Co-reporter:Li-Hua Gan;Rui Wu;Jian-Lei Tian;Patrick W. Fowler
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 1) pp:419-425
Publication Date(Web):2016/12/21
DOI:10.1039/C6CP07370K
Structural identification is a difficult task in the study of metallofullerenes, but understanding of the mechanism of formation of these structures is a pre-requisite for new high-yield synthetic methods. Here, systematic density functional theory calculations demonstrate that metal sulfide fullerenes Sc2S@Cn have similar cage geometries from C70 to C84 and form a close-knit family of structures related by Endo–Kroto insertion/extrusion of C2 units and Stone–Wales isomerization transformations. The stabilities predicted for favoured isomers by DFT calculations are in good agreement with available experimental observations, have implications for the formation of metallofullerenes, and will aid structural identification from within the combinatorially vast pool of conceivable isomers.
Co-reporter:Li-Hua Gan, Dan Lei and Chong Zhao
RSC Advances 2015 vol. 5(Issue 39) pp:30409-30415
Publication Date(Web):30 Mar 2015
DOI:10.1039/C5RA02915E
In order to predict the structures of the detected and assumed endohedral metallofullerene Sc2S@C68, and Sc2O2@C68, and provide insights into their structures and properties, we have studied all of the isomers of C68 and tens of candidate isomers of Sc2S@C68 and Sc2O2@C68. The results show that Sc2S@C68 shares the same parent cage as Sc2C2@C68:6073, however Sc2O2 is ready to be encaged inside C68:6094. The calculations demonstrate that the transferred electrons from the encaged Sc2S and Sc2O2 clusters stabilize the active cages. Sc2S is V-shaped inside the cage whereas Sc2O2 is square-like inside the cage. Interestingly, the two Sc atoms of Sc2O2@C68 have no tendency to bond with the two fused pentagons of C68:6094 and unusually form a Sc–Sc single bond. The calculations show that encagement of Sc2O2 in C68:6094 is more favourable than that of Sc2S inside C68:6073, and that the HOMO–LUMO gap of Sc2O2@C68 is evidently broader than that of Sc2S@C68. These results suggest that there is a distinct possibility that the detected compound is Sc2O2@C68 and at least Sc2O2@C68 is much easier to synthesize under similar experimental conditions. The MS and UV spectra are provided to help their structural identification in the future.
Co-reporter:Li-Hua Gan, Dingrong Deng, Yanjun Zhang, Gen Li, Xueyun Wang, Li Jiang and Chun-Ru Wang
Journal of Materials Chemistry A 2014 vol. 2(Issue 8) pp:2461-2466
Publication Date(Web):16 Dec 2013
DOI:10.1039/C3TA14242F
Novel Zn3V2O8 hexagon nanosheets were synthesized by a simple hydrothermal method and characterized by various spectroscopic techniques. The Zn3V2O8 nanosheets were revealed to show a high performance as an anode material for lithium-ion batteries with good rate capacity, high cycling stability, and excellent discharge capacity up to 1103 mA h g−1 at a 200 mA g−1 current density.
Co-reporter:Li-Hua Gan, Dan Lei, Chong Zhao, Xiao Guo
Chemical Physics Letters 2014 Volume 604() pp:101-104
Publication Date(Web):3 June 2014
DOI:10.1016/j.cplett.2014.03.042
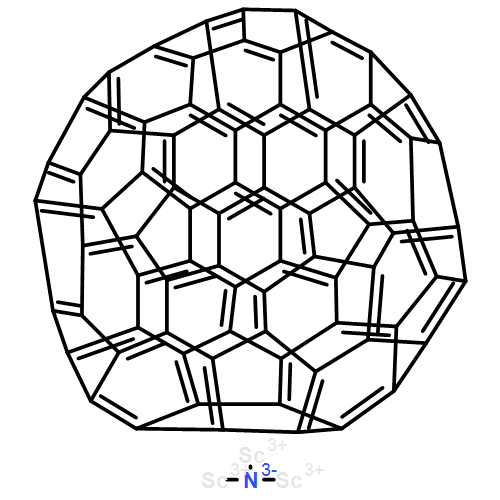
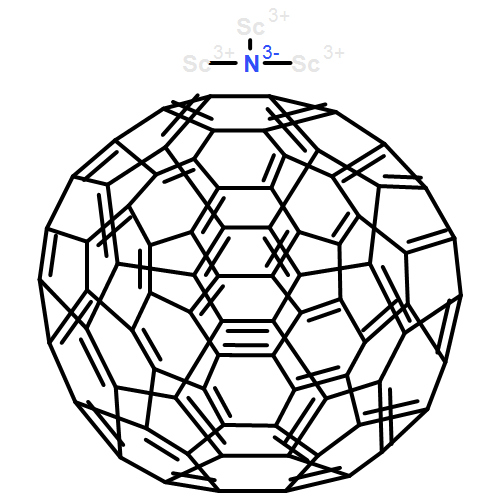
•Sc2S prefers to be encapsulated inside D5h-C80 and C2v-C80, instead of the well-known Ih-C80.•The two lowest-energy isomers of Sc2S@C80 may coexist in the soot.•Sc2S@C80 may have different properties from those usual C80-based metallofullerenes.Sc2S@C80 has been detected but not been isolated and characterized. To investigate the structures and properties of Sc2S@C80, a density functional theory study on fullerene C80 and metallofullerene Sc2S@C80 was carried out. The calculations demonstrate that Sc2S prefers to be encapsulated inside D5h-C80 and C2v-C80, instead of the well-known Ih-C80. The two lowest-energy isomers of Sc2S@C80 may coexist in the soot. The calculations reveal that there exists strong covalent interaction between the cage and Sc2S cluster, suggesting Sc2S@C80 may have different properties from those usual C80-based metallofullerenes. Raman spectra are provided to help future experimental identification of Sc2S@C80.Sc2S@C80 has been detected but not been isolated and characterized. DFT calculations demonstrate that Sc2S prefers to be encapsulated inside D5h-C80 and C2v-C80, and the two lowest-energy isomers of Sc2S@C80 may coexist in the soot. The calculations reveal that Sc2S@C80 may have different properties from those usual C80-based metallofullerenes.
