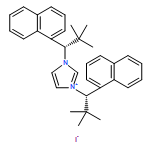
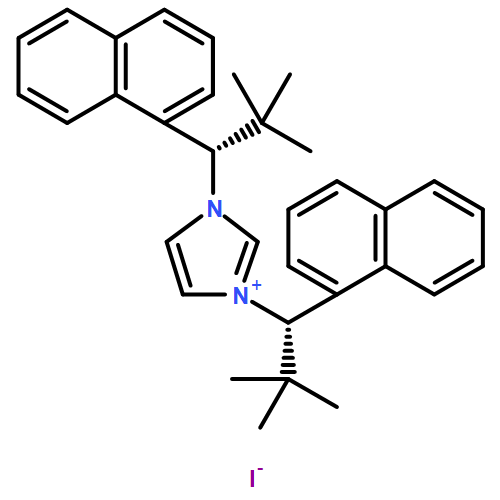
Co-reporter:Julia Pedroni and Nicolai Cramer
Journal of the American Chemical Society September 13, 2017 Volume 139(Issue 36) pp:12398-12398
Publication Date(Web):August 23, 2017
DOI:10.1021/jacs.7b07024
An enantioselective C–H functionalization route to perfluoroalkyl-containing 3-azabicyclo[3.1.0]hexanes is disclosed. A modular and bench-stable diazaphospholane ligand enables highly enantioselective Pd(0)-catalyzed cyclopropane C–H functionalization using trifluoroacetimidoyl chlorides as electrophilic partners. In turn, the resulting cyclic ketimine products react smoothly with a broad variety of nucleophiles in one-pot processes enabling the rapid and modular construction of heavily substituted pyrrolidines.
Co-reporter:Daria Grosheva and Nicolai Cramer
ACS Catalysis November 3, 2017 Volume 7(Issue 11) pp:7417-7417
Publication Date(Web):September 26, 2017
DOI:10.1021/acscatal.7b02783
Enantioselective Pd(0)-catalyzed C–H functionalizations of ketene aminal phosphates provide isoindoline scaffolds with high enantioselectivity at ambient temperature. The high level of enantiocontrol is enabled by a tailored monodentate electron-rich phosphine ligand featuring a point-chiral phospholane module and a bulky atropchiral binaphthyl backbone.Keywords: asymmetric catalysis; C−H functionalization; enantioselectivity; ligand design; palladium;
Co-reporter:Christopher G. Newton, Shou-Guo Wang, Caio C. Oliveira, and Nicolai Cramer
Chemical Reviews July 12, 2017 Volume 117(Issue 13) pp:8908-8908
Publication Date(Web):February 17, 2017
DOI:10.1021/acs.chemrev.6b00692
The development of new methods for the direct functionalization of unactivated C–H bonds is ushering in a paradigm shift in the field of retrosynthetic analysis. In particular, the catalytic enantioselective functionalization of C–H bonds represents a highly atom- and step-economic approach toward the generation of structural complexity. However, as a result of their ubiquity and low reactivity, controlling both the chemo- and stereoselectivity of such processes constitutes a significant challenge. Herein we comprehensively review all asymmetric transition-metal-catalyzed methodologies that are believed to proceed via an inner-sphere-type mechanism, with an emphasis on the nature of stereochemistry generation. Our analysis serves to document the considerable and rapid progress within in the field, while also highlighting limitations of current methods.
Co-reporter:D. Kossler;N. Cramer
Chemical Science (2010-Present) 2017 vol. 8(Issue 3) pp:1862-1866
Publication Date(Web):2017/02/28
DOI:10.1039/C6SC05092A
Cyclopentadienyl ruthenium(II) complexes with a large number of available coordination sites are frequently used catalysts for a broad range of transformations. To be able to render these transformations enantioselective, we have designed a chiral neutral CpxRu(II)Cl complex basing on an atropchiral cyclopentadienyl (Cpx) ligand which is accessed in a streamlined C–H functionalisation approach. The catalyst displays excellent levels of reactivity and enantioselectivity for enantioselective [2+2]-cycloadditions leading to strained chiral cyclobutenes, allowing for catalyst loadings as low as 1 mol%. A very strong counterion effect of a bound chloride anion transforms the corresponding unselective cationic complex into a highly enantioselective neutral version. Moreover, by adding norbornadiene at the end of the reaction the catalyst can be recovered and subsequently reused.
Co-reporter:G. Smits;B. Audic;M. D. Wodrich;C. Corminboeuf;N. Cramer
Chemical Science (2010-Present) 2017 vol. 8(Issue 10) pp:7174-7179
Publication Date(Web):2017/09/25
DOI:10.1039/C7SC02986A
The electronic and steric properties of tailored cyclopentadienyl (Cp) ligands are powerful handles to modulate the catalytic properties of their metal complexes. This requires the individual preparation, purification and storage of each ligand/metal combination. Alternative, ideally in situ, complexation protocols would be of high utility. We disclose a new approach to access Cp metal complexes. Common metal precursors rapidly react with cyclopentadienyl carbinols via β-carbon eliminations to directly give the Cp-metal complexes. An advantage of this is the direct and flexible use of storable pre-ligands. No auxiliary base is required and the Cp complexes can be prepared in situ in the reaction vessel for subsequent catalytic transformations.
Co-reporter:Yun-Suk Jang;Dr. Michael Dieckmann; Dr. Nicolai Cramer
Angewandte Chemie 2017 Volume 129(Issue 47) pp:15284-15288
Publication Date(Web):2017/11/20
DOI:10.1002/ange.201708440
AbstractAn enantioselective C−H amidation of phosphine oxides by using an iridium(III) catalyst bearing an atropchiral cyclopentadienyl (Cpx) ligand is reported. A very strong cooperative effect between the chiral Cpx ligand and a phthaloyl tert-leucine enabled the transformation. Matched–mismatched cases of the different acid enantiomers are shown. The amidated P-chiral arylphosphine oxides are formed in yields of up to 95 % and with excellent enantioselectivities of up to 99:1 er. Enantiospecific reduction provides access to valuable P-chiral phosphorus(III) compounds.
Co-reporter:David Kossler;Florian G. Perrin;Abdusalom A. Suleymanov;Dr. Gregor Kiefer;Dr. Rosario Scopelliti; Dr. Kay Severin; Dr. Nicolai Cramer
Angewandte Chemie 2017 Volume 129(Issue 38) pp:11648-11651
Publication Date(Web):2017/09/11
DOI:10.1002/ange.201706013
AbstractVinyl triazenes were obtained by enantioselective [2+2] cycloaddition reactions of bicyclic alkenes with 1-alkynyl triazenes in the presence of a RuII catalyst with a chiral cyclopentadienyl ligand. These triazenes serve as unique vinyl cation surrogates. Under acidic conditions, the triazene functionality can be replaced with a variety of groups, including halides, alkoxides, sulfoxides, amides, arenes, and heteroarenes, thus providing efficient access to a pool of chiral polycyclic compounds.
