Co-reporter:Oliver Masur, Martin Schütz, Lorenzo Maschio, and Denis Usvyat
Journal of Chemical Theory and Computation 2016 Volume 12(Issue 10) pp:5145-5156
Publication Date(Web):August 24, 2016
DOI:10.1021/acs.jctc.6b00651
We present a periodic/finite-cluster interface for fragment-based direct local ring-coupled-cluster doubles (d-LrCCD) calculations embedded in the periodic mean field. The fragment is defined by a set of Wannier functions (WFs), obtained from a periodic Hartree–Fock calculation. The pair-specific virtual space is spanned by projected atomic orbitals (PAOs) truncated to pair domains. The computational procedure is initiated by a periodic local Møller–Plesset (LMP2) calculation. A subset of the WF pairs is then subsequently subjected to a finite-cluster d-LrCCD treatment using the local coupled cluster program of Molpro; this subset is specified by an interorbital cutoff distance. The orbital, pair, and domain lists, as well as other essential quantities needed for d-LrCCD such as the Fock and overlap matrices, and the electron repulsion integrals (ERIs) in the basis of WFs and PAOs are evaluated in the periodic framework and passed to Molpro via an interface. These periodic quantities provide the correct periodic mean-field embedding for the fragment d-LrCCD. Moreover, no expensive orbital transformations involving orbital coefficients related to large supporting clusters are necessary. ERIs appearing in the d-LrCCD diagrams are factorized via density fitting, which enables an efficient processing of the corresponding terms via three-index intermediates. The corresponding 3-index and the metric 2-index ERIs involving auxiliary functions are also computed and transformed to the WF-PAO basis (the 3-index ERI) on the periodic side. Although the direct ring-CCD method itself is not generally more accurate than MP2, it is more stable in the case of small band gap systems, as it sums up the ring diagrams to infinite order. Furthermore, this interface is a first step toward a high-level fragment-based quantum chemical treatment such as local CCSD(T) within a periodic embedding that is treated at a lower level. As two test examples we study the physisorption of H2 and argon on graphane.
Co-reporter:Thomas Merz, Matthias Wenninger, Michael Weinberger, Eberhard Riedle, Hans-Achim Wagenknecht and Martin Schütz
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 42) pp:18607-18619
Publication Date(Web):11 Sep 2013
DOI:10.1039/C3CP52344F
Charge transfer in DNA cannot be understood without addressing the complex conformational flexibility, which occurs on a wide range of timescales. In order to reduce this complexity four dinucleotide models 1X consisting of benzophenone linked by a phosphodiester to one of the natural nucleosides X = A, G, T, C were studied in water and methanol. The theoretical work focuses on the dynamics and electronic structure of 1G. Predominant conformations in the two solvents were obtained by molecular dynamics simulations. 1G in MeOH adopts mainly an open geometry with a distance of 12–16 Å between the two aromatic parts. In H2O the two parts of 1G form primarily a stacked conformation yielding a distance of 5–6 Å. The low-lying excited states were investigated by electronic structure theory in a QM/MM environment for representative snapshots of the trajectories. Photo-induced intramolecular charge transfer in the S1 state occurs exclusively in the stacked conformation. Ultrafast transient absorption spectroscopy with 1X reveals fast charge transfer from S1 in both solvents with varying yields. Significant charge transfer from the T1 state is only found for the nucleobases with the lowest oxidation potential: in H2O, charge transfer occurs with 3.2 × 109 s−1 for 1A and 6.0 × 109 s−1 for 1G. The reorganization energy remains nearly unchanged going from MeOH to the more polar H2O. The electronic coupling is rather low even for the stacked conformation with HAB = 3 meV and explains the moderate charge transfer rates. The solvent controls the conformational distribution and therefore gates the charge transfer due to differences in distance and stacking.
