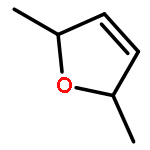
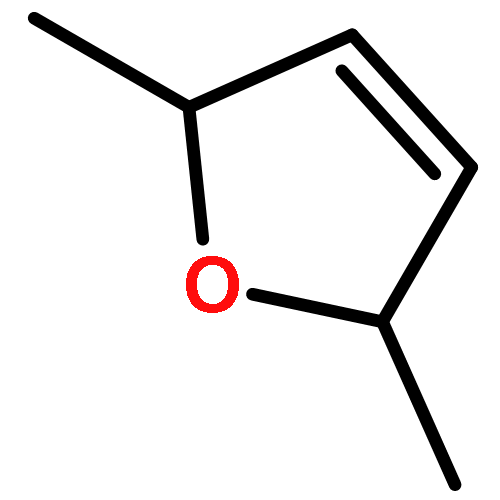
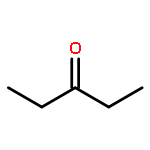
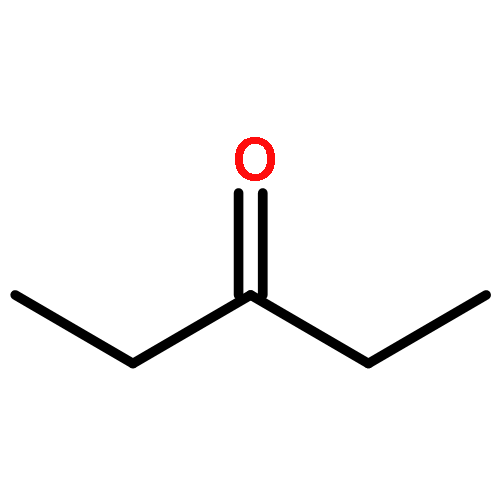
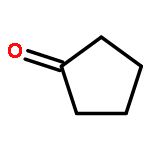
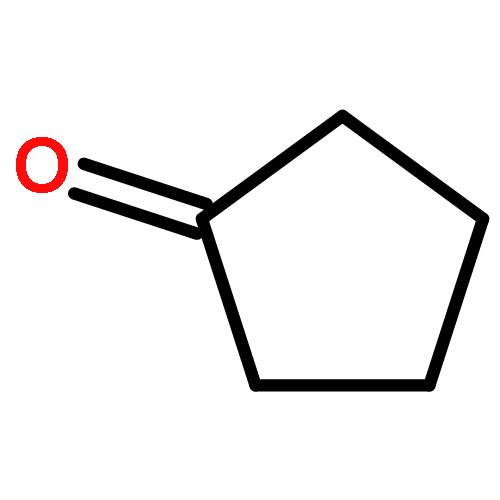
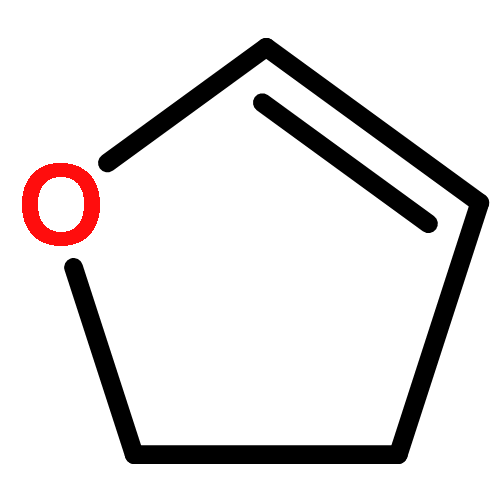
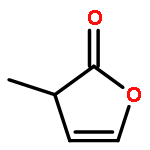
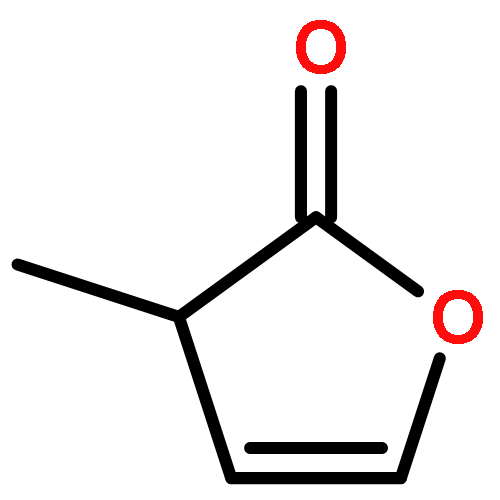
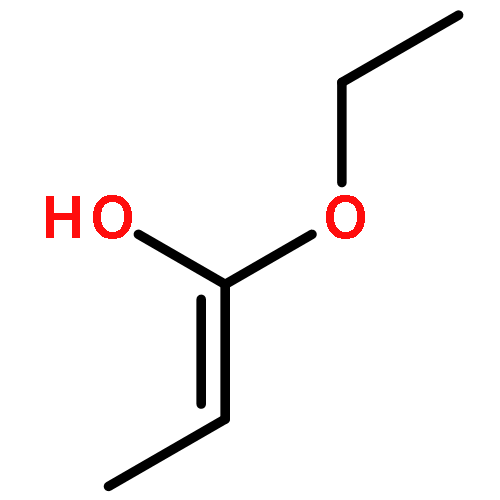
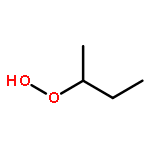
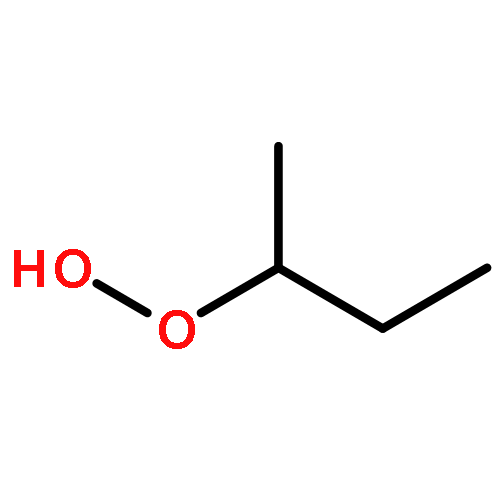
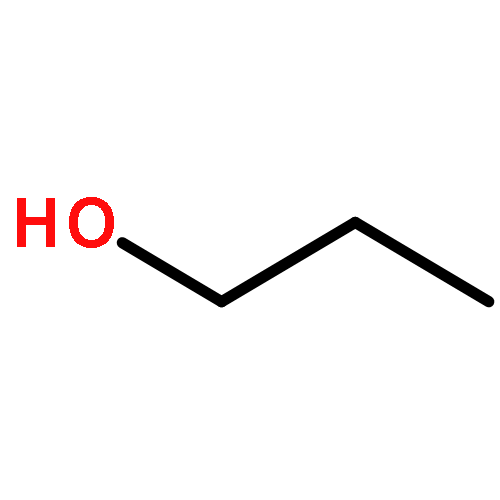
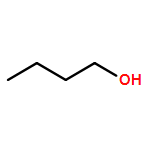
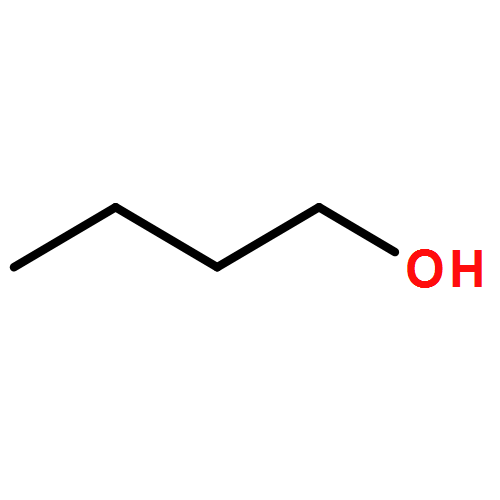
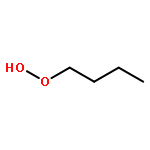
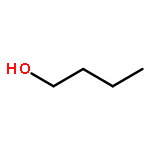
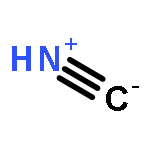
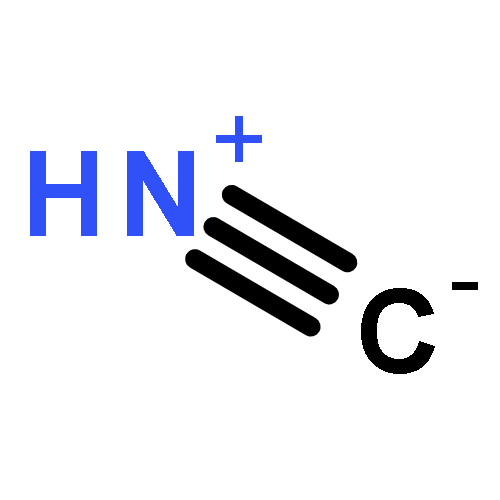
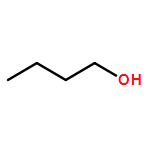
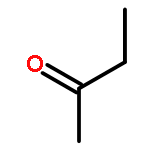
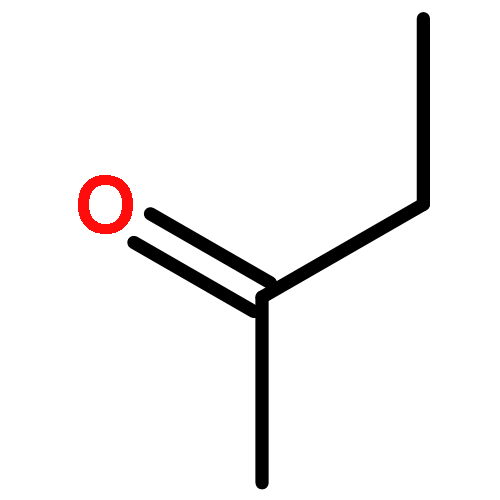
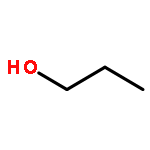
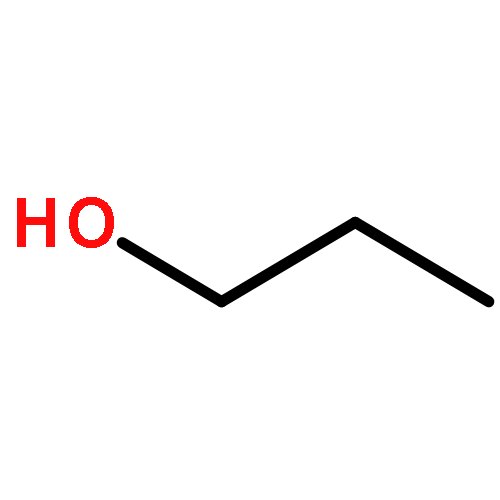
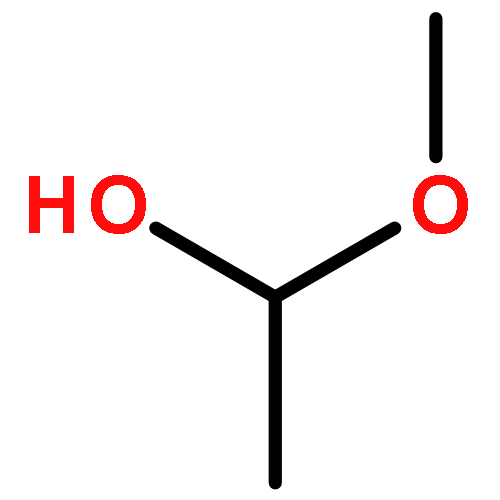
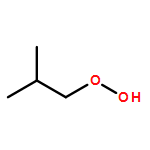
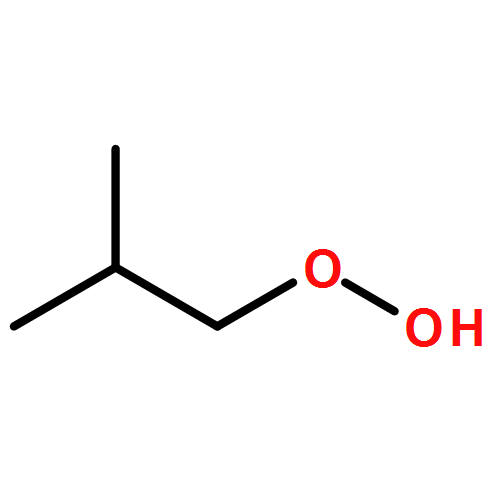
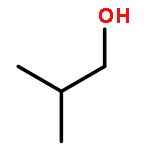
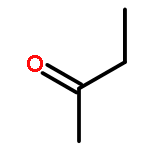
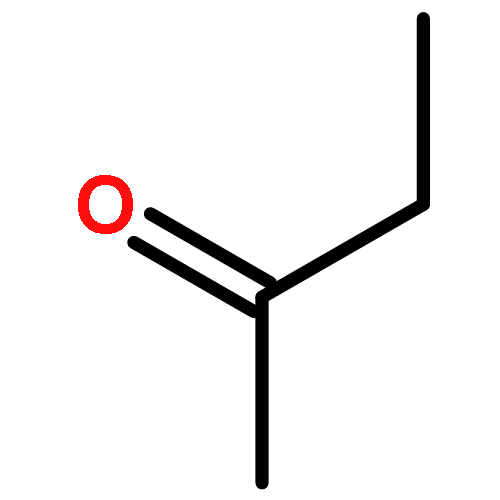