Co-reporter:Dr. Christopher G. Newton;Dr. Duc N. Tran;Dr. Matthew D. Wodrich; Dr. Nicolai Cramer
Angewandte Chemie 2017 Volume 129(Issue 44) pp:13964-13968
Publication Date(Web):2017/10/23
DOI:10.1002/ange.201708333
AbstractA gram-scale synthesis of psiguadial B, a purported inhibitor of human hepatoma cell growth, has been achieved in one step by a biomimetic three-component coupling of caryophyllene, benzaldehyde, and diformylphloroglucinol. This cascade reaction is catalyzed by N,N′-dimethylethylenediamine, and proceeds at ambient temperature to generate four stereocenters, two rings, one C−O bond, and three C−C bonds. Combined computational and experimental investigations suggest the biosynthesis of the natural product is non-enzyme mediated, and is the result of a Michael addition between caryophyllene and a reactive ortho-quinone methide, followed by two sequential intramolecular cationic cyclization events.
Co-reporter:Yun-Suk Jang;Dr. Michael Dieckmann; Dr. Nicolai Cramer
Angewandte Chemie International Edition 2017 Volume 56(Issue 47) pp:15088-15092
Publication Date(Web):2017/11/20
DOI:10.1002/anie.201708440
AbstractAn enantioselective C−H amidation of phosphine oxides by using an iridium(III) catalyst bearing an atropchiral cyclopentadienyl (Cpx) ligand is reported. A very strong cooperative effect between the chiral Cpx ligand and a phthaloyl tert-leucine enabled the transformation. Matched–mismatched cases of the different acid enantiomers are shown. The amidated P-chiral arylphosphine oxides are formed in yields of up to 95 % and with excellent enantioselectivities of up to 99:1 er. Enantiospecific reduction provides access to valuable P-chiral phosphorus(III) compounds.
Co-reporter:Christopher G. Newton; David Kossler
Journal of the American Chemical Society 2016 Volume 138(Issue 12) pp:3935-3941
Publication Date(Web):February 10, 2016
DOI:10.1021/jacs.5b12964
Application of chiral derivatives of the versatile and ubiquitous cyclopentadienyl ligand has long remained an underdeveloped area in asymmetric catalysis. In this Perspective we highlight recent exciting results that demonstrate their enormous potential. In particular, we provide a comparative analysis of the available ligand families, an overview of their complexation chemistry, and an examination of their application in catalytic enantioselective reactions. We also discuss current limitations and speculate on the developments that are necessary to advance the field further.
Co-reporter:Joachim S. E. Ahlin and Nicolai Cramer
Organic Letters 2016 Volume 18(Issue 13) pp:3242-3245
Publication Date(Web):June 21, 2016
DOI:10.1021/acs.orglett.6b01492
An enantioselective nickel(0)-catalyzed reductive three-component coupling between aromatic aldehydes, norbornenes, and silanes affords directly silyl-protected indanol derivatives. A new bulky chiral C2-symmetric NHC (NHC = N-heterocyclic carbene) ligand basing on the 1,2-di(napthalen-1-yl)ethylene diamine backbone allows accessing the annulated products as single diastereoisomers in high enantioselectivity.
Co-reporter:Julia Pedroni and Nicolai Cramer
Organic Letters 2016 Volume 18(Issue 8) pp:1932-1935
Publication Date(Web):April 7, 2016
DOI:10.1021/acs.orglett.6b00795
Perfluoroalkylated indoles are valuable compounds in drug discovery. A Pd(0)-catalyzed C(sp3)–H functionalization enables access to 2-(trifluoromethyl)indoles from trifluoroacetimidoyl chlorides. These are stable compounds, easily obtained from anilines. The cyclization operates with catalyst loadings as low as 1 mol % and accommodates a variety of substituents.
Co-reporter:Manh V. Pham ;Dr. Nicolai Cramer
Chemistry - A European Journal 2016 Volume 22( Issue 7) pp:2270-2273
Publication Date(Web):
DOI:10.1002/chem.201504998
Abstract
Chiral spirocyclic sultams are a valuable compound class in organic and medicinal chemistry. A rapid entry to this structural motif involves a [3+2] annulation of an N-sulfonyl ketimine and an alkyne. Although the directing-group properties of the imino group for C−H activation have been exploited, the developments of related asymmetric variants have remained very challenging. The use of rhodium(III) complexes equipped with a suitable atropchiral cyclopentadienyl ligand, in conjunction with a carboxylic acid additive, enables an enantioselective and high yielding access to such spirocyclic sultams.
Co-reporter:Laetitia Souillart and Nicolai Cramer
Chemical Reviews 2015 Volume 115(Issue 17) pp:9410
Publication Date(Web):June 5, 2015
DOI:10.1021/acs.chemrev.5b00138
Co-reporter:Baihua Ye and Nicolai Cramer
Accounts of Chemical Research 2015 Volume 48(Issue 5) pp:1308
Publication Date(Web):April 17, 2015
DOI:10.1021/acs.accounts.5b00092
Transition-metal catalyzed C–H functionalizations became a complementary and efficient bond-forming strategy over the past decade. In this respect, Cp*Rh(III) complexes have emerged as powerful catalysts for a broad spectrum of reactions giving access to synthetically versatile building blocks. Despite their high potential, the corresponding catalytic enantioselective transformations largely lag behind. The targeted transformations require all the remaining three coordination sites of the central rhodium atom of the catalyst. In consequence, the chiral information on a competent catalyst can only by stored in the cyclopentadienyl unit. The lack of suitable enabling chiral cyclopentadienyl (Cpx) ligands is the key hurdle preventing the development of such asymmetric versions. In this respect, an efficient set of chiral Cpx ligands useable with a broad variety of different transition-metals can unlock substantial application potential. This Account provides a description of our developments of two complementary classes of C2-symmetric Cpx derivatives. We have introduced a side- and back-wall concept to enforce chirality transfer onto the central metal atom. The first generation consists of a fused cyclohexane unit having pseudo axial methyl groups as chiral selectors and a rigidifying acetal moiety. The second ligand generation derives from an atrop-chiral biaryl-backbone and which possesses adjustable substituents at its 3,3′-positions. Both ligand families can be modulated in their respective steric bulk to adjust for the specific needs of the targeted application. The cyclopentadienes can be metalated under standard conditions. The corresponding chiral rhodium(I) ethylene complexes are relatively air and moisture and represent storable stable precatalysts for the targeted asymmetric Rh(III)-catalyzed C–H functionalizations. These complexes are then conveniently oxidized in situ by dibenzoyl peroxide to give the reactive CpxRh(III)(OBz)2 species. For instance, this catalyst is used for directed C–H activations of aryl hydroxamates and the subsequent enantioselective trapping with olefins, providing dihydroisoquinolones in very high enantioselectivities. In addition, we have established highly selective intramolecular trapping reactions with tethered higher substituted alkenes giving dihydrobenzofurans with quaternary stereogenic centers. Concerning intermolecular reactions, allene coupling partners allow for an enantioselective hydroarylation yielding substituted allylated compounds. A trapping process of the cyclometalated intermediate with diazo reactants enables the enantioselective construction of isoindolinones. Moreover, the catalysts can be used for the construction of atropchiral biaryl motives using a dehydrogenative Heck-type reaction. The development of flexibly adjustable chiral Cpx ligands is described in this Account showcasing their applicability for a variety of Rh(III) catalyzed C–H functionalization reactions. These Cpx derivatives hold promise as powerful steering ligands for further transition-metals used in asymmetric catalysis.