Co-reporter:Chong Zhao;Dan Lei; Li-Hua Gan;Dr. Zhu-Xia Zhang; Chun-Ru Wang
ChemPhysChem 2014 Volume 15( Issue 13) pp:2780-2784
Publication Date(Web):
DOI:10.1002/cphc.201402225
Abstract
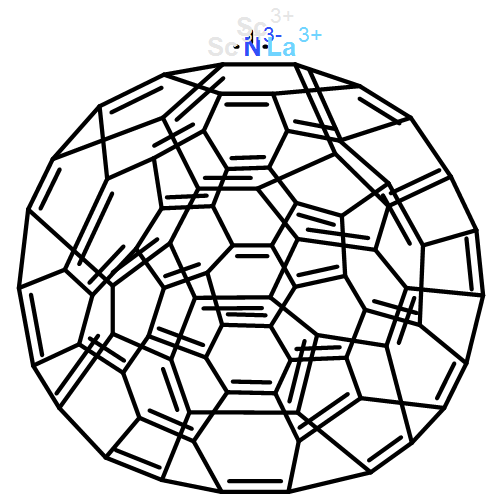
Sc2S@C84 has recently been detected but not structurally characterized.1 Density functional theory calculations on C84 and Sc2S@C84 show that the favored isomer of Sc2S@C84 shares the same parent cage as Sc2C2@C84, whereas Sc2S@C84:51383, which violates the isolated-pentagon rule, is the second lowest energy isomer with the widest HOMO–LUMO gap and shows high kinetic stability. The analysis shows that Sc2S@C84:51575 is favored when the temperature exceeds 2 800 K and it can transform into the most favorable isomer Sc2S@C84:51591. Molecular orbital analysis indicates that both Sc2S and Sc2C2 formally transfer four electrons to the cage, and quantum theory of atoms in molecules analysis demonstrates that there is a covalent interaction between Sc2S and C84:51591. The IR spectra of Sc2S@C84 are provided to aid future structural identification.
Co-reporter:Dingrong Deng;Dr. Yanjun Zhang;Gen Li;Xueyun Wang;Dr. Li-Hua Gan;Dr. Li Jiang; Chun-Ru Wang
Chemistry – An Asian Journal 2014 Volume 9( Issue 5) pp:1265-1269
Publication Date(Web):
DOI:10.1002/asia.201301632
Abstract
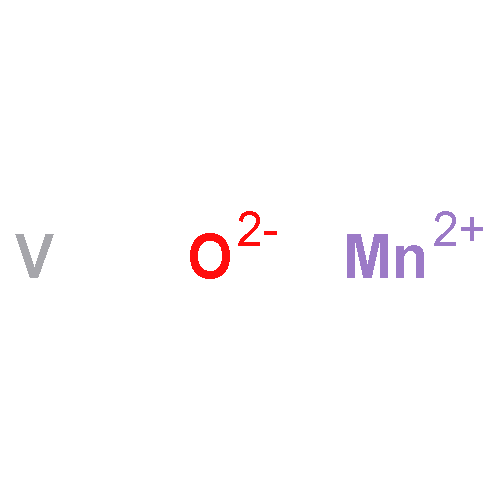
Nanometer-sized flakes of MnV2O6 were synthesized by a hydrothermal method. No surfactant, expensive metal salt, or alkali reagent was used. These MnV2O6 nanoflakes present a high discharge capacity of 768 mA h g−1 at 200 mA g−1, good rate capacity, and excellent cycling stability. Further investigation demonstrates that the nanoflake structure and the specific crystal structure make the prepared MnV2O6 a suitable material for lithium-ion batteries.
Co-reporter:Yong-Tao Shen, Li Guan, Xue-Mei Zhang, Shuai Wang, Li-Hua Gan, Qing-Dao Zeng and Chen Wang
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 30) pp:12475-12479
Publication Date(Web):28 Mar 2013
DOI:10.1039/C3CP50371B
2D porous networks have attracted great attention as they can be used to immobilize functional units as guest molecules in a spatially ordered arrangement. In this work, a novel molecular hybrid network with two kinds of cavities was fabricated. Several kinds of guest molecules, such as coronene, copper(II) phthalocyanine (CuPc), triphenylene, heptanoic acid and fullerene molecules, can be immobilized into this template. Site- and size-selective effects can be observed. Furthermore, we have also fabricated interesting 2D crystal architecture with complex four-component structure at the liquid–solid interface, following investigation by scanning tunnelling microscopy (STM). The current findings provide a convenient approach towards the formation of more complex and functionalized surface nanopatterns, which can benefit the study of host–guest assembly behaviour within a monolayer composed by several components at interfaces.
