Co-reporter:Sheng Liu, Wenshuai Dai, Lijuan Zhang, Min Cheng, Yikui Du, Qihe Zhu
Journal of Molecular Structure 2017 Volume 1146(Volume 1146) pp:
Publication Date(Web):15 October 2017
DOI:10.1016/j.molstruc.2017.05.101
•The structures of cis and trans 2FNMA are optimized.•The relative stability of cis and trans 2FNMA are discussed.•REMPI and MATI spectra of 2FNMA are measured.•The E1 and IE of trans 2FNMA are obtained.Theoretical calculations predicted that there are only two stable conformers, trans and cis, for 2-fluoro-N-methylaniline (2FNMA) in the S0, S1 and D0 states. Compared to the cis conformer, the trans one is more stable, and has a population more than 99% at room temperature. The optimized molecular skeleton of trans and cis 2FNMA are both non-planar in the S0 state, but planar in the S1 and D0 states. The one-dimensional potential energy surface of 2FNMA in the S0 state is obtained. The Resonance-enhanced two-photon ionization (R2PI) and Mass-analyzed threshold ionization (MATI) spectra of trans 2FNMA are obtained. The first electronic excitation energy (E1) and the adiabatic ionization energy (IE) of trans 2FNMA are determined. The substitution effect on the molecular structures, transition energies and vibrations of 2FNMA are discussed.
Co-reporter:Sheng Liu, Wenshuai Dai, Dan Lin, Min Cheng, YiKui Du, Qihe Zhu
Journal of Molecular Spectroscopy 2017 Volume 338(Volume 338) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.jms.2017.04.019
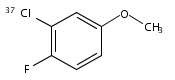
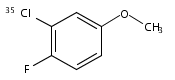
•REMPI and MATI spectra of cis and trans 3F4ClA are obtained.•We measured the E1s and IEs and the relative stability of two conformers.•Theoretical calculations were performed to interpret the spectroscopic data.The Resonance-enhanced two-photon ionization (R2PI) and Mass-analyzed threshold ionization (MATI) spectra of 4-chloro-3-fluoroanisole (4Cl3FA) were recorded in detail. The experimental and calculated results revealed that cis and trans 4Cl3FA are the only two stable conformers in each of the S0, S1 and D0 states. The first electronic excitation energies (E1’s) of cis and trans 35Cl-4Cl3FA were measured to be 35,326 ± 3 and 35,443 ± 3 cm−1 by the R2PI spectroscopy and the corresponding adiabatic ionization energies (IEs) were determined to be 67,330 ± 5 and 67,591 ± 5 cm−1 by the MATI spectroscopy. The E1’s and IEs of cis and trans 37Cl-4Cl3FA were also obtained and found to be identical to that of the 35Cl-4Cl3FA conformers. Compared with the trans 4Cl3FA, the cis 4Cl3FA is more stable in the S0, S1 and D0 states. The vibrational bands observed in the R2PI and MATI spectra are related to the vibrations involving the in-plane deformation of the benzene ring, the CH3 torsion and the OCH3 inversion. The substituent effects on the molecular structures, transition energies and vibrations of 4Cl3FA are discussed.Download high-res image (127KB)Download full-size image
Co-reporter:Sheng Liu, Lijuan Zhang, Wenshuai Dai, Min Cheng, Yikui Du, Qihe Zhu
Journal of Molecular Spectroscopy 2017 Volume 336(Volume 336) pp:
Publication Date(Web):1 June 2017
DOI:10.1016/j.jms.2017.03.013
•REMPI and MATI spectra of cis and trans 3ClNMA are obtained.•We measured the E1s and IEs and the relative stability of two rotamers.•We found a correlation between relative stability of rotamers and rigidity of ring.•Theoretical calculations were performed to interpret the spectroscopic data.The Resonance-enhanced two-photon ionization (R2PI) and Mass-analyzed threshold ionization (MATI) spectra of 3-Chloro-N-methylaniline (3ClNMA) were recorded in detail. The experimental and calculated results revealed that cis and trans 3ClNMA are the only two stable conformers for each of the S0, S1 and D0 states. The first electronic excitation energies (E1s) of cis and trans 35Cl-3ClNMA were measured to be 33,003 ± 3 and 32,886 ± 3 cm−1 by R2PI spectroscopy. The adiabatic ionization energies (IEs) of cis and trans 