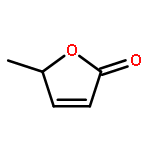
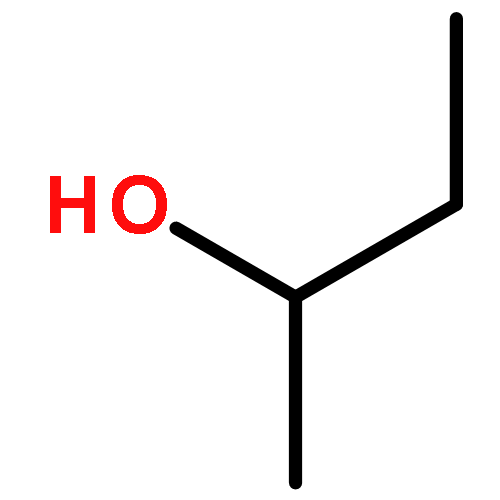
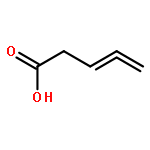
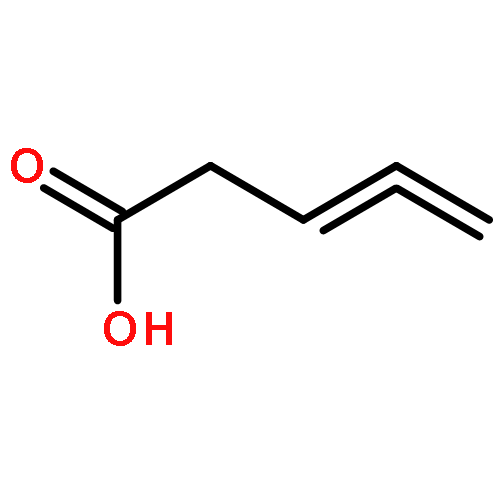
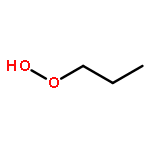
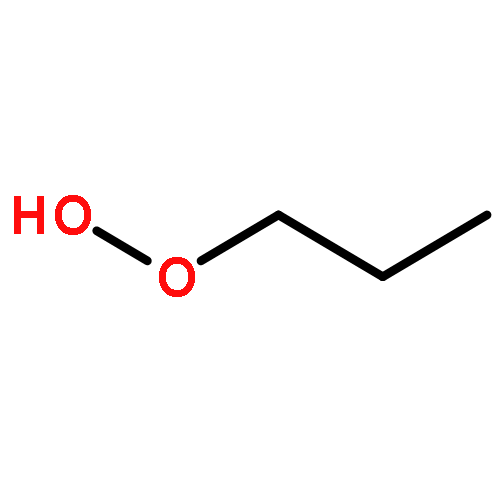
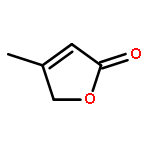
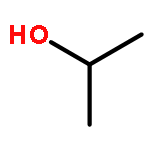
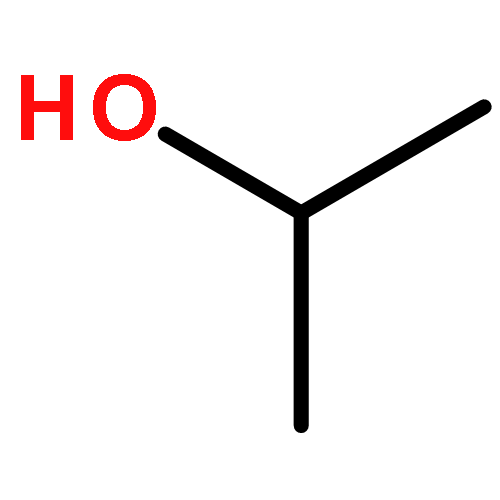
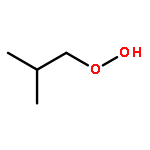
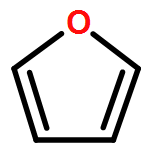
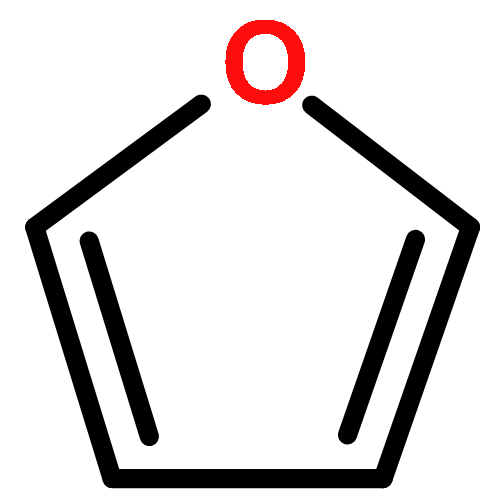
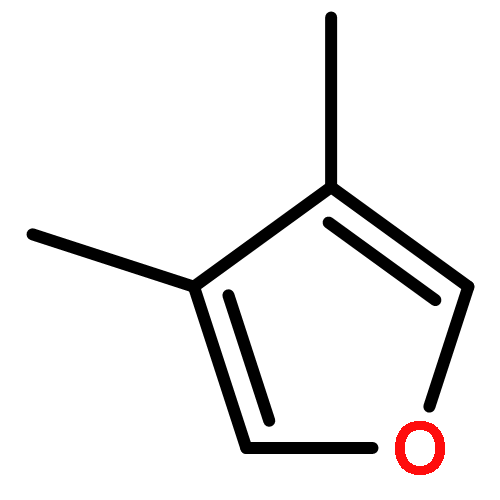
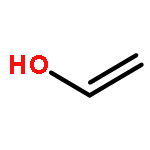
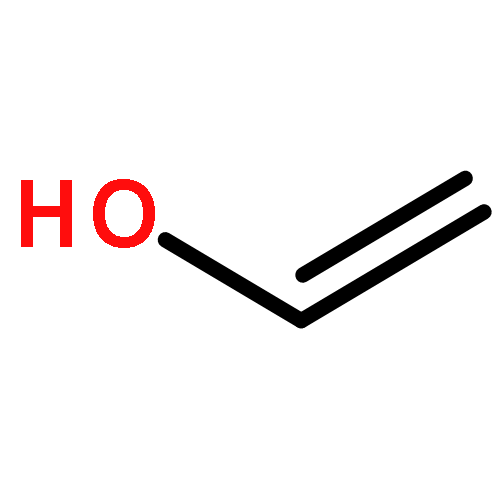
Co-reporter:J. M. Simmie and J. N. Sheahan
The Journal of Physical Chemistry A 2016 Volume 120(Issue 37) pp:7370-7384
Publication Date(Web):September 1, 2016
DOI:10.1021/acs.jpca.6b07503
In order to test new procedures for the calculation of basic molecular properties, a properly validated database and computational method appropriate to the range of species at hand is essential. Here formation enthalpies of chemical species CmHnNpOq from their constituent atoms are computed by midlevel composite model chemistries in order to check the contents of the best established and most