Co-reporter:Jin Zhou, Qing Chang, Li-Hua Gan and Yun-Gui Peng
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 33) pp:6732-6739
Publication Date(Web):21 Jun 2012
DOI:10.1039/C2OB25970B
The roles of benzoic acid and water on the Michael reaction of pentanal and nitrostyrene catalyzed by diarylprolinol silyl ether are revealed by density functional theory calculations. The calculations demonstrate that the benzoic acid is ready to attack the catalysts and form a hydrogen bond between the hydrogen atom of the COOH of benzoic acid and one of the N atoms of the catalyst. The complex formed from pentanal, catalyst and benzoic acid attacks nitroalkene and forms transition states. Finally, the transition states hydrolyze and the products are formed. The calculations demonstrate that the stereoselectivity is dominated by the steric hindrance of the 2-substituent groups, and the benzoic acid can increase the reaction rate evidently by decreasing the activation energies; however, H3O+ or strong acid may prevent the formation of the transition states between enamines and nitroalkenes. The employed solvent can decrease the activation energies and promote the proton transfer from benzoic acid onto the catalyst 2. The calculated enantiomeric excess values are in good agreement with the experimental results. These calculations also reveal that the role of benzoic acid is dependent on the sophisticated structures of the catalysts and provide a valuable index for the structural design of new catalysts and selection of additives or co-catalysts.
Co-reporter:Li-Hua Gan, Rui Li and Jie An
RSC Advances 2012 vol. 2(Issue 32) pp:12466-12473
Publication Date(Web):12 Oct 2012
DOI:10.1039/C2RA21720A
The structures and stability of (BN)n clusters with alternate B and N atoms containing squares, hexagons and octagons ((BN)n-F4F6F8) are investigated by using density functional theory. The results demonstrate that the isomers of (BN)n-F4F6F8 clusters generally satisfy the isolated-square rule (ISR) and the square adjacency penalty rule (SAPR). The energetically favorable isomers generally have fewer square–square bonds, larger HOMO–LUMO gaps, lower sphericity and asphericity, as well as lower pyramidalization of B and N atoms than other structures. As a whole, the stability of (BN)n-F4F6F8 clusters decreases with the number of octagons. However, four isomers containing one or two octagons in four isomeric clusters (i.e. (BN)n-F4F6F8 (n = 19, 20, 23, and 24) are more thermodynamically stable than their (BN)n-F4F6 counterparts. Further structural analysis demonstrates that octagon(s) of (BN)n-F4F6F8 clusters can release the strain energy by decreasing the pyramidalization angles of the corresponding vertex. Finally, the entropy effect is examined to evaluate the relative stability of (BN)n-F4F6F8 (n = 19, 20, 23, and 24) clusters at high temperatures.
Co-reporter:Qing Chang;Jin Zhou
Journal of Physical Organic Chemistry 2012 Volume 25( Issue 8) pp:667-673
Publication Date(Web):
DOI:10.1002/poc.2898
The mechanisms of the single and double Mannich reactions between acetaldehyde and N-Boc imines are clarified by density functional theory calculations. For single addition of Mannich reaction, the energy difference between the transition states of different configurations correspond to an enantiomeric excess value of 90.58% (without solvent) and 98.46% (in acetonitrile) in favor of the (S)-configuration product. For bis-addition of Mannich reaction, the calculated enantiomeric excess value is 95.02% (without solvent) and 98.57% (in acetonitrile) in favor of the (S, S)-configuration product. These calculated results are in good agreement with the experimental results. The calculations clearly demonstrate that the hydrogen-bonding determine the stereochemistry of the reactions. Copyright © 2012 John Wiley & Sons, Ltd.
Co-reporter:Li-Hua Gan, Qing Chang, Li Xu, Xing-Liang Huang, Chun-Ying Shu, Chun-Ru Wang
Chemical Physics Letters 2012 Volume 532() pp:68-71
Publication Date(Web):12 April 2012
DOI:10.1016/j.cplett.2012.01.082
Abstract
Classical isomers of fullerenes usually obey the pentagon adjacent penalty rule. However, density functional theory calculations demonstrate that an isomer of fullerene C60 with 15 pentagon–pentagon fusions is lower in energy than any isomer of C60 with 14 pentagon–pentagon fusions and thus violates the established pentagon adjacent penalty rule. Further examination demonstrates that this isomer follows the spherical rule; however it is anti-aromatic with positive nucleus-independent chemical shifts at the cage center and most ring centers. The calculations show that hydrogenation and cluster encapsulation are both highly exothermic and demonstrate that this isomer may be captured in the form of its derivatives.
Co-reporter:Li Xu, Shu-Fei Li, Li-Hua Gan, Chun-Ying Shu, Chun-Ru Wang
Chemical Physics Letters 2012 Volume 521() pp:81-85
Publication Date(Web):10 January 2012
DOI:10.1016/j.cplett.2011.11.011
Abstract
DFT calculations demonstrated that the parent cage of both Lu3N@C88 and Gd3N@C88 is D2 symmetrical C88:81 738 and their structures and stability are mainly determined by electron transfer interaction between the encaged cluster and the parent cage. However, for La3N@C88, the parent cage is Cs symmetric C88:81 734, La3N cluster are pyramidal in most screened cages, and the structure and stability of La3N@C88 is determined by both electron transfer interaction and size effect. The calculations can rationalize the recent experimental observations and clearly demonstrate that the size of encaged cluster play a substantial role in determining the structures and stability of M3N@C88.