35Cl-3ClNMA were determined to be 61,531 ± 5 and 61,625 ± 5 cm−1 by MATI spectroscopy. The E1s and IEs of cis and trans 37Cl-3ClNMA were also obtained and found to be identical to that of the 35Cl-3ClNMA rotamers. Compared with the trans rotamer, the cis rotamer is less stable by 50 cm−1 in the S1 state, but more stable by 67 and 161 cm−1 in S0 and D0 states. Most of the active vibration modes observed in the R2PI and MATI spectra are in-plane ring deformation and substituent-sensitive modes. Detailed analysis reveals a correlation between the typical ring vibration frequencies and the relative stability of the two rotamers, suggesting that the conformation effect on the ring rigidity plays a key role in determining the relative stability of the conformers. The ab initio and density functional theory (DFT) calculations were performed to analyze the substitution and conformation effects on the properties of 3ClNMA rotamers.Download high-res image (158KB)Download full-size image
Co-reporter:Lijuan Zhang, Sheng Liu, Min Cheng, Yikui Du, and Qihe Zhu
The Journal of Physical Chemistry A 2016 Volume 120(Issue 1) pp:81-94
Publication Date(Web):December 15, 2015
DOI:10.1021/acs.jpca.5b11991
The mass-analyzed threshold ionization spectra of jet-cooled cis- and trans-3-fluoro-N-methylaniline (3FNMA) were recorded by ionizing via the vibrationless 00 and various vibrational levels of the S1 state. The adiabatic ionization energies of cis- and trans-3FNMA are determined to be 61 742 ± 5 and 61 602 ± 5 cm–1, respectively. In the 0–1800 cm–1 region, most of the observed vibrations in the D0 state result from the in-plane ring deformation and substituent-sensitive modes. For the high-frequency vibration region, the infrared–ultraviolet double-resonance and autoionization-detected infrared spectroscopies were applied to investigate the N–H and C–H stretching vibrations of bare 3FNMA in the S0 and D0 states. The C–H stretching vibrational information, which we failed to obtain for the bare 3FNMA cation, is complemented by recording the infrared–photodissociation spectra of its Ar cluster cation. It is revealed that a red-shifted frequency and an enhanced intensity are observed for the N–H stretch, while blue-shifted frequencies and greatly decreased intensities are found for both aromatic and the methyl C–H stretches. The blue shift of the C–H stretches is first explained by the balance of two factors, namely, the hyperconjugative interaction and the rehybridization effect. Analysis of the vibrational frequencies reveals a correlation between the relative stability of two rotamers in different electronic states and the relative rigidity of aromatic ring, indicating a mechanism of the long-range interactions “through bond” between the substituents. The density functional theory calculations can well reproduce the vibrational spectra in both S0 and D0 states. With the experimental and theoretical data, the substitution and conformation effects on the properties of 3FNMA in the S0 and D0 states, including the molecular structures, the reactive sites of electrophilic attack, and the vibrational behaviors, were discussed in detail.
Co-reporter:Lijuan Zhang, Sheng Liu, Changwu Dong, Min Cheng, Yikui Du, Qihe Zhu, Cunhao Zhang
Journal of Molecular Spectroscopy 2014 Volume 296() pp:28-35
Publication Date(Web):February 2014
DOI:10.1016/j.jms.2013.12.005
