Co-reporter:Patrick J. Kelleher, Fabian S. Menges, Joseph W. DePalma, Joanna K. Denton, and Mark A. Johnson, Gary H. Weddle , Barak Hirshberg and R. Benny Gerber
The Journal of Physical Chemistry Letters October 5, 2017 Volume 8(Issue 19) pp:4710-4710
Publication Date(Web):September 12, 2017
DOI:10.1021/acs.jpclett.7b02120
The heterogeneous reaction of N2O5 with sea spray aerosols yields the ClNO2 molecule, which is postulated to occur through water-mediated charge separation into NO3– and NO2+ ions followed by association with Cl–. Here we address an alternative mechanism where the attack by a halide ion can yield XNO2 by direct insertion in the presence of water. This was accomplished by reacting X–(D2O)n (X = Cl, Br, I) cluster ions with N2O5 to produce ions with stoichiometry [XN2O5]−. These species were cooled in a 20 K ion trap and structurally characterized by vibrational spectroscopy using the D2 messenger tagging technique. Analysis of the resulting band patterns with DFT calculations indicates that they all correspond to exit channel ion–molecule complexes based on the association of NO3– with XNO2, with the NO3– constituent increasingly perturbed in the order I > Br > Cl. These results establish that XNO2 can be generated even when more exoergic reaction pathways involving hydrolysis are available and demonstrate the role of the intermediate [XN2O5]− in the formation of XNO2.
Co-reporter:Musleh U. Munshi, Stephanie M. Craig, Giel Berden, Jonathan Martens, Andrew F. DeBlase, David J. Foreman, Scott A. McLuckey, Jos Oomens, and Mark A. Johnson
The Journal of Physical Chemistry Letters October 19, 2017 Volume 8(Issue 20) pp:5047-5047
Publication Date(Web):September 29, 2017
DOI:10.1021/acs.jpclett.7b02223
Gas-phase ion chemistry methods that capture and characterize the degree of activation of small molecules in the active sites of homogeneous catalysts form a powerful new tool to unravel how ligand environments affect reactivity. A key roadblock in this development, however, is the ability to generate the fragile metal oxidation states that are essential for catalytic activity. Here we demonstrate the preparation of the key Ni(I) center in the widely used cyclam scaffold using ion–ion recombination as a gas-phase alternative to electrochemical reduction. The singly charged Ni+(cyclam) coordination complex is generated by electron transfer from fluoranthene and azobenzene anions to doubly charged Ni2+(cyclam), using the electron-transfer dissociation protocol in a commercial quadrupole ion trap instrument and in a custom-built octopole RF ion trap. The successful preparation of the Ni+(cyclam) cation is verified through analysis of its vibrational spectrum obtained using the infrared free electron laser FELIX.
Co-reporter:Joseph W. DePalmaPatrick J. Kelleher, Laís C. Tavares, Mark A. Johnson
The Journal of Physical Chemistry Letters January 19, 2017 Volume 8(Issue 2) pp:
Publication Date(Web):January 6, 2017
DOI:10.1021/acs.jpclett.6b02964
We explore the intramolecular distortions present in divalent metal ion–carboxylate ion pairs using vibrational spectroscopy of the cryogenically cooled, mass-selected species isolated in the gas phase. The spectral signatures of the C–O stretching modes are identified using the perdeutero isotopologues of the acetate and propionate anions to avoid congestion arising from the CH2 fundamentals. Both Ca2+ and Mg2+ are observed to bind in a symmetrical, so-called “bidentate” arrangement to the −CO2¯ group. The very strong deformations of the head groups displayed by the binary complexes dramatically relax when either neutral water molecules or counterions are attached to the Mg2+RCO2¯ cation. These results emphasize the critical role that local coordination plays when using the RCO2¯ bands to deduce the metal ion complexation motif in condensed media.
Co-reporter:Nan Yang, Chinh H. Duong, Patrick J. Kelleher, Mark A. Johnson, Anne B. McCoy
Chemical Physics Letters 2017 Volume 690(Volume 690) pp:
Publication Date(Web):16 December 2017
DOI:10.1016/j.cplett.2017.09.042
•Tag-free IR2PD linear spectrum.•Excited molecule spectrum.•Identification of local OH oscillators.•Intermolecular and intramolecular coupling between H2O molecules.•Multiple resonance laser spectroscopy.We reveal the microscopic mechanics of iodide ion microhydration by recording the isotopomer-selective vibrational spectra of the I−·(H2O)·(D2O), I−·(HOD)·(D2O), and I−·(DOH)·(H2O) isotopologues using a new class of ion spectrometer that is optimized to carry out two-color, IR-IR photodissociation in a variety of pump-probe schemes. Using one of these, we record the linear absorption spectrum of a cryogenically cooled cluster without the use of a messenger “tag”. In another protocol, we reveal the spectra of individual H2O and D2O molecules embedded in each of the two possible binding sites in the iodide dihydrate, as well as the bands due to individual OH and OD groups in each of the four local binding environments. Finally, we demonstrate how temperature dependent isotopic scrambling among the spectral features can be used to monitor the onset of large amplitude motion, heretofore inferred from changes in the envelope of the OH stretching vibrational manifold.Download high-res image (61KB)Download full-size image
Co-reporter:Dr. Fabian S. Menges;Stephanie M. Craig;Niklas Tötsch;Dr. Aaron Bloomfield;Subrata Ghosh;Dr. Hans-Jörg Krüger;Dr. Mark A. Johnson
Angewandte Chemie 2016 Volume 128( Issue 4) pp:
Publication Date(Web):
DOI:10.1002/ange.201680461
Co-reporter:Dr. Fabian S. Menges;Stephanie M. Craig;Niklas Tötsch;Dr. Aaron Bloomfield;Subrata Ghosh;Dr. Hans-Jörg Krüger;Dr. Mark A. Johnson
Angewandte Chemie International Edition 2016 Volume 55( Issue 4) pp:
Publication Date(Web):
DOI:10.1002/anie.201680461
Co-reporter:Conrad T. Wolke;Matias R. Fagiani;Tuguldur T. Odbadrakh;Joseph A. Fournier;Laura C. Dzugan;Knut R. Asmis;Kenneth D. Jordan;Anne B. McCoy;Harald Knorke
Science 2016 Volume 354(Issue 6316) pp:1131-1135
Publication Date(Web):02 Dec 2016
DOI:10.1126/science.aaf8425
A view of acidic proton transport emerges in vibrational spectra of deuterated water clusters bound to a succession of bases.
Co-reporter:Dr. Fabian S. Menges;Stephanie M. Craig;Niklas Tötsch;Dr. Aaron Bloomfield;Subrata Ghosh;Dr. Hans-Jörg Krüger;Dr. Mark A. Johnson
Angewandte Chemie International Edition 2016 Volume 55( Issue 4) pp:1282-1285
Publication Date(Web):
DOI:10.1002/anie.201507965
Abstract
We describe a systematic method for the preparation and spectroscopic characterization of a CO2 molecule coordinated to an activated bisphenoidal nickel(I) compound containing a tetraazamacrocyclic ligand in the gas phase. The resulting complex was then structurally characterized by using mass-selected vibrational predissociation spectroscopy. The results indicate that a highly distorted CO2 molecule is bound to the metal center in an η2-C,O coordination mode, thus establishing an efficient and rational method for the preparation of metal-activated CO2 for further studies using ion chemistry techniques.
Co-reporter:Dr. Fabian S. Menges;Stephanie M. Craig;Niklas Tötsch;Dr. Aaron Bloomfield;Subrata Ghosh;Dr. Hans-Jörg Krüger;Dr. Mark A. Johnson
Angewandte Chemie 2016 Volume 128( Issue 4) pp:1304-1307
Publication Date(Web):
DOI:10.1002/ange.201507965
Abstract
We describe a systematic method for the preparation and spectroscopic characterization of a CO2 molecule coordinated to an activated bisphenoidal nickel(I) compound containing a tetraazamacrocyclic ligand in the gas phase. The resulting complex was then structurally characterized by using mass-selected vibrational predissociation spectroscopy. The results indicate that a highly distorted CO2 molecule is bound to the metal center in an η2-C,O coordination mode, thus establishing an efficient and rational method for the preparation of metal-activated CO2 for further studies using ion chemistry techniques.
Co-reporter:Kenny Hanke, Matin Kaufmann, Gerhard Schwaab, Martina Havenith, Conrad T. Wolke, Olga Gorlova, Mark A. Johnson, Bishnu Prasad Kar, Wolfram Sander and Elsa Sanchez-Garcia
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 13) pp:8518-8529
Publication Date(Web):12 Feb 2015
DOI:10.1039/C5CP00116A
This study explores the interactions underlying the IR spectra of the ionic liquid [NC4111][NTf2] and its deuterated isotopomer [d9-NC4111][NTf2] by first isolating the spectra of charged ionic building blocks using mass-selective CIVP spectroscopy and then following the evolution of these bands upon sequential assembly of the ionic constituents. The spectra of the (1,1) and (2,2) neutral ion pairs are recorded using superfluid helium droplets as well as a solid neon matrix, while those of the larger charged aggregates are again obtained with CIVP. In general, the cluster spectra are similar to that of the bulk, with the (2,2) system displaying the closest resemblance. Analysis of the polarization-dependent band intensities of the neutral ion pairs in liquid droplets as a function of external electric field yields dipole moments of the neutral aggregates. This information allows a coarse assessment of the packing structure of the neutral pairs to be antiparallel at 0.37 K, in contrast to the parallel arrangement found for the assembly of small, high-dipole neutral molecules with large rotational constants (e.g., HCN). The role of an extra anion or cation attached to both the (1,1) and the (2,2) ion pairs to form the charged clusters is discussed in the context of an additional remote, more unfavorable binding site intrinsic to the nature of the charged IL clusters and as such not anticipated in the bulk phase. Whereas for the anion itself only the lowest energy trans conformer was observed, the higher clusters showed an additional population of the cis conformer. The interactions are found to be consistent with a minimal role of hydrogen bonding.