accurate database, ATcT. Once discrepancies are identified alternative independent procedures and/or higher level model chemistries, which include CCSDT(Q) calculations, are employed to resolve the problems. Shortcomings of the midlevel methods used are signaled where these occur. In addition a more visual statistical analysis than is usual is presented which highlights the outliers and identifies the bias of each method together with associated error bars and the 95% limits of agreement and its error bars.
Co-reporter:John M. Simmie
The Journal of Physical Chemistry A 2015 Volume 119(Issue 42) pp:10511-10526
Publication Date(Web):September 30, 2015
DOI:10.1021/acs.jpca.5b06054
Accurate thermochemical data for compounds containing C/H/N/O are required to underpin kinetics simulation and modeling of the reactions of these species in different environments. There is a dearth of experimental data so computational quantum chemistry has stepped in to fill this breach and to verify whether particular experiments are in need of revision. A number of composite model chemistries (CBS-QB3, CBS-APNO, G3, and G4) are used to compute theoretical atomization energies and hence enthalpies of formation at 0 and 298.15 K, and these are benchmarked against the best available compendium of values, the Active Thermochemical Tables or ATcT. In general the agreement is very good for some 28 species with the only discrepancy being for hydrazine. It is shown that, although individually the methods do not perform that well, collectively the mean unsigned error is <1.7 kJ mol–1; hence, this approach provides a useful tool to screen published values and validate new experimental results. Using multiple model chemistries does have some drawbacks but can produce good results even for challenging molecules like HOON and CN2O2. The results for these smaller validated molecules are then used as anchors for determining the formation enthalpies of larger species such as methylated hydrazines and diazenes, five- and six-membered heterocyclics via carefully chosen isodesmic working reactions with the aim of resolving some discrepancies in the literature and establishing a properly validated database. This expanded database could be useful in testing the performance of computationally less-demanding density function methods with newer functionals that have the capacity to treat much larger systems than those tested here.