Co-reporter:Li-Hua Gan;Qing Chang;Li Xu;Zuo-Hua Liu;Jun Du;Chang-Yuan Tao
Structural Chemistry 2012 Volume 23( Issue 3) pp:711-715
Publication Date(Web):2012 June
DOI:10.1007/s11224-011-9919-4
The carbon cages composed of pentagons and heptagons (F5F7 isomers) are the analogs of fullerenes composed of pentagons and hexagons (F5F6 isomers). To provide insight into the structures and stability of the hydrides of F5F6 and F5F7 isomers, systematical density functional theory calculations are performed on all the 1,812 F5F6–C60H60 and 56 F5F7–C60H60. The calculated results demonstrate that the isomer with lowest/highest energy has most/fewest fused pentagons for both F5F6 and F5F7 hydrides and the stability of these hydrides increase with the number of fused pentagons roughly. The lowest energy F5F6–C60H60 and F5F7–C60H60 are 237.1 and 152.5 kcal/mol lower in energy than the isolated pentagon rule (IPR) C60H60, respectively; however, these two parent cages are 529.6 and 660.0 kcal/mol higher in energy than the IPR C60. The calculations suggest that heptagon-containing cages, not only those violating the IPR can be the candidate cages for fullerene derivatives and the possible repulsion between the added atoms may play an important role in determining the structures and stability of the hydrides of carbon cages.
Co-reporter:Yongtao Shen, Lijin Zeng, Da Lei, Xuemei Zhang, Ke Deng, Yiyu Feng, Wei Feng, Shengbin Lei, Shufei Li, Lihua Gan, Qingdao Zeng and Chen Wang
Journal of Materials Chemistry A 2011 vol. 21(Issue 24) pp:8787-8791
Publication Date(Web):12 May 2011
DOI:10.1039/C1JM10260E
The nanoporous network formed by 1,3,5-tris(10-carboxydecyloxy) benzene (TCDB) was used as the host nanoporous network. It has been identified that a variety of guest molecules (such as triphenylene, 1-phenyloctane and copper(II) phthalocyanine (CuPc)) can be dispersed in this template to form binary supramolecular architectures, which were studied by scanning tunneling microscopy (STM). It is interesting to observe that the host network can adjust itself in response to the molecular size and shape of the guest, and the guest molecules can be excluded by some other guest molecules. The dynamics of CuPc molecules entrapped in TCDB is reported. The STM images as well as the density-functional theory (DFT) calculations reveal that the guest selectivity depends not only on geometry of guest molecules, but also on their adsorption energy in host networks.
Co-reporter:Jie An, Li-Hua Gan, Xiaoqing Fan, Fusheng Pan
Chemical Physics Letters 2011 Volume 511(4–6) pp:351-355
Publication Date(Web):5 August 2011
DOI:10.1016/j.cplett.2011.06.053
Abstract
A systematic survey is performed on all isomers of fullerene C46 composed of squares, pentagons, hexagons. Density functional calculations demonstrate that the classical isomers generally follow the pentagon adjacency penalty rule. Unexpectedly, a non-classical isomer incorporating a square, i.e., C46-10-1 is predicted to be more energetically favorable than its lowest-energy classical rival. It is interesting that the non-classical structures with one or two squares exhibit unusually large HOMO–LUMO gaps. The relative thermodynamical stability is evaluated for several low-lying isomers in terms of the Gibbs function. The maximum pyramidization angles are also introduced to evaluate their stability.
Co-reporter:He-Li Zhao, Fusheng Pan, Zuo-Hua Liu, Chang-Yuan Tao, Li-Hua Gan
Computational and Theoretical Chemistry 2011 Volume 963(Issue 1) pp:115-118
Publication Date(Web):January 2011
DOI:10.1016/j.comptc.2010.10.007
The structures and stability of fullerene derivatives were obtained from the results of density functional theory calculations that were performed on 6 non-classical and 306 classical isomers of C58X18 (X = H, F, Cl). The calculated results demonstrated that the most energetically stable isomers of C58H18 and C58F18 are heptagon-containing non-classical structures. However, for the X = Cl series of isomers, the classical C58Cl18-1156 isomer is predicted to be over 34 kcal mol−1 energetically stable than the non-classical structures. Structural analysis demonstrates that the size of added atoms plays an important role in determining the structures and stability of C58X18 (X = H, F, Cl).
Co-reporter:Dr. Li-Hua Gan;Jie An; Fu-Sheng Pan;Qing Chang; Zuo-Hua Liu; Chang-Yuan Tao
Chemistry – An Asian Journal 2011 Volume 6( Issue 6) pp:1304-1314
Publication Date(Web):
DOI:10.1002/asia.201100020
Abstract
The discovery of buckminsterfullerene C60 opened up a new scientific area and stimulated the development of nanoscience and nanotechnology directly. Fullerene science has since emerged to include fullerenes, endohedral fullerenes (mainly metallofullerenes), exofullerenes, and carbon nanotubes as well. Herein, we look back at the development of fullerene science from the perspective of epistemology by highlighting the proposed main rules or criteria for understanding and predicting the structures and stability of fullerene-based compounds. We also point out that a rule or criterion may contribute significantly to the corresponding discipline and suggest that two unsolved issues in fullerene science are the addition patterns of fullerene derivatives and the structures and stability of nonclassical fullerenes.
Co-reporter:Li-Xia Gao;Jie An;Fu-Sheng Pan
Structural Chemistry 2011 Volume 22( Issue 4) pp:749-755
Publication Date(Web):2011 August
DOI:10.1007/s11224-011-9744-9
A density functional theory study was performed on fullerene derivatives C60X18 and C70X10 (X = H, F, Cl, and Br). The calculated results show that the lowest energy isomers are IPR-satisfying for C60X18 (X = H, F, Cl, and Br). It is found that the addition patterns of X (X = Cl and Br) are different from those of X (X = H and F) for C60, demonstrating that the stability of fullerene derivatives is partly attributed to the steric repulsion and electronegativity of added atoms. However, the lowest energy isomers are IPR-violating for C70X10 (X = H, F, and Cl), suggesting that many more fullerene derivatives may violate the isolated pentagon rule.