Co-reporter:Cesare Pisani, Martin Schütz, Silvia Casassa, Denis Usvyat, Lorenzo Maschio, Marco Lorenz and Alessandro Erba
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 21) pp:7615-7628
Publication Date(Web):26 Jan 2012
DOI:10.1039/C2CP23927B
CRYSCOR is a periodic post-Hartree–Fock program based on local functions in direct space, i.e., Wannier functions and projected atomic orbitals. It uses atom centered Gaussians as basis functions. The Hartree–Fock reference, as well as symmetry information, is provided by the CRYSTAL program. CRYSCOR presently features an efficient and parallel implementation of periodic local second order Møller–Plesset perturbation theory (MP2), which allows us to study 1D-, 2D- and 3D-periodic systems beyond 1000 basis functions per unit cell. Apart from the correlation energy also the MP2 density matrix, and from that the Compton profile, are available. Very recently, a new module for calculating excitonic band gaps at the uncorrelated Configuration-Interaction-Singles (CIS) level has been added. Other advancements include new extrapolation techniques for calculating surface adsorption on semi-infinite solids. In this paper the diverse features and recent advances of the present CRYSCOR version are illustrated by exemplary applications to various systems: the adsorption of an argon monolayer on the MgO (100) surface, the rolling energy of a boron nitride nanoscroll, the relative stability of different aluminosilicates, the inclusion energy of methane in methane–ice-clathrates, and the effect of electron correlation on charge and momentum density of α-quartz. Furthermore, we present some first tentative CIS results for excitonic band gaps of simple 3D-crystals, and their dependence on the diffuseness of the basis set.
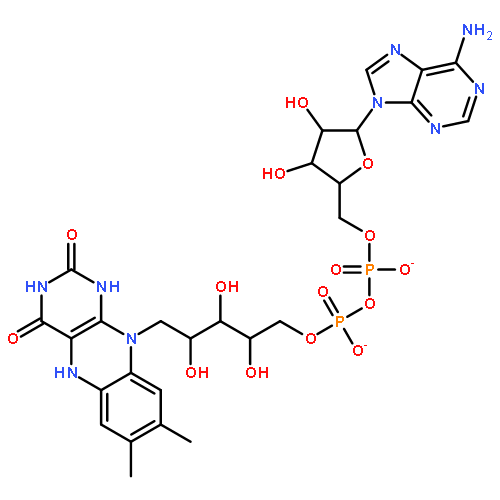
Co-reporter:Thomas Merz, Keyarash Sadeghian and Martin Schütz
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 32) pp:14775-14783
Publication Date(Web):12 Jul 2011
DOI:10.1039/C1CP21386E
The photophysics of roseoflavin in three different environments is investigated by using ab initio and quantum mechanics/molecular mechanics methods. Intramolecular charge transfer is shown to be responsible for the quenching of the fluorescence in the gas phase, and in the water environment. However, for the roseoflavin incorporated into the blue light using flavin (BLUF) protein environment (substituting the native flavin) no such deactivation is found. The conical intersection between the locally excited state of the chromophore and the charge transfer state involving the tyrosine residue, which in the native BLUF domain is responsible for initiating the photocycle, is missing for the roseoflavin substituted protein. This explains the experimental observations of the lack of any photocycle, and the loss of the biological function of the BLUF photoreceptor reported earlier.
Co-reporter:Dr. Keyarash Sadeghian;Dr. Marco Bocola; Dr. Martin Schütz
ChemPhysChem 2011 Volume 12( Issue 7) pp:1251-1254
Publication Date(Web):
DOI:10.1002/cphc.201100160
Abstract
The intermolecular interactions of the photodamaged cyclobutane pyrimidine dimer (CPD) lesion with adjacent nucleobases in the native intrahelical DNA double strand are investigated at the level of density functional theory symmetry-adapted perturbation theory (DFT-SAPT) and compared to the original (or repaired) case with pyrimidines (TpT) instead of CPD. The CPD aggregation is on average destabilized by about 6 kcal mol−1 relative to that involving TpT. The effect of destabilization is asymmetric, that is, it involves a single H-bonding (Watson–Crick (WC) type) base-pair interaction.