Co-reporter:Joseph W. DePalma, Patrick J. Kelleher, Christopher J. Johnson, Joseph A. Fournier, and Mark A. Johnson
The Journal of Physical Chemistry A 2015 Volume 119(Issue 30) pp:8294-8302
Publication Date(Web):July 1, 2015
DOI:10.1021/acs.jpca.5b04612
Elucidation of the molecular-level mechanics underlying the dissolution of salts is one of the long-standing, fundamental problems in electrolyte chemistry. Here we follow the incremental structural changes that occur when water molecules are sequentially added to the ternary [MgSO4Mg]2+ ionic assembly using cryogenic vibrational predissociation spectroscopy of the cold, mass-selected [MgSO4Mg(H2O)n=4–11]2+ cluster ions. Although the bare [MgSO4Mg]2+ ion could not be prepared experimentally, its calculated minimum energy structure corresponds to a configuration where the two Mg2+ ions attach on opposite sides of the central SO42– ion in a bifurcated fashion to yield a D2d symmetry arrangement. Analysis of the observed spectral patterns indicate that water molecules preferentially attach to the flanking Mg2+ ions for the n ≤ 7 hydrates, which results in an incremental weakening of the interaction between the ions. Water molecules begin to interact with the sequestered SO42– anion promptly at n = 8, where changes in the band pattern clearly demonstrate that the intrinsic bifurcated binding motif among the ions evolves into quasilinear Mg2+–O–S arrangements as water molecules H-bond to the now free SO groups. Although condensed-phase MgSO4 occurs with a stable hexahydrate in which water molecules lie between the ion pairs, addition of a sixth water molecule to one of the Mg2+ ions in the n = 11 cluster occurs with the onset of the second hydration shell such that the cation remains coordinated to one of the SO42– oxygen atoms.
Co-reporter:Patrick J. Kelleher, Christopher J. Johnson, Joseph A. Fournier, Mark A. Johnson, and Anne B. McCoy
The Journal of Physical Chemistry A 2015 Volume 119(Issue 18) pp:4170-4176
Publication Date(Web):April 13, 2015
DOI:10.1021/acs.jpca.5b03114
To explore the extent of the molecular cation perturbation induced by complexation with He atoms required for the application of cryogenic ion vibrational predissociation (CIVP) spectroscopy, we compare the spectra of a bare NH4+(H2O) ion (obtained using infrared multiple photon dissociation (IRMPD)) with the one-photon CIVP spectra of the NH4+(H2O)·He1–3 clusters. Not only are the vibrational band origins minimally perturbed, but the rotational fine structures on the NH and OH asymmetric stretching vibrations, which arise from the free internal rotation of the −OH2 and −NH3 groups, also remain intact in the adducts. To establish the location and the quantum mechanical delocalization of the He atoms, we carried out diffusion Monte Carlo (DMC) calculations of the vibrational zero point wave function, which indicate that the barriers between the three equivalent minima for the He attachment are so small that the He atom wave function is delocalized over the entire −NH3 rotor, effectively restoring C3 symmetry for the embedded −NH3 group.
Co-reporter:Joseph A. Fournier, Conrad T. Wolke, and Mark A. Johnson, Tuguldur T. Odbadrakh and Kenneth D. Jordan , Shawn M. Kathmann and Sotiris S. Xantheas
The Journal of Physical Chemistry A 2015 Volume 119(Issue 36) pp:9425-9440
Publication Date(Web):July 9, 2015
DOI:10.1021/acs.jpca.5b04355
We review the role that gas-phase, size-selected protonated water clusters, H+(H2O)n, have played in unraveling the microscopic mechanics responsible for the spectroscopic behavior of the excess proton in bulk water. Because the larger (n ≥ 10) assemblies are formed with three-dimensional cage morphologies that more closely mimic the bulk environment, we report the spectra of cryogenically cooled (10 K) clusters over the size range 2 ≤ n ≤ 28, over which the structures evolve from two-dimensional arrangements to cages at around n = 10. The clusters that feature a complete second solvation shell around a surface-embedded hydronium ion yield spectral signatures of the proton defect similar to those observed in dilute acids. The origins of the large observed shifts in the proton vibrational signature upon cluster growth were explored with two types of theoretical analyses. First, we calculate the cubic and semidiagonal quartic force constants and use these in vibrational perturbation theory calculations to establish the couplings responsible for the large anharmonic red shifts. We then investigate how the extended electronic wave functions that are responsible for the shapes of the potential surfaces depend on the nature of the H-bonded networks surrounding the charge defect. These considerations indicate that, in addition to the sizable anharmonic couplings, the position of the OH stretch most associated with the excess proton can be traced to large increases in the electric fields exerted on the embedded hydronium ion upon formation of the first and second solvation shells. The correlation between the underlying local structure and the observed spectral features is quantified using a model based on Badger’s rule as well as via the examination of the electric fields obtained from electronic structure calculations.
Co-reporter:Conrad T. Wolke, Fabian S. Menges, Niklas Tötsch, Olga Gorlova, Joseph A. Fournier, Gary H. Weddle, and Mark A. Johnson, Nadja Heine, Tim K. Esser, Harald Knorke, and Knut R. Asmis , Anne B. McCoy, Daniel J. Arismendi-Arrieta, Rita Prosmiti and Francesco Paesani
The Journal of Physical Chemistry A 2015 Volume 119(Issue 10) pp:1859-1866
Publication Date(Web):February 3, 2015
DOI:10.1021/jp510250n
The strong temperature dependence of the I–·(H2O)2 vibrational predissociation spectrum is traced to the intracluster dissociation of the ion-bound water dimer into independent water monomers that remain tethered to the ion. The thermodynamics of this process is determined using van’t Hoff analysis of key features that quantify the relative populations of H-bonded and independent water molecules. The dissociation enthalpy of the isolated water dimer is thus observed to be reduced by roughly a factor of three upon attachment to the ion. The cause of this reduction is explored with electronic structure calculations of the potential energy profile for dissociation of the dimer, which suggest that both reduction of the intrinsic binding energy and vibrational zero-point effects act to weaken the intermolecular interaction between the water molecules in the first hydration shell. Additional insights are obtained by analyzing how classical trajectories of the I–·(H2O)2 system sample the extended potential energy surface with increasing temperature.
Co-reporter:Conrad T. Wolke, Andrew F. DeBlase, Christopher M. Leavitt, Anne B. McCoy, and Mark A. Johnson
The Journal of Physical Chemistry A 2015 Volume 119(Issue 52) pp:13018-13024
Publication Date(Web):November 26, 2015
DOI:10.1021/acs.jpca.5b10649
To understand how the D2d oxalate scaffold (C2O4)2– distorts upon capture of a proton, we report the vibrational spectra of the cryogenically cooled HO2CCO2– anion and its deuterated isotopologue DO2CCO2–. The transitions associated with the skeletal vibrations and OH bending modes are sharp and are well described by inclusion of cubic terms in the normal mode expansion of the potential surface through an extended Fermi resonance analysis. The ground state structure features a five-membered ring with an asymmetric intramolecular proton bond. The spectral signatures of the hydrogen stretches, on the contrary, are surprisingly diffuse, and this behavior is not anticipated by the extended Fermi scheme. We trace the diffuse bands to very strong couplings between the high-frequency OH-stretch and the low-frequency COH bends as well as heavy particle skeletal deformations. A simple vibrationally adiabatic model recovers this breadth of oscillator strength as a 0 K analogue of the motional broadening commonly used to explain the diffuse spectra of H-bonded systems at elevated temperatures, but where these displacements arise from the configurations present at the vibrational zero-point level.
Co-reporter:Andrew J. Ingram ; Arron B. Wolk ; Cornelia Flender ; Jialing Zhang ; Christopher J. Johnson ; Ulrich Hintermair ; Robert H. Crabtree ; Mark A. Johnson ;Richard N. Zare
Inorganic Chemistry 2014 Volume 53(Issue 1) pp:423-433
Publication Date(Web):November 14, 2013
DOI:10.1021/ic402390t
Sodium periodate (NaIO4) is added to Cp*IrIII (Cp* = C5Me5–) or (cod)IrI (cod = cyclooctadiene) complexes, which are water and C–H oxidation catalyst precursors, and the resulting aqueous reaction is investigated from milliseconds to seconds using desorption electrospray ionization, electrosonic spray ionization, and cryogenic ion vibrational predissociation spectroscopy. Extensive oxidation of the Cp* ligand is observed, likely beginning with electrophilic C–H hydroxylation of a Cp* methyl group followed by nonselective pathways of further oxidative degradation. Evidence is presented that the supporting chelate ligand in Cp*Ir(chelate) precursors influences the course of oxidation and is neither eliminated from the coordination sphere nor oxidatively transformed. Isomeric products of initial Cp* oxidation are identified and structurally characterized by vibrational spectroscopy in conjunction with density functional theory (DFT) modeling. Less extensive but more rapid oxidation of the cod ligand is also observed in the (cod)IrI complexes. The observations are consistent with the proposed role of Cp* and cod as sacrificial placeholder ligands that are oxidatively removed from the precursor complexes under catalytic conditions.
Co-reporter:Andrew F. DeBlase, Steven R. Kass and Mark A. Johnson
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 10) pp:4569-4575
Publication Date(Web):07 Jan 2014
DOI:10.1039/C3CP54117G
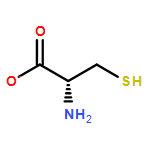
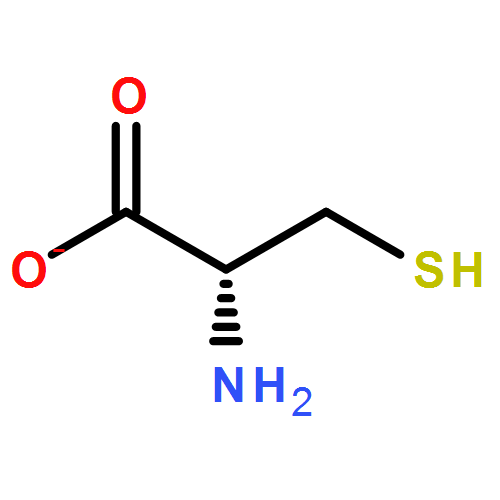
The gas phase structure of deprotonated cysteine (Cys–H+)− has recently gained attention because of its counterintuitive calculated minimum energy structure in which it appears that deprotonation occurs at the –SH moiety rather than at the nominally more acidic carboxylic acid group. Because previous experimental efforts have not yielded to a consensus regarding the structure of the anion, we report the cryogenic ion vibrational predissociation (CIVP) spectra of its cryogenically cooled H/D isotopologues in an effort to clarify the situation. The unexpected isotope dependence of key features in the spectrum and the similarity of the band pattern to that displayed by the intramolecular H-bonded linkage in a deprotonated diacid (HCO2(CH2)10CO2−) indicate that the dominant form of the anion occurs with a strongly shared proton between the thiolate (–S−) and carboxylate (–CO2−) groups. An interesting aspect of this (–S−⋯H+⋯−O2C–) linkage is that, although the global minimum places the shared proton closer to the oxygen atom, the soft potential energy curve calculated for displacement of the bridging proton would likely support sufficient zero-point motion both to blur the distinction between thiolate- and carboxylate-based structures and to account for the unusual isotope effects.