Co-reporter:Li-Hua Gan, Rui Li, Li-Xia Gao
Journal of Molecular Structure: THEOCHEM 2010 Volume 945(1–3) pp:8-11
Publication Date(Web):15 April 2010
DOI:10.1016/j.theochem.2009.12.037
The structures and stabilities of (BN)n(BN)n polyhedrons composed of squares, hexagons and octagons with the alternation of B and N atoms are studied with density functional theory method. The calculated results demonstrate that the lowest energy isomers are the structures without octagon; the second lowest energy isomers of (BN)10(BN)10, (BN)12(BN)12 and (BN)14(BN)14 contain one, one and two octagons, respectively. The isomers with octagon(s) also satisfy the square adjacency penalty rule, and their energies markedly increase with the number of octagons. Usually, the lowest energy isomers with different number of octagons have approximate sphericity, fewest B44 bonds and large HOMO–LUMOHOMO–LUMO gaps. Structural analysis demonstrates that the pyramidalization of B and N atoms determines the stability of (BN)n(BN)n polyhedrons.
Co-reporter:Jian-Qiang Zhao
European Journal of Organic Chemistry 2009 Volume 2009( Issue 16) pp:2661-2665
Publication Date(Web):
DOI:10.1002/ejoc.200900051
Abstract
The asymmetric Michael reactions of aldehydes and nitroalkenes catalyzed by trimethylsilyl-protected diphenylprolinol were investigated by using density functional theory calculations. As a result of the stereospecific blockade of the bulky diphenylsiloxymethyl group on the pyrrolidine ring, the Re face of the enamine double bond is effectively shielded. For acetaldehyde, there are two different conformers of the enamine intermediate. On the basis of the two conformers of the enamine intermediate, four different reaction pathways were considered and four different transition states were searched for the enantioselective asymmetric Michael reaction of acetaldehyde and nitroalkene. The lowest- and second-lowest-energy transition states are both formed via the same intermediate IM2. The enantiomeric excess, calculated to be 96 % ee, is in good agreement with the experimental value. For propanal, on the basis of the four different conformers of the prolinol–enamine intermediate, eight different reaction pathways were considered and eight transition states were searched for the enantioselective asymmetric Michael reaction. The calculated ee value is 99.5 %, which is in good agreement with the experimental ee value of 99 %. The lowest- and second-lowest-energy transition states are formed via different enamine intermediates, which is different from the case of acetaldehyde. The calculations also reveal that the intermediates play an important role in the reactions.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2009)
Co-reporter:Rui Li, Li-Hua Gan, Qian Li, Jie An
Chemical Physics Letters 2009 Volume 482(1–3) pp:121-124
Publication Date(Web):6 November 2009
DOI:10.1016/j.cplett.2009.09.094
Abstract
The structures and stabilities of B13N13 polyhedrons with alternant B and N atoms formed by squares, hexagons and octagons are studied with DFT method. It is found that the isomers with octagon(s) also satisfy the square adjacency penalty rule, and their relative energies markedly increase with the number of octagons generally. However, an isomer with one octagon in C1 symmetry is thermodynamically more stable than other isomers and it has approximate sphericity and fewest B44 bonds. These findings suggest that the isomers with octagon(s) should be considered during the search for the lowest energy isomer of (BN)n polyhedrons.
Co-reporter:Li-Hua Gan, Jian Liu, Qun Hui, Song-Qing Shao, Zuo-Hua Liu
Chemical Physics Letters 2009 Volume 472(4–6) pp:224-227
Publication Date(Web):20 April 2009
DOI:10.1016/j.cplett.2009.03.028
Abstract
Density functional theory calculations are performed on several classes of carbon polyhedra with square, pentagonal, hexagonal and heptagonal faces. The calculated results demonstrate that the polyhedra obey the isolated square rule, square–pentagon adjacency penalty rule, isolated pentagon rule and pentagon adjacency penalty rule in turn. Thus the carbon polyhedra obey the isolated strain rule in general, and the well-known isolated pentagon rule and pentagon adjacency penalty rule are subordinated to it. The binding energy is fitted to the numbers of edges shared by different types of faces and a model is proposed for predicting the relative stability of these polyhedral molecules.
Co-reporter:LiHua Gan
Science China Chemistry 2009 Volume 52( Issue 5) pp:584-589
Publication Date(Web):2009 May
DOI:10.1007/s11426-009-0070-7
Based on the calculated findings that the sizes of encaged clusters determine the structures and the stability of C80-based trimetallic nitride fullerenes (TNFs), more extensive density functional theory calculations were performed on M3N@C68, M3N@C78 and M3N@C80 (M=Sc, Y and La). The calculated results demonstrated that the structures and stability undergo a transition with the increasing of the sizes of the cages and clusters. Sc3N is planar inside the three considered cages, Y3N is slightly pyramidal inside C68-6140 and C78-5 and planar inside Ih C80-7, however, La3N is pyramidal inside all the three cages. Those cages with pyramidal clusters inside deformed considerably, compared with their parent cages. In these cases, the bonding of metallic atoms toward the cages does not play an important role, and the encaged cluster tends to be located inside the cages with the largest M-M and M-C distances so that the strain energy can be released mostly. These calculations revealed the size effect of fullerene cages and encaged clusters, and can explain the position priority of M3N inside fullerene cages and the differences in yield of M3N@C2n.
