Co-reporter:Hongjin Lv, T. Purnima A. Ruberu, Valerie E. Fleischauer, William W. Brennessel, Michael L. Neidig, and Richard Eisenberg
Journal of the American Chemical Society 2016 Volume 138(Issue 36) pp:11654-11663
Publication Date(Web):September 1, 2016
DOI:10.1021/jacs.6b05040
The development of active, robust systems for light-driven hydrogen production from aqueous protons based on catalysts and light absorbers composed solely of earth abundant elements remains a challenge in the development of an artificial photosynthetic system for water splitting. Herein, we report the synthesis and characterization of four closely related Fe bis(benzenedithiolate) complexes that exhibit catalytic activity for hydrogen evolution when employed in systems with water-soluble CdSe QDs as photosensitizer and ascorbic acid as a sacrificial electron source under visible light irradiation (520 nm). The complex with the most electron-donating dithiolene ligand exhibits the highest activity, the overall order of activity correlating with the reduction potential of the formally Fe(III) dimeric dianions. Detailed studies of the effect of different capping agents and the extent of surface coverage of these capping agents on the CdSe QD surfaces reveal that they affect system activity and provide insight into the continued development of such systems containing QD light absorbers and molecular catalysts for H2 formation.
Co-reporter:Po-Yu Ho, Bo Zheng, Daniel Mark, Wai-Yeung Wong, David W. McCamant, and Richard Eisenberg
Inorganic Chemistry 2016 Volume 55(Issue 17) pp:8348-8358
Publication Date(Web):August 17, 2016
DOI:10.1021/acs.inorgchem.6b00496
Two new dyads have been synthesized and studied as photosensitizers for the light-driven generation of H2 from aqueous protons. One of the dyads, Dy-1, consists of a strongly absorbing Bodipy (dipyrromethene-BF2) dye and a platinum diimine benzenedithiolate (bdt) charge transfer (CT) chromophore, denoted as PtN2S2. The two components are connected through an amide linkage on the bdt side of the PtN2S2 complex. The second dyad, Dy-2, contains a diketopyrrolopyrrole dye that is linked directly to the acetylide ligands of a Pt diimine bis(arylacetylide) CT chromophore. The two dyads, as well as the Pt diimine bis(arylacetylide) CT chromophore, were attached to platinized TiO2 via phosphonate groups on the diimine through sonication of the corresponding esters, and each system was examined for photosensitizer effectiveness in photochemical generation of H2 from aqueous protons and electrons supplied by ascorbic acid. Of the three photosensitizers, Dy-1 is the most active under 530 nm radiation with an initial turnover frequency of 260 h–1 and a total of 6770 turnovers over 60 h of irradiation. When a “white” LED light source is used, samples with Dy-2 and the Pt diimine bis(arylacetylide) chromophore, while not as effective as Dy-1, perform relatively better. A key conclusion is that the presence of a strongly absorbing organic dye increases dyad photosensitizer effectiveness only if the energy of the CT excited state lies below that of the organic dye’s lowest excited state; if not, the organic dye does not improve the effectiveness of the CT chromophore for promoting electron transfer and the light-driven generation of H2. The nature of the spacer between the organic dye and the charge transfer chromophore also plays a role in the effectiveness of using dyads to improve light-driven energy-storing reactions.
Co-reporter:Randy P. Sabatini, Brian Lindley, Theresa M. McCormick, Theodore Lazarides, William W. Brennessel, David W. McCamant, and Richard Eisenberg
The Journal of Physical Chemistry B 2016 Volume 120(Issue 3) pp:527-534
Publication Date(Web):January 5, 2016
DOI:10.1021/acs.jpcb.5b11035
A series of Boron-dipyrromethene (Bodipy) dyes were used as photosensitizers for photochemical hydrogen production in conjunction with [CoIII(dmgH)2pyCl] (where dmgH = dimethylglyoximate, py = pyridine) as the catalyst and triethanolamine (TEOA) as the sacrificial electron donor. The Bodipy dyes are fully characterized by electrochemistry, X-ray crystallography, quantum chemistry calculations, femtosecond transient absorption, and time-resolved fluorescence, as well as in long-term hydrogen production assays. Consistent with other recent reports, only systems containing halogenated chromophores were active for hydrogen production, as the long-lived triplet state is necessary for efficient bimolecular electron transfer. Here, it is shown that the photostability of the system improves with Bodipy dyes containing a mesityl group versus a phenyl group, which is attributed to increased electron donating character of the mesityl substituent. Unlike previous reports, the optimal ratio of chromophore to catalyst is established and shown to be 20:1, at which point this bimolecular dye/catalyst system performs 3–4 times better than similar chemically linked systems. We also show that the hydrogen production drops dramatically with excess catalyst concentration. The maximum turnover number of ∼700 (with respect to chromophore) is obtained under the following conditions: 1.0 × 10–4 M [Co(dmgH)2pyCl], 5.0 × 10–6 M Bodipy dye with iodine and mesityl substituents, 1:1 v:v (10% aqueous TEOA):MeCN (adjusted to pH 7), and irradiation by light with λ > 410 nm for 30 h. This system, containing discrete chromophore and catalyst, is more active than similar linked Bodipy–Co(dmg)2 dyads recently published, which, in conjunction with our other measurements, suggests that the nominal dyads actually function bimolecularly.
Co-reporter:T. Purnima A. Ruberu, Yuming Dong, Amit Das, and Richard Eisenberg
ACS Catalysis 2015 Volume 5(Issue 4) pp:2255
Publication Date(Web):February 6, 2015
DOI:10.1021/cs5021035
The present study reports photelectrochemical H2 evolution using a water-solubilized S3-cap-CdSe quantum dot-sensitized NiO as the photocathode and either [Co(bdt)2]− (bdt =1,2-benzenedithiolate) or Ni(DHLA)x (DHLA= the anion of dihydrolipoic acid) complex as the H2-forming catalyst. The NiO-S3-cap-CdSe/[Co(bdt)2]− system produces H2 with a turnover frequency of 3000 per CdSe mol·h. Faradaic efficiency for this system is essentially quantitative. Both systems are stable for more than 16 h.Keywords: hydrogen; molecular catalyst; photocathodes; photoelectrochemistry; quantum dots; solar energy
Co-reporter:Amit Das, Zhiji Han, William W. Brennessel, Patrick L. Holland, and Richard Eisenberg
ACS Catalysis 2015 Volume 5(Issue 3) pp:1397
Publication Date(Web):January 14, 2015
DOI:10.1021/acscatal.5b00045
A series of nickel bis(chelate) complexes having square planar coordination are studied for light-driven and electrocatalytic hydrogen production from water. The complexes Ni(abt)2 (abt = 2-aminobenzenethiolate), Ni(mp)2 (mp = 2-mercaptophenolate) and Ni(mpo)2 (mpo = 2-mercaptopyridyl-N-oxide) are found to be active catalysts under light-driven conditions, using fluorescein (Fl) as the photosensitizer (PS) and triethanolamine (TEOA) as the sacrificial electron donor in water under basic pH (pH = 9.8). These molecular systems achieve a turnover number (TON) of ∼6000 (relative to catalyst) and are stable for more than 100 h under H2-generating conditions. When water-soluble CdSe quantum dots with tripodal S-donor capping agents are employed as PS and ascorbic acid (AA) is used as the sacrificial electron donor at pH 4.5, an active and robust system is obtained for the light-driven generation of H2 from aqueous protons. A TON of over 280 000 is achieved for the three active catalysts. These complexes are also examined electrochemically in organic solvents with weak organic acids as the proton source and in aqueous and aqueous/organic media for proton reduction. The most active photochemical catalysts also show excellent electrocatalytic activity in neutral pH water, achieving Faradaic yields close to 100% under anaerobic conditions and ∼80% under aerobic conditions.Keywords: CdSe quantum dots; fluorescein; HER mechanism; heterocoupling; hydrogen evolution; molecular HER catalysis; Ni catalysts with redox active ligands; photochemistry
Co-reporter:Kara L. Bren;Harry B. Gray
PNAS 2015 112 (43 ) pp:13123-13127
Publication Date(Web):2015-10-27
DOI:10.1073/pnas.1515704112
Two articles published by Pauling and Coryell in PNAS nearly 80 years ago described in detail the magnetic properties of oxy-
and deoxyhemoglobin, as well as those of closely related compounds containing hemes. Their measurements revealed a large difference
in magnetism between oxygenated and deoxygenated forms of the protein and, along with consideration of the observed diamagnetism
of the carbonmonoxy derivative, led to an electronic structural formulation of oxyhemoglobin. The key role of hemoglobin as
the main oxygen carrier in mammalian blood had been established earlier, and its allosteric behavior had been described in
the 1920s. The Pauling–Coryell articles on hemoglobin represent truly seminal contributions to the field of bioinorganic chemistry
because they are the first to make connections between active site electronic structure and the function of a metalloprotein.