Co-reporter:Christopher J. Johnson, Laura C. Dzugan, Arron B. Wolk, Christopher M. Leavitt, Joseph A. Fournier, Anne B. McCoy, and Mark A. Johnson
The Journal of Physical Chemistry A 2014 Volume 118(Issue 35) pp:7590-7597
Publication Date(Web):May 29, 2014
DOI:10.1021/jp504139j
Vibrational predissociation spectra of D2-“tagged” Mg2+OH–(H2O)n=1–6 and Ca2+OH–(H2O)n=1–5 clusters are reported to explore how the M2+OH– contact ion pairs respond to stepwise formation of the first hydration shell. In both cases, the hydroxide stretching frequency is found to red-shift strongly starting with addition of the third water molecule, quickly becoming indistinguishable from nonbonded OH groups associated with solvent water molecules by n = 5. A remarkably broad feature centered around 3200 cm–1 and spanning up to ∼1000 cm–1 appears for the n ≥ 4 clusters that we assign to a single-donor ionic hydrogen bond between a proximal first solvent shell water molecule and the embedded hydroxide ion. The extreme broadening is rationalized with a theoretical model that evaluates the range of local OH stretching frequencies predicted for the heavy particle configurations available in the zero-point vibrational wave function describing the low-frequency modes. The implication of this treatment is that extreme broadening in the vibrational spectrum need not arise from thermal fluctuations in the ion ensemble, but can rather reflect combination bands based on the OH stretching fundamental that involve many quanta of low-frequency modes whose displacements strongly modulate the OH stretching frequency.
Co-reporter:Florian Schinle, Christoph R. Jacob, Arron B. Wolk, Jean-François Greisch, Matthias Vonderach, Patrick Weis, Oliver Hampe, Mark A. Johnson, and Manfred M. Kappes
The Journal of Physical Chemistry A 2014 Volume 118(Issue 37) pp:8453-8463
Publication Date(Web):June 2, 2014
DOI:10.1021/jp501772d
Although the sequencing of protonated proteins and peptides with tandem mass spectrometry has blossomed into a powerful means of characterizing the proteome, much less effort has been directed at their deprotonated analogues, which can offer complementary sequence information. We present a unified approach to characterize the structure and intermolecular interactions present in the gas-phase pentapeptide leucine-enkephalin anion by several vibrational spectroscopy schemes as well as by ion-mobility spectrometry, all of which are analyzed with the help of quantum-chemical computations. The picture emerging from this study is that deprotonation takes place at the C terminus. In this configuration, the excess charge is stabilized by strong intramolecular hydrogen bonds to two backbone amide groups and thus provides a detailed picture of a potentially common charge accommodation motif in peptide anions.
Co-reporter:Joseph A. Fournier;Conrad T. Wolke;Nadja Heine;Sandy Gewinner;Wieland Schöllkopf;Matias R. Fagiani;Tim K. Esser;Christopher J. Johnson;Knut R. Asmis;Harald Knorke
PNAS 2014 Volume 111 (Issue 51 ) pp:18132-18137
Publication Date(Web):2014-12-23
DOI:10.1073/pnas.1420734111
Theoretical models of proton hydration with tens of water molecules indicate that the excess proton is embedded on the surface
of clathrate-like cage structures with one or two water molecules in the interior. The evidence for these structures has been
indirect, however, because the experimental spectra in the critical H-bonding region of the OH stretching vibrations have
been too diffuse to provide band patterns that distinguish between candidate structures predicted theoretically. Here we exploit
the slow cooling afforded by cryogenic ion trapping, along with isotopic substitution, to quench water clusters attached to
the H3O+ and Cs+ ions into structures that yield well-resolved vibrational bands over the entire 215- to 3,800-cm−1 range. The magic H3O+(H2O)20 cluster yields particularly clear spectral signatures that can, with the aid of ab initio predictions, be traced to specific
classes of network sites in the predicted pentagonal dodecahedron H-bonded cage with the hydronium ion residing on the surface.
Co-reporter:Conrad T. Wolke;Arron B. Wolk;Gary H. Weddle;Joseph A. Fournier;Christopher J. Johnson
Science 2014 Volume 344(Issue 6187) pp:1009-1012
Publication Date(Web):30 May 2014
DOI:10.1126/science.1253788
Blackjack water cluster detected
Spectroscopy of protonated water clusters has played a pivotal role in elucidating the molecular arrangement of acid solutions. Whereas bulk liquids manifest broad spectral features, the cluster bands tend to be sharper. The 21-membered water cluster has for decades inspired particular interest on account of its stability and its place in the transition from two-dimensional to three-dimensional hydrogen-bonding network motifs, but the spectral signature of its bound proton has proved elusive. Fournier et al. have now detected this long-sought vibrational feature by applying an innovative ion cooling technique.
Science, this issue p. 1009
Co-reporter:Joseph A. Fournier, Arron B. Wolk, and Mark A. Johnson
Analytical Chemistry 2013 Volume 85(Issue 15) pp:7339
Publication Date(Web):June 16, 2013
DOI:10.1021/ac401228y
Cryogenic ion vibrational predissociation (CIVP) spectroscopy is used to structurally characterize electrochemically (EC)-generated oxidation products of the benchmark compound reserpine. Ionic products were isolated using EC-electrospray ionization (ESI) coupled to a 25 K ion trap prior to injection into a double-focusing, tandem time-of-flight photofragmentation mass spectrometer. Vibrational predissociation spectroscopy was carried out by photoevaporation of weakly bound N2 adducts over the range 800–3800 cm–1 in a linear (i.e., single photon) action regime, thus enabling direct comparison of the experimental vibrational pattern with harmonic calculations. The locations of the NH and OH stretching fundamentals are most consistent with formation of 9-hydroxyreserpine, which is a different isomer than considered previously. This approach thus provides a powerful structural dimension for the analysis of electrochemical processes detected with the sensitivity of mass spectrometry.
Co-reporter:Andrew F. DeBlase, Michael T. Scerba, Thomas Lectka, Mark A. Johnson
Chemical Physics Letters 2013 Volumes 568–569() pp:9-13
Publication Date(Web):1 May 2013
DOI:10.1016/j.cplett.2013.03.002
•We obtain vibrational predissociation spectra of Ar-tagged silyl cations.•The spectra are free from perturbations from solvents or counterions.•Vibrational spectra are in close agreement with ab initio harmonic level calculations.•Binding of the Ar tag has a minor perturbation on the spectra of the bare ions.Vibrational predissociation spectra of the (CH3)2RSi+·Arn, (R = H and CH3, n = 1 and 2) ions are compared with harmonic calculations to structurally characterize these putative reactive intermediates. Although the vibrational photofragmentation behavior indicates that the Ar–Si bond is quite strong relative to that found in closed shell ions, formation of the Ar adducts is calculated to cause only minor perturbations to the intrinsic vibrational band patterns of the isolated ions. In both (R = H and CH3) cases, the vibrational spectra are very simple, consisting entirely of sharp features readily assigned to fundamentals anticipated by their harmonic spectra.Graphical abstract
Co-reporter:Arron B. Wolk, Christopher M. Leavitt, Joseph A. Fournier, Michael Z. Kamrath, Gayan B. Wijeratne, Timothy A. Jackson, Mark A. Johnson
International Journal of Mass Spectrometry 2013 Volumes 354–355() pp:33-38
Publication Date(Web):15 November 2013
DOI:10.1016/j.ijms.2013.04.022
•We present the infrared predissociation spectrum of peroxo manganese intermediate.•The species were electrosprayed and cooled in a cryogenically cooled ion trap.•Action spectra on N2 adducts give a side on binding motif of the O2 adduct.•Comparison with 18O2 substitution and harmonic spectra confirms assignment.•Isolation of reactive intermediates will pave way for cluster mediated chemistry.We apply cryogenic ion vibrational predissociation (CIVP) spectroscopy to characterize the O2 attachment motif in a prototypical peroxo manganese (III) reaction intermediate. In this approach, species are extracted from solution using electrospray ionization and cooled in a 30 K ion trap. The infrared spectrum is then obtained by monitoring the photoinduced evaporation of a single, weakly bound N2 molecule as a function of laser wavelength. Because the resulting CIVP action spectrum is linear in laser fluence, the pattern of well-resolved transitions can be directly compared with harmonic spectra calculated for predicted local minima using density functional theory (DFT). The assignment of the OO stretching band derived from the activated O2 ligand is established by following the evolution of the bands with 18O2 substitution, and the energy of this transition indicates that O2 is bound side-on to the Mn center. The successful application of CIVP to this class of compounds opens the way for sensitive spectroscopic characterization of weakly abundant species in complex solution environments.
Co-reporter:Christopher M. Leavitt, Andrew F. DeBlase, Christopher J. Johnson, Michael van Stipdonk, Anne B. McCoy, and Mark A. Johnson
The Journal of Physical Chemistry Letters 2013 Volume 4(Issue 20) pp:3450-3457
Publication Date(Web):September 20, 2013
DOI:10.1021/jz401681y
Survey vibrational predissociation spectra of several representative protonated peptides and model compounds reveal very diffuse absorptions near 2500 cm–1 that are traced to pentagonal cyclic ionic hydrogen bonds (C5 interactions) involving the excess charge centers. This broadening occurs despite the fact that the ions are cooled close to their vibrational zero-point levels and their spectra are obtained by predissociation of weakly bound adducts (H2, N2, CO2) prepared in a cryogenic ion trap. The C5 band assignments are based on H/D isotopic substitution, chemical derivatization, solvation behavior, and calculated spectra. We evaluate the extent to which this broadening is caused by anharmonic coupling in the isolated molecules by including cubic coupling terms in the normal mode expansion of the potential energy surface. This analysis indicates that the harmonic H-bonded stretching vibration is mixed with dark background states over much of the energy range covered by the observed features. The difficulty with identifying these features in earlier studies of dipeptides is traced to both the breadth and the fact they are calculated to be intrinsically weaker than cases involving linear variations of the N···H+···O motif.Keywords: cold ions; cryogenic ion vibrational predissociation spectroscopy; hydrogen bonding; vibrational spectroscopy;
Co-reporter:Arron B. Wolk, Etienne Garand, Ian M. Jones, Andrew D. Hamilton, and Mark A. Johnson
The Journal of Physical Chemistry A 2013 Volume 117(Issue 29) pp:5962-5969
Publication Date(Web):January 10, 2013
DOI:10.1021/jp3111925
We report how two flexible diphenylacetylene (DPA) derivatives distort to accommodate both cationic and anionic partners in the binary X±·DPA series with X = TMA+ (tetramethylammonium), Na+, Cl–, Br–, and I–. This is accomplished through theoretical analysis of X±·DPA·2D2 vibrational spectra, acquired by predissociation of the weakly bound D2 adducts formed in a 10 K ion trap. DPA binds the weakly coordinating TMA+ ion with an arrangement similar to that of the neutral compound, whereas the smaller Na+ ion breaks all intramolecular H-bonds yielding a structure akin to the transition state for interconversion of the two conformations in neutral DPA. Halides coordinate to the urea NH donors in a bidentate H-bonded configuration analogous to the single intramolecular H-bonded motif identified at high chloride concentrations in solution. Three positions of the “switch” are thus identified in the intrinsic ion accommodation profile that differ by the number of intramolecular H-bonds (0, 1, or 2) at play.