Co-reporter:John M. Simmie and Kieran P. Somers
The Journal of Physical Chemistry A 2015 Volume 119(Issue 28) pp:7235-7246
Publication Date(Web):January 12, 2015
DOI:10.1021/jp511403a
The theoretical atomization energies of some 45 CxHyOz molecules present in the Active Thermochemical Tables compilation and of particular interest to the combustion chemistry community have been computed using five composite model chemistries as titled. The species contain between 1–8 “heavy” atoms, and a few are conformationally diverse with up to nine conformers. The enthalpies of formation at 0 and 298.15 K are then derived via the atomization method and compared against the recommended values. In general, there is very good agreement between our averaged computed values and those in the ATcT; those for 1,3-cyclopentadiene exceptionally differ considerably, and we show from isodesmic reactions that the true value for 1,3-cyclopentadiene is closer to 134 kJ mol–1 than the reported 101 kJ mol–1. If one is restricted to using a single method, statistical measures indicate that the best methods are in the rank order G3 ≈ G4 > W1BD > CBS-APNO > CBS-QB3. The CBS-x methods do on average predict ΔfH⊖(298.15 K) within ≈5 kJ mol–1 but are prone to occasional lapses. There are statistical advantages to be gained from using a number of methods in tandem, and all possible combinations have been tested. We find that the average formation enthalpy coming from using CBS-APNO/G4, CBS-APNO/G3, and G3/G4 show lower mean signed and mean unsigned errors, and lower standard and root-mean-squared deviations, than any of these methods in isolation. Combining these methods also leads to the added benefit of providing an uncertainty rooted in the chemical species under investigation. In general, CBS-APNO and W1BD tend to underestimate the formation enthalpies of target species, whereas CBS-QB3, G3, and G4 have a tendency to overestimate the same. Thus, combining CBS-APNO with a G3/G4 combination leads to an improvement in all statistical measures of accuracy and precision, predicting the ATcT values to within 0.14 ± 4.21 kJ mol–1, thus rivalling “chemical accuracy” (±4.184 kJ mol–1) without the excessive cost associated with higher-level methods such as W1BD.
Co-reporter:Ian Barnes, Stefan Kirschbaum, and John M. Simmie
The Journal of Physical Chemistry A 2014 Volume 118(Issue 27) pp:5013-5019
Publication Date(Web):June 19, 2014
DOI:10.1021/jp502489k
The total rates of reaction between four cyclic esters (β-butyro-, γ-butyro-, γ-valero- and δ-valero-lactones) and the OH radical have been measured relative to the rate of reaction of a reference compound, ethene, at room temperatures. The measurements show that the rates increase with increasing ring size. Theoretical calculations on the four lactones with the inclusion of a fifth, α-methyl-γ-butyrolactone, are broadly in agreement with this picture but provide a more insightful view of the sites at which hydrogen atom abstraction occurs in each molecule.
Co-reporter: John M. Simmie;Dr. Judith Würmel
ChemSusChem 2013 Volume 6( Issue 1) pp:36-41
Publication Date(Web):
DOI:10.1002/cssc.201200738
Abstract
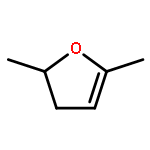
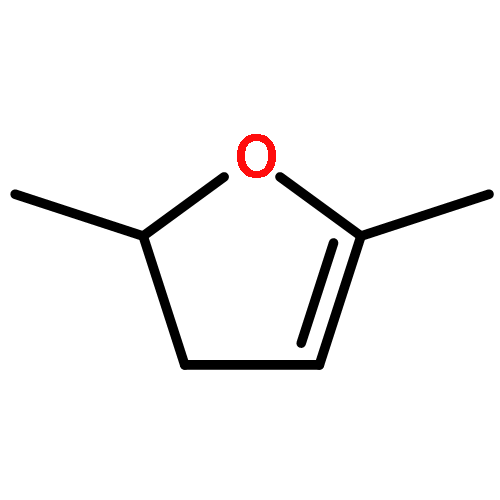
The rapid development in methods for transforming non-edible biomass into platform chemicals and fuels has accelerated over recent years. However, the determination of whether these ‘next-generation’ biofuels perform in a satisfactory manner in engines, turbines and burners has lagged behind. The evaluation of the ecological and toxicological aspects has also been unable to keep up. We show, by using 2,5-dimethylfuran (DMF) as a concrete example, how a range of studies is needed to establish the benefits and risks of using a particular biofuel. In this regard, the variable with the largest impact about which little is known is probably the behaviour of DMF when it is accidentally introduced into groundwater. A primary consideration is to avoid a repetition of the methyl tert-butyl ether (MTBE) fiasco.
Co-reporter:David Feller and John M. Simmie
The Journal of Physical Chemistry A 2012 Volume 116(Issue 47) pp:11768-11775
Publication Date(Web):November 2, 2012
DOI:10.1021/jp3095984
A high-level ab initio thermochemical technique, known as the Feller–Petersen–Dixon method, is used to calculate the total atomization energies and hence the enthalpies of formation of 2,5-dimethylfuran, 2-methylfuran, and furan itself as a means of rationalizing significant discrepancies in the literature. In order to avoid extremely large standard coupled cluster theory calculations, the explicitly correlated CCSD(T)-F12b variation was used with basis sets up to cc-pVQZ-F12. After extrapolating to the complete basis set limit and applying corrections for core/valence, scalar relativistic, and higher order effects, the final ΔfH° (298.15 K) values, with the available experimental values in parentheses are furan −34.8 ± 3 (−34.7 ± 0.8), 2-methylfuran −80.3 ± 5 (−76.4 ± 1.2), and 2,5-dimethylfuran −124.6 ± 6 (−128.1 ± 1.1) kJ mol–1. The theoretical results exhibit a compelling internal consistency.