Co-reporter:Bo Zheng;Randy P. Sabatini;Wen-Fu Fu;Min-Sik Eum;William W. Brennessel;Lidong Wang;David W. McCamant
PNAS 2015 Volume 112 (Issue 30 ) pp:E3987-E3996
Publication Date(Web):2015-07-28
DOI:10.1073/pnas.1509310112
New dyads consisting of a strongly absorbing Bodipy (dipyrromethene-BF2) dye and a platinum diimine dithiolate (PtN2S2) charge transfer (CT) chromophore have been synthesized and studied in the context of the light-driven generation of H2 from aqueous protons. In these dyads, the Bodipy dye is bonded directly to the benzenedithiolate ligand of the PtN2S2 CT chromophore. Each of the new dyads contains either a bipyridine (bpy) or phenanthroline (phen) diimine with an attached
functional group that is used for binding directly to TiO2 nanoparticles, allowing rapid electron photoinjection into the semiconductor. The absorption spectra and cyclic voltammograms
of the dyads show that the spectroscopic and electrochemical properties of the dyads are the sum of the individual chromophores
(Bodipy and the PtN2S2 moieties), indicating little electronic coupling between them. Connection to TiO2 nanoparticles is carried out by sonication leading to in situ attachment to TiO2 without prior hydrolysis of the ester linking groups to acids. For H2 generation studies, the TiO2 particles are platinized (Pt-TiO2) so that the light absorber (the dyad), the electron conduit (TiO2), and the catalyst (attached colloidal Pt) are fully integrated. It is found that upon 530 nm irradiation in a H2O solution (pH 4) with ascorbic acid as an electron donor, the dyad linked to Pt-TiO2 via a phosphonate or carboxylate attachment shows excellent light-driven H2 production with substantial longevity, in which one particular dyad [4(bpyP)] exhibits the highest activity, generating ∼40,000
turnover numbers of H2 over 12 d (with respect to dye).
Co-reporter:Zhiji Han and Richard Eisenberg
Accounts of Chemical Research 2014 Volume 47(Issue 8) pp:2537-2544
Publication Date(Web):June 26, 2014
DOI:10.1021/ar5001605
A major new development in the work described is the use of water-soluble CdSe quantum dots (QDs) as light absorbers for H2 generation in water. Both activity and robustness of the most successful systems are impressive with turnover numbers (TONs) approaching 106, activity maintained over 15 days, and a quantum yield for H2 of 36% with 520 nm light. The water solubilizing capping agent for the first system examined was dihydrolipoic acid (DHLA) anion, and the catalyst was determined to be a DHLA complex of Ni(II) formed in situ. Dissociation of DHLA from the QD surface proved problematic in assessing other catalysts and stimulated the synthesis of tridentate trithiolate (S3) capping agents that are inert to dissociation. In this way, CdSe QD’s having these S3 capping agents were used in systems for the photogeneration of H2 that allowed meaningful comparison of the relative activity of different catalysts for the light-driven production of H2 from water. This new chemistry also points the way to the development of new photocathodes based on S3-capped QDs for removal of the chemical sacrificial electron donor and its replacement electrochemically in photoelectrosynthetic cells.
Co-reporter:Randy Pat Sabatini ; William T. Eckenhoff ; Alexandra Orchard ; Kacie R. Liwosz ; Michael R. Detty ; David F. Watson ; David W. McCamant
Journal of the American Chemical Society 2014 Volume 136(Issue 21) pp:7740-7750
Publication Date(Web):May 6, 2014
DOI:10.1021/ja503053s
A series of chalcogenorhodamine dyes with oxygen, sulfur, and selenium atoms in the xanthylium core was synthesized and used as chromophores for solar hydrogen production with a platinized TiO2 catalyst. Solutions containing the selenorhodamine dye generate more hydrogen [181 turnover numbers (TONs) with respect to chromophore] than its sulfur (30 TONs) and oxygen (20 TONs) counterparts. This differs from previous work incorporating these dyes into dye-sensitized solar cells (DSSCs), where the oxygen- and selenium-containing species perform similarly. Ultrafast transient absorption spectroscopy revealed an ultrafast electron transfer under conditions for dye-sensitized solar cells and a slower electron transfer under conditions for hydrogen production, making the chromophore’s triplet yield an important parameter. The selenium-containing species is the only dye for which triplet state population is significant, which explains its superior activity in hydrogen evolution. The discrepancy in rates of electron transfer appears to be caused by the presence or absence of aggregation in the system, altering the coupling between the dye and TiO2. This finding demonstrates the importance of understanding the differences between, as well as the effects of the conditions for DSSCs and solar hydrogen production.
Co-reporter:Ali Han, Pingwu Du, Zijun Sun, Haotian Wu, Hongxing Jia, Rui Zhang, Zhenning Liang, Rui Cao, and Richard Eisenberg
Inorganic Chemistry 2014 Volume 53(Issue 7) pp:3338-3344
Publication Date(Web):March 11, 2014
DOI:10.1021/ic402624u
Reversible mechanochromic luminescence in cationic platinum(II) terpyridyl complexes is described. The complexes [Pt(Nttpy)Cl]X2 (Nttpy = 4′-(p-nicotinamide-N-methylphenyl)-2,2′:6′,2″-terpyridine, X = PF6 (1), SbF6 (2), Cl (3), ClO4 (4), OTf (5), BF4 (6)) exhibit different colors under ambient light in the solid state, going from red to orange to yellow. All of these complexes are brightly luminescent at both room temperature and 77 K. Upon gentle grinding, the yellow complexes (4–6) turn orange and exhibit bright red luminescence. The red luminescence can be changed back to yellow by the addition of a few drops of acetonitrile to the sample. Crystallographic studies of the yellow and red forms of complex 5 suggest that the mechanochromic response is likely the result of a change in intermolecular Pt···Pt distances upon grinding.
Co-reporter:William T. Eckenhoff, William W. Brennessel, and Richard Eisenberg
Inorganic Chemistry 2014 Volume 53(Issue 18) pp:9860-9869
Publication Date(Web):August 27, 2014
DOI:10.1021/ic501440a
Homogeneous light-driven systems employing molecular molybdenum catalysts for hydrogen production are described. The specific Mo complexes studied are six-coordinate bis(benzenedithiolate) derivatives having two additional isocyanide or phosphine ligands to complete the coordination sphere. Each of the complexes possesses a trigonal prismatic coordination geometry. The complexes were investigated as proton reduction catalysts in the presence of [Ru(bpy)3]2+, ascorbic acid, and visible light. Over 500 TON are obtained over 24 h. Electrocatalysis occurs between the MoIV/MoIII and MoIII/MoII redox couples, around 1.0 V vs SCE. Mechanistic studies by 1H NMR spectroscopy show that upon two-electron reduction the Mo(CNR)2(bdt)2 complex dissociates the isocyanide ligands, followed by addition of acid to result in the formation of molecular hydrogen and the Mo(bdt)2 complex.
Co-reporter:Randy Pat Sabatini, Bo Zheng, Wen-Fu Fu, Daniel J. Mark, Michael F. Mark, Emily Anne Hillenbrand, Richard Eisenberg, and David W. McCamant
The Journal of Physical Chemistry A 2014 Volume 118(Issue 45) pp:10663-10672
Publication Date(Web):September 4, 2014
DOI:10.1021/jp508283d
The effects of solvent and substituents on a multichromophoric complex containing a boron-dipyrromethene (Bodipy) chromophore and Pt(bpy)(bdt) (bpy = 2,2′-bipyridine, bdt =1,2-benzenedithiolate) were studied using steady-state absorption, emission, and ultrafast transient absorption spectroscopy. When the Bodipy molecule is connected to either the bpy or bdt in acetonitrile, excitation ultimately leads to the dyad undergoing triplet energy transfer (TEnT) from the redox-active Pt triplet mixed−metal-ligand−to−ligand′ charge transfer (3MMLL′CT) state to the Bodipy 3ππ* state in 8 and 160 ps, respectively. This is disadvantageous for solar energy applications. Here, we investigate two methods to lower the energy of the 3MMLL′CT state, thereby making TEnT unfavorable. By switching to a low dielectric constant solvent, we are able to extend the lifetime of the 3MMLL′CT state to over 1 ns, the time frame of our experiment. Additionally, electron-withdrawing groups, such as carboxylate and phosphonate esters, on the bpy lower the energy of the 3MMLL′CT state such that the photoexcited dyad remains in that state and avoids TEnT to the Bodipy 3ππ* state. It is also shown that a single methylene spacer between the bpy and phosphonate ester is sufficient to eliminate this effect, raising the energy of the 3MMLL′CT state and inducing relaxation to the 3ππ*.