Co-reporter:David Kossler
Journal of the American Chemical Society 2015 Volume 137(Issue 39) pp:12478-12481
Publication Date(Web):September 15, 2015
DOI:10.1021/jacs.5b08232
The cyclopentadienyl (Cp) group is a ligand of great importance for many transition-metal complexes used in catalysis. Cationic CpRuII complexes with three free coordination sites are highly versatile catalysts for many atom-economic transformations. We report the synthesis of a family of CpxRuII complexes with chiral Cp ligands keeping the maximum number of available coordination sites. The cationic members are efficient and selective catalysts for yne-enone cyclizations via formal hetero-Diels–Alder reactions. The transformation proceeds in <1 h at −20 °C and provides pyrans in up to 99:1 er. Unsaturated ester or Weinreb-amide substrates directly yield the iridoid skeleton.
Co-reporter:Christoph Heinz
Journal of the American Chemical Society 2015 Volume 137(Issue 35) pp:11278-11281
Publication Date(Web):August 24, 2015
DOI:10.1021/jacs.5b07964
Fijiolide A is a secondary metabolite isolated from a marine-derived actinomycete and displays inhibitory activity against TNF-α-induced activation of NFκB, an important transcription factor and a potential target for the treatment of different cancers and inflammation related diseases. Fijiolide A is a glycosylated complex paracyclophane, which is structurally closely related to the Bergman-aromatization product of enediyne C-1027. We report an enantioselective synthesis of fijiolide A demonstrating the power of fully intermolecular ruthenium-catalyzed [2 + 2 + 2]-cyclotrimerizations with three different alkynes to assemble the heavily substituted central arene core. The characteristic strained [2.6]paracyclophane structure is accessed by a templated atropselective macroetherification reaction.
Co-reporter:J. Pedroni, T. Saget, P. A. Donets and N. Cramer
Chemical Science 2015 vol. 6(Issue 9) pp:5164-5171
Publication Date(Web):17 Jun 2015
DOI:10.1039/C5SC01909E
Taddol-based phosphoramidite ligands enable enantioselective palladium(0)-catalyzed C–H arylation of cyclopropanes. The cyclized products are obtained in high yields and enantioselectivities. The reported method provides efficient access to a broad range of synthetically attractive cyclopropyl containing dihydroquinolones and dihydroisoquinolones as well as allows for an efficient enantioselective construction of the 7-membered ring of the cyclopropyl indolobenzazepine core of BMS-791325.
Co-reporter:Julia Pedroni and Nicolai Cramer
Chemical Communications 2015 vol. 51(Issue 100) pp:17647-17657
Publication Date(Web):26 Oct 2015
DOI:10.1039/C5CC07929B
Monodentate TADDOL-derived phosphoramidites and phosphonites are versatile chiral ligands for enantioselective Pd(0)-catalysed C–H functionalisations. They enable highly selective cyclisations to access a wide range of chiral carbo- and heterocycles. The high attractiveness of this ligand class consists in their modular structure, allowing for a quick assembly of a library with variable steric properties. Asymmetric C–H functionalisation methods utilising catalytic systems based on Pd(0) complexes and TADDOL-type ligands are presented and aspects of selectivity are discussed.
Co-reporter:Dr. Pavel A. Donets ;Dr. Nicolai Cramer
Angewandte Chemie 2015 Volume 127( Issue 2) pp:643-647
Publication Date(Web):
DOI:10.1002/ange.201409669
Abstract
The 1,6-annulated 2-pyridone motif is found in many biologically active compounds and its close relation to the indolizidine and quinolizidine alkaloid core makes it an attractive building block. A nickel-catalyzed CH functionalization of 2-pyridones and subsequent cyclization affords 1,6-annulated 2-pyridones by selective intramolecular olefin hydroarylation. The switch between the exo- and endo-cyclization modes is controlled by two complementary sets of ligands. Irrespective of the ring size, the regioselectivity during the cyclization is under full catalyst control. Simple cyclooctadiene promotes an exo-selective cyclization, whereas a bulky N-heterocyclic carbene ligand results in an endo-selective mode. The method was further applied in the synthesis of the lupin alkaloid cytisine.
Co-reporter:Dr. Michael Dieckmann;Yun-Suk Jang ;Dr. Nicolai Cramer
Angewandte Chemie 2015 Volume 127( Issue 41) pp:12317-12320
Publication Date(Web):
DOI:10.1002/ange.201506483
Abstract
The cyclopentadienyl (Cp) group is a very important ligand for many transition-metal complexes which have been applied in catalysis. The availability of chiral cyclopentadienyl ligands (Cpx) lags behind other ligand classes, thus hampering the investigation of enantioselective processes. We report a library of chiral CpxIrIII complexes equipped with an atropchiral Cp scaffold. A robust complexation procedure reliably provides CpxIrIII complexes with tunable counterions. In a proof-of-concept application, the iodide-bearing members are shown to be highly selective for enyne cycloisomerization reactions. The dehydropiperidine-fused cyclopropane products are formed in good yields and enantioselectivities.
Co-reporter:Julia Pedroni ;Dr. Nicolai Cramer
Angewandte Chemie 2015 Volume 127( Issue 40) pp:11992-11995
Publication Date(Web):
DOI:10.1002/ange.201505916
Abstract
Cyclopropanes fused to pyrrolidines are important structural features found in a number of marketed drugs and development candidates. Typically, their synthesis involves the cyclopropanation of a dihydropyrrole precursor. Reported herein is a complementary approach which employs a palladium(0)-catalyzed CH functionalization of an achiral cyclopropane to close the pyrrolidine ring in an enantioselective manner. In contrast to aryl–aryl couplings, palladium(0)-catalyzed CH functionalizations involving the formation of C(sp3)C(sp3) bonds of saturated heterocycles are very scarce. The presented strategy yields cyclopropane-fused γ-lactams from chloroacetamide substrates. A bulky Taddol phosphonite ligand in combination with adamantane-1-carboxylic acid as a cocatalyst provides the γ-lactams in excellent yields and enantioselectivities.
Co-reporter:Dr. Michael Dieckmann;Yun-Suk Jang ;Dr. Nicolai Cramer
Angewandte Chemie International Edition 2015 Volume 54( Issue 41) pp:12149-12152
Publication Date(Web):
DOI:10.1002/anie.201506483
Abstract
The cyclopentadienyl (Cp) group is a very important ligand for many transition-metal complexes which have been applied in catalysis. The availability of chiral cyclopentadienyl ligands (Cpx) lags behind other ligand classes, thus hampering the investigation of enantioselective processes. We report a library of chiral CpxIrIII complexes equipped with an atropchiral Cp scaffold. A robust complexation procedure reliably provides CpxIrIII complexes with tunable counterions. In a proof-of-concept application, the iodide-bearing members are shown to be highly selective for enyne cycloisomerization reactions. The dehydropiperidine-fused cyclopropane products are formed in good yields and enantioselectivities.