Co-reporter:Christopher J. Johnson and Mark A. Johnson
The Journal of Physical Chemistry A 2013 Volume 117(Issue 50) pp:13265-13274
Publication Date(Web):July 23, 2013
DOI:10.1021/jp404244y
Particles consisting of ammonia and sulfuric acid are widely regarded as seeds for atmospheric aerosol nucleation, and incorporation of alkylamines has been suggested to substantially accelerate their growth. Despite significant efforts, little direct experimental evidence exists for the structures and chemical processes underlying multicomponent particle nucleation. Here we are concerned with the positively charged clusters of ammonia and sulfuric acid with compositions H+(NH3)m(H2SO4)n (2 ≤ m ≤ 5, 1 ≤ n ≤ 4), for which equilibrium geometry structures have been reported in recent computational searches. The computed harmonic vibrational spectra of such minimum energy structures can be directly compared with the experimental spectra of each cluster composition isolated in the laboratory using cryogenic ion chemistry methods. We present one-photon (i.e., linear) infrared action spectra of the isolated gas phase ions cryogenically cooled to 10 K, allowing us to resolve the characteristic vibrational signatures of these clusters. Because the available calculated spectra for different structural candidates have been obtained using different levels of theory, we reoptimized the previously reported structures with several common electronic structure methods and find excellent agreement can be achieved for the (m = 3, n = 2) cluster using CAM-B3LYP with only minor structural differences from the previously identified geometries. At the larger sizes, the experimental spectra strongly resemble that observed for 180 nm ammonium bisulfate particles. The characteristic ammonium- and bisulfate-localized bands are clearly evident at all sizes studied, indicating that the cluster structures are indeed ionic in nature. With the likely (3,2) structure in hand, we then explore the spectral and structural changes caused when methylamine is substituted for ammonia. This process is found to occur with minimal perturbation of the unsubstituted cluster. The thermal decomposition pathways were also evaluated using multiple-photon induced dissociation and are, in all cases, dominated (>100:1) by evaporation of a neutral ammonia molecule rather than methylamine. Spectra obtained for the product cluster ions resulting from this evaporation are consistent with the formation of a single hydrogen bond between two neighboring bisulfate ions, partially regenerating a sulfuric acid molecule. These results provide critical experimental benchmarks for ongoing theoretical efforts to understand the early stages of aerosol growth.
Co-reporter: Dr. Gautam R. Desiraju; Dr. Mark A. Johnson; Dr. Wolfram Ser
ChemPhysChem 2013 Volume 14( Issue 4) pp:631-633
Publication Date(Web):
DOI:10.1002/cphc.201200980
No abstract is available for this article.
Co-reporter:Etienne Garand, Joseph A. Fournier, Michael Z. Kamrath, Nathan D. Schley, Robert H. Crabtree and Mark A. Johnson
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 29) pp:10109-10113
Publication Date(Web):08 Jun 2012
DOI:10.1039/C2CP41490B
We report the vibrational predissociation spectra of two related organometallic half-sandwich iridium species which have been recently reported as activated intermediates in the context of homogenous water oxidation. These compounds are extracted from solution into a cryogenic photofragmentation mass spectrometer and “tagged” with weakly bound H2 molecules that do not significantly perturb the intrinsic structures of the ions. The resulting spectra display very sharp (∼5 cm−1), well-resolved bands that provide a stringent test for electronic structure calculations, and are accurately recovered by harmonic predictions for the bare species. The spectra reveal subtle distortions of the ligand structure when a solvent molecule (acetonitrile) is directly coordinated with the metal center.
Co-reporter:Anne B. McCoy, Timothy L. Guasco, Christopher M. Leavitt, Solveig G. Olesen and Mark A. Johnson
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 20) pp:7205-7214
Publication Date(Web):30 Jan 2012
DOI:10.1039/C2CP24110B
The harmonic approximation provides a powerful approach for interpreting vibrational spectra. In this treatment, the energy and intensity of the 3N − 6 normal modes are calculated using a quadratic expansion of the potential energy and a linear expansion of the dipole moment surfaces, respectively. In reality, transitions are often observed that are not accounted for by this approach (e.g. combination bands, overtones, etc.), and these transitions arise from inherent anharmonicities present in the system. One interesting example occurs in the vibrational spectrum of H2O(l), where a band is observed near 2000 cm−1 that is commonly referred to as the “association band”. This band lies far from the expected bend and stretching modes of the water molecule, and is not recovered at the harmonic level. In a recent study, we identified a band in this spectral region in gas-phase clusters involving atomic and molecular adducts to the H3O+ ion. In the current study we probe the origins of this band through a systematic analysis of the argon-predissociation spectra of H3O+·X3 where X = Ar, CH4, N2 or H2O, with particular attention to the contributions from the non-linearities in the dipole surfaces, often referred to as non-Condon effects. The spectra of the H3O+ clusters all display strong transitions between 1900–2100 cm−1, and theoretical modeling indicates that they can be assigned to a combination band involving the HOH bend and frustrated rotation of H3O+ in the solvent cage. This transition derives its oscillator strength entirely from strong non-Condon effects, and we discuss its possible relationship to the association band in the spectrum of liquid water.
Co-reporter:Kristin J. Breen, Andrew F. DeBlase, Timothy L. Guasco, Vamsee K. Voora, Kenneth D. Jordan, Takashi Nagata, and Mark A. Johnson
The Journal of Physical Chemistry A 2012 Volume 116(Issue 3) pp:903-912
Publication Date(Web):December 6, 2011
DOI:10.1021/jp209493v
The transition states of a chemical reaction in solution are generally accessed through exchange of thermal energy between the solvent and the reactants. As such, an ensemble of reacting systems approaches the transition state configuration of reactant and surrounding solvent in an incoherent manner that does not lend itself to direct experimental observation. Here we describe how gas-phase cluster chemistry can provide a detailed picture of the microscopic mechanics at play when a network of six water molecules mediates the trapping of a highly reactive “hydrated electron” onto a neutral CO2 molecule to form a radical anion. The exothermic reaction is triggered from a metastable intermediate by selective excitation of either the reactant CO2 or the water network, which is evidenced by the evaporative decomposition of the product cluster. Ab initio molecular dynamics simulations of energized CO2·(H2O)6– clusters are used to elucidate the nature of the network deformations that mediate intracluster electron capture, thus revealing the detailed solvent fluctuations implicit in the Marcus theory for electron-transfer kinetics in solution.
Co-reporter:Michael T. Scerba, Andrew F. DeBlase, Steven Bloom, Travis Dudding, Mark A. Johnson, and Thomas Lectka
The Journal of Physical Chemistry A 2012 Volume 116(Issue 14) pp:3556-3560
Publication Date(Web):March 6, 2012
DOI:10.1021/jp211688v
We characterize a highly unusual, charged NH–O hydrogen bond formed within esters of 8-(dimethylamino)naphthalen-1-ol in which an ammonium ion serves as an intramolecular hydrogen bond donor to spatially proximate ester ether oxygen atoms. Infrared spectroscopic analysis of the ester carbonyl frequencies demonstrates significant blue-shifting when ether hydrogen bonding is possible, in stark contrast to the more commonly observed red shift that occurs upon hydrogen bonding to the ester carbonyl oxygen. The intrinsic behavior of the linkage (i.e., in which counterions and solvent effects are eliminated) is provided by vibrational predissociation spectroscopy of the isolated gas-phase cations complexed with weakly bound D2 molecules.
Co-reporter:Etienne Garand;Michael Z. Kamrath;Peter A. Jordan;Arron B. Wolk;Christopher M. Leavitt;Anne B. McCoy;Scott J. Miller
Science 2012 Volume 335(Issue 6069) pp:694-698
Publication Date(Web):10 Feb 2012
DOI:10.1126/science.1214948
Co-reporter:Christopher M. Leavitt, Arron B. Wolk, Joseph A. Fournier, Michael Z. Kamrath, Etienne Garand, Michael J. Van Stipdonk, and Mark A. Johnson
The Journal of Physical Chemistry Letters 2012 Volume 3(Issue 9) pp:1099-1105
Publication Date(Web):April 9, 2012
DOI:10.1021/jz3003074
Isomer-specific vibrational predissociation spectra are reported for the gas-phase GlySarH+ and SarSarH+ [Gly = glycine; Sar = sarcosine] ions prepared by electrospray ionization and tagged with weakly bound D2 adducts using a cryogenic ion trap. The contributions of individual isomers to the overlapping vibrational band patterns are completely isolated using a pump–probe photochemical hole-burning scheme involving two tunable infrared lasers and two stages of mass selection (hence IR2MS2). These patterns are then assigned by comparison with harmonic (MP2/6-311+G(d,p)) spectra for various possible conformers. Both systems occur in two conformations based on cis and trans configurations with respect to the amide bond. In addition to the usual single intramolecular hydrogen bond motif between the protonated amine and the nearby amide oxygen atom, cis-SarSarH+ adopts a previous unreported conformation in which both amino NH's act as H-bond donors. The correlated red shifts in the NH donor and C═O acceptor components of the NH···O═C linkage to the acid group are unambiguously assigned in the double H-bonded conformer.Keywords: cryogenic ion trap; double resonance; isomers; peptides; vibrational spectroscopy;
Co-reporter:Michael Z. Kamrath ; Etienne Garand ; Peter A. Jordan ; Christopher M. Leavitt ; Arron B. Wolk ; Michael J. Van Stipdonk ; Scott J. Miller
Journal of the American Chemical Society 2011 Volume 133(Issue 16) pp:6440-6448
Publication Date(Web):March 30, 2011
DOI:10.1021/ja200849g
We present infrared photodissociation spectra of two protonated peptides that are cooled in a ∼10 K quadrupole ion trap and “tagged” with weakly bound H2 molecules. Spectra are recorded over the range of 600−4300 cm−1 using a table-top laser source, and are shown to result from one-photon absorption events. This arrangement is demonstrated to recover sharp (Δν ∼6 cm−1) transitions throughout the fingerprint region, despite the very high density of vibrational states in this energy range. The fundamentals associated with all of the signature N−H and C═O stretching bands are completely resolved. To address the site-specificity of the C═O stretches near 1800 cm−1, we incorporated one 13C into the tripeptide. The labeling affects only one line in the complex spectrum, indicating that each C═O oscillator contributes a single distinct band, effectively “reporting” its local chemical environment. For both peptides, analysis of the resulting band patterns indicates that only one isomeric form is generated upon cooling the ions initially at room temperature into the H2 tagging regime.