Co-reporter:LiHua Gan;Jian Liu;Jian Zou;ZuoHua Liu;Li Li
Science China Chemistry 2009 Volume 52( Issue 8) pp:1085-1089
Publication Date(Web):2009 August
DOI:10.1007/s11426-009-0139-3
To gain insight into the structures and stability of F4F6 polyhedrons formed by squares and hexagons, a density functional theory study was performed on all isomers of F4F6 polyhedrons with sizes from 8 to 60. The calculated results demonstrate that the six squares tend to isolate from each other, i.e. these F4F6 polyhedrons obey the isolated square rule. Those isomers with fewer B44 bonds (square-square adjacencies) are more stable than those with more B44 bonds, i.e. they obey square adjacency penalty rule. Both of the two rules in F4F6 polyhedrons are in the same status as the isolated pentagon rule and pentagon adjacency penalty rule in F5F6 fullerenes and can be used to screen the lowest energy isomers of F4F6 polyhedrons as IPR and PAPR do in classic fullerenes. Structural analysis demonstrates that the pyramidalization of carbon atoms at the square-square adjacencies determines the stability of corresponding structures.
Co-reporter:Rui Li, Li-Hua Gan, Li Lin, Jian-Qiang Zhao, Jian Liu
Journal of Molecular Structure: THEOCHEM 2009 Volume 911(1–3) pp:75-80
Publication Date(Web):15 October 2009
DOI:10.1016/j.theochem.2009.06.044
To gain an insight into the structures and stability of F4F6-(BN)n polyhedrons with alternation of B and N atoms, a density functional theory study was performed on all isomers of F4F6-(BN)n polyhedrons with n between 10 and 22. The calculation results demonstrate that the lowest energy isomers do not contain B44 bonds (the bonds shared by two squares) and the energies of those isomers containing B44 bonds increase with the number of B44 bonds linearly, indicating that the energetically favored structures of F4F6-(BN)n polyhedrons satisfy the isolated square rule and square adjacency penalty rule. The structural analysis reveals that the stability is determined by the pyramidalization of B and N atoms at the square–square fusion. The binding energy is fitted to the numbers of edges and a model is proposed for predicting the relative stability of these B–N polyhedral molecules.
Co-reporter:Li-Hua Gan Dr.;Ruo Yuan
ChemPhysChem 2006 Volume 7(Issue 6) pp:1306-1310
Publication Date(Web):4 MAY 2006
DOI:10.1002/cphc.200500666
To provide insight into the influence of encaged clusters on the structures and stability of trimetallic nitride fullerenes (TNFs), extensive density functional theory calculations were performed on Sc3N@C80, Y3N@C80, and La3N@C80 as well as their encaged clusters. The calculated results demonstrated that both Sc3N and Y3N units are planar, whereas La3N units are pyramidal inside C80-Ih, and that both of the Y3N@C80 and La3N@C80 cages deform considerably in the planes of Y3 and La3. The calculated results suggest that M–cage attraction/repulsion and M–M repulsion interactions determine the geometries of these three complex molecules and the dynamics of the corresponding encaged clusters. These calculated findings distinctly reveal the influence of the size of the encaged clusters on the structures and stability of TNFs and may rationalize their significant differences in yields and chemical reactivity.
Co-reporter:Dong Wang, Rui Shi, Li-Hua Gan
Chemical Physics Letters (February 2017) Volume 669() pp:
Publication Date(Web):February 2017
DOI:10.1016/j.cplett.2016.12.028
•t-C8B2N2 is a potential superhard material with I-4m2 space group and its theoretical bulk modulus, shear modules, Young’s modulus, Poisson’s ratio and Vickers hardness are almost same to those of well-known superhard material c-BN.A potential superhard material C8B2N2 with I-4m2 space group is found and confirmed to be stable with first-principles calculations. The results show that its structure is highly incompressible with bulk modulus of 383.4 GPa and shear modulus of 383.0 GPa. It shows that this material is nearly isotropy with universal anisotropy index of 0.056, and its fractional anisotropy ratio of shear modulus and bulk modulus are 0.0055 and 0.0, respectively. Interestingly, its theoretical bulk modulus, shear modules, Young’s modulus, Poisson’s ratio and Vickers hardness are almost same to those of well-known superhard material c-BN.DFT calculations show that C8B2N2 with I-4m2 space group is a stable superhard material with bulk modulus of 383.4 GPa and shear modulus of 383.0 GPa, almost same to those of well-known superhard material c-BN.