Co-reporter:Julia Pedroni ;Dr. Nicolai Cramer
Angewandte Chemie International Edition 2015 Volume 54( Issue 40) pp:11826-11829
Publication Date(Web):
DOI:10.1002/anie.201505916
Abstract
Cyclopropanes fused to pyrrolidines are important structural features found in a number of marketed drugs and development candidates. Typically, their synthesis involves the cyclopropanation of a dihydropyrrole precursor. Reported herein is a complementary approach which employs a palladium(0)-catalyzed CH functionalization of an achiral cyclopropane to close the pyrrolidine ring in an enantioselective manner. In contrast to aryl–aryl couplings, palladium(0)-catalyzed CH functionalizations involving the formation of C(sp3)C(sp3) bonds of saturated heterocycles are very scarce. The presented strategy yields cyclopropane-fused γ-lactams from chloroacetamide substrates. A bulky Taddol phosphonite ligand in combination with adamantane-1-carboxylic acid as a cocatalyst provides the γ-lactams in excellent yields and enantioselectivities.
Co-reporter:Laetitia Souillart ;Dr. Nicolai Cramer
Chemistry - A European Journal 2015 Volume 21( Issue 5) pp:1863-1867
Publication Date(Web):
DOI:10.1002/chem.201406135
Abstract
The exploitation of strain release in small rings as driving force to enable complex transformations is a powerful synthetic tool. Among them, cyclobutanones are particularly versatile substrates that can be elaborated in a wide variety of structurally diverse building blocks. Herein, Lewis acid catalyzed rearrangement reactions are presented that provide selective access to two structurally distinct polycyclic scaffolds, that is, indenylacetic acid derivatives and benzoxabicyclo[3.2.1]octan-3-ones. The choice of the Lewis acid fully controls the reaction pathway and the regioselectivity of the cyclobutanone CC bond cleavage site.
Co-reporter:Dr. Pavel A. Donets ;Dr. Nicolai Cramer
Angewandte Chemie International Edition 2015 Volume 54( Issue 2) pp:633-637
Publication Date(Web):
DOI:10.1002/anie.201409669
Abstract
The 1,6-annulated 2-pyridone motif is found in many biologically active compounds and its close relation to the indolizidine and quinolizidine alkaloid core makes it an attractive building block. A nickel-catalyzed CH functionalization of 2-pyridones and subsequent cyclization affords 1,6-annulated 2-pyridones by selective intramolecular olefin hydroarylation. The switch between the exo- and endo-cyclization modes is controlled by two complementary sets of ligands. Irrespective of the ring size, the regioselectivity during the cyclization is under full catalyst control. Simple cyclooctadiene promotes an exo-selective cyclization, whereas a bulky N-heterocyclic carbene ligand results in an endo-selective mode. The method was further applied in the synthesis of the lupin alkaloid cytisine.
Co-reporter:Laetitia Souillart and Nicolai Cramer
Chemical Science 2014 vol. 5(Issue 2) pp:837-840
Publication Date(Web):30 Oct 2013
DOI:10.1039/C3SC52753K
Rhodium(I)-catalyzed β-carbon eliminations of tert-cyclobutanols followed by oxidative addition give benzorhoda(III)cyclopentenes. These key intermediates trigger intramolecular C–H arylations leading to β-tetralones with quaternary stereogenic centers in excellent enantioselectivity. The versatility of the rhoda(III)cyclic species is further shown in formal intramolecular [4+2]-cycloadditions providing access to benzobicyclo[2.2.2]octanones.
Co-reporter:Laetitia Souillart ;Dr. Nicolai Cramer
Angewandte Chemie 2014 Volume 126( Issue 36) pp:9794-9798
Publication Date(Web):
DOI:10.1002/ange.201405834
Abstract
The lactone motif is ubiquitous in natural products and pharmaceuticals. The Tishchenko disproportionation of two aldehydes, a carbonyl hydroacylation, is an efficient and atom-economic access to lactones. However, these reaction types are limited to the transfer of a hydride to the accepting carbonyl group. The transfer of alkyl groups enabling the formation of CC bonds during the ester formation would be of significant interest. Reported herein is such asymmetric carbonyl carboacylation of aldehydes and ketones, thus affording complex bicyclic lactones in excellent enantioselectivities. The rhodium(I)-catalyzed transformation is induced by an enantiotopic CC bond activation of a cyclobutanone and the formed rhodacyclic intermediate reacts with aldehyde or ketone groups to give highly functionalized lactones.
Co-reporter:Julia Pedroni;Michele Boghi;Dr. Tanguy Saget ;Dr. Nicolai Cramer
Angewandte Chemie 2014 Volume 126( Issue 34) pp:9210-9213
Publication Date(Web):
DOI:10.1002/ange.201405508
Abstract
β-Lactams are very important structural motifs because of their broad biological activities as well as their propensity to engage in ring-opening reactions. Transition-metal-catalyzed CH functionalizations have emerged as strategy enabling yet uncommon highly efficient disconnections. In contrast to the significant progress of Pd0-catalyzed CH functionalization for aryl–aryl couplings, related reactions involving the formation of saturated C(sp3)C(sp3) bonds are elusive. Reported here is an asymmetric CH functionalization approach to β-lactams using readily accessible chloroacetamide substrates. Important aspects of this transformation are challenging C(sp3)C(sp3) and strain-building reductive eliminations to for the four-membered ring. In general, the β-lactams are formed in excellent yields and enantioselectivities using a bulky taddol phosphoramidite ligand in combination with adamantyl carboxylic acid as cocatalyst.
Co-reporter:Manh V. Pham ;Dr. Nicolai Cramer
Angewandte Chemie 2014 Volume 126( Issue 13) pp:3552-3555
Publication Date(Web):
DOI:10.1002/ange.201310723
Abstract
Larger condensed arenes are of interest owing to their electro- and photochemical properties. An efficient synthesis is the catalyzed aromatic annulation of a smaller arene with two alkyne molecules. Besides difunctionalized starting materials, directed CH functionalization can be used for such aromatic homologation. However, thus far the requirement of either pre-functionalized substrates or suitable directing groups were limiting this approach. Herein, we describe a rhodium(III)-catalyzed method allowing the use of completely unbiased arenes and internal alkynes. The reaction works best with copper(II) 2-ethylhexanoate and decabromodiphenyl ether as the oxidant combination. This aromatic annulation tolerates a variety of functional groups and delivers homologated condensed arenes. Aside from simple benzenes, naphthalenes and higher condensed arenes provide access to highly substituted and highly soluble acenes structures having important electronic and photophysical properties.
