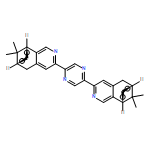
Co-reporter:Feng-Yang Bai, Shuang Lv, Yuan Ma, Chun-Yu Liu, Chun-Fang He, Xiu-Mei Pan
Chemosphere 2017 Volume 171(Volume 171) pp:
Publication Date(Web):1 March 2017
DOI:10.1016/j.chemosphere.2016.12.037
•We studied mechanism of Cl-initiated oxidation of (CH3)3CC(O)X (X = F, Cl, and Br).•The total rate constants are computed for (CH3)3CC(O)X (X = F, Cl, and Br) at 298 K.•Atmospheric lifetimes and GWPs for (CH3)3CC(O)X (X = F, Cl, and Br) are estimated.•The subsequent reactions may produce organic aerosol in the presence of O2/NO.In this work, the density functional and high-level ab initio theories are adopted to investigate the mechanisms and kinetics of reaction of (CH3)3CC(O)X (X = F, Cl, and Br) with atomic chlorine. Rate coefficients for the reactions of chlorine atom with (CH3)3CC(O)F (k1), (CH3)3CC(O)Cl (k2), and (CH3)3CC(O)Br (k3) are calculated using canonical variational transition state theory coupled with small curvature tunneling method over a wide range of temperatures from 250 to 1000 K. The dynamic calculations are performed by the variational transition state theory with the interpolated single-point energies method at the CCSD(T)/aug-cc-pVDZ//B3LYP/6-311++G(d,p) level of theory. Computed rate constant is in good line with the available experimental value. The rate constants for the title reactions are in this order: k1
Co-reporter:Chunyu Liu, Yanling Si, Shaoqing Shi, Guochun Yang and Xiumei Pan
Dalton Transactions 2016 vol. 45(Issue 17) pp:7285-7293
Publication Date(Web):15 Mar 2016
DOI:10.1039/C6DT00089D
Chiral transition metal complexes not only have large nonlinear optical (NLO) response but also meet the non-centrosymmetric requirement of second-order NLO materials. Therefore, chiral transition metal complexes become very active in the NLO area. Recently, the second-order NLO response of chiral dinuclear Re(I) complex 2 has been found to be 1.5 times larger than that of KH2PO4 (KDP) based on experimental measurement. However, its NLO origin has not been determined and a structure–property relationship has not been established at the microscopic level, which are very important to further improve the performance. It is found that charge transfer from metal to ligand is mainly responsible for its NLO origin. Based on complex 2, the designed complexes have remarkably large second-order NLO activity. For instance, the designed complex 9 has a very large second-order NLO response value (115.81 × 10−30 esu), which is about 668 times larger than the organic molecule urea. Moreover, time-dependent density functional theory (TDDFT) calculations have been used to investigate their UV-Vis/CD spectra. The simulated circular dichroism (CD) spectra of the complex 2 are in good agreement with the experimental ones, which can be used to assign the absolute configurations (ACs) of chiral dinuclear Re(I) complexes with high confidence. The electronic absorption wavelengths, electron transition properties, and the second-order NLO responses strongly depend on the nature of substituent, different ligands (pyridine and isoquinoline) and their combinations. Based on NBO analysis, the interactions between [Re(CO)3Cl] fragments and ligands are of n → σ* character.
Co-reporter:Jin-Ting Ye;Feng-Yang Bai
Environmental Science and Pollution Research 2016 Volume 23( Issue 23) pp:23467-23484
Publication Date(Web):2016 December
DOI:10.1007/s11356-016-7505-4
Multichannel gas-phase reactions of CH3OCH2CH2Cl/CH3CH2OCH2CH2Cl with chlorine atom and hydroxyl radical have been investigated using ab initio method and canonical variational transition-state dynamic computations with the small-curvature tunneling correction. Further energetic information is refined by the coupled-cluster calculations with single and double excitations (CCSD)(T) method. Both hydrogen abstraction and displacement processes are carried out at the same level. Our results reveal that H-abstraction from the –OCH2– group is the dominant channel for CH3OCH2CH2Cl by OH radical or Cl atom, and from α-CH2 of the group CH3CH2– is predominate for the reaction CH3CH2OCH2CH2Cl with Cl/OH. The contribution of displacement processes may be unimportant due to the high barriers. The values of the calculated rate constants reproduce remarkably well the available experiment data. Standard enthalpies of formation for reactants and product radicals are calculated by isodesmic reactions. The Arrhenius expressions are given within 220–1200 K. The atmospheric lifetime, ozone depleting potential (ODP), ozone formation potential (OFP), and global warming potential (GWP) of CH3OCH2CH2Cl/CH3CH2OCH2CH2Cl are investigated. Meanwhile, the atmospheric fate of the alkoxy radicals are also researched using the same level of theory. To shed light on the atmospheric degradation, a mechanistic study is obtained, which indicates that reaction with O2 is the dominant path for the decomposition of CH3OCH(O•)CH2Cl, the C–C bond scission reaction is the primary reaction path in the consumption of CH3CH(O•)OCH2CH2Cl in the atmosphere.Ab initio method and canonical variational transition-state theory are employed to study the kinetic nature of hydrogen abstraction reactions of CH3OCH2CH2Cl/CH3CH2OCH2CH2Cl with Cl atom and OH radical and fate of alkoxy radicals (CH3OCH(O•)CH2Cl/CH3CH(O•)OCH2CH2Cl).