Co-reporter:Li-Hua Gan, Ding-Rong Deng, Jin Zhou, Zuo-Hua Liu, Chang-Yuan Tao
Computational and Theoretical Chemistry (15 November 2012) Volume 1000() pp:
Publication Date(Web):15 November 2012
DOI:10.1016/j.comptc.2012.08.030
The carbon polyhedrons only composed of pentagons and heptagons (F5F7–Cn) are the analogues of classical fullerenes (F5F6–Cn), but they have been neglected for a long time. Very recently, we have performed a systematical study on classical C60H60 and F5F7–C60H60 without endo C–H bonds and found that the stability of F5F7–C60H60 is comparable to their classical ones. To put further insight into the derivatives of F5F7 polyhedrons, we here performed a systematical density functional theory study on F5F7–C60H60 with 1–10 endo C–H bonds. The calculations demonstrate that some isomers of F5F7–C60H60 are lower in energy than the C60H60 obeying the isolated pentagon rule (IPR) even the parent cages of them are all higher in energy than the IPR one by as much as 350 kcal/mol. Meanwhile, the F5F7–C60H60 with six endo C–H bonds is lower in energy than the IPR C60H60 without endo C–H bond by over 300 kcal/mol. These results demonstrate that F5F7 polyhedrons should not be neglected at all during the search for the favorable derivatives of carbon polyhedrons and that the repulsion between the added atoms plays an important role in determining the stability of the derivatives of carbon polyhedrons.Graphical abstractDFT calculations demonstrate that the F5F7–C60H60 with six endo C–H bonds is lower in energy than the IPR C60H60 without endo C–H bond by over 300 kcal/mol and demonstrate that the repulsion between the added atoms plays an important role in determining the stability of the derivatives of carbon polyhedrons.Highlights► A systematical DFT study was performed on F5F7–C60H60 with 1–10 endo C–H bonds. ► F5F7–H6@70C60H54 is lower in energy than the IPR C60H60 without endo C–H bond by over 300 kcal/mol. ► The repulsion between the added atoms plays an important role in cage-like CnHn.
Co-reporter:Li-Hua Gan, Rui Wu, Jian-Lei Tian and Patrick W. Fowler
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 1) pp:NaN425-425
Publication Date(Web):2016/11/22
DOI:10.1039/C6CP07370K
Structural identification is a difficult task in the study of metallofullerenes, but understanding of the mechanism of formation of these structures is a pre-requisite for new high-yield synthetic methods. Here, systematic density functional theory calculations demonstrate that metal sulfide fullerenes Sc2S@Cn have similar cage geometries from C70 to C84 and form a close-knit family of structures related by Endo–Kroto insertion/extrusion of C2 units and Stone–Wales isomerization transformations. The stabilities predicted for favoured isomers by DFT calculations are in good agreement with available experimental observations, have implications for the formation of metallofullerenes, and will aid structural identification from within the combinatorially vast pool of conceivable isomers.
Co-reporter:Yong-Tao Shen, Li Guan, Xue-Mei Zhang, Shuai Wang, Li-Hua Gan, Qing-Dao Zeng and Chen Wang
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 30) pp:NaN12479-12479
Publication Date(Web):2013/03/28
DOI:10.1039/C3CP50371B
2D porous networks have attracted great attention as they can be used to immobilize functional units as guest molecules in a spatially ordered arrangement. In this work, a novel molecular hybrid network with two kinds of cavities was fabricated. Several kinds of guest molecules, such as coronene, copper(II) phthalocyanine (CuPc), triphenylene, heptanoic acid and fullerene molecules, can be immobilized into this template. Site- and size-selective effects can be observed. Furthermore, we have also fabricated interesting 2D crystal architecture with complex four-component structure at the liquid–solid interface, following investigation by scanning tunnelling microscopy (STM). The current findings provide a convenient approach towards the formation of more complex and functionalized surface nanopatterns, which can benefit the study of host–guest assembly behaviour within a monolayer composed by several components at interfaces.
Co-reporter:Li-Hua Gan, Dingrong Deng, Yanjun Zhang, Gen Li, Xueyun Wang, Li Jiang and Chun-Ru Wang
Journal of Materials Chemistry A 2014 - vol. 2(Issue 8) pp:NaN2466-2466
Publication Date(Web):2013/12/16
DOI:10.1039/C3TA14242F
Novel Zn3V2O8 hexagon nanosheets were synthesized by a simple hydrothermal method and characterized by various spectroscopic techniques. The Zn3V2O8 nanosheets were revealed to show a high performance as an anode material for lithium-ion batteries with good rate capacity, high cycling stability, and excellent discharge capacity up to 1103 mA h g−1 at a 200 mA g−1 current density.
Co-reporter:Yongtao Shen, Lijin Zeng, Da Lei, Xuemei Zhang, Ke Deng, Yiyu Feng, Wei Feng, Shengbin Lei, Shufei Li, Lihua Gan, Qingdao Zeng and Chen Wang
Journal of Materials Chemistry A 2011 - vol. 21(Issue 24) pp:NaN8791-8791
Publication Date(Web):2011/05/12
DOI:10.1039/C1JM10260E
The nanoporous network formed by 1,3,5-tris(10-carboxydecyloxy) benzene (TCDB) was used as the host nanoporous network. It has been identified that a variety of guest molecules (such as triphenylene, 1-phenyloctane and copper(II) phthalocyanine (CuPc)) can be dispersed in this template to form binary supramolecular architectures, which were studied by scanning tunneling microscopy (STM). It is interesting to observe that the host network can adjust itself in response to the molecular size and shape of the guest, and the guest molecules can be excluded by some other guest molecules. The dynamics of CuPc molecules entrapped in TCDB is reported. The STM images as well as the density-functional theory (DFT) calculations reveal that the guest selectivity depends not only on geometry of guest molecules, but also on their adsorption energy in host networks.