•Resonant two-photon ionization spectrum of 3-fluoro-N-methylaniline was obtained.•The S1 ← S0 electronic transition energy and the ionization energy were determined.•The relative stability of two rotamers in the S0, S1 and D0 states was derived.•The substitution and conformation effects on 3FNMA were discussed in detail.•The calculated results compare well with the experimental observations.The ab initio and density functional theory (DFT) calculations of 3-fluoro-N-methylaniline (3FNMA) reveal that two rotamers, cis and trans 3FNMA, are stable for each of the S0, S1, and D0 states. The vibronic spectra of the two rotamers in the S1 state have been recorded by the one-color resonant two-photon ionization (R2PI) spectroscopy. The band origins of the S1 ← S0 electronic transition of cis and trans 3FNMA are found to be 33 816 ± 3 and 34 023 ± 3 cm−1. The two rotamers display similar vibrational frequencies, and the slight energy difference in some modes reflects the conformation effect due to the relative orientation of the NHCH3 group. Besides, the trans rotamer displays more vibronic features in the low-frequency region, which are active modes mainly involving the CH3 and the NHCH3 groups. By the two-color R2PI spectroscopy, the adiabatic ionization energies (IEs) of cis and trans 3FNMA are determined to be 61 742 ± 10 and 61 602 ± 10 cm−1, respectively. It is derived from the R2PI spectroscopic data that, compared with the trans rotamer, the cis rotamer is more stable by 302 ± 25 cm−1 in the S1 state, but less stable by 45 ± 25 cm−1 in the D0 state. With the aid of theoretical calculations, the substitution and conformation effects on the properties of 3FNMA, including the molecular structures, vibrational frequencies, and the relative stability of the two rotamers, were discussed in detail.Graphical abstract
Co-reporter:Changwu Dong, Lijuan Zhang, Sheng Liu, Lili Hu, Min Cheng, Yikui Du, Qihe Zhu, Cunhao Zhang
Journal of Molecular Structure 2014 Volume 1058() pp:205-212
Publication Date(Web):24 January 2014
DOI:10.1016/j.molstruc.2013.10.068
•The REMPI spectra of rotamers were obtained.•The S1 ← S0 excitation energies and ionization energies were determined.•The relative stability of two rotamers in each of the S0, S1 and D0 states was evaluated by experimental method.•La/Lb mixing transition was predicted by theoretical calculation.•The calculated results compared well with experimental observations.The ab initio and DFT calculations predict that two rotamers, cis and trans m-aminostyrene, are stable for each of the S0, S1 and D0 states. In the one-color resonant two-photon ionization (1C-R2PI) spectra, the band origins of the S1 ← S0 electronic transitions (00 band) of cis and trans m-aminostyrene appear respectively at 30,937 ± 3 and 31,141 ± 3 cm−1. The electronic transition energies (E1’s) of both rotamers of m-aminostyrene are lower than that of p-aminostyrene, which is contrary to the prediction according to the previous studies. The redshift of E1’s may be related to the mixing character of “1Lb” and “1La” in the S1 states of the two rotamers. The 2C-R2PI spectra give the adiabatic ionization energies of 61,278 ± 15 and 61,495 ± 15 cm−1 for cis and trans rotamers. The observed active modes of rotamers in the S1 states involve mainly the in-plane ring deformation and substituent-sensitive bending vibration. It is derived from the 1C- and 2C-R2PI spectroscopic data that the cis rotamer of m-aminostyrene is more stable than the trans rotamer by 30 ± 30, 234 ± 30 and 247 ± 30 cm−1 for the S0, S1 and D0 states, respectively. This is different from the m-aminophenol and m-aminoanisole that have a more stable trans rotamer.Graphical abstract
Co-reporter:Changwu Dong, Lijuan Zhang, Sheng Liu, Lili Hu, Min Cheng, Yikui Du, Qihe Zhu, Cunhao Zhang
Journal of Molecular Spectroscopy 2014 Volume 295() pp:62
Publication Date(Web):January 2014
DOI:10.1016/j.jms.2013.11.005
Co-reporter:Changwu Dong, Lijuan Zhang, Sheng Liu, Lili Hu, Min Cheng, Yikui Du, Qihe Zhu, Cunhao Zhang
Journal of Molecular Spectroscopy 2013 Volume 292() pp:35-46
Publication Date(Web):October 2013
DOI:10.1016/j.jms.2013.09.008