Co-reporter:Zhiji Han ; Luxi Shen ; William W. Brennessel ; Patrick L. Holland
Journal of the American Chemical Society 2013 Volume 135(Issue 39) pp:14659-14669
Publication Date(Web):September 4, 2013
DOI:10.1021/ja405257s
A series of mononuclear nickel(II) thiolate complexes (Et4N)Ni(X-pyS)3 (Et4N = tetraethylammonium; X = 5-H (1a), 5-Cl (1b), 5-CF3 (1c), 6-CH3 (1d); pyS = pyridine-2-thiolate), Ni(pySH)4(NO3)2 (2), (Et4N)Ni(4,6-Y2-pymS)3 (Y = H (3a), CH3 (3b); pymS = pyrimidine-2-thiolate), and Ni(4,4′-Z-2,2′-bpy)(pyS)2 (Z = H (4a), CH3 (4b), OCH3 (4c); bpy = bipyridine) have been synthesized in high yield and characterized. X-ray diffraction studies show that 2 is square planar, while the other complexes possess tris-chelated distorted-octahedral geometries. All of the complexes are active catalysts for both the photocatalytic and electrocatalytic production of hydrogen in 1/1 EtOH/H2O. When coupled with fluorescein (Fl) as the photosensitizer (PS) and triethylamine (TEA) as the sacrificial electron donor, these complexes exhibit activity for light-driven hydrogen generation that correlates with ligand electron donor ability. Complex 4c achieves over 7300 turnovers of H2 in 30 h, which is among the highest reported for a molecular noble metal-free system. The initial photochemical step is reductive quenching of Fl* by TEA because of the latter’s greater concentration. When system concentrations are modified so that oxidative quenching of Fl* by catalyst becomes more dominant, system durability increases, with a system lifetime of over 60 h. System variations and cyclic voltammetry experiments are consistent with a CECE mechanism that is common to electrocatalytic and photocatalytic hydrogen production. This mechanism involves initial protonation of the catalyst followed by reduction and then additional protonation and reduction steps to give a key Ni–H–/N–H+ intermediate that forms the H–H bond in the turnover-limiting step of the catalytic cycle. A key to the activity of these catalysts is the reversible dechelation and protonation of the pyridine N atoms, which enable an internal heterocoupling of a metal hydride and an N-bound proton to produce H2.
Co-reporter:Gabriel García-Herbosa, William R. McNamara, William W. Brennessel, José V. Cuevas, Sandip Sur, Richard Eisenberg
Polyhedron 2013 Volume 58() pp:39-46
Publication Date(Web):13 July 2013
DOI:10.1016/j.poly.2012.08.017
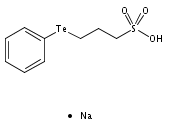
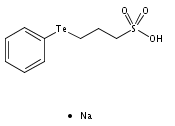
The unexpectedly aqueous stable bis-tellurium cationic complex [Co(dmgH)2{PhTe(CH2)3SO3Na}2]+(1+) (dmgH− = dimethylglyoximate) has been fully characterized in the solid state and in solution. The UV–Vis spectrum of 1+ differs from that of earlier cobaloximes, exhibiting a new low energy band centered at λmax = 425 nm (ε ∼ 26155 dm3 mol−1 cm−1) and assigned with DFT calculations to allowed LMCT transitions. 1H, 13C, and 125Te NMR spectra show unequivocally that two diastereomers exist equally in water, 50% of the achiral C2h meso-[Co(dmgH)2{R,S-PhTe(CH2)3SO3Na}2]+ and 50% of the chiral C2rac-[Co(dmgH)2{PhTe(CH2)3SO3Na}2]+ (25% of each enantiomer [Co(dmgH)2{R,R-PhTe(CH2)3SO3Na}2]+ and [Co(dmgH)2{S,S-PhTe(CH2)3SO3Na}2]+). The solid-state structure of the meso diastereomer has been solved. Neutral mono-tellurium complexes [CoCl(dmgH)2{Te(p-MeO–C6H4)2}](2) and [CoCl(dpgH)2{Te(p-MeO–C6H4)2}](3) (dpgH− = diphenylglyoximate) have also been characterized in the solid (3) state and in solution (2 and 3). The structural results provide the first examples of Co(III)–Te bonds. The solid-state structure of the related cobaloxime salt [Co(dmgH)2(py)2][CoCl2(dmgH)2] obtained during one synthesis is also reported.
Co-reporter:Amit Das;Mohsen Golbon Haghighi;Zhiji Han
PNAS 2013 Volume 110 (Issue 42 ) pp:18346-18348
Publication Date(Web):2013-10-15
DOI:10.1073/pnas.1316755110
Unique tripodal S-donor capping agents with an attached carboxylate are found to bind tightly to the surface of CdSe nanocrystals
(NCs), making the latter water soluble. Unlike that in similarly solubilized CdSe NCs with one-sulfur or two-sulfur capping
agents, dissociation from the NC surface is greatly reduced. The impact of this behavior is seen in the photochemical generation
of H2 in which the CdSe NCs function as the light absorber with metal complexes in aqueous solution as the H2-forming catalyst and ascorbic acid as the electron donor source. This precious-metal–free system for H2 generation from water using [Co(bdt)2]− (bdt, benzene-1,2-dithiolate) as the catalyst exhibits excellent activity with a quantum yield for H2 formation of 24% at 520 nm light and durability with >300,000 turnovers relative to catalyst in 60 h.
Co-reporter:Pingwu Du and Richard Eisenberg
Energy & Environmental Science 2012 vol. 5(Issue 3) pp:6012-6021
Publication Date(Web):06 Jan 2012
DOI:10.1039/C2EE03250C
This article reviews recent significant advances in the field of water splitting. Catalysts play very important roles in two half reactions of water splitting - water reduction and water oxidation. Considering potential future applications, catalysts made of cheap and earth abundant element(s) are especially important for economically viable energy conversion. This article focuses only on catalysts made of cobalt (Co), nickel (Ni) and iron (Fe) elements for water reduction and water oxidation. Different series of catalysts that can be applied in electrocatalytic and photocatalytic water spitting are discussed in detail and their catalytic mechanisms are introduced. Finally, the future outlook and perspective of catalysts made of earth abundant elements will be discussed.
Co-reporter:William T. Eckenhoff and Richard Eisenberg
Dalton Transactions 2012 vol. 41(Issue 42) pp:13004-13021
Publication Date(Web):30 May 2012
DOI:10.1039/C2DT30823A
Recent work towards the production of hydrogen via reduction of protons is described. Most of the systems examined in this perspective use a molecular chromophore for harvesting visible light, a catalyst, which is reduced by the excited (or reduced) chromophore, and finally a sacrificial electron source to oxidatively or reductively quench the chromophore. The reduced catalyst is then responsible for the reduction of protons resulting in hydrogen evolution. Relevant mechanistic work on this topic is also discussed.
Co-reporter:Zhiji Han;Dr. William R. McNamara;Dr. Min-Sik Eum; Patrick L. Holl; Richard Eisenberg
Angewandte Chemie International Edition 2012 Volume 51( Issue 7) pp:1667-1670
Publication Date(Web):
DOI:10.1002/anie.201107329
Co-reporter:Zhiji Han;Patrick L. Holland;Fen Qiu;Todd D. Krauss
Science 2012 Volume 338(Issue 6112) pp:1321-1324
Publication Date(Web):07 Dec 2012
DOI:10.1126/science.1227775
Co-reporter:Zhiji Han;William R. McNamara;William W. Brennessel;Chih-Juo (Madeline) Yin;Patrick L. Holland
PNAS 2012 Volume 109 (Issue 39 ) pp:
Publication Date(Web):2012-09-25
DOI:10.1073/pnas.1120757109
Artificial photosynthesis (AP) is a promising method of converting solar energy into fuel (H2). Harnessing solar energy to generate H2 from H+ is a crucial process in systems for artificial photosynthesis. Widespread application of a device for AP would rely on the
use of platinum-free catalysts due to the scarcity of noble metals. Here we report a series of cobalt dithiolene complexes
that are exceptionally active for the catalytic reduction of protons in aqueous solvent mixtures. All catalysts perform visible-light-driven
reduction of protons from water when paired with as the photosensitizer and ascorbic acid as the sacrificial donor. Photocatalysts with electron withdrawing groups exhibit
the highest activity with turnovers up to 9,000 with respect to catalyst. The same complexes are also active electrocatalysts
in 1∶1 acetonitrile/water. The electrocatalytic mechanism is proposed to be ECEC, where the Co dithiolene catalysts undergo
rapid protonation once they are reduced to . Subsequent reduction and reaction with H+ lead to H2 formation. Cobalt dithiolene complexes thus represent a new group of active catalysts for the reduction of protons.