Co-reporter:S.G. Olesen, T.L. Guasco, J.R. Roscioli, M.A. Johnson
Chemical Physics Letters 2011 Volume 509(4–6) pp:89-95
Publication Date(Web):14 June 2011
DOI:10.1016/j.cplett.2011.04.060
Abstract
The Zundel ion, H2O·H+·H2O, provides a versatile scaffold with which to explore the quantum structure of the intermolecular proton bond (IPB). This information is encoded in the vibrational frequencies adopted by the shared proton, νsp, which are observed to follow a remarkably similar trend as the exterior OH groups are sequentially solvated or are replaced by methyl substituents. In effect, solvents H-bonding to exterior OH groups act to increase the proton affinity of the water to which they are bound in a roughly additive fashion. We discuss this behavior in the context of the extreme sensitivity of IPBs to their solvation environments.
Co-reporter:Helen K. Gerardi, George H. Gardenier, Usha Viswanathan, Scott M. Auerbach, Mark A. Johnson
Chemical Physics Letters 2011 Volume 501(4–6) pp:172-178
Publication Date(Web):7 January 2011
DOI:10.1016/j.cplett.2010.10.062
Abstract
We report vibrational predissociation spectra and theoretical analysis of the Ar-tagged cluster ions of imidazole, Im1–3H+·Ar. The frequencies of the external N–H stretches are observed to incrementally blue-shift toward that of neutral imidazole upon addition of the second and third Im molecules, consistent with the calculated behavior of the Im3H+ complex in which the excess charge is symmetrically shared by two internal N–H–N hydrogen bonds. A very strong, symmetrical doublet near 1000 cm−1 is observed for the Im2H+ complex and attributed to the parallel bridging proton displacement based on anharmonic frequency calculations.
Co-reporter:Helen K. Gerardi, Andrew F. DeBlase, Xiaoge Su, Kenneth D. Jordan, Anne B. McCoy, and Mark A. Johnson
The Journal of Physical Chemistry Letters 2011 Volume 2(Issue 19) pp:2437-2441
Publication Date(Web):August 26, 2011
DOI:10.1021/jz200937v
Reductive activation offers an attractive synthetic route for conversion of CO2 to transportable fuels, a process that often involves creation of the formate ion as an intermediate. We carry out an Ar-tagging infrared spectroscopic study of isolated HCO2¯ and its first hydrate, HCO2¯·H2O, and analyze the resulting band patterns with electronic structure and vibrationally anharmonic calculations. Strong vibronic interactions and intramolecular mode couplings are identified that are responsible for the deceptively complex solvation behavior of this familiar ion. In particular, the CH stretch fundamental is found to be anomalously low in energy in the isolated ion and to dramatically blue shift (by hundreds of cm–1) upon solvation. These two effects are traced to the large dependence of the electronic wave function on the CH bond length, reminiscent of the classic curve-crossings that dominate the dissociation behavior of neutral salt molecules.Keywords: Fermi resonance; monohydrate; reductive activation; solvatochromic shift; vibronic interaction;
Co-reporter:Christopher M. Leavitt;Arron B. Wolk
Journal of The American Society for Mass Spectrometry 2011 Volume 22( Issue 11) pp:
Publication Date(Web):2011 November
DOI:10.1007/s13361-011-0228-3
We report vibrational predissociation spectra of the four protonated dipeptides derived from glycine and sarcosine, GlyGlyH+•(H2)1,2, GlySarH+•(D2)2, SarGlyH+•(H2)2, and SarSarH+•(D2)2, generated in a cryogenic ion trap. Sharp bands were recovered by monitoring photoevaporation of the weakly bound H2 (D2) molecules in a linear action regime throughout the 700–4200 cm–1 range using a table-top laser system. The spectral patterns were analyzed in the context of the low energy structures obtained from electronic structure calculations. These results indicate that all four species are protonated on the N-terminus, and feature an intramolecular H-bond involving the amino group. The large blue-shift in the H-bonded N–H fundamental upon incorporation of a methyl group at the N-terminus indicates that this modification significantly lowers the strength of the intramolecular H-bond. Methylation at the amide nitrogen, on the other hand, induces a significant rotation (~110o) about the peptide backbone.
Co-reporter:Timothy L. Guasco and Mark A. Johnson and Anne B. McCoy
The Journal of Physical Chemistry A 2011 Volume 115(Issue 23) pp:5847-5858
Publication Date(Web):January 7, 2011
DOI:10.1021/jp109999b
The nature of anharmonic couplings in the H5O2+ “Zundel” ion and its deuterated isotopologues is investigated through comparison of their measured and calculated vibrational spectra. This follows a recent study in which we reported spectra for H5O2+, D5O2+, and D4HO2+ from ∼600 to 4000 cm−1, as well as H4DO2+ in the OH and OD stretching regions [ J. Phys. Chem. B 2008, 112, 321]. While the assignments of the higher-energy transitions associated with the fundamentals of the exterior OH and OD motions are relatively straightforward, difficulties arise in the assignment of the lower-frequency regions that involve displacement of the bridging proton, especially for the isotopically mixed species. Here we revisit the Ar-tagged isotopomers, and report the low energy action spectrum of H4DO2+ for the first time, as well as present significantly improved spectra for the D4HO2+ and D5O2+ systems. Band assignments are clarified in several cases using IR−IR hole-burning. We then investigate the physical origin of the anharmonic effects encoded in these spectra using a recently developed technique in which the anharmonic frequencies and intensities of transitions (involving up to two quanta of excitation) are evaluated using the ground state probability amplitudes [ J. Phys. Chem. A 2009, 113, 7346] obtained from diffusion Monte Carlo simulations. This approach has the advantage that it is applicable to low-symmetry systems [such as (HDO)H+(OH2)] that are not readily addressed using highly accurate methods such as the multiconfigurational time-dependent Hartree (MCTDH) approach. Moreover, it naturally accommodates an intuitive evaluation of the types of motion that contribute oscillator strength in the various regions of the spectrum, even when the wave function is intrinsically not separable as a product of low-dimensional approximate solutions. Spectra for H5O2+, D5O2+, H4DO2+, and D4HO2+ that are calculated by this approach are shown to be in excellent agreement with the measured spectra for these species, leading to reassignments of two of the bands in the intramolecular bending region of D4HO2+.
Co-reporter:Michael Z. Kamrath, Rachael A. Relph, Timothy L. Guasco, Christopher M. Leavitt, Mark A. Johnson
International Journal of Mass Spectrometry 2011 300(2–3) pp: 91-98
Publication Date(Web):
DOI:10.1016/j.ijms.2010.10.021
Co-reporter:Michael Z. Kamrath ; Rachael A. Relph
Journal of the American Chemical Society 2010 Volume 132(Issue 44) pp:15508-15511
Publication Date(Web):October 19, 2010
DOI:10.1021/ja1073036
We report the vibrational predissociation spectrum of C5H5N-CO2−, a radical anion which is closely related to the key intermediates postulated to control activation of CO2 in photoelectrocatalysis with pyridine (Py). The anion is prepared by the reaction of Py vapor with (CO2)m− clusters carried out in an ionized, supersonic entrainment ion source. Comparison with the results of harmonic frequency calculations establishes that this species is a covalently bound molecular anion derived from the corresponding carbamate, C5H5N-CO2− (H+). These results confirm the structural assignment inferred in an earlier analysis of the cluster distributions and photoelectron spectra of the mixed Pym(CO2)n− complexes [J. Chem. Phys. 2000, 113 (2), 596−601]. The spectra of the (CO2)m− (m = 5 and 7) clusters are presented for the first time in the lower energy range (1000−2400 cm−1), which reveal several of the fundamental modes that had only been characterized previously by their overtones and combination bands. Comparison of these new spectra with those displayed by Py(CO2)n− suggests that a small fraction of the Py(CO2)n− ions are trapped entrance channel reaction intermediates in which the charge remains localized on the (CO2)m− part of the cluster.
Co-reporter:Timothy L. Guasco, Ben M. Elliott and Mark A. Johnson, Jing Ding and Kenneth D. Jordan
The Journal of Physical Chemistry Letters 2010 Volume 1(Issue 15) pp:2396-2401
Publication Date(Web):July 20, 2010
DOI:10.1021/jz100730q
We report the spectral signatures of water molecules occupying individual sites in an extended H-bonding network using mass-selective, double-resonance vibrational spectroscopy of isotopomers. The scheme is demonstrated on the water heptamer anion, (H2O)7¯, where we first randomly incorporate a single, intact D2O molecule to create an ensemble of isotopomers. The correlation between the two OD stretching frequencies and that of the intramolecular DOD bending transition is then revealed by photochemical modulation of the isotopomer population responsible for particular features in the vibrational spectrum. The observed patterns confirm the assignment of the dominant doublet, appearing most red-shifted from the free OD stretch, to a single water molecule attached to the network in a double H-bond acceptor (AA) arrangement. The data also reveal the unanticipated role of accidentally overlapping transitions, where the highest-energy OD stretch, for example, occurs with its companion OD stretch obscured by the much stronger AA feature.Keywords (keywords): dip-infrared spectroscopy; gas phase; hole burning; hydrated electron; supersonic;
Co-reporter:Timothy L. Guasco;Ben M. Elliott;Rachael A. Relph;Michael Z. Kamrath;Anne B. McCoy;Ryan P. Steele;Daniel P. Schofield;Kenneth D. Jordan;Albert A. Viggiano;Eldon E. Ferguson
Science 2010 Volume 327(Issue 5963) pp:
Publication Date(Web):
DOI:10.1126/science.1177118
It's the Network
Numerous reactions of small molecules and ions in the atmosphere take place in the confines of watery aerosols. Relph et al. (p. 308; see the Perspective by Siefermann and Abel) explored the specific influence of a water cluster's geometry on the transformation of solvated nitrosonium (NO+) to nitrous acid (HONO). The reaction involves (O)N–O(H) bond formation with one water molecule, concomitant with proton transfer to additional, surrounding water molecules. Vibrational spectroscopy and theoretical simulations suggest that certain arrangements of the surrounding water network are much more effective than others in accommodating this charge transfer, and thus facilitating the reaction.