•The REMPI and MATI spectra of rotamers and isotopomers were obtained.•The S1 ← S0 excitation energies and ionization energies were determined.•The relative stability of two rotamers in each of the S0, S1 and D0 states was evaluated by experimental method.•The conformation effect on the frequencies is greater than the isotope effect.•The calculated results compared well with experimental observations.The ab initio and DFT calculations predict two stable rotamers, cis and trans, for 3-chlorostyrene in each of the S0, S1 and D0 states. In the two-color resonant two-photon ionization (2C-R2PI) spectra, the band origins of S1 ← S0 electronic transition of cis and trans 3-chlorostyrene appears respectively at 33 766 ± 3 and 34 061 ± 3 cm−1. The mass-analyzed threshold ionization (MATI) spectra give the adiabatic ionization energies of 69 701 ± 4 cm−1 and 69 571 ± 4 cm−1 for cis and trans rotamers. Within the experimental detection limit, the measured transition energies are the same for both the 35Cl and 37Cl isotopomers. The observed active modes of rotamers in the S1 and D0 states involve mainly the in-plane ring deformation and substituent-sensitive bending vibration. It is derived from the 2C-R2PI and MATI spectroscopic data that the cis rotamer is more stable than the trans rotamer by 218 ± 30, 513 ± 30 and 88 ± 30 cm−1 for the S0, S1 and D0 states, respectively.Graphical abstract
Co-reporter:Lili Hu, Zhimin Zhou, Changwu Dong, Lijuan Zhang, Yikui Du, Min Cheng, and Qihe Zhu
The Journal of Physical Chemistry A 2013 Volume 117(Issue 21) pp:4352-4357
Publication Date(Web):April 24, 2013
DOI:10.1021/jp401310g
From the photofragment translational spectra of C–H symmetric stretch excited CH3I [v1 = 1, v2 = 0] photodissociatioin at 277.5 nm, the vibrational distribution of photofragments CH3 (v1 = 0, v2 = 0), (0,1), (1,0), (1,1) in the I* channel are measured to be 0.02, 0.02, 0.47, 0.25, and those of CH3 (1,0), (1,1) in the I channel are 0.04, 0.05, respectively. It shows that most of the dissociated CH3I [1,0] retain the C–H symmetric stretch vibration v1 = 1 in the photofragments CH3, and the vibrational distribution in umbrella bending mode is not seriously affected by the original C–H symmetric stretch excitation. The photodissociation of CH3I [1,0] mainly follows the vibrationally adiabatic process. The original vibrational excitation [v1 = 1] of CH3I is quite like a spectator, and the intramolecular vibrational-energy redistribution (IVR) does not play obvious part during photodissociation.
Co-reporter:Lijuan Zhang, Dan Yu, Changwu Dong, Min Cheng, Lili Hu, Zhimin Zhou, Yikui Du, Qihe Zhu, Cunhao Zhang
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2013 Volume 104() pp:235-242
Publication Date(Web):March 2013
DOI:10.1016/j.saa.2012.11.049
The ab initio and density functional theory (DFT) calculations reveal that two rotamers, denoted by cis and trans 3-chloro-5-fluoroanisole (3C5FA), are stable for each of the S0, S1, and D0 states. In the one-color resonant two-photon ionization (R2PI) spectra, the band origins of the S1 ← S0 electronic transition (00 bands) of cis35Cl-3C5FA and cis37Cl-3C5FA are both located at 36,468 ± 3 cm−1, while the 00 bands of trans35Cl-3C5FA and trans37Cl-3C5FA are found to be 36,351 ± 3 and 36,354 ± 3 cm−1. The two rotamers display very similar vibrational frequencies in the S1 state, and the observed active modes mainly involve the in-plane ring deformation vibrations. By the two-color R2PI spectroscopy, the adiabatic ionization energies (IEs) of both isotopomers of 3C5FA are determined to be 69,720 ± 15 cm−1 for the cis rotamer and 69,636 ± 15 cm−1 for the trans rotamer. The substitution, conformation, and isotope effects on the properties of 3C5FA, including the molecular structures, vibrational frequencies, and electronic transition and ionization energies, were also discussed in detail.Graphical abstract1C-R2PI spectra of 35Cl and 37Cl isotopomers of cis and trans 3-chloro-5-fluoroanisole.Highlights► The REMPI spectra of the rotamers and isotopomers were obtained. ► The S1 ← S0 excitation energies and the ionization energies were determined. ► The conformation effect on the frequencies is greater than the isotope effect. ► The di-halogen substituents give a cumulative effect on the shift of E1s and IEs. ► The calculated results compared well with the experimental results.