Co-reporter:Richard Eisenberg
Coordination Chemistry Reviews 2011 Volume 255(7–8) pp:825-836
Publication Date(Web):April 2011
DOI:10.1016/j.ccr.2010.09.003
The discovery of trigonal prismatic (TP) coordination in tris(dithiolene) complexes is recounted. The research was stimulated by the efforts of Gray, Schrauzer, Holm and Davison in the 1960s on the chemistry of dithiolene complexes that showed multiple reversible electron transfer processes and challenged conventional oxidation state assignments. The structures of Re(S2C2Ph2)3, V(S2C2Ph2)3 and Mo(S2C2H2)3 were reported at that time. Bonding pictures based on semiempirical molecular orbital calculations were presented and the basis for stability of TP coordination was put forward based on partial oxidation of the unsaturated 1,2-dithiolate ligands. The structures of M(L)3n complexes for M = Groups 5–7 and n = 0, −1, −2, −3 from the Cambridge structural database are tabulated. The results show that for any M(L)3 system as the magnitude of n increases, the coordination geometry twists to intermediate between TP and octahedral. The notion of redox-non-innocence in the dithiolene ligands is revisited through the recent work of Wieghardt including two studies that focus on the molecular and electronic structures of Re(L)3n and V(L)3n complexes. New experimental work is briefly summarized and the bonding in these systems is reanalyzed. A comparison is given between the early studies of the 1960s and the experimentally and computationally more complete studies recently published.
Co-reporter:William R. McNamara ; Zhiji Han ; Paul J. Alperin ; William W. Brennessel ; Patrick L. Holland
Journal of the American Chemical Society 2011 Volume 133(Issue 39) pp:15368-15371
Publication Date(Web):August 24, 2011
DOI:10.1021/ja207842r
The complex [Co(bdt)2]− (where bdt = 1,2-benzenedithiolate) is an active catalyst for the visible light driven reduction of protons from water when employed with Ru(bpy)32+ as the photosensitizer and ascorbic acid as the sacrificial electron donor. At pH 4.0, the system exhibits very high activity, achieving >2700 turnovers with respect to catalyst and an initial turnover rate of 880 mol H2/mol catalyst/h. The same complex is also an active electrocatalyst for proton reduction in 1:1 CH3CN/H2O in the presence of weak acids, with the onset of a catalytic wave at the reversible redox couple of −1.01 V vs Fc+/Fc. The cobalt–dithiolene complex [Co(bdt)2]− thus represents a highly active catalyst for both the electrocatalytic and photocatalytic reduction of protons in aqueous solutions.
Co-reporter:Tulaza Vaidya ; Gerald F. Manbeck ; Sylvia Chen ; Alison J. Frontier
Journal of the American Chemical Society 2011 Volume 133(Issue 10) pp:3300-3303
Publication Date(Web):February 15, 2011
DOI:10.1021/ja111317q
In contrast to 2-substituted pyrrole enones, furyl and benzofuryl enones do not undergo the Nazarov electrocyclization. Instead, these furyl and benzofuryl enones exhibit unusual rearrangement sequences in the presence of catalytic amounts of [IrBr(CO)(DIM)((R)-(+)-BINAP)](SbF6)2 (1; DIM = diethylisopropylidene malonate) and AgSbF6 (1:1). A 1,2-H shift followed by intramolecular Friedel−Crafts alkylation leads to synthetically valuable cyclohexanones with furanylic quaternary centers. The electrophilicity of 1 is essential for this rearrangement.
Co-reporter:Matthew P. McLaughlin, Theresa M. McCormick, Richard Eisenberg and Patrick L. Holland
Chemical Communications 2011 vol. 47(Issue 28) pp:7989-7991
Publication Date(Web):17 Jun 2011
DOI:10.1039/C1CC12347E
Light-driven H2 production is catalyzed by [Ni(P2PhN2Ph)2](BF4)2 when irradiated with visible light in water/acetonitrile mixed solvent in the presence of a photosensitizer (PS) and ascorbate. The catalyst gives over 2700 turnovers over 150 h, and does not degrade despite photodecomposition of the PS.
Co-reporter:Theresa M. McCormick ; Zhiji Han ; David J. Weinberg ; William W. Brennessel ; Patrick L. Holland
Inorganic Chemistry 2011 Volume 50(Issue 21) pp:10660-10666
Publication Date(Web):October 7, 2011
DOI:10.1021/ic2010166
Ligand exchange on the Co(dmgH)2(py)Cl water reduction catalyst was explored under photocatalytic conditions. The photosensitizer fluorescein was connected to the catalyst through the axially coordinated pyridine. While this two-component complex produces H2 from water under visible light irradiation in the presence of triethanolamine (TEOA), it is less active than a system containing separate fluorescein and [CoIII(dmgH)2(py)Cl] components. NMR and photolysis experiments show that the Co catalyst undergoes pyridine exchange. Interestingly, glyoximate ligand exchange was also observed photocatalytically and by NMR spectroscopy, thereby showing that integrated systems in which the photosensitizer is linked directly to the Co(dmgH)2(py)Cl catalyst may not remain intact during H2 photogeneration. These studies have also given rise to insights into the catalyst decomposition mechanism.
Co-reporter:Richard Eisenberg and Harry B. Gray
Inorganic Chemistry 2011 Volume 50(Issue 20) pp:9741-9751
Publication Date(Web):September 13, 2011
DOI:10.1021/ic2011748
Noninnocence in inorganic chemistry traces its roots back half a century to work that was done on metal complexes containing unsaturated dithiolate ligands. In a flurry of activity in the early 1960s by three different research groups, homoleptic bis and tris complexes of these ligands, which came to be known as dithiolenes, were synthesized, and their structural, electrochemical, spectroscopic, and magnetic properties were investigated. The complexes were notable for facile one-electron transfers and intense colors in solution, and conventional oxidation-state descriptions could not account for their electronic structures. The bis complexes were, in general, found to be square-planar, including the first examples of this geometry for paramagnetic complexes and different formal dn configurations. Several of the neutral and monoanionic tris complexes were found to have trigonal-prismatic coordination, the first time that this geometry had been observed in molecular metal complexes. Electronic structural calculations employing extended Hückel and other semiempirical computational methods revealed extensive ligand–metal mixing in the frontier orbitals of these systems, including the observation of structures in which filled metal-based orbitals were more stable than ligand-based orbitals of the same type, suggesting that the one-electron changes upon oxidation or reduction were occurring on the ligand rather than on the metal center. A summary of this early work is followed with a brief section on the current interpretations of these systems based on more advanced spectroscopic and computational methods. The take home message is that the early work did indeed provide a solid foundation for what was to follow in investigations of metal complexes containing redox-active ligands.
Co-reporter:Marco G. Crestani, Gerald F. Manbeck, William W. Brennessel, Theresa M. McCormick, and Richard Eisenberg
Inorganic Chemistry 2011 Volume 50(Issue 15) pp:7172-7188
Publication Date(Web):June 29, 2011
DOI:10.1021/ic2007588
Heteroleptic copper(I) complexes of the types [Cu(N,N)(P,P)] and [Cu(N,O)(P,P)], where (P,P) = phosphine (PPh3) or diphosphine (dppb, DPEPHOS, XANTPHOS), (N,N) = pyrrole-2-phenylcarbaldimine, 2PyN: [Cu(2PyN)(PPh3)2] (1), [Cu(2PyN) (dppb)] (2), [Cu(2PyN)(DPEPHOS)] (3), and [Cu(2PyN)(XANTPHOS)] (4), (N,N) = indole-2-phenylcarbaldimine, 2IndN: [Cu(2IndN)(DPEPHOS)] (8), and (N,O) = pyrrole-2-carboxaldehyde, 2PyO: [Cu(2PyO)(DPEPHOS)] (5), [Cu(2PyO)(XANTPHOS)] (6), or (N,O) = indole-2-carboxaldehyde, 2IndO: [Cu(2IndO)(DPEPHOS)] (7), were synthesized and characterized by multinuclear NMR spectroscopy, electronic absorption spectroscopy, fluorescence spectroscopy, and X-ray crystallography (1–3, 5–8). The complexes with aldimine ligands are thermally stable, and sublimation of 2–4 was possible at T = 230–250 °C under vacuum. All complexes exhibit long-lived emission in solution, in the solid state, and in frozen glasses. The excited states have been assigned as mixed intraligand and metal-to-ligand charge transfer 3(MLCT + π–π*) transitions through analysis of the photophysical properties and DFT calculations on representative examples.
