Abstract
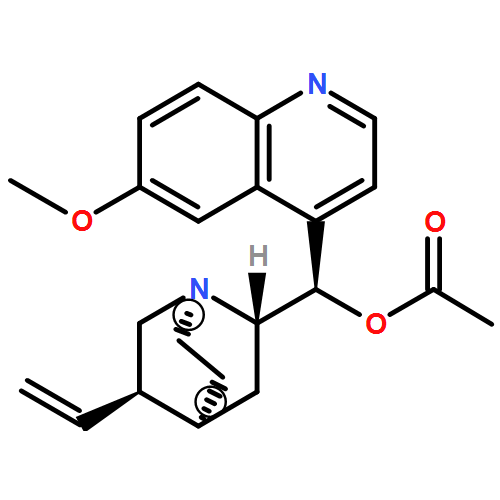
Cinchona alkaloids catalyze the oxa-Michael cyclization of 4-(2-hydroxyphenyl)-2-butenoates to benzo-2,3-dihydrofuran-2-yl acetates and related substrates in up to 99 % yield and 91 % ee (ee=enantiomeric excess). Catalyst and substrate variation studies reveal an important role of the alkaloid hydroxy group in the reaction mechanism, but not in the sense of a hydrogen-bonding activation of the carbonyl group of the substrate as assumed by the Hiemstra–Wynberg mechanism of bifunctional catalysis. Deuterium labeling at C-2 of the substrate shows that addition of RO![[BOND]](http://onlinelibrarystatic.wiley.com/undisplayable_characters/00f8ff.gif) H to the alkenoate occurs with syn diastereoselectivity of ≥99:1, suggesting a mechanism-based specificity. A concerted hydrogen-bond network mechanism is proposed, in which the alkaloid hydroxy group acts as a general acid in the protonation of the α-carbanionic center of the product enolate. The importance of concerted hydrogen-bond network mechanisms in organocatalytic reactions is discussed. The relative stereochemistry of protonation is proposed as analytical tool for detecting concerted addition mechanisms, as opposed to ionic 1,4-additions.
H to the alkenoate occurs with syn diastereoselectivity of ≥99:1, suggesting a mechanism-based specificity. A concerted hydrogen-bond network mechanism is proposed, in which the alkaloid hydroxy group acts as a general acid in the protonation of the α-carbanionic center of the product enolate. The importance of concerted hydrogen-bond network mechanisms in organocatalytic reactions is discussed. The relative stereochemistry of protonation is proposed as analytical tool for detecting concerted addition mechanisms, as opposed to ionic 1,4-additions.