Co-reporter:Lijuan Zhang, Changwu Dong, Min Cheng, Lili Hu, Yikui Du, Qihe Zhu, Cunhao Zhang
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2012 Volume 96() pp:578-585
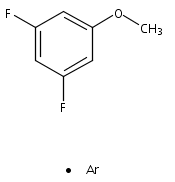
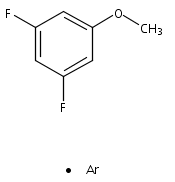
Publication Date(Web):October 2012
DOI:10.1016/j.saa.2012.07.022
The structure and vibrations of 3,5-difluoroanisole (3,5-DFA) in the first electronically excited (S1) state were studied by mass-analyzed resonant two-photon ionization (R2PI) technique as well as the quantum chemical calculations. The ab initio and density functional theory (DFT) calculations reveal that only one structure is stable for each of the S0, S1, and D0 states. In the one color R2PI spectrum, the band origin of the S1 ← S0 electronic transition (00 band) of 3,5-DFA is found to be 37,595 ± 3 cm−1. In the S1 state, most of the bands observed are related to the in-plane ring deformation and out-of-plane bending vibrations. The adiabatic ionization energy (IE) of 3,5-DFA is determined to be 70,096 ± 15 cm−1 by the two color R2PI technique, in agreement with the values predicted by the DFT approaches. The dihalogen-substitution effects on the molecular structure, vibrational frequencies, and electronic transition and ionization energies were discussed in detail. The van der Waals complex of 3,5-DFA with argon (3,5-DFA···Ar) was also observed and studied. The 00 band of 3,5-DFA···Ar complex is red-shifted by about 9 cm−1 with respect to that of 3,5-DFA. Both the experimental data and the calculated results indicate that the formation of 3,5-DFA···Ar complex gives only a weak influence on the properties of 3,5-DFA moiety.Graphical abstract1C-R2PI spectrum of 3,5-difluoroanisole. The inset shows the band origin of its complex with one Ar atom.Highlights► Resonant two-photon ionization spectrum of 3,5-difluoroanisole was firstly obtained. ► The S1 ← S0 electronic transition energy and the ionization energy were determined. ► Formation of Ar complex gives a weak influence on the properties of 3,5-DFA moiety. ► The calculated results compared well with the experimental results. ► The dihalogen-substitution effect was discussed in detail.
Co-reporter:Dan Yu, Changwu Dong, Min Cheng, Lili Hu, Yikui Du, Qihe Zhu, Cunhao Zhang
Journal of Molecular Spectroscopy 2011 Volume 265(Issue 2) pp:86-91
Publication Date(Web):February 2011
DOI:10.1016/j.jms.2010.11.006
The geometric structures and vibrations of p-chloroanisole isotopomers in the first electronically excited state were studied by mass-analyzed resonance-enhanced two-photon ionization spectroscopy and by theoretical calculations. The band origins of the S1 ← S0 electronic transitions of 35Cl and 37Cl isotopomers were found to be equivalent at 34 859 ± 3 cm−1. Assignments of the observed vibrational bands of the two isotopomers were made mainly based on the CIS/cc-PVDZ calculations and on conformity with the available data in the literature. Although the general spectral features of these two isotopomers are similar, the frequencies of some vibrational modes are different. This frequency shift partially depends on the degree of involvement of the chlorine atom in the molecular vibrations.Graphical abstract1C-R2PI spectra of 35Cl and 37Cl isotopomers of p-chloroanisole in the energy range of −100∼900 cm−1 (a) and 850–1800 cm−1 (b) from the band origin.Research highlights► Resonance-enhanced multiphoton ionization spectra of the 35Cl and 37Cl isotopomers of p-chloroanisole have been firstly obtained. ► The S1 ← S0 electronic transition energies of 35Cl and 37Cl isotopomers were found to be equivalent at 34 859 ± 3 cm−1. ► Assignments of the observed vibrational bands of the two isotopomers were made mainly based on the CIS/cc-PVDZ calculations and on conformity with the available data in the literature. ► The isotope effect on vibrational frequency partially depends on the degree of involvement of the chlorine atom in the molecular vibrations.
Co-reporter:Dan Yu, Changwu Dong, Lijuan Zhang, Min Cheng, Lili Hu, Yikui Du, Qihe Zhu, Cunhao Zhang
Journal of Molecular Structure 2011 1000(1–3) pp: 92-98
Publication Date(Web):
DOI:10.1016/j.molstruc.2011.05.058
Co-reporter:Min Cheng, Zijun Yu, Lili Hu, Dan Yu, Changwu Dong, Yikui Du, and Qihe Zhu
The Journal of Physical Chemistry A 2011 Volume 115(Issue 7) pp:1153-1160
Publication Date(Web):January 27, 2011
DOI:10.1021/jp106624q
The photodissociation dynamics of CH3I from 277 to 304 nm is studied with our mini-TOF photofragment translational spectrometer. A single laser beam is used for both photodissociation of CH3I and REMPI detection of iodine. Many resolved peaks in each photofragment translational spectrum reveal the vibrational states of the CH3 fragment. There are some extra peaks showing the existence of the hot-band states of CH3I. After careful simulation with consideration of the hot-band effect, the distribution of vibrational states of the CH3 fragment is determined. The fraction σ of photofragments produced from the hot-band CH3I varies from 0.07 at 277.38 nm to 0.40 at 304.02 nm in the I* channel and from 0.05 at 277.87 nm to 0.16 at 304.67 nm in the I channel. Eint/Eavl of photofragments from ground-state CH3I remains at about 0.03 in the I* channel for all four wavelengths, but Eint/Eavl decreases from 0.09 at 277.87 nm to 0.06 at 304.67 nm in the I channel. From the ground-state CH3I, the quantum yield Φ(I*) is determined to be 0.59 at 277 nm and 0.05 at 304 nm. The curve-crossing probability Pcc from the hot-band CH3I is lower than that from the ground-state CH3I. The potential energy at the curve-crossing point is determined to be 32 740 cm−1.