Co-reporter:Gerald F. Manbeck ; William W. Brennessel
Inorganic Chemistry 2011 Volume 50(Issue 8) pp:3431-3441
Publication Date(Web):March 21, 2011
DOI:10.1021/ic102338g
A series of heteroleptic copper(I) complexes incorporating amido-triazole and diphosphine ligands, [CuI(N-phenyl-2-(1-phenyl-1H-1,2,3-triazol-4-yl)aniline)(dppb)] (1), [CuI(N-(4-methylphenyl)-2-(1-phenyl-1H-1,2,3-triazol-4-yl)aniline)(dppb)] (2), [CuI(N-(4-methoxyphenyl)-2-(1-phenyl-1H-1,2,3-triazol-4-yl)aniline)(dppb)] (3), [CuI(N-(4-chlorophenyl)-2-(1-phenyl-1H-1,2,3-triazol-4-yl)aniline)(dppb)] (4), [CuI(2,6-dimethyl-N-[2-(1-phenyl-1H-1,2,3-triazol-4-yl)phenyl]aniline)(dppb)] (5), [CuI(2,6-dimethyl-N-[2-(1-benzyl-1H-1,2,3-triazol-4-yl)phenyl]aniline)(dppb)] (6), (dppb = 1,2-bis(diphenylphosphino)benzene), have been prepared. The complexes adopt a distorted tetrahedral geometry in the solid state with the amido-triazole ligand forming a six-member ring with the Cu(I) ion. The complexes exhibit long-lived photoluminescence with colors ranging from yellow to red-orange in the solid state, in frozen glass at 77 K, and in fluid solution with modest quantum yields of up to 0.022. Electrochemically, complexes 1−4 show irreversible oxidation waves while 5 and 6 are characterized by quasi-reversible oxidations as determined by cyclic voltammetry. For 1−4, the emission energy and oxidation potential are found to vary linearly with the Hammett parameter σp of the substituent in the para position of the amido ligand, while in 5 and 6, large differences in emission are observed because of the nature of N3 substitution in the triazole ring. Density functional theory calculations have been performed on the singlet ground states (So) of all complexes at the BP86/6-31G(d) level to assist in assignment of the excited states. On the basis of both experimental and computational results, we have assigned the excited states as intraligand + metal-to-ligand charge transfer 3(ILCT+MLCT) or ligand-to-ligand charge transfer mixed with MLCT 3(MLCT +LLCT) in these complexes.
Co-reporter:Randy Pat Sabatini, Theresa M. McCormick, Theodore Lazarides, Kristina C. Wilson, Richard Eisenberg, and David W. McCamant
The Journal of Physical Chemistry Letters 2011 Volume 2(Issue 3) pp:223-227
Publication Date(Web):January 19, 2011
DOI:10.1021/jz101697y
A series of halogenated boron-dipyrromethene (Bodipy) chromophores with potential applications in solar energy conversion were synthesized and characterized by steady state and ultrafast laser spectroscopy. The ultrafast dynamics of the chromophores were compared between a series containing H, Br, or I at the 2,6 positions of the Bodipy dye. The parent Bodipy has a fluorescent lifetime (τfl) of 3−5 ns, a fluorescence quantum yield (Φfl) of 0.56, and negligible triplet state yield. Bromination enhances the intersystem crossing (ISC) such that τfl and Φfl decrease to ∼1.2 ns and 0.11, respectively, while iodination further accelerates ISC so that τfl is only ∼130 ps and Φfl is 0.011. Transient absorption experiments lead to the observation of excited state absorption bands from the singlet (S1) and triplet (T1) states at ∼345 and 447 nm, respectively, and characterization of ISC via the dynamics of these bands and the decay of S1 stimulated emission.Keywords (keywords): femtosecond transient absorption; sensitizers; solar energy; triplet; ultrafast;
Co-reporter:Tulaza Vaidya; Richard Eisenberg; Alison J. Frontier
ChemCatChem 2011 Volume 3( Issue 10) pp:1531-1548
Publication Date(Web):
DOI:10.1002/cctc.201100137
Co-reporter:Theodore Lazarides ; Theresa M. McCormick ; Kristina C. Wilson ; Soohyun Lee ; David W. McCamant
Journal of the American Chemical Society 2010 Volume 133(Issue 2) pp:350-364
Publication Date(Web):December 22, 2010
DOI:10.1021/ja1070366
The dyads 3, 4, and 6, combining the Bodipy chromophore with a Pt(bpy)(bdt) (bpy = 2,2′-bipyridine, bdt = 1,2-benzenedithiolate, 3 and 6) or a Pt(bpy)(mnt) (mnt = maleonitriledithiolate, 4) moiety, have been synthesized and studied by UV−vis steady-state absorption, transient absorption, and emission spectroscopies and cyclic voltammetry. Comparison of the absorption spectra and cyclic voltammograms of dyads 3, 4, and 6 and those of their model compounds 1a, 2, 5, and 7 shows that the spectroscopic and electrochemical properties of the dyads are essentially the sum of their constituent chromophores, indicating negligible interaction of the constituent chromophores in the ground state. However, emission studies on 3 and 6 show a complete absence of both Bodipy-based fluorescence and the characteristic luminescence of the Pt(bpy)(bdt) unit. Dyad 4 shows a weak Pt(mnt)-based emission. Transient absorption studies show that excitation of the dyads into the Bodipy-based 1ππ* excited state is followed by singlet energy transfer (SEnT) to the Pt(dithiolate)-based 1MMLL′CT (mixed metal-ligand to ligand charge transfer) excited state ( = 0.6 ps, = 0.5 ps, and = 1.6 ps), which undergoes rapid intersystem crossing to the 3MMLL′CT state due to the heavy Pt(II) ion. The 3MMLL′CT state is then depopulated by triplet energy transfer (TEnT) to the low-lying Bodipy-based 3ππ* excited state ( = 8.2 ps, = 5 ps, and = 160 ps). The transition assignments are supported by TD-DFT calculations. Both energy-transfer processes are shown to proceed via a Dexter electron exchange mechanism. The much longer time constants for dyad 6 relative to 3 are attributed to the significantly poorer coupling and resonance of charge-separated species that are intermediates in the electron exchange process.
Co-reporter:Theresa M. McCormick ; Brandon D. Calitree ; Alexandra Orchard ; Nadine D. Kraut ; Frank V. Bright ; Michael R. Detty
Journal of the American Chemical Society 2010 Volume 132(Issue 44) pp:15480-15483
Publication Date(Web):October 14, 2010
DOI:10.1021/ja1057357
Rhodamine photosensitizers (PSs) substituting S or Se for O in the xanthene ring give turnover numbers (TONs) as high as 9000 for the generation of hydrogen via the reduction of water using [CoIII(dmgH)2(py)Cl] (where dmgH = dimethylglyoximate and py = pyridine) as the catalyst and triethanolamine as the sacrificial electron donor. The turnover frequencies were 0, 1700, and 5500 mol H2/mol PS/h for O, S, and Se derivatives, respectively (ΦH2 = 0%, 12.2%, and 32.8%, respectively), which correlates well with relative triplet yields estimated from quantum yields for singlet oxygen generation. Phosphorescence from the excited PS was quenched by the sacrificial electron donor. Fluorescence lifetimes were similar for the O- and S-containing rhodamines (∼2.6 ns) and shorter for the Se analog (∼0.1 ns). These data suggest a reaction pathway involving reductive quenching of the triplet excited state of the PS giving the reduced PS− that then transfers an electron to the Co catalyst. The longer-lived triplet state is necessary for effective bimolecular electron transfer. While the cobalt/rhodamine/triethanolamine system gives unprecedented yields of hydrogen for the photoreduction of water, mechanistic insights regarding the overall reaction pathway as well as system degradation offer significant guidance to developing even more stable and efficient photocatalytic systems.
Co-reporter:Gerald F. Manbeck ; William W. Brennessel ; Robert A. Stockland ; Jr.