Co-reporter:Dr. Keyarash Sadeghian;Dr. Marco Bocola; Dr. Martin Schütz
ChemPhysChem 2011 Volume 12( Issue 7) pp:
Publication Date(Web):
DOI:10.1002/cphc.201190035
Co-reporter:Keyarash Sadeghian ; Marco Bocola ; Thomas Merz ;Martin Schütz
Journal of the American Chemical Society 2010 Volume 132(Issue 45) pp:16285-16295
Publication Date(Web):October 26, 2010
DOI:10.1021/ja108336t
UV irradiation of DNA can lead to the formation of mutagenic (6−4) pyrimidine−pyrimidone photolesions. The (6−4) photolyases are the enzymes responsible for the photoinduced repair of such lesions. On the basis of the recently published crystal structure of the (6−4) photolyase bound to DNA [Maul et al. 2008] and employing quantum mechanics/molecular mechanics techniques, a repair mechanism is proposed, which involves two photoexcitations. The flavin chromophore, initially being in its reduced anionic form, is photoexcited and donates an electron to the (6−4) form of the photolesion. The photolesion is then protonated by the neighboring histidine residue and forms a radical intermediate. The latter undergoes a series of energy stabilizing hydrogen-bonding rearrangements before the electron back transfer to the flavin semiquinone. The resulting structure corresponds to the oxetane intermediate, long thought to be formed upon DNA−enzyme binding. A second photoexcitation of the flavin promotes another electron transfer to the oxetane. Proton donation from the same histidine residue allows for the splitting of the four-membered ring, hence opening an efficient pathway to the final repaired form. The repair of the lesion by a single photoexcitation was shown not to be feasible.
Co-reporter:Keyarash Sadeghian, Marco Bocola and Martin Schütz
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 31) pp:8840-8846
Publication Date(Web):08 Jun 2010
DOI:10.1039/B925908B
Based on QM/MM calculations using a combination of time-dependent Hartree–Fock and coupled cluster response theory a mechanism is proposed for the photocycle of blue light using flavin (BLUF) domains in the signaling/light adapted conformation. In analogy to the dark-adapted form, a charge transfer state drives proton transfer from the highly conserved tyrosine residue to the flavin chromophore. The latter step is mediated by the adjacent glutamine residue, which, in the light adapted conformation, exists as its imidic tautomer. However, before the proton transfer is even halfway completed, a conical intersection seam between the charge transfer and ground state is reached. Two channels for the decay back to the initial light-adapted conformation are open, a rapid one leading directly through the funnel of the conical intersection, bypassing the formation of the biradical intermediate, and a slower one via the biradical intermediate. The mechanism as proposed here: (i) explains the very rapid photocycle; and (ii) confirms the concept of photoirreversibility, both of which have been experimentally observed for BLUF domains in their light-adapted conformations.
Co-reporter:Keyarash Sadeghian, Marco Bocola and Martin Schütz
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 31) pp:NaN8846-8846
Publication Date(Web):2010/06/08
DOI:10.1039/B925908B
Based on QM/MM calculations using a combination of time-dependent Hartree–Fock and coupled cluster response theory a mechanism is proposed for the photocycle of blue light using flavin (BLUF) domains in the signaling/light adapted conformation. In analogy to the dark-adapted form, a charge transfer state drives proton transfer from the highly conserved tyrosine residue to the flavin chromophore. The latter step is mediated by the adjacent glutamine residue, which, in the light adapted conformation, exists as its imidic tautomer. However, before the proton transfer is even halfway completed, a conical intersection seam between the charge transfer and ground state is reached. Two channels for the decay back to the initial light-adapted conformation are open, a rapid one leading directly through the funnel of the conical intersection, bypassing the formation of the biradical intermediate, and a slower one via the biradical intermediate. The mechanism as proposed here: (i) explains the very rapid photocycle; and (ii) confirms the concept of photoirreversibility, both of which have been experimentally observed for BLUF domains in their light-adapted conformations.