Co-reporter:Zijun Yu, Min Cheng, Xiling Xu, Dan Yu, Yikui Du, Qihe Zhu
Chemical Physics Letters 2010 Volume 488(4–6) pp:158-161
Publication Date(Web):22 March 2010
DOI:10.1016/j.cplett.2010.02.044
The photodissociation of CF3I has been studied near 304 nm. The partially resolved vibrational peaks in the I* and I channels reveal that the ν2 umbrella mode of CF3 is preferentially excited. Besides, in the I* channel, the extra vibrational peaks assigned to the umbrella mode of CF3 from the hot band ν3′=1,2 states of CF3I are also partially resolved. The Eint/Eavl shows a gradual increase as the state of CF3I changes from ν3′=0 to ν3′=2. The anisotropy parameter is also obtained and discussed with the interrelation of the 3Q1, 3Q0, and 1Q1 potential energy surfaces.Resolved photofragment translational spectra of the I* channel from CF3I photodissociation at 304.02 nm.
Co-reporter:Dan Yu, Changwu Dong, Zijun Yu, Min Cheng, Yikui Du, Qihe Zhu, Cunhao Zhang
Journal of Molecular Structure 2010 Volume 984(1–3) pp:307-315
Publication Date(Web):15 December 2010
DOI:10.1016/j.molstruc.2010.09.047
Based on the theoretical and REMPI spectroscopic study, a comparison between 1-methylphenylhydrazine and phenylhydrazine in the excited S1 state is conducted to reveal the formation and influence of p–p–π conjugation. The ab initio and DFT calculations predict that phenylhydrazine and 1-methylphenylhydrazine respectively have three stable conformers in the S1 state. However, only the conformer with p–p–π conjugation for phenylhydrazine and the conformer with p–π conjugation for 1-methylphenylhydrazine in the S1 state can match the experimental results well. In the one color resonant two photon ionization (1C-R2PI) spectrum of 1-methylphenylhydrazine, the band origin of the S1 ← S0 transition (E1) is determined to be 33292 ± 3 cm−1, which is red-shifted by 318 cm−1 with respect to that of phenylhydrazine, but is just the same as that of N-methylaniline, showing no amino substitution effect on E1. While the S1 ← S0 transition energy of phenylhydrazine is red-shifted by 419 cm−1 with respect to that of aniline, showing a remarkable amino substitution effect on E1. For the S1 state, some benzene ring vibration frequencies of 1-methylphenylhydrazine are similar to those of N-methylaniline, but different from those of phenylhydrazine. With 2C-R2PI technique, the lifetimes of the S100 state are measured to be about 9 ± 6 ns for 1-methylphenylhydrazine and 15 ± 6 ns for phenylhydrazine. The structural difference between phenylhydrazine and 1-methylphenylhydrazine in the S1 state might account for their differences in transitional energy, vibrational frequencies and the lifetime of the S100 state.