Co-reporter:Chunyu Liu, Yanling Si, Xiumei Pan and Guochun Yang
RSC Advances 2015 vol. 5(Issue 89) pp:72907-72915
Publication Date(Web):13 Aug 2015
DOI:10.1039/C5RA12577D
Helicene and its derivatives have received considerable attention as candidates for organic photoelectronic materials. Recently, novel quinoxaline-fused [7]carbohelicene derivatives have exhibited unique structural and photophysical properties, especially in the crystal state. However, their structure–property relationships have not been fully understood from their micromechanisms, which is also important to further improve their performance. Herein, the electronic transitions, electronic circular dichroism (CD), second-order nonlinear optical (NLO) responses and charge transport properties of five quinoxaline-fused [7]carbohelicene derivatives have been investigated based on density functional theory calculations. The experimental UV-Vis/CD spectra of the studied compounds were reproduced well by our calculations. Thus, we can assign their electron transition properties and absolution configurations (ACs) with high confidence. It is found that the CD bands of quinoxaline-fused [7]carbohelicene derivatives mainly originate from exciton coupling between quinoxaline, phenyl or 4-methoxyphenyl groups and [7]carbohelicene, which is in sharp contrast to [7]carbohelicene. More interestingly, these derivatives possess large first hyperpolarizability values. For example, the βHRS value of compound 6 is 32.96 × 10−30 esu, which is about 190 times larger than that of the organic urea molecule. The bandwidth of the valence band of compound 2 is comparable to that of the conduction band and slightly larger than that of tris(8-hydroxyquinolinato)aluminium. This means that compound 2 is a potential candidate as an ambipolar charge transport material.
Co-reporter:Feng-Yang Bai, Xu Wang, Yan-Qiu Sun and Xiu-Mei Pan
RSC Advances 2015 vol. 5(Issue 107) pp:88087-88095
Publication Date(Web):30 Sep 2015
DOI:10.1039/C5RA16215G
The gas-phase reactions of CH3I and C2H5I with NO3 radicals have been studied using a dual-level direct kinetics method. The minimum energy paths have been refined by CCSD(T) and QCISD(T) methods. One displacement and two hydrogen abstraction processes were found for the reaction of CH3I + NO3. For the reaction of C2H5I + NO3, three hydrogen abstraction and one displacement channel were found. The hydrogen abstraction from the –CH2– group was found to be the dominant channel. The displacement channel of the title reactions may be negligible because of the high barrier. The rate constants for the individual reaction channels were followed by means of the canonical variational transition state with the small-curvature tunneling correction. The calculated rate constants were in reasonable agreement with the available data from experiments. The Arrhenius expressions for the title reactions are given as follows: ka = 8.62 × 10−32T6.66exp(1324.23/T), kb = 9.48 × 10−27T5.75exp(−655.34/T) cm3 per molecule per s. The atmospheric lifetimes of CH3I and C2H5I determined by reaction with the NO3 radical were about 3.07 and 5.86 h, which indicate that they can be degraded in the gas phase within a short time to serve as a source of reactive iodine compounds at night-time.
Co-reporter:Feng-Yang Bai;Xiao-Le Zhu;Zi-Man Jia;Xu Wang;Yan-Qiu Sun; Rong-Shun Wang ; Xiu-Mei Pan
ChemPhysChem 2015 Volume 16( Issue 8) pp:1768-1776
Publication Date(Web):
DOI:10.1002/cphc.201402799
Abstract
The mechanism and kinetics of the reactions of CF3COOCH2CH3, CF2HCOOCH3, and CF3COOCH3 with Cl and OH radicals are studied using the B3LYP, MP2, BHandHLYP, and M06-2X methods with the 6-311G(d,p) basis set. The study is further refined by using the CCSD(T) and QCISD(T)/6-311++G(d,p) methods. Seven hydrogen-abstraction channels are found. All the rate constants, computed by a dual-level direct method with a small-curvature tunneling correction, are in good agreement with the experimental data. The tunneling effect is found to be important for the calculated rate constants in the low-temperature range. For the reaction of CF3COOCH2CH3+Cl, H-abstraction from the CH2 group is found to be the dominant reaction channel. The standard enthalpies of formation for the species are also calculated. The Arrhenius expressions are fitted within 200–1000 K as kT(1)=8.4×10−20T 2.63exp(381.28/T), kT(2)=2.95×10−21T 3.13exp(−103.21/T), kT(3)=1.25×10−23T 3.37exp(791.98/T), and kT(4)=4.53×10−22T 3.07exp(465.00/T).