Co-reporter:Laetitia Souillart;Dr. Evelyne Parker ;Dr. Nicolai Cramer
Angewandte Chemie 2014 Volume 126( Issue 11) pp:3045-3049
Publication Date(Web):
DOI:10.1002/ange.201311009
Abstract
The selective functionalization of carbon–carbon σ bonds is a synthetic strategy that offers uncommon retrosynthetic disconnections. Despite progress in CC activation and its great importance, the development of asymmetric reactions lags behind. Rhodium(I)-catalyzed selective oxidative additions into enantiotopic CC bonds in cyclobutanones are reported. Even operating at a reaction temperature of 130 °C, the process is characterized by outstanding enantioselectivity with the e.r. generally greater than 99.5:0.5. The intermediate rhodacycle is shown to react with a wide variety of tethered olefins to deliver complex bicyclic ketones in high yields.
Co-reporter:Baihua Ye;Dr. Pavel A. Donets ;Dr. Nicolai Cramer
Angewandte Chemie 2014 Volume 126( Issue 2) pp:517-521
Publication Date(Web):
DOI:10.1002/ange.201309207
Abstract
Metal-catalyzed functionalizations at the ortho position of a directing group have become an efficient bond-forming strategy. A wide range of transformations that employ Cp*RhIII catalysts have been described, but despite their synthetic potential, enantioselective variants that use chiral versions of the Cp* ligand remain scarce (Cp*=pentamethyl cyclopentadienyl). Cyclopentadienyl compounds with an atropchiral biaryl backbone are shown to be suitable ligands for the efficient intramolecular enantioselective hydroarylation of aryl hydroxamates. Dihydrofurans that bear methyl-substituted quaternary stereocenters are thus obtained by CH functionalization under mild conditions.
Co-reporter:Baihua Ye ;Dr. Nicolai Cramer
Angewandte Chemie 2014 Volume 126( Issue 30) pp:8030-8033
Publication Date(Web):
DOI:10.1002/ange.201404895
Abstract
Directed Cp*RhIII-catalyzed carbon–hydrogen (CH) bond functionalizations have evolved as a powerful strategy for the construction of heterocycles. Despite their high value, the development of related asymmetric reactions is largely lagging behind due to a limited availability of robust and tunable chiral cyclopentadienyl ligands. Rhodium complexes comprising a chiral Cp ligand with an atropchiral biaryl backbone enables an asymmetric synthesis of isoindolones from arylhydroxamates and weakly alkyl donor/acceptor diazo derivatives as one-carbon component under mild conditions. The complex guides the substrates with a high double facial selectivity yielding the chiral isoindolones in good yields and excellent enantioselectivities.
Co-reporter:Joachim S. E. Ahlin;Dr. Pavel A. Donets ;Dr. Nicolai Cramer
Angewandte Chemie International Edition 2014 Volume 53( Issue 48) pp:13229-13233
Publication Date(Web):
DOI:10.1002/anie.201408364
Abstract
Cyclopentenones are versatile structural motifs of natural products as well as reactive synthetic intermediates. The nickel-catalyzed reductive [3+2] cycloaddition of α,β-unsaturated aromatic esters and alkynes constitutes an efficient method for their synthesis. Here, nickel(0) catalysts comprising a chiral bulky C1-symmetric N-heterocyclic carbene ligand were shown to enable an efficient asymmetric synthesis of cyclopentenones from mesityl enoates and internal alkynes under mild conditions. The bulky NHC ligand provided the cyclopentenone products in very high enantioselectivity and led to a regioselective incorporation of unsymmetrically substituted alkynes.
Co-reporter:Baihua Ye;Dr. Pavel A. Donets ;Dr. Nicolai Cramer
Angewandte Chemie International Edition 2014 Volume 53( Issue 2) pp:507-511
Publication Date(Web):
DOI:10.1002/anie.201309207
Abstract
Metal-catalyzed functionalizations at the ortho position of a directing group have become an efficient bond-forming strategy. A wide range of transformations that employ Cp*RhIII catalysts have been described, but despite their synthetic potential, enantioselective variants that use chiral versions of the Cp* ligand remain scarce (Cp*=pentamethyl cyclopentadienyl). Cyclopentadienyl compounds with an atropchiral biaryl backbone are shown to be suitable ligands for the efficient intramolecular enantioselective hydroarylation of aryl hydroxamates. Dihydrofurans that bear methyl-substituted quaternary stereocenters are thus obtained by CH functionalization under mild conditions.
Co-reporter:Manh V. Pham ;Dr. Nicolai Cramer
Angewandte Chemie International Edition 2014 Volume 53( Issue 13) pp:3484-3487
Publication Date(Web):
DOI:10.1002/anie.201310723
Abstract
Larger condensed arenes are of interest owing to their electro- and photochemical properties. An efficient synthesis is the catalyzed aromatic annulation of a smaller arene with two alkyne molecules. Besides difunctionalized starting materials, directed CH functionalization can be used for such aromatic homologation. However, thus far the requirement of either pre-functionalized substrates or suitable directing groups were limiting this approach. Herein, we describe a rhodium(III)-catalyzed method allowing the use of completely unbiased arenes and internal alkynes. The reaction works best with copper(II) 2-ethylhexanoate and decabromodiphenyl ether as the oxidant combination. This aromatic annulation tolerates a variety of functional groups and delivers homologated condensed arenes. Aside from simple benzenes, naphthalenes and higher condensed arenes provide access to highly substituted and highly soluble acenes structures having important electronic and photophysical properties.
Co-reporter:Laetitia Souillart;Dr. Evelyne Parker ;Dr. Nicolai Cramer
Angewandte Chemie International Edition 2014 Volume 53( Issue 11) pp:3001-3005
Publication Date(Web):
DOI:10.1002/anie.201311009
Abstract
The selective functionalization of carbon–carbon σ bonds is a synthetic strategy that offers uncommon retrosynthetic disconnections. Despite progress in CC activation and its great importance, the development of asymmetric reactions lags behind. Rhodium(I)-catalyzed selective oxidative additions into enantiotopic CC bonds in cyclobutanones are reported. Even operating at a reaction temperature of 130 °C, the process is characterized by outstanding enantioselectivity with the e.r. generally greater than 99.5:0.5. The intermediate rhodacycle is shown to react with a wide variety of tethered olefins to deliver complex bicyclic ketones in high yields.
Co-reporter:Duc N. Tran ;Dr. Nicolai Cramer
Chemistry - A European Journal 2014 Volume 20( Issue 34) pp:10654-10660
Publication Date(Web):
DOI:10.1002/chem.201403082
Abstract
(+)-Bicyclogermacrene is a strained bicyclic and common sesquiterpene found in several essential oils. A short and good yielding synthesis of bicyclogermacrene proceeding in seven steps is reported. This terpene is used as key platform intermediate for a biomimetic access to several aromadendrene sesquiterpenoids, such as ledene, viridiflorol, palestrol, and spathulenol. Furthermore, bicyclogermacrene is shown to be the terpene component in the synthesis of the meroterpenoids psiguadial A, C, and D.