Co-reporter:Helen K. Gerardi, Kristin J. Breen, Timothy L. Guasco, Gary H. Weddle, George H. Gardenier, Jennifer E. Laaser and Mark A. Johnson
The Journal of Physical Chemistry A 2010 Volume 114(Issue 3) pp:1592-1601
Publication Date(Web):January 21, 2010
DOI:10.1021/jp9095419
We report predissociation spectra of Ar-tagged C2H2− and C2D2− anions, and explore vibrationally mediated photodetachment from various vibrational levels of the bare C2H2− ion using velocity-map imaging. Intense photodetachment resonances are observed in the C−H stretching region that are strongly correlated with vibrational hot bands in the anion photoelectron spectra, indicating that one-color, resonant two-photon photodetachment (R2PD) is complicated by excitation of vibrationally excited states with autodetaching upper levels embedded in the continuum. Isolation of the R2PD spectrum was achieved using a two-color, IR−IR scheme in which vibrational excitation and photodetachment were carried out in two separate laser interaction regions.
Co-reporter:Joseph C. Bopp, Anastassia N. Alexandrova, Ben M. Elliott, Tobias Herden, Mark A. Johnson
International Journal of Mass Spectrometry 2009 Volume 283(1–3) pp:94-99
Publication Date(Web):1 June 2009
DOI:10.1016/j.ijms.2009.02.003
We report vibrational predissociation spectra of the On−, n = 3–10, 12 cluster ions in the 700–2400 cm−1 range. The odd numbered clusters are consistent with their identification as O3−⋅(O2)n. The even numbered clusters are based on the O4− core ion, where the first two O2 molecules add to O4− in locations that individually break the symmetry of the core ion, but together restore this symmetry. Beyond O4−⋅(O2)2, subsequent O2 attachment yields bands close to that of neutral O2, indicating that the special character of the O4−⋅(O2)2 cluster is retained.Anionic clusters of oxygen (On−), formed in a supersonic free jet, build basically neutral solvent shells upon an ionic core.
Co-reporter:Rachael A. Relph, Ben M. Elliott, Gary H. Weddle and Mark A. Johnson, Jing Ding and Kenneth D. Jordan
The Journal of Physical Chemistry A 2009 Volume 113(Issue 6) pp:975-981
Publication Date(Web):January 16, 2009
DOI:10.1021/jp808283r
We introduce a method based on sequential application of vibrational predissociation spectroscopy to explore the high-amplitude rearrangements available in a small H-bonded complex that is vibrationally excited within a larger Ar cluster. The weakly bound Ar atoms play the role of a solvent in mediating the energy content of the embedded system, ultimately quenching it into local minima through evaporation. We demonstrate the approach on the NO2−·H2O binary hydrate, which is known to occur in two nearly isoenergetic isomeric forms. The scheme involves three stages of mass separation to select a particular NO2−·H2O·Arm parent ion cluster prior to vibrational excitation and then isolate the NO2−·H2O·Ar fragment ions for interrogation using resonant vibrational predissociation with a second infrared laser. The initial vibrational excitation selectively energizes one of the isomers through one of its characteristic resonances while the predissociation spectrum of the NO2−·H2O·Ar fragment encodes the distribution of isomers present after Ar evaporation. Isomerization from the front- to backside form is found to occur upon excitation of the NO stretch near 1200 cm−1; although the reverse reaction is not observed upon excitation of the NO stretch, it is observed upon excitation of the higher-energy OH stretching fundamental near 3000 cm−1. We discuss these observations in the context of the calculated isomerization energetics, which focus on the minimum energy structures for the isomers as well as the transition states for their interconversion.
Co-reporter:George H. Gardenier and Mark. A. Johnson, Anne B. McCoy
The Journal of Physical Chemistry A 2009 Volume 113(Issue 16) pp:4772-4779
Publication Date(Web):March 19, 2009
DOI:10.1021/jp811493s
The primary event in the ionization of water involves rapid proton transfer, leading to charge localization on H3O+ and the creation of a hydroxyl radical. We trap the nascent [H3O+·•OH] exit channel intermediate in the bimolecular reaction by Ar-mediated ionization of the neutral water dimer and characterize the nature of this ion−radical complex using vibrational predissociation spectroscopy of the Ar-tagged species. The resulting bands involving the displacement of the bridging proton are broad and appear as a strong triplet centered around 2000 cm−1. The observed band pattern is analyzed with theoretical calculations to identify the origin of the anhamonic effects evident in the spectrum. In the course of this work, expressions were derived for treating the coupling terms within a sinc-DVR. Although this level of treatment did not reveal the assignment of the triplet structure, its characteristic ∼100 cm−1 spacing suggests activity involving the frustrated rotation of the hydroxyl radical upon excitation of the bridging-proton vibration parallel to the heavy atom axis. The behavior of this system is considered in the context of that reported previously for the related H5O2+, H3O2−, and F−·H2O complexes.
Co-reporter:Azusa Muraoka, Yoshiya Inokuchi, Nathan I. Hammer, Joong-Won Shin, Mark A. Johnson and Takashi Nagata
The Journal of Physical Chemistry A 2009 Volume 113(Issue 31) pp:8942-8948
Publication Date(Web):July 15, 2009
DOI:10.1021/jp903578e
The [(CO2)n(H2O)]− cluster anions are studied using infrared photodissociation (IPD) spectroscopy in the 2800−3800 cm−1 range. The observed IPD spectra display a drastic change in the vibrational band features at n = 4, indicating a sharp discontinuity in the structural evolution of the monohydrated cluster anions. The n = 2 and 3 spectra are composed of a series of sharp bands around 3600 cm−1, which are assignable to the stretching vibrations of H2O bound to C2O4− in a double ionic hydrogen-bonding (DIHB) configuration, as was previously discussed (J. Chem. Phys. 2005, 122, 094303). In the n ≥ 4 spectrum, a pair of intense bands additionally appears at ≈3300 cm−1. With the aid of ab initio calculations at the MP2/6-31+G* level, the 3300 cm−1 bands are assigned to the bending overtone and the hydrogen-bonded OH vibration of H2O bound to CO2− via a single O−H···O linkage. Thus, the structures of [(CO2)n(H2O)]− evolve with cluster size such that DIHB to C2O4− is favored in the smaller clusters with n = 2 and 3 whereas CO2− is preferentially stabilized via the formation of a single ionic hydrogen-bonding (SIHB) configuration in the larger clusters with n ≥ 4.
Co-reporter:Laura R. McCunn, Jeffrey M. Headrick and Mark A. Johnson
Physical Chemistry Chemical Physics 2008 vol. 10(Issue 21) pp:3118-3123
Publication Date(Web):10 Apr 2008
DOI:10.1039/B801372A
We report the results of an experimental study designed to establish whether, once formed, one of the isomer classes of the hydrated electron clusters, (H2O)n−, can interconvert with others when a water molecule is added by condensation. This is accomplished in an Ar-cluster mediated approach where a single intact D2O molecule is collisionally incorporated into argon-solvated water hexamer anions, creating the isotopically labeled D2O·(H2O)6−·Arn heptamer anion. Photoelectron and infrared predissociation spectroscopies are employed both to characterize the isomers generated in the condensation event and to track the position that the D2O label adopts within these isomeric structures. Despite the fact that the water hexamer anion precursor clusters initially exist in the isomer I form, incorporation of D2O produces mostly isomers I′ and II in the labeled heptamer, which bind the electron more (I′) or less (II) strongly than does the isomer I class. Isomers I and I′ are known to feature electron binding primarily onto a single water molecule that resides in an AA (A = H-bond acceptor) site in the network. Surprisingly, the D2O molecule can displace this special electron-binding H2O molecule such that the anionic cluster retains the high binding arrangement. In the more weakly binding isomer II clusters, the D2O molecule fractionates preferentially to sites that give rise to the vibrational signature of isomer II.
Co-reporter:George H. Gardenier, Joseph R. Roscioli and Mark A. Johnson
The Journal of Physical Chemistry A 2008 Volume 112(Issue 47) pp:12022-12026
Publication Date(Web):November 4, 2008
DOI:10.1021/jp800948s
We report Ar-predissociation vibrational spectra of the binary proton-bound hydrates of acetonitrile (AN), AN·H+·OH2 and AN·D+·OD2, in the 600−3800 cm−1 energy range. This complex was specifically chosen to explore the nature of the intermolecular proton bond when there is a large difference between the electric dipole moments of the two tethered molecules. Sharp, isotope-dependent bands in the vicinity of 1000 cm−1 are traced to AN·H+·OH2 vibrations involving the parallel displacement of the shared proton along the heavy atom axis, νsp(∥). These transitions lie much lower in energy than anticipated by a recently reported empirical trend which found the νsp(∥) fundamentals to be strongly correlated with the difference in proton affinities (ΔPA) between the two tethered molecules (Roscioli et al., Science, 2007, 316, 249). The different behavior of the AN·H+·OH2 complex is discussed in the context of the recent theoretical prediction (Fridgen, J. Phys. Chem A., 2006, 110, 6122) that a large disparity in dipole moments would lead to such a deviation from the reported (ΔPA) trend.