Co-reporter:Feng-Yang Bai, Gang Sun, Xu Wang, Yan-Qiu Sun, Rong-Shun Wang, and Xiu-Mei Pan
The Journal of Physical Chemistry A 2015 Volume 119(Issue 8) pp:1256-1266
Publication Date(Web):January 28, 2015
DOI:10.1021/jp5125553
Reactions of (CF3)2CFOCH3 and (CF3)2CFOCHO with hydroxyl radical and chlorine atom are studied at the B3LYP and BHandHLYP/6-311+G(d,p) levels along with the geometries and frequencies of all stationary points. This study is further refined by CCSD(T) and QCISD(T)/6-311+G(d,p) methods in the minimum energy paths. For the reaction (CF3)2CFOCH3 + OH, two hydrogen abstraction channels are found. The total rate constants for the reactions (CF3)2CFOCH3 + OH/Cl and (CF3)2CFOCHO + Cl are followed by means of the canonical variational transition state with the small-curvature tunneling correction. The comparison between the hydrogen abstraction rate constants by hydroxyl and chlorine atom is discussed. Calculated rate constants are in reasonable agreement with the available experiment data. The standard enthalpies of formation for the reactants, (CF3)2CFOCH3 and (CF3)2CFOCHO, and two products, (CF3)2CFOCH2 and (CF3)2CFOCO, are evaluated by a series of isodesmic reactions. The Arrhenius expressions for the title reactions are given as follows: k1= 1.08 × 10–22 T3.38 exp(−213.31/T), k2= 3.55 × 10–22 T3.61 exp(−240.26/T), and k3= 3.00 × 10 –19 T2.58 exp(−1294.34/T) cm3 molecule–1 s–1.
Co-reporter:Li-Jie Wang, Shi-Ling Sun, Rong-Lin Zhong, Yan Liu, Dong-Lai Wang, Heng-Qing Wu, Hong-Liang Xu, Xiu-Mei Pan and Zhong-Min Su
RSC Advances 2013 vol. 3(Issue 32) pp:13348-13352
Publication Date(Web):10 May 2013
DOI:10.1039/C3RA40909K
Recently, C60Cl8 (C2v) has been experimentally synthesized (Y.-Z. Tan, et al., Nat. Mater., 2008, 7, 790) by the addition of eight chlorine atoms to C60 (C2v), which is associated with a Stone–Wales transformation of C60 (Ih). In this work, the first hyperpolarizabilities (βtot) of C60 (C2v) and C60Cl8 (C2v) are investigated. After the Stone–Wales transformation and chlorine addition reaction, the βtot values slightly increase from 0 for C60 (Ih) to 60 au for C60 (C2v) and 502 au for C60Cl8 (C2v), respectively. To further enhance the first hyperpolarizability, the endohedral fullerene derivative, Li@C60Cl8, formed by encapsulating a lithium (Li) atom inside the C60Cl8, has been designed. Interestingly, the electron transfer between Li and C60Cl8 leads to an extremely large βtot value of 25569 au, which is considerably larger (51 times) than the 502 au of C60Cl8. It shows that the encapsulated Li effect plays an important role in enhancing the first hyperpolarizability, so the Li@C60Cl8 can be considered as a candidate for high-performance nonlinear optical materials.
Co-reporter:Hao Chen, Fang Wang, Shanshan Tong, Shuangling Guo, Xiumei Pan
Applied Surface Science 2012 Volume 258(Issue 16) pp:6097-6102
Publication Date(Web):1 June 2012
DOI:10.1016/j.apsusc.2012.03.009
Abstract
A series of porous carbon samples as electric double layer capacitor electrode materials were prepared by a pyrolysis process using phenol formaldehyde resin (PF) as precursors and KOH/ZnCl2 as activation agents. Porous carbon samples were characterized by thermogravimetric analysis, X ray diffraction, nitrogen adsorption/desorption isotherms and transmission electron microscopy. The results showed that the KOH/ZnCl2/PF mass mixing ratio and activation temperature had a remarkable effect on the porosity, the specific surface area and the pore size of the carbons. The prepared carbon material PC-6 exhibits a high specific capacitance of 141.56 F/g and a average specific energy of 74.13 Wh/kg at a current density of 120 mA/g in the electrolyte of 1 M Et3MeNBF4/PC, and the average specific energy still remained 49.48 Wh/kg even at a high current density of 2000 mA/g. The excellent electrochemical behavior of PC-6 can be attributed to the highly development pore structure.