Co-reporter:John M. Simmie
The Journal of Physical Chemistry A 2012 Volume 116(Issue 18) pp:4528-4538
Publication Date(Web):April 11, 2012
DOI:10.1021/jp301870w
The enthalpies of formation, entropies, specific heats at constant pressure, enthalpy functions, and all carbon–hydrogen and carbon–methyl bond dissociation energies have been computed using high-level methods for the cyclic ethers (oxolanes) tetrahydrofuran, 2-methyltetrahydrofuran, and 2,5-dimethyltetrahydrofuran. Barrier heights for hydrogen-abstraction reactions by hydrogen atoms and the methyl radical are also computed and shown to correlate with reaction energy change. The results show a pleasing consistency and considerably expands the available data for these important compounds. Abstraction by ȮH is accompanied by formation of both pre- and postreaction weakly bound complexes. The resulting radicals formed after abstraction undergo ring-opening reactions leading to readily recognizable intermediates, while competitive H-elimination reactions result in formation of dihydrofurans. Formation enthalpies of all 2,3- and 2,5-dihydrofurans and associated radicals are also reported. It is probable that the compounds at the center of this study will be relatively clean-burning biofuels, although formation of intermediate aldehydes might be problematic.
Co-reporter:John M. Simmie and Wayne K. Metcalfe
The Journal of Physical Chemistry A 2011 Volume 115(Issue 32) pp:8877-8888
Publication Date(Web):June 16, 2011
DOI:10.1021/jp2039477
The initial steps in the thermal decomposition of 2,5-dimethylfuran are identified as scission of the C–H bond in the methyl side chain and formation of β- and α-carbenes via 3,2-H and 2,3-methyl shifts, respectively. A variety of channels are explored which prise the aromatic ring open and lead to a number of intermediates whose basic properties are essentially unknown. Once the furan ring is opened demethylation to yield highly unsaturated species such as allenylketenes appears to be a feature of this chemistry. The energetics of H abstraction by the hydroxyl radical (and other abstracting species) from a number of mono- and disubstituted methyl furans has been studied. H-atom addition to 2,5-dimethylfuran followed by methyl elimination is shown to be the most important route to formation of the less reactive 2-methylfuran. Identification of 2-ethenylfuran as an C6H6O intermediate in 2,5-dimethylfuran flames is probably not correct and is more likely the isomeric 2,5-dimethylene-2,5-dihydrofuran for which credible formation channels exist.
Co-reporter:John M. Simmie and Henry J. Curran
The Journal of Physical Chemistry A 2009 Volume 113(Issue 27) pp:7834-7845
Publication Date(Web):June 11, 2009
DOI:10.1021/jp903244r
Although enols have been identified in alcohol and other flames and in interstellar space and have been implicated in the formation of carboxylic acids in the urban troposphere in the past few years, the reactions that give rise to them are virtually unknown. To address this data deficit, particularly with regard to biobutanol combustion, we have carried out a number of ab initio calculations with the multilevel methods CBS-QB3 and CBS-APNO to determine the activation enthalpies for methyl addition to the CH2 group of CH2═CHX where X = H, OH, and CH3. These average at 26.3 ± 1.0 kJ mol−1 and are not influenced by the nature of X; addition to the CHX end is energetically costlier and does show the influence of group X = OH and CH3. Replacing the attacking methyl radical by ethyl makes very little difference to addition at CH2 and follows the same trend of a higher barrier for addition to the CH(OH) end. In the case of H-addition it is more problematic to draw general conclusions since the DFT-based methodology, CBS-QB3, struggles to locate transition states for some reactions. However, the increase in barrier heights in reaction at the CHX end in comparison to addition at the methylene end is evident. For hydrogen atom reaction with the carbonyl group in the compounds methanal, ethanal, propanal, and butanal we see that for addition at the O-center the barrier heights of ca. 38 kJ mol−1 are not influenced by the nature of the alkyl group whereas addition at the C-center is different on going from H → alkyl but seems to be invariant at 20 kJ mol−1 once alkylated. Rate constants for H-atom elimination from 1-hydroxyethyl, 1-hydroxypropyl, and 1-hydroxybutyl radicals, valid over the range 800−2000 K, are reported. These demonstrate that enols are more prevalent than previously suspected and that 1-buten-1-ol should be almost as abundant as its isomeric aldehyde 1-butanal during the combustion of 1-butanol and that this will also be the case for other alcohols provided that the appropriate structural features are present. Since the toxicity of enols is not known experiments and further theoretical studies are clearly desirable before the large-scale usage of alcohol biofuels commences. An enthalpy of formation for butanal of ΔfH(298.15 K) = −204.4 ± 1.4 kJ mol−1 [Buckley, E.; Cox, J. D. Trans. Faraday Soc. 1967, 63, 895−901] is recommended, the uncertainty surrounding that for the 2-hydroxypropyl radical has been markedly reduced, and new values for 1-buten-1-ol, 1-propen-1-ol, and 2-propen-2-ol of −171.8 ± 1.6, −151.8 ± 1.7, and −169.9 ± 1.5 kJ mol−1, respectively, are proposed.