Co-reporter:Cesare Pisani, Martin Schütz, Silvia Casassa, Denis Usvyat, Lorenzo Maschio, Marco Lorenz and Alessandro Erba
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 21) pp:NaN7628-7628
Publication Date(Web):2012/01/26
DOI:10.1039/C2CP23927B
CRYSCOR is a periodic post-Hartree–Fock program based on local functions in direct space, i.e., Wannier functions and projected atomic orbitals. It uses atom centered Gaussians as basis functions. The Hartree–Fock reference, as well as symmetry information, is provided by the CRYSTAL program. CRYSCOR presently features an efficient and parallel implementation of periodic local second order Møller–Plesset perturbation theory (MP2), which allows us to study 1D-, 2D- and 3D-periodic systems beyond 1000 basis functions per unit cell. Apart from the correlation energy also the MP2 density matrix, and from that the Compton profile, are available. Very recently, a new module for calculating excitonic band gaps at the uncorrelated Configuration-Interaction-Singles (CIS) level has been added. Other advancements include new extrapolation techniques for calculating surface adsorption on semi-infinite solids. In this paper the diverse features and recent advances of the present CRYSCOR version are illustrated by exemplary applications to various systems: the adsorption of an argon monolayer on the MgO (100) surface, the rolling energy of a boron nitride nanoscroll, the relative stability of different aluminosilicates, the inclusion energy of methane in methane–ice-clathrates, and the effect of electron correlation on charge and momentum density of α-quartz. Furthermore, we present some first tentative CIS results for excitonic band gaps of simple 3D-crystals, and their dependence on the diffuseness of the basis set.
Co-reporter:Thomas Merz, Matthias Wenninger, Michael Weinberger, Eberhard Riedle, Hans-Achim Wagenknecht and Martin Schütz
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 42) pp:NaN18619-18619
Publication Date(Web):2013/09/11
DOI:10.1039/C3CP52344F
Charge transfer in DNA cannot be understood without addressing the complex conformational flexibility, which occurs on a wide range of timescales. In order to reduce this complexity four dinucleotide models 1X consisting of benzophenone linked by a phosphodiester to one of the natural nucleosides X = A, G, T, C were studied in water and methanol. The theoretical work focuses on the dynamics and electronic structure of 1G. Predominant conformations in the two solvents were obtained by molecular dynamics simulations. 1G in MeOH adopts mainly an open geometry with a distance of 12–16 Å between the two aromatic parts. In H2O the two parts of 1G form primarily a stacked conformation yielding a distance of 5–6 Å. The low-lying excited states were investigated by electronic structure theory in a QM/MM environment for representative snapshots of the trajectories. Photo-induced intramolecular charge transfer in the S1 state occurs exclusively in the stacked conformation. Ultrafast transient absorption spectroscopy with 1X reveals fast charge transfer from S1 in both solvents with varying yields. Significant charge transfer from the T1 state is only found for the nucleobases with the lowest oxidation potential: in H2O, charge transfer occurs with 3.2 × 109 s−1 for 1A and 6.0 × 109 s−1 for 1G. The reorganization energy remains nearly unchanged going from MeOH to the more polar H2O. The electronic coupling is rather low even for the stacked conformation with HAB = 3 meV and explains the moderate charge transfer rates. The solvent controls the conformational distribution and therefore gates the charge transfer due to differences in distance and stacking.
Co-reporter:Thomas Merz, Keyarash Sadeghian and Martin Schütz
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 32) pp:NaN14783-14783
Publication Date(Web):2011/07/12
DOI:10.1039/C1CP21386E
The photophysics of roseoflavin in three different environments is investigated by using ab initio and quantum mechanics/molecular mechanics methods. Intramolecular charge transfer is shown to be responsible for the quenching of the fluorescence in the gas phase, and in the water environment. However, for the roseoflavin incorporated into the blue light using flavin (BLUF) protein environment (substituting the native flavin) no such deactivation is found. The conical intersection between the locally excited state of the chromophore and the charge transfer state involving the tyrosine residue, which in the native BLUF domain is responsible for initiating the photocycle, is missing for the roseoflavin substituted protein. This explains the experimental observations of the lack of any photocycle, and the loss of the biological function of the BLUF photoreceptor reported earlier.

![(7,8-dimethyl-2,4-dioxo-3,4-dihydrobenzo[g]pteridin-10(2H)-yl)acetaldehyde](http://img.cochemist.com/ccimg/4300/4250-90-2.png)
![(7,8-dimethyl-2,4-dioxo-3,4-dihydrobenzo[g]pteridin-10(2H)-yl)acetaldehyde](http://img.cochemist.com/ccimg/4300/4250-90-2_b.png)