Co-reporter:Y. Zhang, L.L. Zhang, R.S. Wang, X.M. Pan
Journal of Molecular Graphics and Modelling 2012 Volume 34() pp:46-56
Publication Date(Web):April 2012
DOI:10.1016/j.jmgm.2011.12.007
Molecules with D-π-A structures are drawing increased attention for applications in organic electronic devices due to their distinct optoelectronic properties. A study of a new series of bipolar fluorophores that have been chemically modified for use as highly efficient nondoped blue organic light-emitting diodes (OLEDs) has been carried out based on existing molecular structures and a literature survey. The aim of this study is to provide a profound interpretation of the optical and electronic properties and the structure–property relationships of a series of new bipolar fluorophores. The study also aims to predict the photophysical and optoelectronic properties of the new fluorophores. The density functional theory (DFT) has been confirmed as reliable, especially in predicting the properties of unknown products. The geometry and the electronic structure of these molecules in the ground state were studied with DFT and ab initio HF, whereas the lowest singlet excited-state geometries were optimized by ab initio singlet configuration interaction (CIS). The absorption and emission spectra, both in the gas phase and in THF, and the lowest singlet excited energies were calculated by employing the time-dependent density functional theory (TDDFT) and the polarizable continuum model (PCM). To precisely predict the charge-transporting and charge-confining properties of the new fluorophores, three-layered devices have been simulated. The results show that the molecular geometries, HOMOs, LUMOs, energy gaps, ionization potentials (IP), electron affinities (EA), radiative lifetimes (τ), absorption and emission spectra are all tuned by chemical modifications with different π-conjugated bridges. The results also show that these molecular materials could be used as bipolar light-emitting materials for blue and deep-blue OLEDs.Graphical abstract. The configuration of simulated three-layered OLEDs and theoretical energy (in THF) diagrams of the title compounds.Highlights► Some bipolar fluorophores for highly efficient nondoped blue OLEDs have been emerged. ► DFT calculation is reliable especially in predicting the properties of unknown products. ► These molecular materials could be used as bipolar blue or deep-blue light-emitting materials.
Co-reporter:He Ren, Lingling Zhang, Rongshun Wang, and Xiumei Pan
The Journal of Physical Chemistry A 2012 Volume 116(Issue 44) pp:10647-10655
Publication Date(Web):October 17, 2012
DOI:10.1021/jp3064905
A dual-level direct dynamic method is employed to study the reaction mechanism of hydroxyl radical with (CH3)3COOH and (CH3)2CHOOH. Eight hydrogen abstraction channels are found for title reactions. The energy paths are optimized at the BH&H-HLYP/6-311G(d,p) level, and the energy profiles are further refined by interpolated single-point energies method at the CCSD(T) and QCISD(T) theories. Rate coefficients for the reactions of the OH with (CH3)3COOH/(CH3)2CHOOH are computed by the canonical variational transition-state theory with the small-curvature tunneling correction between 200 and 2000 K. The Arrhenius expressions k1 (T) = 1.49 × 10–26 T4.71 exp(1981.92/T) and k2 (T) = 1.58 × 10–20 T3.32 exp(210.59/T) over 200–2000 K are obtained.
Co-reporter:Yang Gao;Xiu-Juan Jia;Sha Li;Yan-Bo Yu;Rong-Shun Wang
Theoretical Chemistry Accounts 2010 Volume 127( Issue 1-2) pp:81-94
Publication Date(Web):2010 September
DOI:10.1007/s00214-009-0707-9
Both the singlet and triplet potential energy surfaces (PESs) of the NH (X3Σ−) + HCNO reaction have been investigated at the BMC-CCSD level based on the UB3LYP/6-311++G(d, p) structures. The results show that the title reaction is more favorable through the singlet potential energy surface than the triplet one. For the singlet potential energy surface of the NH (X3Σ−) + HCNO reaction, the most feasible association of NH (X3Σ−) with HCNO is found to be a non-barrier nitrogen-to-carbon attack forming the adduct a (trans-HNCHNO), which can isomerize to the adduct b (cis-HNCHNO). The most feasible channel is that the 1, 3-H shift with N2–H2 and C–N1 bonds cleavage associated with the N1–H2 bond formation of adduct a leads to the product P1 (HCN + HNO). Moreover, P2 (HNC + HNO) should be the competitive product. The other products, including P3 (NH2 + NCO) and P4 (N2H2 + CO), are minor products. The product P1 can be obtained through two competitive channels Path 1: R → a → P1 and Path 3: R → b → d → P1, whereas the product P2 can be formed through Path 2: R → b → d → P2. At high temperatures, the nitrogen-to-nitrogen approach may become feasible. For the triplet potential energy surface of the NH (X3Σ−) + HCNO reaction, the Path 10: R → 3a → 3a1 → P1 should be the most feasible pathway due to the less reaction steps and lower barriers. These conclusions will have impacts on further experimental investigations.