Co-reporter:Daoqing Xiao, Dan Yu, Xiling Xu, Zijun Yu, Min Cheng, Yikui Du, Weijun Zheng, Qihe Zhu and Cunhao Zhang
Physical Chemistry Chemical Physics 2009 vol. 11(Issue 18) pp:3532-3538
Publication Date(Web):27 Feb 2009
DOI:10.1039/B818688J
Phenylhydrazine and its van der Waals complexes with one or two argon atoms were investigated with theoretical calculations and resonant two photon ionization (R2PI) spectroscopy. The ab initio and DFT calculations found a conversion of the orbital hybridization of the Nβ atom from sp3-like in the S0 state to sp2-like in the S1 state, suggesting that the lone pair electrons of the Nβ atom are involved in a super p-p–π conjugation over the skeleton of phenylhydrazine in the S1 state. The structural change of the hydrazino group in the S1← S0 electronic transition was reflected by the vibrational excitations of the hydrazino group observed in the 1C-R2PI spectrum. The band origin of the S1← S0 transition is determined to be 33610 cm−1 and the adiabatic ionization energy (IE) of phenylhydrazine, measured by 2C-R2PI spectroscopy, is 62829 ± 15 cm−1. The S1← S0 electronic transitions of phenylhydrazine–Ar and phenylhydrazine–Ar2 complexes were also observed in the 1C-R2PI spectrum, and their band origins are, respectively, red-shifted by 39 and 80 cm−1 from that of phenylhydrazine.
Co-reporter:Daoqing Xiao, Dan Yu, Xiling Xu, Zijun Yu, Yikui Du, Zhen Gao, Qihe Zhu, Cunhao Zhang
Journal of Molecular Structure 2009 Volume 918(1–3) pp:154-159
Publication Date(Web):29 January 2009
DOI:10.1016/j.molstruc.2008.07.032
The structures and vibrations of p-methoxystyrene in the first electronically excited state (S1) were studied by mass analyzed resonance-enhanced two photon ionization (R2PI) spectroscopy and theoretical calculations. The ab initio and the density functional theory (DFT) calculations reveal that two rotamers, cis and trans, are stable in both the ground S0 state and the excited S1 state, and their optimized molecular geometries are achieved. The band origins of the S1 ← S0 electronic transition of cis and trans p-methoxystyrene are measured to be 33242 and 33324 cm−1, respectively. Assignment of the observed spectral bands of the two rotamers in the excited S1 state was made mainly based on the ab initio calculations and the conformity with the available data in the literature.
Co-reporter:Daoqing Xiao, Dan Yu, Xiling Xu, Zijun Yu, Yikui Du, Zhen Gao, Qihe Zhu, Cunhao Zhang
Journal of Molecular Structure 2008 Volume 882(1–3) pp:56-62
Publication Date(Web):30 June 2008
DOI:10.1016/j.molstruc.2007.09.012
The vibronic structure of p-fluoroanisole in the first excited state (S1) has been investigated with mass selected resonance-enhanced two photon ionization spectroscopy. The band origin of S1 ← S0 transition of p-fluoroanisole is measured to be 35149 cm−1, which is red-shifted by 1234 cm−1 with respect to that of anisole. Combining with the ab initio calculations, the measured frequencies 397, 487, 559, 840 and 1150 cm−1 in the S1 state are assigned as the in-plane ring vibrational mode 9b, 6a, 6b, 1 and 9a, respectively. The optimized molecular geometries and vibrational frequencies of p-fluoroanisole in the ground state (S0) and cation ground state (D0) are also achieved from DFT calculations.
Co-reporter:Daoqing Xiao, Dan Yu, Xiling Xu, Zijun Yu, Min Cheng, Yikui Du, Weijun Zheng, Qihe Zhu and Cunhao Zhang
Physical Chemistry Chemical Physics 2009 - vol. 11(Issue 18) pp:NaN3538-3538
Publication Date(Web):2009/02/27
DOI:10.1039/B818688J
Phenylhydrazine and its van der Waals complexes with one or two argon atoms were investigated with theoretical calculations and resonant two photon ionization (R2PI) spectroscopy. The ab initio and DFT calculations found a conversion of the orbital hybridization of the Nβ atom from sp3-like in the S0 state to sp2-like in the S1 state, suggesting that the lone pair electrons of the Nβ atom are involved in a super p-p–π conjugation over the skeleton of phenylhydrazine in the S1 state. The structural change of the hydrazino group in the S1← S0 electronic transition was reflected by the vibrational excitations of the hydrazino group observed in the 1C-R2PI spectrum. The band origin of the S1← S0 transition is determined to be 33610 cm−1 and the adiabatic ionization energy (IE) of phenylhydrazine, measured by 2C-R2PI spectroscopy, is 62829 ± 15 cm−1. The S1← S0 electronic transitions of phenylhydrazine–Ar and phenylhydrazine–Ar2 complexes were also observed in the 1C-R2PI spectrum, and their band origins are, respectively, red-shifted by 39 and 80 cm−1 from that of phenylhydrazine.