Co-reporter:John M. Simmie and Henry J. Curran
The Journal of Physical Chemistry A 2009 Volume 113(Issue 17) pp:5128-5137
Publication Date(Web):March 30, 2009
DOI:10.1021/jp810315n
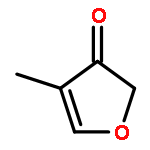
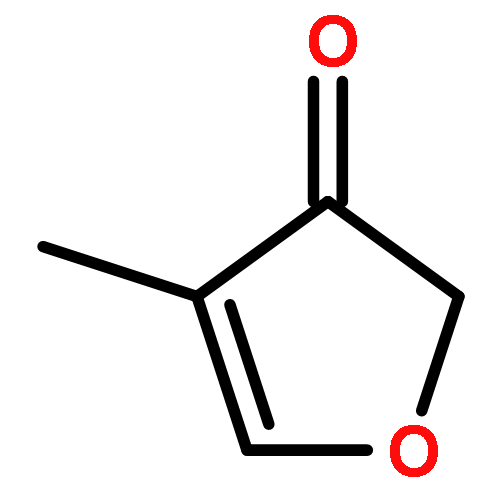
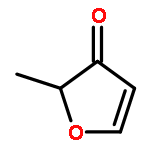
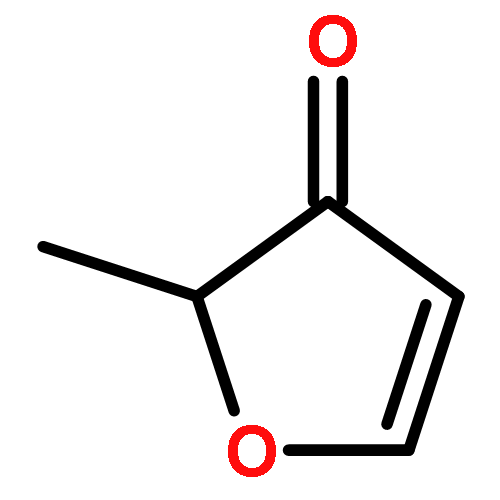
Enthalpies of formation, ΔHf(298.15 K), of 2-methyl-, 3-methyl-, 2-ethyl-, 2-vinyl-, 2,3-dimethyl-, 2,4-dimethyl-, and 3,4-dimethylfurans are computed with three compound quantum chemical methods, CBS-QB3, CBS-APNO, and G3, via a number of isodesmic reactions. We show that previously experimentally determined enthalpies of formation of furan itself, 2,5-dimethyl-, 2-tert-butyl-, and 2,5-di-tert-butylfurans are self-consistent but that for 2-vinylfuran is most probably in error. The formation enthalpies of over 20 furyl and furfuryl radicals have also been determined and consequently the bond dissociation energies of a number of C−H, C−CH3, C−F, C−Cl, and C−OH bonds. The ring-carbon−H bonds in alkylfurans are much stronger than previously thought and are among the strongest ever C−H bonds recorded exceeding 500 kJ mol−1. The relative thermodynamic instability of the various furyl radicals means that bonds to methyl, fluorine, and chlorine are also unusually strong. This is as a consequence of the inability of the radical to effectively delocalize the unpaired electron and the geometrical inflexibility of the five-membered heterocyclic ring. By way of contrast the furfuryl radicals are more stable than similar benzyl radicals which results in weaker side-chain C−H bonds than the corresponding toluene derivatives (although stronger than the corresponding cyclopentadiene analogue). These results have implications for the construction of detailed chemical kinetic models to account for the thermal decomposition and oxidation of alkylfurans either in engines or in the atmosphere.
Co-reporter:Ahmed M. El-Nahas, John M. Simmie, Maria V. Navarro, Joseph W. Bozzelli, Gráinne Black and Henry J. Curran
Physical Chemistry Chemical Physics 2008 vol. 10(Issue 47) pp:7139-7149
Publication Date(Web):20 Oct 2008
DOI:10.1039/B810853F
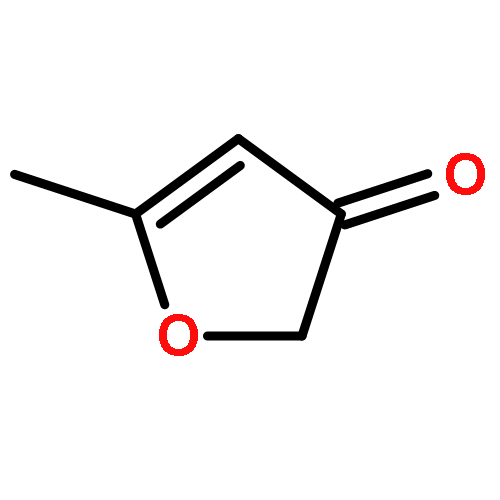
CBS-QB3 calculations have been used to determine thermochemical and kinetic parameters of the isomerisation and decomposition reactions of the acetonylperoxy radical, CH3C(O)CH2OO˙, which has been formed via the reaction of acetonyl radical with O2 leading to the formation of an energised peroxy adduct with a calculated well depth of near 111 kJ mol−1. This species can undergo subsequent 1,5 and 1,3 H-shifts to give the primary and secondary radicals: C˙H2C(O)CH2OOH and CH3C(O)C˙HOOH, respectively, or rearrange to give a 3-methyl-1,2-dioxetan-3-yloxy radical. Rate constants for isomerisation and subsequent decomposition have been estimated using canonical variational transition state theory with small curvature tunneling CVT/SCT. The variational effect for the isomerisation channels is only moderate but the tunneling correction is significant at temperatures up to 1000 K; the formation of a primary radical by a 1,5-shift is the main reaction channel and the competition with the secondary one starts only at around 1500 K. The fate of the primary acetonylhydroperoxy radical is predominantly to form oxetan-3-one while the ketene and 1-oxy-3-hydroxyacetonyl radical channels only compete with the formation of oxetan-3-one at temperatures >1200 K. In addition, consistent and reliable enthalpies of formation have been computed for the molecules acetonylhydroperoxide, 1,3-dihydroxyacetone, methylglyoxal and cyclobutanone, and for some related radicals.