Co-reporter:Xiu-Juan Jia;You-Jun Liu;Jing-Yu Sun;Hao Sun
Theoretical Chemistry Accounts 2010 Volume 127( Issue 1-2) pp:49-56
Publication Date(Web):2010 September
DOI:10.1007/s00214-009-0702-1
The mechanisms of CH2I with NO2 reaction were investigated on the singlet and triplet potential energy surfaces (PESs) by the UB3LYP method. The energetic information is further refined at the UCCSD(T) and UQCISD(T) levels of theory. Our results indicated that the title reaction is more favorable on the singlet PES thermodynamically, and less competitive on the triplet one. On the singlet PES, the title reaction is most likely to be initiated by the carbon-to-oxygen approach forming the adduct IM1 (H2ICONO-trans) without any transition state, which can isomerizes to IM2 (H2ICNO2) and IM3 (H2ICONO-cis), respectively. The most feasible pathway is the 1, 3-I shift with C–I and O–N bonds cleavage along with the N–I bond formation of IM1 lead to the product P1 (CH2O + INO), which can further dissociate to give P3 (CH2O + I + NO). The competitive pathway is 1, 3-H shift associated with O–N bond rupture of IM1 to form P2 (CHIO + HNO). The theoretically obtained major product CH2O and adducts IM1 and IM2 are in good agreement with the kinetic detection in experiment. The similarities and discrepancies between CH2I + NO2 and CH2Br + NO2 reactions are discussed in terms of the electronegativity of halogen atom and the barrier height of the rate-determining process. The present study may be helpful for further experimental investigation of the title reaction.
Co-reporter:Li-Li Liu, Xiu-Juan Jia, Yan Zhang, Rong-Shun Wang, Xiu-Mei Pan
Journal of Molecular Structure: THEOCHEM 2010 Volume 960(1–3) pp:106-114
Publication Date(Web):30 November 2010
DOI:10.1016/j.theochem.2010.08.032
Theoretical investigation has been carried out for the electronic structures, optical properties and electron transition mechanism of R-substituted 2-phenylbenzoxazole complexes (nb) and derived phenolic Schiff bases (na) (n = 1, R = CH3; n = 2, R = N(CH3)2; n = 3, R = Cl; n = 4, R = NO2). In the gas phase, the ground and excited states were fully optimized at the B3LYP/cc-pVDZ, HF/cc-pVDZ and CIS/cc-pVDZ, respectively. For each derivative, two conformations are available, one with a downward hydrogen (H2) and the other with an upward hydrogen, and the former one is more stable than the latter one. Absorption and emission spectra of all species were calculated by the time-dependent density functional theory (TD-DFT) based on the ground and excited states geometries. The absorption and emission spectra were consistently blue shifted in going from na to nb. The solvent effects on molecular geometries and optical properties were characterized in several solvents from B3LYP/cc-pVDZ and HF/cc-pVDZ calculations employing the Onsager model within the framework of the self-consistent reaction field (SCRF) theory. The SCRF calculations provide reliable information regarding the solvent effects on the geometries and optical properties of the conjugate compounds.
Co-reporter:L.L. Liu, X.M. Pan, W. Zheng, L.L. Cui, G.C. Yang, Z.M. Su, R.S. Wang
Journal of Molecular Graphics and Modelling 2010 Volume 28(Issue 5) pp:427-434
Publication Date(Web):January 2010
DOI:10.1016/j.jmgm.2009.10.002
A comparative study using density functional theory (DFT) on the molecular structure, electronic structure and relative properties of 1,3,4-oxadiazole dimers 1,3-bis[2-(4-methylphenyl)-1,3,4-oxadiazol-5-yl]benzene (OXD-X) and its derivatives with alkoxy substituents –O(CH2)n−1CH3 (OXD-An, n = 1, 2) and electron-withdrawing substituents –CN (OXD-C), –CF3 (OXD-TFM), –NO2 (OXD-N) added at meta-substitution in the phenyl ring are presented. The ground state structures of the title complexes are optimized at B3LYP/6-31G level of theory. In addition, the time dependent density functional theory (TDDFT) method is applied to calculate the absorption and emission spectra of the derivatives based on the ground state geometries. Comparing with the alkoxy substituents, the results show that the electron-withdrawing substituents have remarkable influences on the energy levels of the frontier molecular orbitals, the spectra and the transition compositions. Especially, –NO2 group plays the prominent role and the fluorine improves the energy level of LUMO further. The experimental absorption wavelengths for OXD-X, OXD-A3 and OXD-A7 are well reproduced by the TDDFT technique. Moreover the absorption wavelengths and the transition compositions can be effectively adjusted through introducing electro-withdrawing groups in the phenyl ring. The reorganization energies for hole and electron are smaller than that of typical hole and electron transport materials.