Journal of the American Chemical Society 2010 Volume 132(Issue 35) pp:12307-12318
Publication Date(Web):August 12, 2010
DOI:10.1021/ja103400e
Gold(I) bis(acetylide) complexes [PPN][AuR2] (1−3) where PPN = bis(triphenylphosphine)iminium) and R = ethisterone (1); 1-ethynylcyclopentanol (2); 1-ethynylcyclohexanol (3) have been prepared. The reaction of 1 with [Cu(MeCN)4][PF6] in a 1:1 or 3:2 ratio provides the octanuclear complex [Au4Cu4(ethisterone)8] (4) or pentanuclear complex [PPN][Au3Cu2(ethisterone)6] (5). Complexes 2 and 3 react with [Cu(MeCN)4][PF6] to form only pentanuclear Au(I)/Cu(I) complexes [PPN][Au3Cu2(1-ethynylcyclopentanol)6] (6) and [PPN][Au3Cu2(1-ethynylcyclohexanol)6] (7). X-ray crystallographic studies of 1−3 reveal nontraditional hydrogen bonding between hydroxyl groups and the acetylide units of adjacent molecules. Complexes 6 and 7 each form polymorphs in which the structures (6 a,b and 7 a,b,c) differ by Au···Au, Au···Cu, and Cu−C distances. The polymorphs exhibit different emission energies with colors ranging from blue to yellow in the solid state. In solution, pentanuclear clusters 5−7 emit with λmax = 570−580 nm and Φ = 0.05−0.19. Complex 4 emits at 496 nm in CH2Cl2 with a quantum yield of 0.65. Complex 5 exists in equilibrium with 1 and 4 in the presence of methanol, ethanol, ethyl acetate, or water. This equilibrium has been probed by X-ray crystallography, NMR spectroscopy, and luminescence experiments. DFT calculations have been performed to analyze the orbitals involved in the electronic transitions of 4, 6, and 7.
Co-reporter:Pingwu Du and Richard Eisenberg
Chemical Science 2010 vol. 1(Issue 4) pp:502-506
Publication Date(Web):09 Jul 2010
DOI:10.1039/C0SC00244E
The platinum(II) terpyridine acetylide complex, [Pt(ttpy)(CCPh)]ClO4 (1, where ttpy = 4′-p-tolylterpyridine), sensitizes the 3π–π* excited state of 9,10-diphenylanthracene (DPA) that in turn produces upconverted fluorescence via triplet–triplet annihilation (TTA). The photoluminescence of 1 is readily quenched by DPA with a rate constant close to the diffusion limit via energy transfer. Selective excitation of 1 leads to the upconverted fluorescence with a near quadratic dependence of the DPA fluorescence intensity on incident light power. This is the first time that the 3MLCT charge transfer excited state of a platinum polypyridine complex has been used to promote photon upconversion.
Co-reporter:Abdurrahman C. Atesin ; Jing Zhang ; Tulaza Vaidya ; William W. Brennessel ; Alison J. Frontier
Inorganic Chemistry 2010 Volume 49(Issue 9) pp:4331-4342
Publication Date(Web):April 2, 2010
DOI:10.1021/ic100300y
The oxidative addition of MeI to the Ir(I) square-planar complex IrI(CO)((R)-(+)-BINAP) where (R)-(+)-BINAP = (R)-(+)-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl)) results in the formation of two diastereomers in a 2:1 ratio of the Ir(III) oxidative addition product IrI2(CO)(Me)((R)-(+)-BINAP) (4a and 4b) in a 85% overall yield. Upon iodide abstraction from the diastereomeric mixture with 2 equiv of AgSbF6 in the presence of diethyl isopropylidene malonate (DIM), two diastereomers of the dicationic complex [Ir(CO)(Me)(DIM)((R)-(+)-BINAP)][SbF6]2 (5) are formed in a 90% yield with a ratio of 9:1. One diastereomer of the diiodide complex 4 and one diastereomer of the dicationic complex 5 have been characterized by X-ray diffraction. An anion exchange reaction of 5 with NaBAr4f− (BAr4f− = B(3,5-(CF3)2C6H3)4) affords [Ir(CO)(Me)(DIM)((R)-(+)-BINAP)][BAr4f]2 (6) in quantitative yield. Both 5 and 6 are active Lewis acid catalysts for the polarized Nazarov cyclization and the Diels−Alder reaction. In the Nazarov cyclization, when NaBAr4f is added as a cofactor for the reaction catalyzed by 5 or 6, the resultant oxyallyl cation intermediate from the 4π conrotation undergoes a Wagner−Meerwein rearrangement to afford spirocyclic cyclopentenones in modest to good yields.
Co-reporter:Gerald F. Manbeck ; William W. Brennessel ; Christopher M. Evans
Inorganic Chemistry 2010 Volume 49(Issue 6) pp:2834-2843
Publication Date(Web):February 16, 2010
DOI:10.1021/ic902236n
A series of tetranuclear Cu4I4(Ln)2 clusters (1−3) supported by the chelating 4,4′-(4,5-diX-1,2-phenylene)bis(1-benzyl-1H-1,2,3-triazole) ligands (L1, X = H; L2, X = CH3; L3, X = F) have been prepared. Crystal structure determinations have shown that the clusters adopt a distorted “step-type” geometry in which the triazole ligands exhibit both chelating and bridging coordination modes to Cu(I) ions. When L3 was employed in a 3:2 CuI/L3 ratio, a crystalline product, 4, was obtained. Product 4 is actually a 1:1 cocrystallization of the tetranuclear cluster 3 (denoted 4a) and the dinuclear Cu2I2(L3)2 complex (4b). X-ray structures of 1, 2, and 4 show the presence of close Cu···Cu contacts in the tetranuclear and dinuclear forms. 1H NMR experiments indicate that rapid exchange occurs in these systems. All clusters are brightly luminescent in the solid state at 77 K and at room temperature with λmax = 495−524 nm. Emission intensity decays fit a single exponential with lifetimes on the order of 10 to 35 microseconds at room temperature and 90 to 140 microseconds at 77 K. The origin of emission is tentatively assigned as a metal-to-ligand phenylene π* charge transfer.
Co-reporter:Jing Zhang, Tulaza Vaidya, William W. Brennessel, Alison J. Frontier and Richard Eisenberg
Organometallics 2010 Volume 29(Issue 15) pp:3341-3349
Publication Date(Web):July 12, 2010
DOI:10.1021/om100274g
Two new bidentate phosphonamidite ligands − 1,2-bis((3αS)-3,3-diphenyltetrahydro-2-oxa-6α-aza-1-phosphapentaleno)ethane, L1, and 1,2-bis((3αS)-3,3-diphenyltetrahydro-2-oxa-6α-aza-1-phosphapentaleno)benzene, L2 − were prepared from (S)-α,α-diphenylprolinol and either 1,2-bis(dichlorophosphino)ethane or 1,2-bis(dichlorophosphino)benzene, respectively. Reactions of L1 and L2 with one equivalent of Pd(PhCN)2Cl2 in dichloromethane result in the formation of the neutral complexes Pd(L1-P,P)Cl2, 2, and Pd(L2-P,P)Cl2, 3, in good yields (85% and 92%, respectively). Complexes 2 and 3 were fully characterized by NMR spectroscopy, elemental analyses, and X-ray crystallography. Chloride abstraction from 1 and 2 using two equivalents of AgSbF6 in the presence of excess diethylisopropylidene malonate (DIM) leads to the formation of the respective dicationic palladium complexes [Pd(L1-P,P)(DIM)][SbF6]2, 4, and [Pd(L2-P,P)(DIM)][SbF6]2, 5. As a result of the cationic nature of 4 and 5 and the labile coordination of DIM, these complexes exhibit high activity as electrophilic catalysts for the Nazarov cyclization of electronically polarized substrates. For those substrates in which a quaternary carbon center is generated upon Nazarov cyclization, spirocycle formation occurs via a Wagner−Meerwein rearrangement following cyclization. In these cases, addition of NaBAr4f as a cofactor to the reaction system improves the selectivity to spirocycle formation relative to the simple Nazarov product.