Co-reporter:Dr. Matthew D. Wodrich;Baihua Ye;Dr. Jérôme F. Gonthier;Dr. Clémence Corminboeuf;Dr. Nicolai Cramer
Chemistry - A European Journal 2014 Volume 20( Issue 47) pp:15409-15418
Publication Date(Web):
DOI:10.1002/chem.201404515
Abstract
RhIII-catalyzed directed CH functionalizations of arylhydroxamates have become a valuable synthetic tool. To date, the regioselectivity of the insertion of the unsaturated acceptor into the common cyclometalated intermediate was dependent solely on intrinsic substrate control. Herein, we report two different catalytic systems that allow the selective formation of regioisomeric 3-aryl dihydroisoquinolones and previously inaccessible 4-aryl dihydroisoquinolones under full catalyst control. The differences in the catalysts are computationally examined using density functional theory and transition state theory of different possible pathways to elucidate key contributing factors leading to the regioisomeric products. The stabilities of the initially formed rhodium complex styrene adducts, as well as activation barrier differences for the migratory insertion, were identified as key contributing factors for the regiodivergent pathways.
Co-reporter:Manh V. Pham ;Dr. Nicolai Cramer
Angewandte Chemie International Edition 2014 Volume 53( Issue 52) pp:14575-14579
Publication Date(Web):
DOI:10.1002/anie.201409450
Abstract
Enol esters are versatile synthetic building blocks which can be elaborated by a wide variety of transformations. The classical synthesis by O-selective enolate acylation often hampers control of the E/Z selectivity with highly substituted substrates. A rhodium(III)/copper(II)-mediated process is reported to provide tetrasubstituted enol esters in a trans-selective fashion. Overall, the reaction consists of a heteroaryl acyloxylation of alkynes. The process is initiated by a rhodium(III)-catalyzed C2-selective activation of electron-rich heteroarenes, such as benzofuran, furan, and thiophene. Upon addition across an alkyne, a transmetalation to copper(II) enables reductive CO bond formation. The transformation allows the three-component couplings of heteroarenes, alkynes, and carboxylic acids. Application of the method in the functionalization of bioactive furocoumarin natural products is also described.
Co-reporter:Laetitia Souillart ;Dr. Nicolai Cramer
Angewandte Chemie International Edition 2014 Volume 53( Issue 36) pp:9640-9644
Publication Date(Web):
DOI:10.1002/anie.201405834
Abstract
The lactone motif is ubiquitous in natural products and pharmaceuticals. The Tishchenko disproportionation of two aldehydes, a carbonyl hydroacylation, is an efficient and atom-economic access to lactones. However, these reaction types are limited to the transfer of a hydride to the accepting carbonyl group. The transfer of alkyl groups enabling the formation of CC bonds during the ester formation would be of significant interest. Reported herein is such asymmetric carbonyl carboacylation of aldehydes and ketones, thus affording complex bicyclic lactones in excellent enantioselectivities. The rhodium(I)-catalyzed transformation is induced by an enantiotopic CC bond activation of a cyclobutanone and the formed rhodacyclic intermediate reacts with aldehyde or ketone groups to give highly functionalized lactones.
Co-reporter:Julia Pedroni;Michele Boghi;Dr. Tanguy Saget ;Dr. Nicolai Cramer
Angewandte Chemie International Edition 2014 Volume 53( Issue 34) pp:9064-9067
Publication Date(Web):
DOI:10.1002/anie.201405508
Abstract
β-Lactams are very important structural motifs because of their broad biological activities as well as their propensity to engage in ring-opening reactions. Transition-metal-catalyzed CH functionalizations have emerged as strategy enabling yet uncommon highly efficient disconnections. In contrast to the significant progress of Pd0-catalyzed CH functionalization for aryl–aryl couplings, related reactions involving the formation of saturated C(sp3)C(sp3) bonds are elusive. Reported here is an asymmetric CH functionalization approach to β-lactams using readily accessible chloroacetamide substrates. Important aspects of this transformation are challenging C(sp3)C(sp3) and strain-building reductive eliminations to for the four-membered ring. In general, the β-lactams are formed in excellent yields and enantioselectivities using a bulky taddol phosphoramidite ligand in combination with adamantyl carboxylic acid as cocatalyst.
Co-reporter:Baihua Ye ;Dr. Nicolai Cramer
Angewandte Chemie International Edition 2014 Volume 53( Issue 30) pp:7896-7899
Publication Date(Web):
DOI:10.1002/anie.201404895
Abstract
Directed Cp*RhIII-catalyzed carbon–hydrogen (CH) bond functionalizations have evolved as a powerful strategy for the construction of heterocycles. Despite their high value, the development of related asymmetric reactions is largely lagging behind due to a limited availability of robust and tunable chiral cyclopentadienyl ligands. Rhodium complexes comprising a chiral Cp ligand with an atropchiral biaryl backbone enables an asymmetric synthesis of isoindolones from arylhydroxamates and weakly alkyl donor/acceptor diazo derivatives as one-carbon component under mild conditions. The complex guides the substrates with a high double facial selectivity yielding the chiral isoindolones in good yields and excellent enantioselectivities.
Co-reporter:Evelyne Parker and Nicolai Cramer
Organometallics 2014 Volume 33(Issue 3) pp:780-787
Publication Date(Web):January 23, 2014
DOI:10.1021/om4011627
The development of unconventional ligand scaffolds is an important aspect to alter reaction pathways of transition-metal-catalyzed reactions. The nature of the counterion of cationic metal complexes plays an important role in the catalyst reactivity. We herein report a chiral anionic bidentate bis-phosphine ligand based on the popular phospholane scaffold. Subsequently, zwitterionic rhodium(I) complexes with no external counterion were synthesized, and their potential was evaluated in asymmetric carbon–carbon bond activation of cyclobutanones. This type of rhodium complex allows for a significantly lower reaction temperature than analogous cationic rhodium complexes and enables, for the first time, asymmetric transformations with up to 93.5:6.5 enantiomeric ratio.
Co-reporter:Manh V. Pham ;Dr. Nicolai Cramer
Angewandte Chemie 2014 Volume 126( Issue 52) pp:14803-14807
Publication Date(Web):
DOI:10.1002/ange.201409450
Abstract
Enol esters are versatile synthetic building blocks which can be elaborated by a wide variety of transformations. The classical synthesis by O-selective enolate acylation often hampers control of the E/Z selectivity with highly substituted substrates. A rhodium(III)/copper(II)-mediated process is reported to provide tetrasubstituted enol esters in a trans-selective fashion. Overall, the reaction consists of a heteroaryl acyloxylation of alkynes. The process is initiated by a rhodium(III)-catalyzed C2-selective activation of electron-rich heteroarenes, such as benzofuran, furan, and thiophene. Upon addition across an alkyne, a transmetalation to copper(II) enables reductive CO bond formation. The transformation allows the three-component couplings of heteroarenes, alkynes, and carboxylic acids. Application of the method in the functionalization of bioactive furocoumarin natural products is also described.