Co-reporter:Guicai Song, Xiujuan Jia, Yang Gao, Jie Luo, Yanbo Yu, Rongshun Wang and Xiumei Pan
The Journal of Physical Chemistry A 2010 Volume 114(Issue 34) pp:9057-9068
Publication Date(Web):July 29, 2010
DOI:10.1021/jp102421g
The mechanisms and dynamics studies of the multichannel reactions of CH2FCF2OCHF2 + OH (R1) and CH2FOCH2F + OH (R2) have been carried out theoretically. Three hydrogen abstraction channels and two displacement processes are found for reaction R1, whereas there are two hydrogen abstraction channels and one displacement process for reaction R2. The minimum energy paths are optimized at the B3LYP/6-311G(d,p) level, and the energy profiles are further refined by interpolated single-point energies (ISPE) method at the BMC-QCISD level of theory. By means of canonical variational transition state theory with small-curvature tunneling correction, the rate constants of reactions R1 and R2 are obtained over the temperature range of 220−2000 K. The rate constants are in good agreement with the experimental data for reaction R1 and estimated data for reaction R2. The Arrhenius expression k1 = 1.62 × 10−20 T2.75 exp(−1011/T) for reaction R1 and k2 = 3.40 × 10−21 T3.04 exp(−384/T) for reaction R2 over 220−2000 K are obtained. Furthermore, to further reveal the thermodynamics properties, the enthalpies of formation of reactants CH2FCF2OCHF2, CH2FOCH2F, and the product radicals CHFCF2OCHF2, CH2FCF2OCF2, and CHFOCH2F are calculated by using isodesmic reactions.
Co-reporter:Xiujuan Jia, Youjun Liu, Jingyu Sun, Hao Sun, Zhongmin Su, Xiumei Pan and Rongshun Wang
The Journal of Physical Chemistry A 2010 Volume 114(Issue 1) pp:417-424
Publication Date(Web):December 1, 2009
DOI:10.1021/jp908228h
A dual-level direct dynamic method is employed to study the reaction mechanisms of CF3CHFOCF3 (HFE-227 mc) with the OH radical and Cl atom. The geometries and frequencies of all the stationary points and the minimum energy paths (MEPs) are calculated at the BH&H-LYP/6-311G(d,p) level, and the energetic information along the MEPs is further refined by MC-QCISD theory. The classical energy profile is corrected by the interpolated single-point energies (ISPE) approach, incorporating the small-curvature tunneling effect (SCT) calculated by the variational transition state theory (VTST). The rate constants are in good agreement with the experimental data and are found to be k1 = 2.87 × 10−21T2.80 exp(−1328.60/T) and k2 = 3.26 × 10−16T1.65 exp(−4642.76/T) cm3 molecule−1 s−1 over the temperature range 220−2000 K. The standard enthalpies of formation for the reactant CF3CHFOCF3 and product radical CF3CFOCF3 are evaluated via group-balanced isodesmic reactions, and the corresponding values are −454.06 ± 0.2 and −402.74 ± 0.2 kcal/mol, respectively, evaluated by MC-QCISD theory based on the BH&H-LYP/6-311G(d, p) geometries. The theoretical studies provide rate constants of the title reactions and the enthalpies of formation of the species, which are important parameters in determining the atmospheric lifetime and the feasible pathways for the loss of HFE-227 mc.
Co-reporter:Xiu-Juan Jia;You-Jun Liu;Jing-Yu Sun;Yi-Zhen Tang
Theoretical Chemistry Accounts 2009 Volume 124( Issue 1-2) pp:105-113
Publication Date(Web):2009 September
DOI:10.1007/s00214-009-0587-z
Theoretical investigations are carried out on the reaction Cl + CH2FCl by means of direct dynamics method. The minimum energy path (MEP) is obtained at the MP2/6-311G(d, p) level. The energetic information is further improved by single-point energy calculations using QCISD(T)/6-311++G(d, p) method. The kinetics of this reaction are calculated by canonical variational transition state theory incorporating with the small-curvature tunneling correction over a wide temperature range of 220–3,000 K, and rate constant expression are found to be k(T) = 1.48 × 10−17T2.04exp(−913.91/T). For the title reaction, H-abstraction reaction channel is the major channel at the lower temperatures. At higher temperatures, the contribution of Cl-abstraction reaction channel should be taken into account.
