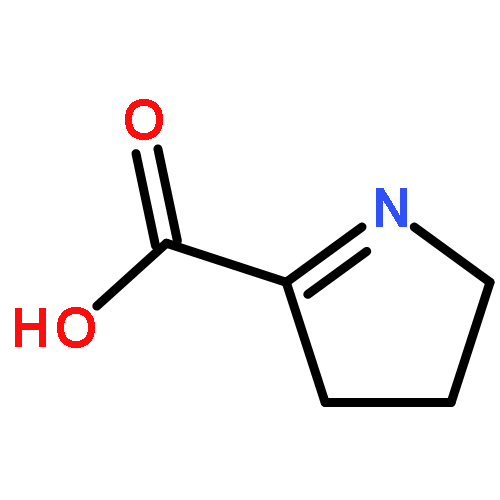
Co-reporter:Salehe Ghasempur, Subramaniam Eswaramoorthy, Brandan S. Hillerich, Ronald D. Seidel, Subramanyam Swaminathan, Steven C. Almo, and John A. Gerlt
Biochemistry 2014 53(20) pp: 3357-3366
Publication Date(Web):May 6, 2014
DOI:10.1021/bi5004298
The l-lyxonate dehydratase (LyxD) in vitro enzymatic activity and in vivo metabolic function were assigned to members of an isofunctional family within the mandelate racemase (MR) subgroup of the enolase superfamily. This study combined in vitro and in vivo data to confirm that the dehydration of l-lyxonate is the biological role of the members of this family. In vitro kinetic experiments revealed catalytic efficiencies of ∼104 M–1 s–1 as previously observed for members of other families in the MR subgroup. Growth studies revealed that l-lyxonate is a carbon source for Pseudomonas aeruginosa PAO1; transcriptomics using qRT-PCR established that the gene encoding LyxD as well as several other conserved proximal genes were upregulated in cells grown on l-lyxonate. The proximal genes were shown to be involved in a pathway for the degradation of l-lyxonate, in which the first step is dehydration by LyxD followed by dehydration of the 2-keto-3-deoxy-l-lyxonate product by 2-keto-3-deoxy-l-lyxonate dehydratase to yield α-ketoglutarate semialdehyde. In the final step, α-ketoglutarate semialdehyde is oxidized by a dehydrogenase to α-ketoglutarate, an intermediate in the citric acid cycle. An X-ray structure for the LyxD from Labrenzia aggregata IAM 12614 with Mg2+ in the active site was determined that confirmed the expectation based on sequence alignments that LyxDs possess a conserved catalytic His-Asp dyad at the end of seventh and sixth β-strands of the (β/α)7β-barrel domain as well as a conserved KxR motif at the end of second β-strand; substitutions for His 316 or Arg 179 inactivated the enzyme. This is the first example of both the LyxD function in the enolase superfamily and a pathway for the catabolism of l-lyxonate.
Co-reporter:Benjamin P. Warlick, Bradley S. Evans, Tobias J. Erb, Udipi A. Ramagopal, Jaya Sriram, Heidi J. Imker, J. Michael Sauder, Jeffrey B. Bonanno, Stephen K. Burley, F. Robert Tabita, Steven C. Almo, Jonathan S. Sweedler, and John A. Gerlt
Biochemistry 2012 Volume 51(Issue 42) pp:
Publication Date(Web):October 4, 2012
DOI:10.1021/bi301215g
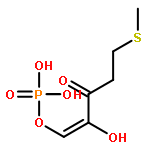
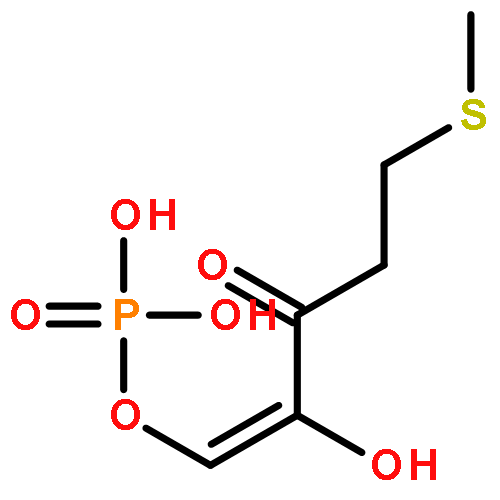
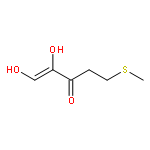
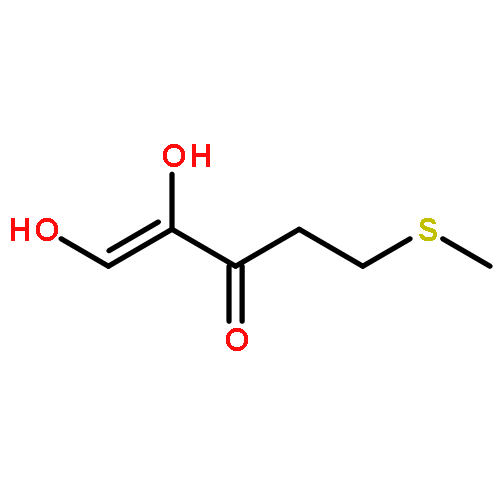
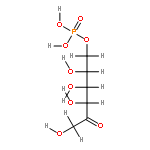
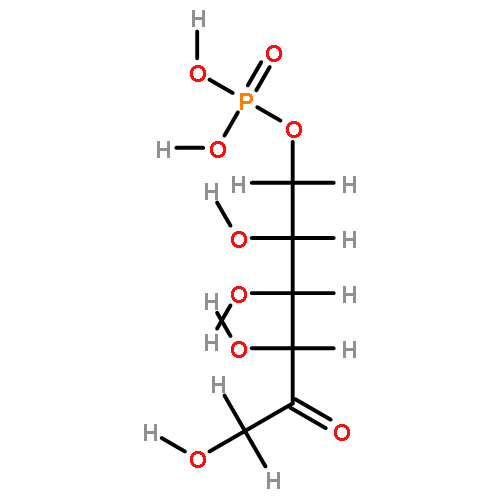
Rhodospirillum rubrum produces 5-methylthioadenosine (MTA) from S-adenosylmethionine in polyamine biosynthesis; however, R. rubrum lacks the classical methionine salvage pathway. Instead, MTA is converted to 5-methylthio-d-ribose 1-phosphate (MTR 1-P) and adenine; MTR 1-P is isomerized to 1-methylthio-d-xylulose 5-phosphate (MTXu 5-P) and reductively dethiomethylated to 1-deoxy-d-xylulose 5-phosphate (DXP), an intermediate in the nonmevalonate isoprenoid pathway [Erb, T. J., et al. (2012) Nat. Chem. Biol., in press]. Dethiomethylation, a novel route to DXP, is catalyzed by MTXu 5-P methylsulfurylase. An active site Cys displaces the enolate of DXP from MTXu 5-P, generating a methyl disulfide intermediate.