Co-reporter:Joachim S. E. Ahlin;Dr. Pavel A. Donets ;Dr. Nicolai Cramer
Angewandte Chemie 2014 Volume 126( Issue 48) pp:13445-13449
Publication Date(Web):
DOI:10.1002/ange.201408364
Abstract
Cyclopentenones are versatile structural motifs of natural products as well as reactive synthetic intermediates. The nickel-catalyzed reductive [3+2] cycloaddition of α,β-unsaturated aromatic esters and alkynes constitutes an efficient method for their synthesis. Here, nickel(0) catalysts comprising a chiral bulky C1-symmetric N-heterocyclic carbene ligand were shown to enable an efficient asymmetric synthesis of cyclopentenones from mesityl enoates and internal alkynes under mild conditions. The bulky NHC ligand provided the cyclopentenone products in very high enantioselectivity and led to a regioselective incorporation of unsymmetrically substituted alkynes.
Co-reporter:Pavel A. Donets
Journal of the American Chemical Society 2013 Volume 135(Issue 32) pp:11772-11775
Publication Date(Web):July 29, 2013
DOI:10.1021/ja406730t
Chiral trivalent phosphorus species are the dominant class of ligands and the key controlling element in asymmetric homogeneous transition-metal catalysis. Here, novel chiral diaminophosphine oxide ligands are described. The arising catalyst system with nickel(0) and trimethylaluminum efficiently activates formamide C–H bonds under mild conditions providing pyrrolidones via intramolecular hydrocarbamoylation in a highly enantioselective manner with as little as 0.25% mol catalyst loading. Mechanistically, the secondary phosphine oxides behave as bridging ligands for the nickel center and the Lewis acidic organoaluminum center to give a heterobimetallic catalyst with superior reactivity.
Co-reporter:Baihua Ye
Journal of the American Chemical Society 2013 Volume 135(Issue 2) pp:636-639
Publication Date(Web):January 3, 2013
DOI:10.1021/ja311956k
The lack of robust and tunable chiral versions of cyclopentadienyl (Cp) ligands hampers progress in the development of catalytic asymmetric versions of a myriad of reactions catalyzed by this ubiquitous ligand. Herein, we describe of a class of chiral Cp ligands with tunable steric parameters. Coordinated to transition metals, the ligand creates a well-defined chiral pocket, able to imprint its chirality onto the metal. The corresponding Rh complexes are shown to be excellent catalysts for enantioselective allylation of N-methoxybenzamides via directed C–H functionalizations at very mild conditions. The obtained enantioselectivities are excellent and demonstrate the viability of chiral Cp complexes as selective transition metal catalysts.
Co-reporter:Tanguy Saget, David Perez, and Nicolai Cramer
Organic Letters 2013 Volume 15(Issue 6) pp:1354-1357
Publication Date(Web):March 1, 2013
DOI:10.1021/ol400380y
The synthesis of cyclopropyl spiroindolines is described using an intramolecular palladium(0)-catalyzed C–H functionalization of a methine C(sp3)–H bond. This transformation can be coupled with intermolecular Suzuki couplings or direct arylations of heteroaromatics to access functionalized indoline scaffolds in a single step.
Co-reporter:Tanguy Saget ;Dr. Nicolai Cramer
Angewandte Chemie International Edition 2013 Volume 52( Issue 30) pp:7865-7868
Publication Date(Web):
DOI:10.1002/anie.201303816
Co-reporter:Tanguy Saget ;Dr. Nicolai Cramer
Angewandte Chemie 2013 Volume 125( Issue 30) pp:8019-8022
Publication Date(Web):
DOI:10.1002/ange.201303816
Co-reporter:Duc N. Tran ;Dr. Nicolai Cramer
Angewandte Chemie 2013 Volume 125( Issue 40) pp:10824-10828
Publication Date(Web):
DOI:10.1002/ange.201304919
Co-reporter:Duc N. Tran ;Dr. Nicolai Cramer
Angewandte Chemie International Edition 2013 Volume 52( Issue 40) pp:10630-10634
Publication Date(Web):
DOI:10.1002/anie.201304919
Co-reporter:Nicolai Cramer;Baihua Ye
Science 2012 Volume 338(Issue 6106) pp:504-506
Publication Date(Web):26 Oct 2012
DOI:10.1126/science.1226938
Co-reporter:Tanguy Saget;Sébastien J. Lemouzy;Dr. Nicolai Cramer
Angewandte Chemie 2012 Volume 124( Issue 9) pp:2281-2285
Publication Date(Web):
DOI:10.1002/ange.201108511
Co-reporter:Tanguy Saget;Dr. Nicolai Cramer
Angewandte Chemie 2012 Volume 124( Issue 51) pp:13014-13017
Publication Date(Web):
DOI:10.1002/ange.201207959
Co-reporter:Manh V. Pham;Baihua Ye ;Dr. Nicolai Cramer
Angewandte Chemie 2012 Volume 124( Issue 42) pp:10762-10766
Publication Date(Web):
DOI:10.1002/ange.201206191
Co-reporter:Manh V. Pham;Baihua Ye ;Dr. Nicolai Cramer
Angewandte Chemie International Edition 2012 Volume 51( Issue 42) pp:10610-10614
Publication Date(Web):
DOI:10.1002/anie.201206191
Co-reporter:Tanguy Saget;Dr. Nicolai Cramer
Angewandte Chemie International Edition 2012 Volume 51( Issue 51) pp:12842-12845
Publication Date(Web):
DOI:10.1002/anie.201207959
Co-reporter:Tanguy Saget;Sébastien J. Lemouzy;Dr. Nicolai Cramer
Angewandte Chemie International Edition 2012 Volume 51( Issue 9) pp:2238-2242
Publication Date(Web):
DOI:10.1002/anie.201108511
Co-reporter:Pavel A. Donets, Tanguy Saget, and Nicolai Cramer
Organometallics 2012 Volume 31(Issue 23) pp:8040-8046
Publication Date(Web):October 17, 2012
DOI:10.1021/om3008772
The development of efficient chiral monodentate phosphine ligands lags behind that of the bidentate congeners. This holds especially true for highly electron rich chiral phosphine analogues able to replace the ubiquitous tricyclohexylphosphine and tri-tert-butylphosphine in catalytic asymmetric transformations. We present a convenient and modular synthesis of a set of chiral monodentate ligands with different steric demands based on the popular phospholane scaffold. Their steric and electronic properties were determined by their corresponding nickel and palladium complexes. They represent good mimics of the popular tricyclohexylphosphine and tri-tert-butylphosphine ligands. Their potential was subsequently evaluated in palladium-catalyzed asymmetric C(sp3)–H functionalization leading to indolines.