Co-reporter:Xiu-Juan Jia;Jing-Yu Sun;Yi-Zhen Tang
Theoretical Chemistry Accounts 2009 Volume 122( Issue 3-4) pp:207-216
Publication Date(Web):2009 March
DOI:10.1007/s00214-008-0500-1
The radical-molecule reaction mechanisms of CH2Br and CHBrCl with NO2 have been explored theoretically at the UB3LYP/6-311G(d, p) level. The single-point energies were calculated using UCCSD(T) and UQCISD(T) methods. The results show that the title reactions are more favorable on the singlet potential energy surface than on the triplet one. For the singlet potential energy surface of CH2Br + NO2 reaction, the association of CH2Br with NO2 is found to be a barrierless carbon-to-oxygen attack forming the adduct IM1 (H2BrCONO-trans), which can isomerize to IM2 (H2BrCNO2), and IM3 (H2BrCONO-cis), respectively. The most feasible pathway is the 1, 3-Br shift with C–Br and O–N bonds cleavage along with the N–Br bond formation of IM1 lead to the product P1 (CH2O + BrNO) which can further dissociate to give P4 (CH2O + Br + NO). The competitive pathway is the 1, 3-H-shift associated with O–N bond rupture of IM1 to form P2 (CHBrO + HNO). For the singlet potential energy surface of CHBrCl + NO2 reaction, there are three important reaction pathways, all of which may have comparable contribution to the reaction of CHBrCl with NO2. The theoretically obtained major products CH2O and CHClO for CH2Br + NO2 and CHBrCl + NO2 reactions, respectively, are in good agreement with the kinetic detection in experiment.
Co-reporter:Y. Liu;X. M. Pan;Z. S. Li;X. J. Jia;S. Li;R. S. Wang
Theoretical Chemistry Accounts 2007 Volume 118( Issue 5-6) pp:869-879
Publication Date(Web):2007 December
DOI:10.1007/s00214-007-0370-y
In gas phase, the hydrations of pentafulvenone to generate three types of cyclopentadienyl carboxylic acids are studied theoretically at the MP2/6-311+G**//B3LYP/6-311+G** level. A water molecule attacking the C=O double bond of pentafulvenone can yield cyclopentadienyl carboxylic acids via the formation of fulvenediols, and attacking the C=C double bond of pentafulvenone can directly yield cyclopentadienyl carboxylic acid. The barriers of rate-determining transition states are 42.2 and 30.4 kcal mol−1, respectively. The barriers of rate-determining transition states for two water molecules system are 20.2 and 19.6 kcal mol−1, respectively. The products can isomerize to each other. In aqueous solvent, the hydrations of pentafulvenone are investigated using PCM-UAHF model at the MP2 (PCM)/6-311+G**// B3LYP (PCM)/6-311+G** and MP2 (PCM)/6-311+G**// B3LYP/6-311+G** levels. The barriers of all rate-determining transition states are decreased. The added water molecule acts as catalyst in both gas phase and aqueous solvent.
Co-reporter:W. Zheng, X.M. Pan, L.L. Cui, Z.M. Su, R.S. Wang
Journal of Molecular Structure: THEOCHEM 2007 Volume 809(1–3) pp:39-43
Publication Date(Web):14 May 2007
DOI:10.1016/j.theochem.2007.01.012
We present a theoretical study using density functional theory (DFT) on molecular structures, electronic structures and absorption characters of –Ph and t-Bu substituted 5-(2-pyridyl) pyrazole boron complexes, namely, BPh2(2-(5-Phenyl-4H-pyrazole-3-yl)-pyridine) (2d), BPh2(2-[5-(2,2,2-Trifluoro-1,1-bis-trifluoromethyl-ethyl)-4H-pyrazole-3-yl]-pyridine) (2e), BPh2(4-Phenyl-2-(4H-pyrazole-3-yl)-pyridine) (2d-1) and BPh2(2-(4H-Pyrazole-3-yl)-4-(2,2,2-trifluoro-1,1-bis-trifluoromethyl-ethyl)-pyridine) (2e-1). The ground state structures of the title complexes are optimized at B3LYP/6-31G level of theory. In addition, time dependent density functional theory (TD-DFT) method is applied to investigate the properties of absorption spectra and electronic transition mechanism based on the ground state geometries. The results show that the chemical bond formed between nitrogen on the pyridyl ring and boron can be attributed to coordination effect and the coordinate bond in 2d-1 is the strongest among the four compounds. The calculated absorption wavelengths for 2d and 2e are in good agreement with the experimental ones. It can be detected that the main transitions of 2d, 2e and 2d-1 correspond to the intraligand π → π∗ character. As the case of 2e-1, the main transition can be assigned as a mixed ligand-to-ligand/interligand charge transfer.
Co-reporter:Zhen Fu, Xiu-mei Pan, Ze-sheng Li, Chia-chung Sun, Rong-shun Wang
Chemical Physics Letters 2006 Volume 430(1–3) pp:13-20
Publication Date(Web):19 October 2006
DOI:10.1016/j.cplett.2006.07.103
The global potential energy surface (PES) of the [CH3,N,C,S] system in singlet and triplet states, involving 16 isomers and 15 transition structures, is studied at DFT(B3LYP), MP2 and QCISD levels. It is shown that the chainlike singlet isomer CH3NCS is the most stable species among all the isomers and the branched-C(CH3)NS has the lowest energy among the triplet species. The stability of these isomers, their isomerizations and dissociations are discussed and theoretical results are consistent with the available experimental ones.The global potential energy surface (PES) of the [CH3,N,C,S] system in singlet and triplet states, involving 16 isomers and 15 transition structures, is studied at DFT(B3LYP), MP2 and QCISD levels. The stability of these isomers, their isomerizations and dissociations are discussed and theoretical results are consistent with the available experimental ones.