Co-reporter:Bogdana Goryanova ; Tina L. Amyes ; John A. Gerlt ;John P. Richard
Journal of the American Chemical Society 2011 Volume 133(Issue 17) pp:6545-6548
Publication Date(Web):April 12, 2011
DOI:10.1021/ja201734z
Orotidine 5′-monophosphate decarboxylase (OMPDC) catalyzes the exchange for deuterium from solvent D2O of the C-6 proton of 1-(β-d-erythrofuranosyl)-5-fluorouracil (FEU), a phosphodianion truncated product analog. The deuterium exchange reaction of FEU is accelerated 1.8 × 104-fold by 1 M phosphite dianion (HPO32−). This corresponds to a 5.8 kcal/mol stabilization of the vinyl carbanion-like transition state, which is similar to the 7.8 kcal/mol stabilization of the transition state for OMPDC-catalyzed decarboxylation of a truncated substrate analog by bound HPO32−. These results show that the intrinsic binding energy of phosphite dianion is used in the stabilization of the vinyl carbanion-like transition state common to the decarboxylation and deuterium exchange reactions.
Co-reporter:John A. Gerlt
Perspectives in Science (December 2016) Volume 9() pp:24-32
Publication Date(Web):1 December 2016
DOI:10.1016/j.pisc.2016.07.001
The number of entries in the sequence databases continues to increase exponentially – the UniProt database is increasing with a doubling time of ∼4 years (2% increase/month). Approximately 50% of the entries have uncertain, unknown, or incorrect function annotations because these are made by automated methods based on sequence homology. If the potential in complete genome sequences is to be realized, strategies and tools must be developed to facilitate experimental assignment of functions to uncharacterized proteins discovered in genome projects. The Enzyme Function Initiative (EFI; previously supported by U54GM093342 from the National Institutes of Health, now supported by P01GM118303) developed web tools for visualizing and analyzing (1) sequence and function space in protein families (EFI-EST) and (2) genome neighbourhoods in microbial and fungal genomes (EFI-GNT) to assist the design of experimental strategies for discovering the in vitro activities and in vivo metabolic functions of uncharacterized enzymes. The EFI developed an experimental platform for large-scale production of the solute binding proteins (SBPs) for ABC, TRAP, and TCT transport systems and their screening with a physical ligand library to identify the identities of the ligands for these transport systems. Because the genes that encode transport systems are often co-located with the genes that encode the catabolic pathways for the transported solutes, the identity of the SBP ligand together with the EFI-EST and EFI-GNT web tools can be used to discover new enzyme functions and new metabolic pathways. This approach is demonstrated with the characterization of a novel pathway for ethanolamine catabolism.