Co-reporter:Tobias Seiser;Tanguy Saget;Duc N. Tran;Dr. Nicolai Cramer
Angewandte Chemie International Edition 2011 Volume 50( Issue 34) pp:7740-7752
Publication Date(Web):
DOI:10.1002/anie.201101053
Abstract
The exploitation of ring strain as a driving force to facilitate chemical reactions is a well-appreciated principle in organic chemistry. The most prominent and most frequently used compound classes in this respect are oxiranes and cyclopropanes. For rather a long time, cyclobutanes lagged behind these three-membered-ring compounds in their development as reactive substrates, but during the past decade an increasing number of useful reactions of four-membered-ring substrates have emerged. This Minireview examines corresponding catalytic reactions ranging from Lewis or Brønsted acid catalyzed processes to enzymatic reactions. The main focus is placed on transition-metal-catalyzed CC bond-insertion and β-carbon-elimination processes, which enable exciting downstream reactions that deliver versatile building blocks.
Co-reporter:Duc N. Tran;Dr. Nicolai Cramer
Angewandte Chemie International Edition 2011 Volume 50( Issue 47) pp:11098-11102
Publication Date(Web):
DOI:10.1002/anie.201105766
Co-reporter:Duc N. Tran;Dr. Nicolai Cramer
Angewandte Chemie 2011 Volume 123( Issue 47) pp:11294-11298
Publication Date(Web):
DOI:10.1002/ange.201105766
Co-reporter:Tobias Seiser;Tanguy Saget;Duc N. Tran;Dr. Nicolai Cramer
Angewandte Chemie 2011 Volume 123( Issue 34) pp:7884-7896
Publication Date(Web):
DOI:10.1002/ange.201101053
Abstract
Ein hoch geschätztes Prinzip in der organischen Chemie ist das Ausnutzen von Ringspannung als Triebkraft, um chemische Reaktionen zu erleichtern. Die bekanntesten und am häufigsten genutzten Verbindungsklassen in dieser Hinsicht sind Epoxide und Cyclopropane. Cyclobutane haben dieser Entwicklung relativ lange hinterhergehinkt, doch in den letzten zehn Jahren wurde eine steigende Zahl an bemerkenswerten Reaktionen entwickelt, die Vierringe als Substrate verwenden. Dieser Kurzaufsatz fasst entsprechende katalytische Reaktionen, von Lewis- über Brønsted-Säure-katalysierte bis hin zu enzymatischen Prozessen, zusammen. Das Hauptaugenmerk liegt dabei auf übergangsmetallkatalysierten C-C Bindungsinsertionen und β-Kohlenstoff-Eliminierungen. Diese Reaktionen ermöglichen faszinierende Folgeprozesse, die zu vielseitigen Synthesebausteinen führen.
Co-reporter:C. M. B. K. Kourra and N. Cramer
Chemical Science (2010-Present) 2016 - vol. 7(Issue 12) pp:NaN7012-7012
Publication Date(Web):2016/07/28
DOI:10.1039/C6SC02285E
Disulfide bridges play a crucial role in defining and rigidifying the three-dimensional structure of peptides. However, disulfides are inherently unstable in reducing environments. Consequently, the development of strategies aiming to circumvent these deficiencies – ideally with little structural disturbance – are highly sought after. Herein, we report a simple protocol converting the disulfide bond of peptides into highly stable methylene thioacetal. The transformation occurs under mild, biocompatible conditions, enabling the conversion of unprotected native peptides into analogues with enhanced stability. The developed protocol is applicable to a range of peptides and selective in the presence of a multitude of potentially reactive functional groups. The thioacetal modification annihilates the reductive lability and increases the serum, pH and temperature stability of the important peptide hormone oxytocin. Moreover, it is shown that the biological activities for oxytocin are retained.
Co-reporter:D. Kossler and N. Cramer
Chemical Science (2010-Present) 2017 - vol. 8(Issue 3) pp:NaN1866-1866
Publication Date(Web):2017/01/19
DOI:10.1039/C6SC05092A
Cyclopentadienyl ruthenium(II) complexes with a large number of available coordination sites are frequently used catalysts for a broad range of transformations. To be able to render these transformations enantioselective, we have designed a chiral neutral CpxRu(II)Cl complex basing on an atropchiral cyclopentadienyl (Cpx) ligand which is accessed in a streamlined C–H functionalisation approach. The catalyst displays excellent levels of reactivity and enantioselectivity for enantioselective [2+2]-cycloadditions leading to strained chiral cyclobutenes, allowing for catalyst loadings as low as 1 mol%. A very strong counterion effect of a bound chloride anion transforms the corresponding unselective cationic complex into a highly enantioselective neutral version. Moreover, by adding norbornadiene at the end of the reaction the catalyst can be recovered and subsequently reused.
Co-reporter:J. Pedroni, T. Saget, P. A. Donets and N. Cramer
Chemical Science (2010-Present) 2015 - vol. 6(Issue 9) pp:NaN5171-5171
Publication Date(Web):2015/06/17
DOI:10.1039/C5SC01909E
Taddol-based phosphoramidite ligands enable enantioselective palladium(0)-catalyzed C–H arylation of cyclopropanes. The cyclized products are obtained in high yields and enantioselectivities. The reported method provides efficient access to a broad range of synthetically attractive cyclopropyl containing dihydroquinolones and dihydroisoquinolones as well as allows for an efficient enantioselective construction of the 7-membered ring of the cyclopropyl indolobenzazepine core of BMS-791325.
Co-reporter:Laetitia Souillart and Nicolai Cramer
Chemical Science (2010-Present) 2014 - vol. 5(Issue 2) pp:NaN840-840
Publication Date(Web):2013/10/30
DOI:10.1039/C3SC52753K
Rhodium(I)-catalyzed β-carbon eliminations of tert-cyclobutanols followed by oxidative addition give benzorhoda(III)cyclopentenes. These key intermediates trigger intramolecular C–H arylations leading to β-tetralones with quaternary stereogenic centers in excellent enantioselectivity. The versatility of the rhoda(III)cyclic species is further shown in formal intramolecular [4+2]-cycloadditions providing access to benzobicyclo[2.2.2]octanones.
Co-reporter:Julia Pedroni and Nicolai Cramer
Chemical Communications 2015 - vol. 51(Issue 100) pp:NaN17657-17657
Publication Date(Web):2015/10/26
DOI:10.1039/C5CC07929B
Monodentate TADDOL-derived phosphoramidites and phosphonites are versatile chiral ligands for enantioselective Pd(0)-catalysed C–H functionalisations. They enable highly selective cyclisations to access a wide range of chiral carbo- and heterocycles. The high attractiveness of this ligand class consists in their modular structure, allowing for a quick assembly of a library with variable steric properties. Asymmetric C–H functionalisation methods utilising catalytic systems based on Pd(0) complexes and TADDOL-type ligands are presented and aspects of selectivity are discussed.

![2-Cyclopentene-1-methanol, 1-[(phenylmethoxy)methyl]-](http://img.cochemist.com/ccimg/61200/61111-55-5.png)
![2-Cyclopentene-1-methanol, 1-[(phenylmethoxy)methyl]-](http://img.cochemist.com/ccimg/61200/61111-55-5_b.png)