Co-reporter:Tulaza Vaidya;AbdurrahmanC. Atesin Dr.;IldikoR. Herrick Dr.;AlisonJ. Frontier
Angewandte Chemie International Edition 2010 Volume 49( Issue 19) pp:3363-3366
Publication Date(Web):
DOI:10.1002/anie.201000100
Co-reporter:Theodore Lazarides ; Theresa McCormick ; Pingwu Du ; Gengeng Luo ; Brian Lindley
Journal of the American Chemical Society 2009 Volume 131(Issue 26) pp:9192-9194
Publication Date(Web):June 16, 2009
DOI:10.1021/ja903044n
A photocatalytic noble metal-free system for the generation of hydrogen has been constructed using Eosin Y (1) as a photosensitizer, the complex [Co(dmgH)2pyCl]2+ (5, dmgH = dimethylglyoximate, py = pyridine) as a molecular catalyst, and triethanolamine (TEOA) as a sacrificial reducing agent. The system produces H2 with an initial rate of ∼100 turnovers per hour upon irradiation with visible light (λ > 450 nm). Addition of free dmgH2 greatly increases the durability of the system addition of 12 equiv of dmgH2 (vs cobalt) to the system produces ∼900 turnovers of H2 after 14 h of irradiation. The rate of H2 evolution is maximum at pH = 7 and decreases sharply at more acidic or basic pH. Spectroscopic study of photolysis solutions suggests that hydrogen production occurs through protonation of a CoI species to give a CoIII hydride, which then reacts further by reduction and protolysis to give CoII and molecular hydrogen.
Co-reporter:Paul Jarosz, Pingwu Du, Jacob Schneider, Soo-Hyun Lee, David McCamant and Richard Eisenberg
Inorganic Chemistry 2009 Volume 48(Issue 20) pp:9653-9663
Publication Date(Web):September 17, 2009
DOI:10.1021/ic9001913
New platinum(II) terpyridyl acetylide complexes having the ability to bind to TiO2 have been synthesized and assayed in their ability to sensitize platinized titanium dioxide for the photogeneration of H2 using visible light (λ > 410 nm). Specifically, the complexes [Pt(tpy-phen-COOH)(C≡C−C6H5)]Cl (1), where tpy-phen-COOH = 4′-(4-carboxyphenyl)-[2,2′;6′,2′′]terpyridine and C≡C−C6H5 = phenylacetylide, and [Pt(tpy-COOH)(C≡C−C6H5)]Cl (2), where tpy-COOH = 4′-carboxy-2,2′;6′,2′′-terpyridine, were prepared to investigate the effectiveness of attachment and proximity to the TiO2 surface on hydrogen yield. Both complexes 1 and 2 sensitize the photogeneration of hydrogen, but produce fewer turnovers than the unbound chromophore, [Pt(ttpy)(C≡C−C6H5)]PF6 (5). On the basis of these observations and electrochemical data, a major limitation to the effectiveness of these chromophores is their instability upon oxidation. To attempt to remedy this problem, two donor-chromophore (D-C) dyads, [Pt(tpy-phen-COOH)(C≡C−C6H4CH2−PTZ)]PF6 (3), where C≡C−C6H4CH2−PTZ = N-(4-ethynylbenzyl)-phenothiazine and [Pt(tpy-COOH)(C≡C−C6H4CH2−PTZ)]Cl (4) were prepared to function as TiO2-attached sensitizers. Transient absorption measurements have shown that the PTZ moiety reductively quenches the Pt center in several picoseconds. While the resultant PTZ+ radical cation is capable of oxidizing rapidly the triethanolamine sacrificial electron donor, dyads 3 and 4 attached to platinized TiO2 do not function to generate hydrogen upon irradiation, in contrast with results seen for 1 and 2.
Co-reporter:Pingwu Du, Jacob Schneider, Genggeng Luo, William W. Brennessel and Richard Eisenberg
Inorganic Chemistry 2009 Volume 48(Issue 11) pp:4952-4962
Publication Date(Web):April 27, 2009
DOI:10.1021/ic900389z
A series of cobaloxime complexes—([Co(dmgH)2pyCl] (1), [Co(dmgH)2(4-COOMe-py)Cl] (2), [Co(dmgH)2(4-Me2N-py)Cl] (3), [Co(dmgH)(dmgH2)Cl2] (4), [Co(dmgH)2(py)2](PF6) (5), [Co(dmgH)2(P(n-Bu)3)Cl] (6), and [Co(dmgBF2)2(OH2)2] (7), where dmgH = dimethylglyoximate monoanion, dmgH2 = dimethylglyoxime, dmgBF2 = (difluoroboryl)dimethylglyoximate anion, and py = pyridine—were synthesized and studied as molecular catalysts for the photogeneration of hydrogen from systems containing a Pt terpyridyl acetylide chromophore and triethanolamine (TEOA) as a sacrificial donor in aqueous acetonitrile. All cobaloxime complexes 1−7 are able to quench the luminescence of the Pt(II) chromophore [Pt(ttpy)(C≡CPh)]ClO4 (C1) (ttpy = 4′-p-tolyterpyridine). The most effective electron acceptor for hydrogen evolution is found to be complex 2, which provides the fastest luminescence quenching rate constant for C1 of 1.7 × 109 M−1 s−1. The rate of hydrogen evolution depends on many factors, including the stability of the catalysts, the driving force for proton reduction, the relative and absolute concentrations of system components (TEOA, Co molecular catalyst, and sensitizer), and the ratio of MeCN/water in the reaction medium. For example, when the concentration of TEOA increases, the rate of H2 photogeneration is faster and the induction period is shorter. Colloidal cobalt experiments and mercury tests were run to verify that the system is homogeneous and that catalysis does not occur from in situ generated colloidal particles during photolysis. The most effective system examined to date consists of the chromophore C1 (1.1 × 10−5 M), TEOA (0.27 M), and catalyst complex 1 (2.0 × 10−4 M) in a MeCN/water mixture (24:1 v/v, total 25 mL); this system has produced ∼2150 turnovers of H2 after only 10 h of photolysis with λ > 410 nm.
Co-reporter:Jacob Schneider, Pingwu Du, Paul Jarosz, Theodore Lazarides, Xiaoyong Wang, William W. Brennessel and Richard Eisenberg
Inorganic Chemistry 2009 Volume 48(Issue 10) pp:4306-4316
Publication Date(Web):April 24, 2009
DOI:10.1021/ic801947v
Three cyclometalated 6-phenyl-4-(p-R-phenyl)-2,2′-bipyridyl (CNN-Ph-R) Pt(II) acetylide complexes, Pt(CNN-Ph-R)(CCPh), where R = Me (1), COOMe (2), and P(O)(OEt)2 (3), have been synthesized and studied. Compounds 1 and 3 have been structurally characterized by single crystal X-ray crystallography and are found to exhibit distorted square planar geometries about the Pt(II) ions. The electrochemical properties of the compounds, as determined by cyclic voltammetry, have also been examined. Complexes 1−3 are brightly emissive in fluid CH2Cl2 solution and in the solid state with λemmax of ca. 600 nm and lifetimes on the order of ca. 500 ns in fluid solution. The emissions are assigned to a 3MLCT transition. The complexes undergo oxidative quenching by MV2+ with quenching rates near the diffusion-controlled limit (kq ∼ 1.4 × 1010 M−1 s−1) in CH2Cl2 solution. Reductive-quenching experiments of complexes 1−3 by the amine donors N,N,N′,N′-tetramethylphenylenediamine (TMPD), phenothiazine (PTZ), and N,N,N′,N′-tetramethylbenzidine (TMB) follow Stern−Volmer behavior, with very fast quenching rates on the order of 109−1010 M−1 s−1 in CH2Cl2 solution. When the complexes are employed as the sensitizer in multiple component systems containing MV2+, TEOA, and colloidal Pt in aqueous media, approximately one turnover of H2 (TN vs mol of chromophore) is produced per hour upon irradiation with λ > 410 nm but only after at least a 2 h induction period.
Co-reporter:Paul Jarosz, Kenneth Lotito, Jacob Schneider, Duraisamy Kumaresan, Russell Schmehl and Richard Eisenberg
Inorganic Chemistry 2009 Volume 48(Issue 6) pp:2420-2428
Publication Date(Web):February 11, 2009
DOI:10.1021/ic801769v
Four new Pt(II) terpyridyl acetylide complexes which possess a covalently linked nitrophenyl moiety were prepared and studied. Specifically, the chromophore−acceptor (C−A) dyads reported here include [Pt(ptpy-ph-p-NO2)(C≡C−C6H5)](PF6)3 (1), where ptpy-ph-p-NO2 = 4′-{4-(4-nitrophenyl)-phenyl}-[2,2′;6′,2′′]terpyridine, and C≡C−C6H5 = phenylacetylide and [Pt(ptpy-ph-m-NO2)(C≡C−C6H5)](PF6)2 (2), where ptpy-ph-m-NO2 = 4′-(4-m-nitrophenyl-phenyl)-2,2′;6′,2′′-terpyridine, as well as the related donor−chromophore−acceptor (D−C−A) triads [Pt(ptpy-ph-p-NO2)(C≡C−C6H4CH2−PTZ)]PF6 (3), where C≡C−C6H4CH2−PTZ = 4-ethynylbenzyl-N-phenothiazine, and [Pt(ptpy-ph-m-NO2)(C≡C−C6H4CH2−PTZ)]PF6 (4). Transient absorption spectroscopy and electrochemical analyses were used to characterize these compounds. In contrast to previous observations for closely related multicomponent systems, it appears that, in the current systems, the nitrophenyl group is not an effective quencher of the excited state. The luminescence and transient absorption properties of the C−A dyads are virtually identical to those of the parent chromophore, [Pt(ttpy)(C≡C−C6H5)]PF6 (5), where ttpy = 4′-p-tolyl-[2,2′;6′,2′′]terpyridine.