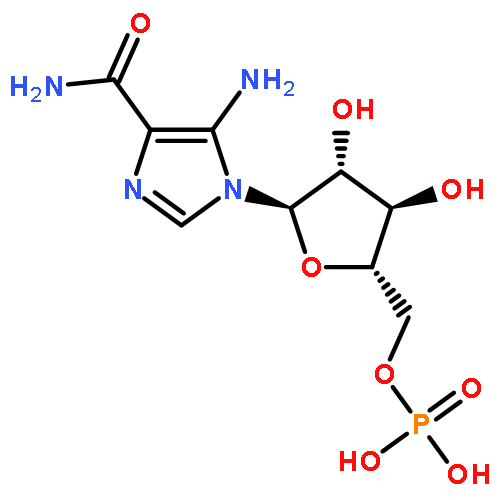
Co-reporter:Hua Huang; Michael S. Carter; Matthew W. Vetting; Nawar Al-Obaidi; Yury Patskovsky; Steven C. Almo
Journal of the American Chemical Society () pp:
Publication Date(Web):November 11, 2015
DOI:10.1021/jacs.5b08968
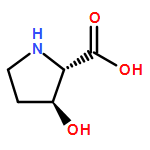
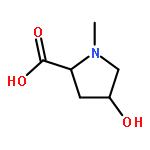
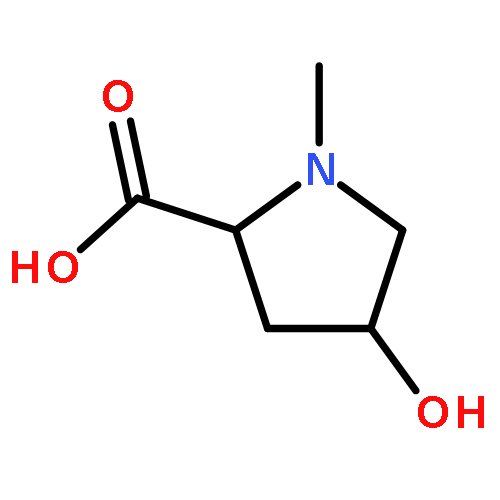
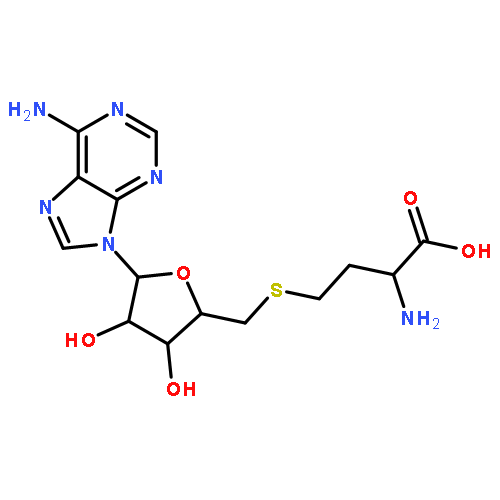
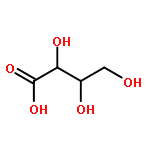
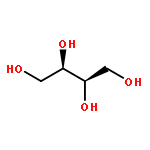
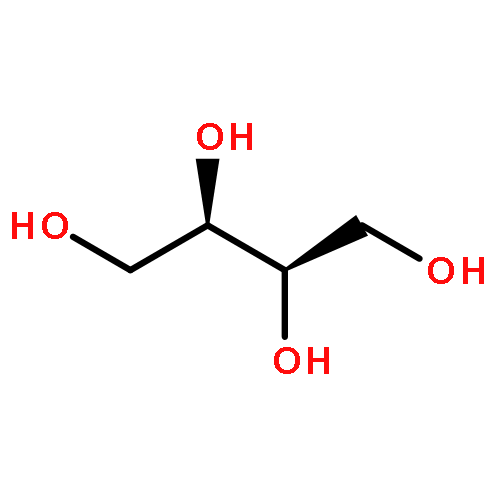
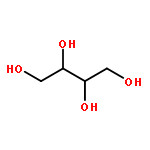
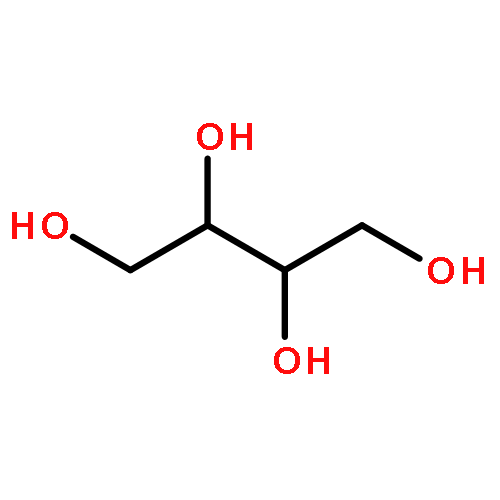
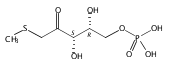
We describe a general integrated bioinformatic and experimental strategy to discover the in vitro enzymatic activities and in vivo functions (metabolic pathways) of uncharacterized enzymes discovered in microbial genome projects using the ligand specificities of the solute binding proteins (SBPs) for ABC transporters. Using differential scanning fluorimetry, we determined that the SBP for an ABC transporter encoded by the genome of Mycobacterium smegmatis is stabilized by d-threitol. Using sequence similarity networks and genome neighborhood networks to guide selection of target proteins for pathway enzymes, we applied both in vitro and in vivo experimental approaches to discover novel pathways for catabolism of d-threitol, l-threitol, and erythritol.
.png)