Co-reporter:Samantha Horvath and Anne B. McCoy, Joseph R. Roscioli and Mark A. Johnson
The Journal of Physical Chemistry A 2008 Volume 112(Issue 48) pp:12337-12344
Publication Date(Web):November 7, 2008
DOI:10.1021/jp805616m
Vibrational predissociation spectra of the F−(H2O)·Ar and F−(D2O)·Ar complexes are observed over a range of 600 to 3800 cm−1, which include bands attributed to the fundamentals as well as the first two overtones of the vibrations primarily associated with the shared hydrogen. This information allows us to characterize both the extended potential surface confining the anionic H-bonded hydrogen and the degree to which this motion is coupled to the motions of other atoms in the complex. We analyze these new data with reduced dimensional treatments using explicit potential energy and electric dipole moment surfaces. The often employed one-dimensional treatment with fixed OF distance does not even qualitatively account for the observed isotope dependent level structures, but a simple extension to two dimensions, corresponding to the OF distance and the shared proton position, accurately recovers the observed spectra. The resulting two-dimensional wave functions are used to evaluate the extent of proton transfer in each vibrational level. The main conclusion of this work is that vibrational excitation of the shared proton can be regarded as optically driven, intracluster proton transfer.
Co-reporter:Laura R. McCunn, Joseph R. Roscioli, Ben M. Elliott, Mark A. Johnson and Anne B. McCoy
The Journal of Physical Chemistry A 2008 Volume 112(Issue 27) pp:6074-6078
Publication Date(Web):June 18, 2008
DOI:10.1021/jp802172q
Recently, we reported the spectrum of Ar·D 4HO 2 + [McCunn; et, al. J. Phys. Chem. B 2008, 112, 321], and here, we extend that work to include the Ar·H 4DO 2 + isotopologue in order to explore why the Ar atom has a much greater propensity for attachment to a dangling OD group than it does for OH, even when many more of the latter binding sites are available. Calculated (MP2/6-311+G(d,p) level of theory/basis) harmonic frequencies reproduce the observed multiplet patterns of OH and OD stretches and confirm the presence of various isomers arising from the different Ar binding sites. The preferential bonding of Ar to OD is traced to changes in the frequencies of the wag and rock modes of the H 5O 2 + moiety rather than to shifts in the oscillator that directly binds the Ar atom.
Co-reporter:Knut R. Asmis Dr.;Yonggang Yang;Gabriele Santambrogio Dr.;Mathias Brümmer Dipl.-Ing.;J. Robert Roscioli;Laura R. McCunn;Mark A. Johnson ;Oliver Kühn Dr.
Angewandte Chemie 2007 Volume 119(Issue 45) pp:
Publication Date(Web):5 OCT 2007
DOI:10.1002/ange.200702607
Auf die Nullpunktsenergie kommt es an: In starken Wasserstoffbrücken mit niedriger Energiebarriere kommt es aufgrund quantenmechanischer Effekte zu einer strukturellen Symmetrisierung (siehe Bild). Dies führt gemeinsam mit der ausgeprägten Anharmonizität der Schwingungsbewegung zu charakteristischen IR-Spektren im Bereich unterhalb von 2000 cm−1. Durch Kombination zweier experimenteller Techniken und anharmonischer quantenmechanischer Rechnungen wird hier das IR-Spektrum für die Protonenbewegung in N2H7+ aufgeklärt.
Co-reporter:Knut R. Asmis Dr.;Yonggang Yang;Gabriele Santambrogio Dr.;Mathias Brümmer Dipl.-Ing.;J. Robert Roscioli;Laura R. McCunn;Mark A. Johnson ;Oliver Kühn Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 45) pp:
Publication Date(Web):5 OCT 2007
DOI:10.1002/anie.200702607
Zero-point energy matters: In strong, low-barrier hydrogen bonds, quantum effects cause a structural symmetrization (see picture). Together with the pronounced anharmonicity of vibrational motion, this situation gives rise to peculiar infrared (IR) spectral signatures in the region below 2000 cm−1. For the shared proton in N2H7+, the IR spectrum is elucidated by combining two experimental techniques with anharmonic quantum calculations.
Co-reporter:J. R. Roscioli;L. R. McCunn;M. A. Johnson
Science 2007 Volume 316(Issue 5822) pp:249-254
Publication Date(Web):13 Apr 2007
DOI:10.1126/science.1138962
Abstract
A proton shared between two closed-shell molecules, [A·H+·B], constitutes a ubiquitous soft binding motif in biological processes. The vibrational transitions associated with the shared proton, which provide a direct probe of this interaction, have been extensively studied in the condensed phase but have yielded only limited detailed information because of their diffuse character. We exploited recent advances in gas-phase ion spectroscopy to identify sharp spectral features that can be assigned to both the shared proton and the two tethered molecules in a survey of 18 cold, isolated [A·H+·B] ions. These data yield a picture of the intermolecular proton bond at a microscopic scale, facilitating analysis of its properties within the context of a floppy polyatomic molecule.
Co-reporter:Jeffrey M. Headrick;Eric G. Diken;Richard S. Walters;Nathan I. Hammer;Richard A. Christie;Jun Cui;Evgeniy M. Myshakin;Michael A. Duncan;Kenneth D. Jordan
Science 2005 Vol 308(5729) pp:1765-1769
Publication Date(Web):17 Jun 2005
DOI:10.1126/science.1113094
Abstract
The ease with which the pH of water is measured obscures the fact that there is presently no clear molecular description for the hydrated proton. The mid-infrared spectrum of bulk aqueous acid, for example, is too diffuse to establish the roles of the putative Eigen (H3O+) and Zundel (H5O2+) ion cores. To expose the local environment of the excess charge, we report how the vibrational spectrum of protonated water clusters evolves in the size range from 2 to 11 water molecules. Signature bands indicating embedded Eigen or Zundel limiting forms are observed in all of the spectra with the exception of the three- and five-membered clusters. These unique species display bands appearing at intermediate energies, reflecting asymmetric solvation of the core ion. Taken together, the data reveal the pronounced spectral impact of subtle changes in the hydration environment.
Co-reporter:J.-W. Shin;N. I. Hammer;E. G. Diken;M. A. Johnson;R. S. Walters;T. D. Jaeger;M. A. Duncan;R. A. Christie;K. D. Jordan
Science 2004 Vol 304(5674) pp:1137-1140
Publication Date(Web):21 May 2004
DOI:10.1126/science.1096466
Abstract
We report the OH stretching vibrational spectra of size-selected H+(H2O)n clusters through the region of the pronounced “magic number” at n = 21 in the cluster distribution. Sharp features are observed in the spectra and assigned to excitation of the dangling OH groups throughout the size range 6 ≤ n ≤ 27. A multiplet of such bands appears at small cluster sizes. This pattern simplifies to a doublet at n = 11, with the doublet persisting up to n = 20, but then collapsing to a single line in the n = 21 and n = 22 clusters and reemerging at n = 23. This spectral simplification provides direct evidence that, for the magic number cluster, all the dangling OH groups arise from water molecules in similar binding sites.
Co-reporter:Nathan I. Hammer;Joong-Won Shin;Jeffrey M. Headrick;Eric G. Diken;Joseph R. Roscioli;Gary H. Weddle
Science 2004 Vol 306(5696) pp:675-679
Publication Date(Web):22 Oct 2004
DOI:10.1126/science.1102792
Abstract
The arrangement of water molecules around a hydrated electron has eluded explanation for more than 40 years. Here we report sharp vibrational bands for small gas-phase water cluster anions, (H2O)4-6– and (D2O)4-6–. Analysis of these bands reveals a detailed picture of the diffuse electron-binding site. The electron is closely associated with a single water molecule attached to the supporting network through a double H-bond acceptor motif. The local OH stretching bands of this molecule are dramatically distorted in the pentamer and smaller clusters because the excited vibrational levels are strongly coupled to the electron continuum. The vibration–to–electronic energy transfer rates, as revealed by line shape analysis, are mode-specific and remarkably fast, with the symmetric stretching mode surviving for less than 10 vibrational periods [50 fs in (H2O)4–].
Co-reporter:M.A Johnson, T.D Märk
International Journal of Mass Spectrometry 2002 Volume 220(Issue 2) pp:97-98
Publication Date(Web):1 October 2002
DOI:10.1016/S1387-3806(02)00825-4
Co-reporter:J.M. Weber, J. Kim, E.A. Woronowicz, G.H. Weddle, I. Becker, O. Cheshnovsky, M.A. Johnson
Chemical Physics Letters 2001 Volume 339(5–6) pp:337-342
Publication Date(Web):18 May 2001
DOI:10.1016/S0009-2614(01)00295-0
Abstract
Photoexcitation of the (H2O)−n (n=20–100) clusters with 100 fs pulses at 800 nm results in an increasing propensity for two-photon electron photoejection with increasing cluster size. This increase correlates with the size range (n≈30) where the first excited electronic state drops below the electron continuum, and the electronic absorption band approaches the energy of the 800 nm pump photon. No above-threshold, two-photon detachment is observed for n=20. Differences in the shape of the resonant two-photon photoelectron spectrum compared to that arising from direct (high energy) photodetachment are interpreted in terms of the vibrational state selection created in the resonant step.
Co-reporter:Jun Kim, Jude A. Kelley, Patrick Ayotte, Steen B. Nielsen, Gary H. Weddle, Mark A. Johnson
Journal of the American Society for Mass Spectrometry 1999 Volume 10(Issue 9) pp:810-814
Publication Date(Web):September 1999
DOI:10.1016/S1044-0305(99)00057-4
We report the preparation of the bare and argon-solvated anion of CH3I, and characterize this species using negative ion photoelectron spectroscopy at 3.495 eV. The photoelectron spectrum consists of a narrow band appearing 0.11 ± 0.02 eV above the binding energy of isolated iodide. Such behavior is similar to that displayed by iodide-(closed shell) solvent molecule complexes, indicating that photodetachment does not access the bound region of the CH3I potential. These observations suggest that CH3I− rearranges (after electron capture) to an ion–radical complex. We advance the hypothesis that this complex adopts a C2v structure where the ion is hydrogen bonded to the methyl radical.