Co-reporter:Jin Zhou, Qing Chang, Li-Hua Gan and Yun-Gui Peng
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 33) pp:NaN6739-6739
Publication Date(Web):2012/06/21
DOI:10.1039/C2OB25970B
The roles of benzoic acid and water on the Michael reaction of pentanal and nitrostyrene catalyzed by diarylprolinol silyl ether are revealed by density functional theory calculations. The calculations demonstrate that the benzoic acid is ready to attack the catalysts and form a hydrogen bond between the hydrogen atom of the COOH of benzoic acid and one of the N atoms of the catalyst. The complex formed from pentanal, catalyst and benzoic acid attacks nitroalkene and forms transition states. Finally, the transition states hydrolyze and the products are formed. The calculations demonstrate that the stereoselectivity is dominated by the steric hindrance of the 2-substituent groups, and the benzoic acid can increase the reaction rate evidently by decreasing the activation energies; however, H3O+ or strong acid may prevent the formation of the transition states between enamines and nitroalkenes. The employed solvent can decrease the activation energies and promote the proton transfer from benzoic acid onto the catalyst 2. The calculated enantiomeric excess values are in good agreement with the experimental results. These calculations also reveal that the role of benzoic acid is dependent on the sophisticated structures of the catalysts and provide a valuable index for the structural design of new catalysts and selection of additives or co-catalysts.

![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bS)-](/data/chemimg/103700/874948-63-7.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bS)-](/data/chemimg/103700/874948-63-7_b.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bR)-](/data/chemimg/115800/791616-63-2.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis[2,4,6-tris(1-methylethyl)phenyl]-, 4-oxide, (11bR)-](/data/chemimg/115800/791616-63-2_b.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,2,6-bis[3,5-bis(trifluoromethyl)phenyl]-4-hydroxy-, 4-oxide, (11bR)-](/data/chemimg/114800/791616-62-1.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,2,6-bis[3,5-bis(trifluoromethyl)phenyl]-4-hydroxy-, 4-oxide, (11bR)-](/data/chemimg/114800/791616-62-1_b.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis(triphenylsilyl)-, 4-oxide, (11bR)-](/data/chemimg/116500/791616-55-2.png)
![Dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin,4-hydroxy-2,6-bis(triphenylsilyl)-, 4-oxide, (11bR)-](/data/chemimg/116500/791616-55-2_b.png)
![Carbamic acid, [(4-nitrophenyl)methylene]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/877100/877031-93-1.png)
![Carbamic acid, [(4-nitrophenyl)methylene]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/877100/877031-93-1_b.png)
![Carbamic acid, [(3-nitrophenyl)methylene]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/877100/877031-92-0.png)
![Carbamic acid, [(3-nitrophenyl)methylene]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/877100/877031-92-0_b.png)
![CARBAMIC ACID, [(3-METHOXYPHENYL)METHYLENE]-, 1,1-DIMETHYLETHYL ESTER](http://img.cochemist.com/ccimg/877100/877031-91-9.png)
![CARBAMIC ACID, [(3-METHOXYPHENYL)METHYLENE]-, 1,1-DIMETHYLETHYL ESTER](http://img.cochemist.com/ccimg/877100/877031-91-9_b.png)
![Carbamic acid, [(2-chlorophenyl)methylene]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/848200/848194-75-2.png)
![Carbamic acid, [(2-chlorophenyl)methylene]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/848200/848194-75-2_b.png)


![TERT-BUTYL N-[(2-BROMOPHENYL)METHYLIDENE]CARBAMATE](http://img.cochemist.com/ccimg/779400/779342-74-4.png)
![TERT-BUTYL N-[(2-BROMOPHENYL)METHYLIDENE]CARBAMATE](http://img.cochemist.com/ccimg/779400/779342-74-4_b.png)
![CARBAMIC ACID, [(4-FLUOROPHENYL)METHYLENE]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/503000/502929-75-1.png)
![CARBAMIC ACID, [(4-FLUOROPHENYL)METHYLENE]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/503000/502929-75-1_b.png)
![CARBAMIC ACID, [(4-METHOXYPHENYL)METHYLENE]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/503000/502929-73-9.png)
![CARBAMIC ACID, [(4-METHOXYPHENYL)METHYLENE]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/503000/502929-73-9_b.png)






![Carbamic acid, [(4-bromophenyl)methylene]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/479500/479423-43-3.png)
![Carbamic acid, [(4-bromophenyl)methylene]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/479500/479423-43-3_b.png)


![CARBAMIC ACID, [(3-METHYLPHENYL)METHYLENE]-, 1,1-DIMETHYLETHYL ESTER](http://img.cochemist.com/ccimg/479500/479423-38-6.png)
![CARBAMIC ACID, [(3-METHYLPHENYL)METHYLENE]-, 1,1-DIMETHYLETHYL ESTER](http://img.cochemist.com/ccimg/479500/479423-38-6_b.png)





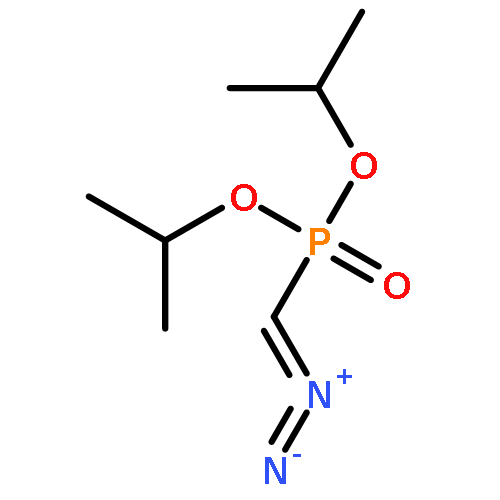
![[(dimethoxyphosphoryl)methylidene]diazenium](http://img.cochemist.com/ccimg/27500/27491-70-9.png)
![[(dimethoxyphosphoryl)methylidene]diazenium](http://img.cochemist.com/ccimg/27500/27491-70-9_b.png)












{kind=link}