Co-reporter:Jacob Schneider ; Pingwu Du ; Xiaoyong Wang ; William W. Brennessel
Inorganic Chemistry 2009 Volume 48(Issue 4) pp:1498-1506
Publication Date(Web):January 13, 2009
DOI:10.1021/ic801767q
Three new cyclometalated 6-phenyl-4-(p-R-phenyl)-2,2′-bipyridyl (C∧N∧N) Pt(II) thiophenolate complexes (R = Me (2a), COOMe (2b), and P(O)(OEt)2 (2c)) have been synthesized and studied. The new C∧N∧N ligands L2 (R = COOMe) and L3 (R = P(O)(OEt)2) undergo cyclometalation with a Pt(II) source to give the Pt(II) chloro complexes 1b and 1c, respectively, which are luminescent in fluid solution with λmax ∼ 575 nm, assigned to a metal-to-ligand charge-transfer (3MLCT) emissive state. Reaction of the chloro complexes 1a (R = Me), 1b, and 1c with sodium thiophenolate gives 2a−2c, respectively, in good yields. The novel thiophenolate complexes have two interesting absorption bands in their electronic spectra tentatively assigned to a charge-transfer to C∧N∧N (1CT) (λabs ∼415 nm) transition and a mixed metal/ligand-to-ligand′ charge-transfer (MMLL′CT, λabs ∼ 555 nm) transition, respectively. The MMLL′CT band is solvatochromic with absorption maxima in the range of 496 nm in MeOH to 590 nm in toluene (ε ∼ 4000 dm3 mol−1 cm−1), which correlate well with an empirical charge-transfer-based solvent scale. Excitation of 2a−2c into the MMLL′CT band gives emission maxima around 680 nm in frozen CH2Cl2 solution, and no emission in fluid solution. Ligand L2 and complexes 1a·MeCN, 1b, and 2b·CH2Cl2 have been characterized by single crystal X-ray crystallography. The electrochemical properties of ligands L1 (R = Me) and L2 and complexes 1a−1c and 2a−2c have been examined by cyclic voltammetry and are shown to exhibit reversible and quasi-reversible reductions and irreversible oxidations.
Co-reporter:Zachary J. Tonzetich, Richard Eisenberg
Inorganica Chimica Acta 2003 Volume 345() pp:340-344
Publication Date(Web):10 March 2003
DOI:10.1016/S0020-1693(02)01276-8
A series of η5-pentamethylcyclopentadienyl tantalum(V) complexes have been prepared and their electronic and emission spectra examined. Compounds of the form Cp*TaX4 (where X=Cl, Br, OC6F5) and Cp*TaCl2X2 (where X=OC6Cl5, 2,6-OC6H3Cl2), as well as CpTaX4, have been found to luminesce at 77 K in frozen glasses (only Cp*TaCl4, reported previously, luminesces at ambient temperature in both solid and fluid solution). The shifts in luminescence maxima with ligand variation lead to the conclusion that the emissive state is a ligand-to-metal charge transfer in which the highest occupied orbital is based on the donor atom of one of the monodentate ligands and the lowest unoccupied orbital is Ta-based.Cp*TaX4 compounds (where X=Cl, Br, OC6F5) and Cp*TaCl2X2 (where X=OC6Cl5, 2,6-OC6H3Cl2), as well as CpTaX4, luminesce at 77 K in frozen glasses (only Cp*TaCl4, reported previously, luminesces at ambient temperature in both solid and fluid solution). The shifts in luminescence maxima with ligand variation suggest that the emissive state is a ligand-to-metal charge transfer with the highest occupied orbital based on the donor atom of one of the monodentate ligands and the lowest unoccupied orbital based on Ta.
Co-reporter:William T. Eckenhoff, William R. McNamara, Pingwu Du, Richard Eisenberg
Biochimica et Biophysica Acta (BBA) - Bioenergetics (August–September 2013) Volume 1827(Issues 8–9) pp:958-973
Publication Date(Web):August–September 2013
DOI:10.1016/j.bbabio.2013.05.003
Co-reporter:Pingwu Du and Richard Eisenberg
Chemical Science (2010-Present) 2010 - vol. 1(Issue 4) pp:NaN506-506
Publication Date(Web):2010/07/09
DOI:10.1039/C0SC00244E
The platinum(II) terpyridine acetylide complex, [Pt(ttpy)(CCPh)]ClO4 (1, where ttpy = 4′-p-tolylterpyridine), sensitizes the 3π–π* excited state of 9,10-diphenylanthracene (DPA) that in turn produces upconverted fluorescence via triplet–triplet annihilation (TTA). The photoluminescence of 1 is readily quenched by DPA with a rate constant close to the diffusion limit via energy transfer. Selective excitation of 1 leads to the upconverted fluorescence with a near quadratic dependence of the DPA fluorescence intensity on incident light power. This is the first time that the 3MLCT charge transfer excited state of a platinum polypyridine complex has been used to promote photon upconversion.
Co-reporter:William T. Eckenhoff and Richard Eisenberg
Dalton Transactions 2012 - vol. 41(Issue 42) pp:NaN13021-13021
Publication Date(Web):2012/05/30
DOI:10.1039/C2DT30823A
Recent work towards the production of hydrogen via reduction of protons is described. Most of the systems examined in this perspective use a molecular chromophore for harvesting visible light, a catalyst, which is reduced by the excited (or reduced) chromophore, and finally a sacrificial electron source to oxidatively or reductively quench the chromophore. The reduced catalyst is then responsible for the reduction of protons resulting in hydrogen evolution. Relevant mechanistic work on this topic is also discussed.
Co-reporter:Matthew P. McLaughlin, Theresa M. McCormick, Richard Eisenberg and Patrick L. Holland
Chemical Communications 2011 - vol. 47(Issue 28) pp:NaN7991-7991
Publication Date(Web):2011/06/17
DOI:10.1039/C1CC12347E
Light-driven H2 production is catalyzed by [Ni(P2PhN2Ph)2](BF4)2 when irradiated with visible light in water/acetonitrile mixed solvent in the presence of a photosensitizer (PS) and ascorbate. The catalyst gives over 2700 turnovers over 150 h, and does not degrade despite photodecomposition of the PS.
.jpg)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)



































![Phosphonic acid, P-[2-(1,3-benzodioxol-5-yl)ethynyl]-, diethyl ester](/data/chemimg/124100/176100-88-2.png)
![Phosphonic acid, P-[2-(1,3-benzodioxol-5-yl)ethynyl]-, diethyl ester](/data/chemimg/124100/176100-88-2_b.png)












![Potassium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate](http://img.cochemist.com/ccimg/105600/105560-52-9.png)
![Potassium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate](http://img.cochemist.com/ccimg/105600/105560-52-9_b.png)
![[2,2'-Bipyridine]-4-carboxaldehyde, 4'-methyl-](/data/chemimg/102400/104704-09-8.png)
![[2,2'-Bipyridine]-4-carboxaldehyde, 4'-methyl-](/data/chemimg/102400/104704-09-8_b.png)





![1-Butanaminium, N,N,N-tributyl-, (SP-4-1)-bis[4-methyl-1,2-benzenedithiolato(2-)-κS1,κS2]cobaltate(1-) (1:1)](/data/chemimg/1259000/52152-15-5.png)
![1-Butanaminium, N,N,N-tributyl-, (SP-4-1)-bis[4-methyl-1,2-benzenedithiolato(2-)-κS1,κS2]cobaltate(1-) (1:1)](/data/chemimg/1259000/52152-15-5_b.png)




]chloro(pyridine)-, (OC-6-42)-](http://img.cochemist.com/ccimg/23300/23295-32-1.png)
]chloro(pyridine)-, (OC-6-42)-](http://img.cochemist.com/ccimg/23300/23295-32-1_b.png)
























![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5.png)
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5_b.png)