Co-reporter:John M. Simmie, Gráinne Black, Henry J. Curran and John P. Hinde
The Journal of Physical Chemistry A 2008 Volume 112(Issue 22) pp:5010-5016
Publication Date(Web):May 8, 2008
DOI:10.1021/jp711360z
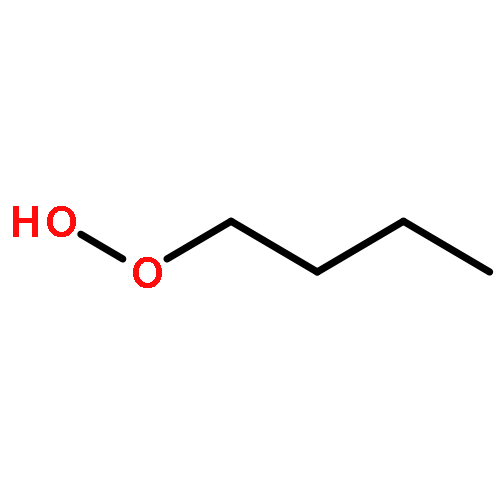
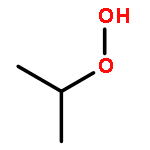
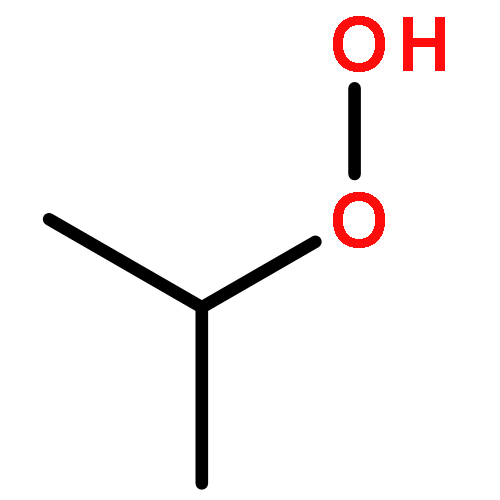
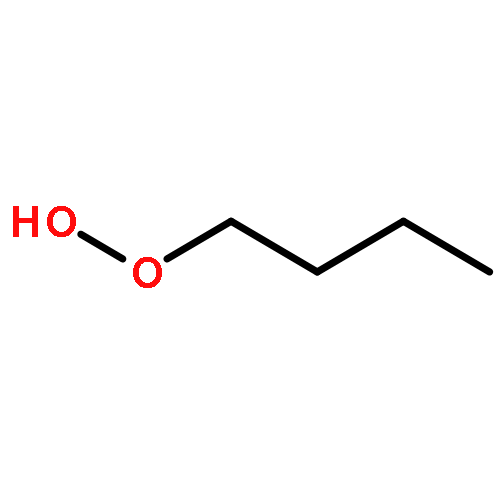
The enthalpies of formation and bond dissociation energies, D(ROO−H), D(RO−OH), D(RO−Ȯ), D(R−Ȯ2) and D(R−OOH) of alkyl hydroperoxides, ROOH, alkyl peroxy, RO, and alkoxide radicals, RȮ, have been computed at CBS-QB3 and APNO levels of theory via isodesmic and atomization procedures for R = methyl, ethyl, n-propyl and isopropyl and n-butyl, tert-butyl, isobutyl and sec-butyl. We show that D(ROO−H) ≈ 357, D(RO−OH) ≈ 190 and D(RO−Ȯ) ≈ 263 kJ mol−1 for all R, whereas both D(R−ȮO) and D(R−OOH) strengthen with increasing methyl substitution at the α-carbon but remain constant with increasing carbon chain length. We recommend a new set of group additivity contributions for the estimation of enthalpies of formation and bond energies.
Co-reporter:John M. Simmie ;Wayne K. Metcalfe ;Henry J. Curran Dr.
ChemPhysChem 2008 Volume 9( Issue 5) pp:700-702
Publication Date(Web):
DOI:10.1002/cphc.200800003
Co-reporter:Ahmed M. El-Nahas, John M. Simmie, Maria V. Navarro, Joseph W. Bozzelli, Gráinne Black and Henry J. Curran
Physical Chemistry Chemical Physics 2008 - vol. 10(Issue 47) pp:NaN7149-7149
Publication Date(Web):2008/10/20
DOI:10.1039/B810853F
CBS-QB3 calculations have been used to determine thermochemical and kinetic parameters of the isomerisation and decomposition reactions of the acetonylperoxy radical, CH3C(O)CH2OO˙, which has been formed via the reaction of acetonyl radical with O2 leading to the formation of an energised peroxy adduct with a calculated well depth of near 111 kJ mol−1. This species can undergo subsequent 1,5 and 1,3 H-shifts to give the primary and secondary radicals: C˙H2C(O)CH2OOH and CH3C(O)C˙HOOH, respectively, or rearrange to give a 3-methyl-1,2-dioxetan-3-yloxy radical. Rate constants for isomerisation and subsequent decomposition have been estimated using canonical variational transition state theory with small curvature tunneling CVT/SCT. The variational effect for the isomerisation channels is only moderate but the tunneling correction is significant at temperatures up to 1000 K; the formation of a primary radical by a 1,5-shift is the main reaction channel and the competition with the secondary one starts only at around 1500 K. The fate of the primary acetonylhydroperoxy radical is predominantly to form oxetan-3-one while the ketene and 1-oxy-3-hydroxyacetonyl radical channels only compete with the formation of oxetan-3-one at temperatures >1200 K. In addition, consistent and reliable enthalpies of formation have been computed for the molecules acetonylhydroperoxide, 1,3-dihydroxyacetone, methylglyoxal and cyclobutanone, and for some related radicals.