Co-reporter:Stephanie M. Craig, Fabian S. Menges, Mark A. Johnson
Journal of Molecular Spectroscopy (February 2017) Volume 332() pp:
Publication Date(Web):February 2017
DOI:10.1016/j.jms.2016.11.015
•Complex formation with two temperature-controlled RF ion traps.•Small molecule activation by docking to an open coordination site of a Ni(I) compound.•Mass selective vibrational predissociation spectroscopy reveals distortions of.•Proof of the asymmetric nature of the CO2 binding motif by isotopic labeling.Recent advances in gas phase ion chemistry, coupled with cryogenic ion vibrational predissociation spectroscopy, provide a powerful way to characterize the structures of small molecules bound to open coordination sites of organometallic compounds. Here we extend our previous measurements on the relatively weakly interacting CO2 molecule with a Ni(I) tetraaza-macrocyclic compound to enable the characterization of more strongly interacting substrates. We first confirm the calculated η2–C,O binding motif of CO2 using isotopic labeling by direct, one photon vibrational predissociation of the Ni(I)-CO2 complex. We then apply this approach to study complexation of N2 at the active site. The generality of the method is then expanded to include application to more strongly bound systems that cannot be photodissociated with one IR photon. This involves implementation of a recently developed scheme (Marsh et al., 2015) involving two temperature-controlled ion traps. The first is optimized to complex the substrate molecule to the active site and the second is cooled to around 10 K to enable condensation of weakly bound “tag” molecules onto the target complex so as to enable its characterization by linear vibrational predissociation spectroscopy. We demonstrate this capability by applying it to the coordination of CO to the active Ni(I) site, as well as to elucidate the nature of the products that are formed upon reaction with N2O.
Co-reporter:Kenny Hanke, Matin Kaufmann, Gerhard Schwaab, Martina Havenith, Conrad T. Wolke, Olga Gorlova, Mark A. Johnson, Bishnu Prasad Kar, Wolfram Sander and Elsa Sanchez-Garcia
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 13) pp:NaN8529-8529
Publication Date(Web):2015/02/12
DOI:10.1039/C5CP00116A
This study explores the interactions underlying the IR spectra of the ionic liquid [NC4111][NTf2] and its deuterated isotopomer [d9-NC4111][NTf2] by first isolating the spectra of charged ionic building blocks using mass-selective CIVP spectroscopy and then following the evolution of these bands upon sequential assembly of the ionic constituents. The spectra of the (1,1) and (2,2) neutral ion pairs are recorded using superfluid helium droplets as well as a solid neon matrix, while those of the larger charged aggregates are again obtained with CIVP. In general, the cluster spectra are similar to that of the bulk, with the (2,2) system displaying the closest resemblance. Analysis of the polarization-dependent band intensities of the neutral ion pairs in liquid droplets as a function of external electric field yields dipole moments of the neutral aggregates. This information allows a coarse assessment of the packing structure of the neutral pairs to be antiparallel at 0.37 K, in contrast to the parallel arrangement found for the assembly of small, high-dipole neutral molecules with large rotational constants (e.g., HCN). The role of an extra anion or cation attached to both the (1,1) and the (2,2) ion pairs to form the charged clusters is discussed in the context of an additional remote, more unfavorable binding site intrinsic to the nature of the charged IL clusters and as such not anticipated in the bulk phase. Whereas for the anion itself only the lowest energy trans conformer was observed, the higher clusters showed an additional population of the cis conformer. The interactions are found to be consistent with a minimal role of hydrogen bonding.
Co-reporter:Laura R. McCunn, Jeffrey M. Headrick and Mark A. Johnson
Physical Chemistry Chemical Physics 2008 - vol. 10(Issue 21) pp:NaN3123-3123
Publication Date(Web):2008/04/10
DOI:10.1039/B801372A
We report the results of an experimental study designed to establish whether, once formed, one of the isomer classes of the hydrated electron clusters, (H2O)n−, can interconvert with others when a water molecule is added by condensation. This is accomplished in an Ar-cluster mediated approach where a single intact D2O molecule is collisionally incorporated into argon-solvated water hexamer anions, creating the isotopically labeled D2O·(H2O)6−·Arn heptamer anion. Photoelectron and infrared predissociation spectroscopies are employed both to characterize the isomers generated in the condensation event and to track the position that the D2O label adopts within these isomeric structures. Despite the fact that the water hexamer anion precursor clusters initially exist in the isomer I form, incorporation of D2O produces mostly isomers I′ and II in the labeled heptamer, which bind the electron more (I′) or less (II) strongly than does the isomer I class. Isomers I and I′ are known to feature electron binding primarily onto a single water molecule that resides in an AA (A = H-bond acceptor) site in the network. Surprisingly, the D2O molecule can displace this special electron-binding H2O molecule such that the anionic cluster retains the high binding arrangement. In the more weakly binding isomer II clusters, the D2O molecule fractionates preferentially to sites that give rise to the vibrational signature of isomer II.
Co-reporter:Anne B. McCoy, Timothy L. Guasco, Christopher M. Leavitt, Solveig G. Olesen and Mark A. Johnson
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 20) pp:NaN7214-7214
Publication Date(Web):2012/01/30
DOI:10.1039/C2CP24110B
The harmonic approximation provides a powerful approach for interpreting vibrational spectra. In this treatment, the energy and intensity of the 3N − 6 normal modes are calculated using a quadratic expansion of the potential energy and a linear expansion of the dipole moment surfaces, respectively. In reality, transitions are often observed that are not accounted for by this approach (e.g. combination bands, overtones, etc.), and these transitions arise from inherent anharmonicities present in the system. One interesting example occurs in the vibrational spectrum of H2O(l), where a band is observed near 2000 cm−1 that is commonly referred to as the “association band”. This band lies far from the expected bend and stretching modes of the water molecule, and is not recovered at the harmonic level. In a recent study, we identified a band in this spectral region in gas-phase clusters involving atomic and molecular adducts to the H3O+ ion. In the current study we probe the origins of this band through a systematic analysis of the argon-predissociation spectra of H3O+·X3 where X = Ar, CH4, N2 or H2O, with particular attention to the contributions from the non-linearities in the dipole surfaces, often referred to as non-Condon effects. The spectra of the H3O+ clusters all display strong transitions between 1900–2100 cm−1, and theoretical modeling indicates that they can be assigned to a combination band involving the HOH bend and frustrated rotation of H3O+ in the solvent cage. This transition derives its oscillator strength entirely from strong non-Condon effects, and we discuss its possible relationship to the association band in the spectrum of liquid water.
Co-reporter:Etienne Garand, Joseph A. Fournier, Michael Z. Kamrath, Nathan D. Schley, Robert H. Crabtree and Mark A. Johnson
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 29) pp:NaN10113-10113
Publication Date(Web):2012/06/08
DOI:10.1039/C2CP41490B
We report the vibrational predissociation spectra of two related organometallic half-sandwich iridium species which have been recently reported as activated intermediates in the context of homogenous water oxidation. These compounds are extracted from solution into a cryogenic photofragmentation mass spectrometer and “tagged” with weakly bound H2 molecules that do not significantly perturb the intrinsic structures of the ions. The resulting spectra display very sharp (∼5 cm−1), well-resolved bands that provide a stringent test for electronic structure calculations, and are accurately recovered by harmonic predictions for the bare species. The spectra reveal subtle distortions of the ligand structure when a solvent molecule (acetonitrile) is directly coordinated with the metal center.
Co-reporter:Andrew F. DeBlase, Steven R. Kass and Mark A. Johnson
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 10) pp:NaN4575-4575
Publication Date(Web):2014/01/07
DOI:10.1039/C3CP54117G
The gas phase structure of deprotonated cysteine (Cys–H+)− has recently gained attention because of its counterintuitive calculated minimum energy structure in which it appears that deprotonation occurs at the –SH moiety rather than at the nominally more acidic carboxylic acid group. Because previous experimental efforts have not yielded to a consensus regarding the structure of the anion, we report the cryogenic ion vibrational predissociation (CIVP) spectra of its cryogenically cooled H/D isotopologues in an effort to clarify the situation. The unexpected isotope dependence of key features in the spectrum and the similarity of the band pattern to that displayed by the intramolecular H-bonded linkage in a deprotonated diacid (HCO2(CH2)10CO2−) indicate that the dominant form of the anion occurs with a strongly shared proton between the thiolate (–S−) and carboxylate (–CO2−) groups. An interesting aspect of this (–S−⋯H+⋯−O2C–) linkage is that, although the global minimum places the shared proton closer to the oxygen atom, the soft potential energy curve calculated for displacement of the bridging proton would likely support sufficient zero-point motion both to blur the distinction between thiolate- and carboxylate-based structures and to account for the unusual isotope effects.
.jpg)
![POTASSIUM, [(3-METHYLPHENYL)METHYL]-](http://img.cochemist.com/ccimg/55400/55301-51-4.png)
![POTASSIUM, [(3-METHYLPHENYL)METHYL]-](http://img.cochemist.com/ccimg/55400/55301-51-4_b.png)




![Chloro(pentamethylcyclopentadienyl)[(2-pyridinyl-kN)phenyl-kC]iridum(III)](http://img.cochemist.com/ccimg/945500/945491-51-0.png)
![Chloro(pentamethylcyclopentadienyl)[(2-pyridinyl-kN)phenyl-kC]iridum(III)](http://img.cochemist.com/ccimg/945500/945491-51-0_b.png)
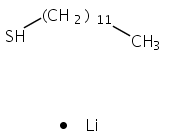
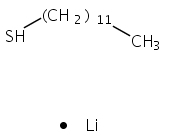




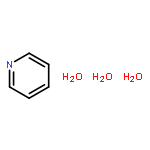
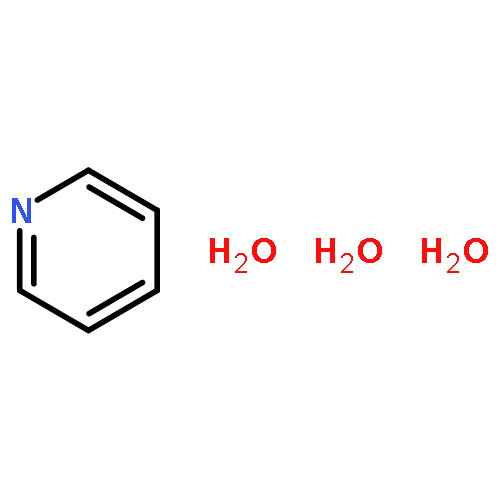




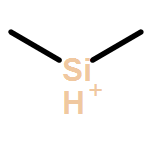
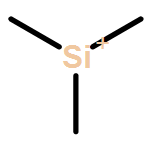


![4,7,13,16,21,24-Hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane](http://img.cochemist.com/ccimg/24000/23978-09-8.png)
![4,7,13,16,21,24-Hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane](http://img.cochemist.com/ccimg/24000/23978-09-8_b.png)