Co-reporter:X.M. Pan, Z. Fu, Z.S. Li, C.C. Sun, H. Sun, Z.M. Su, R.S. Wang
Chemical Physics Letters 2005 Volume 409(1–3) pp:98-104
Publication Date(Web):20 June 2005
DOI:10.1016/j.cplett.2005.04.077
Abstract
The mechanism for reaction of CH3O with NO2 is investigated at DFT (B3LYP), MP2 and QCISD levels with 6-311++G(d,p) basis set. The geometries and vibrational frequencies of stationary points on potential energy surface are obtained. There are six possible reaction paths that involve 8 conformers, 13 transition structures and 4 possible products for the title reaction. The recombination channel for the product CH3ONO2 is dominant. At the adopted levels of theory, the disproportionation channel, which is thought to be possible by simple intermolecular H-atom abstraction conjectured experimentally, has not been found.
Co-reporter:Jin-Ting Ye, Feng-Yang Bai, Shao-Qing Shi, Xiu-Mei Pan
Journal of Molecular Graphics and Modelling (March 2017) Volume 72() pp:156-167
Publication Date(Web):March 2017
DOI:10.1016/j.jmgm.2017.01.002
Co-reporter:Chunyu Liu, Yanling Si, Shaoqing Shi, Guochun Yang and Xiumei Pan
Dalton Transactions 2016 - vol. 45(Issue 17) pp:NaN7293-7293
Publication Date(Web):2016/03/15
DOI:10.1039/C6DT00089D
Chiral transition metal complexes not only have large nonlinear optical (NLO) response but also meet the non-centrosymmetric requirement of second-order NLO materials. Therefore, chiral transition metal complexes become very active in the NLO area. Recently, the second-order NLO response of chiral dinuclear Re(I) complex 2 has been found to be 1.5 times larger than that of KH2PO4 (KDP) based on experimental measurement. However, its NLO origin has not been determined and a structure–property relationship has not been established at the microscopic level, which are very important to further improve the performance. It is found that charge transfer from metal to ligand is mainly responsible for its NLO origin. Based on complex 2, the designed complexes have remarkably large second-order NLO activity. For instance, the designed complex 9 has a very large second-order NLO response value (115.81 × 10−30 esu), which is about 668 times larger than the organic molecule urea. Moreover, time-dependent density functional theory (TDDFT) calculations have been used to investigate their UV-Vis/CD spectra. The simulated circular dichroism (CD) spectra of the complex 2 are in good agreement with the experimental ones, which can be used to assign the absolute configurations (ACs) of chiral dinuclear Re(I) complexes with high confidence. The electronic absorption wavelengths, electron transition properties, and the second-order NLO responses strongly depend on the nature of substituent, different ligands (pyridine and isoquinoline) and their combinations. Based on NBO analysis, the interactions between [Re(CO)3Cl] fragments and ligands are of n → σ* character.
Co-reporter:Chunyu Liu, Guochun Yang, Yanling Si, Youjun Liu and Xiumei Pan
Journal of Materials Chemistry A 2017 - vol. 5(Issue 14) pp:NaN3502-3502
Publication Date(Web):2017/03/10
DOI:10.1039/C7TC00337D
Recently, a chiral conjugated macrocyclic compound containing alternately arranged donor and acceptor motifs exhibited unique advantages (i.e. high absorption coefficient, broad absorption range, and high electron mobility) in organic photovoltaics (OPVs). Understanding the structure–property relationship at the microscopic level is a prerequisite for further performance optimization or improvement. Here, we employed time-dependent density functional theory (TDDFT) to investigate the electronic circular dichroism (CD), UV-vis absorption, charge transport, and second-order nonlinear optical (NLO) properties of the four chiral compounds. The experimental UV-vis/CD spectra of compound 1 were well reproduced by our calculations and could be used to assign the electron transition properties and absolute configuration (AC) with high confidence. The electronic absorption spectra, charge transport properties, and open circuit voltage of compound 1 in OPVs have been rationalized by comparing cyclic and acyclic structures. The designed compounds 2–3 are expected to exhibit excellent performances in OPVs in view of their small energy gaps, large oscillator strengths, and smaller electron reorganization energies. Moreover, the first hyperpolarizability (βtot) of compound 4 is 27 times larger than that of P-nitroaniline. Thus, our studied compounds are also excellent candidates for second-order NLO materials.