Co-reporter:Neal M. Davies
Applied Organometallic Chemistry 2011 Volume 25( Issue 6) pp:
Publication Date(Web):
DOI:10.1002/aoc.1767
No abstract is available for this article.
Co-reporter:Karina R. Vega-Villa;Connie M. Remsberg;Jody K. Takemoto;Yusuke Ohgami;Jaime A. Yáñez;Preston K. Andrews;Neal M. Davies
Chirality 2011 Volume 23( Issue 4) pp:339-348
Publication Date(Web):
DOI:10.1002/chir.20926
Abstract
The chirality of flavonoids has been overlooked in the majority of pharmacokinetic studies of homoeriodictyol, isosakuranetin, and taxifolin. The stereospecific pharmacokinetic disposition of these xenobiotics in male Sprague-Dawley rats is described for the first time. Validated HPLC methods were used to analyze serum and urine samples of rats following intravenous administration of each flavonoid via jugular vein cannulation and to determine their content in selected fruits. The characterization and interpretation of the pharmacokinetic disposition profiles of homoeriodictyol, isosakuranetin, and taxifolin are described. A discrepancy exists between half-lives in serum and urine which may be attributed to low assay sensitivity in serum for the three compounds; thus, a more accurate estimation of the pharmacokinetic parameters was obtained from urine. The pharmacokinetics of homoeriodictyol, isosakuranetin, and taxifolin revealed distribution, metabolism, and elimination that were dependent on the stereochemistry of the stereoisomers. The (−)-(S)-enantiomers of homoeriodictyol and isosakuranetin and the (+)-(2S; 3R)-stereoisomer of taxifolin were predominant in lemon, grapefruit, and tomato. These findings were achieved using chiral methods of analysis; the utility and necessity of developing chiral methods of analysis for chiral xenobiotics are discussed. Chirality, 2011. © 2010 Wiley-Liss, Inc.
Co-reporter:Connie M. Remsberg, Jody K. Takemoto, Rebecca M. Bertram, Neal M. Davies
Journal of Pharmaceutical and Biomedical Analysis 2011 54(4) pp: 878-881
Publication Date(Web):
DOI:10.1016/j.jpba.2010.10.028
Co-reporter:Jody K. Takemoto, Neal M. Davies
Journal of Pharmaceutical and Biomedical Analysis 2011 54(4) pp: 812-816
Publication Date(Web):
DOI:10.1016/j.jpba.2010.11.003
Co-reporter:Antonio M. Bonin;Jaime A. Yáñez;Chie Fukuda;Xiao Wei Teng;Carolyn T. Dillon;Trevor W. Hambley;Peter A. Lay;Neal M. Davies
Cancer Chemotherapy and Pharmacology 2010 Volume 66( Issue 4) pp:755-764
Publication Date(Web):2010/09/01
DOI:10.1007/s00280-009-1220-5
To evaluate, for the first time, the efficacy of copper–indomethacin in the inhibition of aberrant crypt foci formation using the azoxymethane-induced adenocarcinoma model, to examine cell viability in the HCT-116 colorectal cancer cell line, gastrointestinal permeability, mitochondrial oxidative damage, and renal toxicity in rat models.Azoxymethane-induced adenocarcinoma rats were dosed with indomethacin and copper–indomethacin for 28 days and aberrant crypt foci were evaluated. HCT-116 colorectal cancer cells were exposed to indomethacin and copper–indomethacin at 0–250 μg/mL (0–698 μM for indomethacin, and 0–147 μM for copper–indomethacin), and cell viability was measured. Acute gastrointestinal toxicity was measured using gastrointestinal permeability markers, gastrointestinal ulceration and bleeding, and measurement of an acute-phase protein haptoglobin. Effects of acute and chronic administration of indomethacin and copper–indomethacin on urinary electrolyte concentrations were examined.Both indomethacin and copper–indomethacin resulted in a significant reduction in aberrant crypt foci in azoxymethane-treated rats. In parallel, high concentrations of indomethacin and copper–indomethacin also reduced cell viability in HCT-116 colorectal cancer cells. However, copper–indomethacin was considerably safer in all measures of gastrointestinal toxicity compared to indomethacin. In addition, indomethacin reduced urinary electrolytes at an ulcerogenic dose of 10 mg/kg acutely and chronically at 3.0 mg/kg for 28 days, whereas copper–indomethacin at equimolar doses of indomethacin affected urine electrolytes after acute dosing but not after chronic dosing for 28 days.Copper–indomethacin has both gastrointestinal and renal sparing properties while maintaining efficacy in experimental adenocarcinoma.
Co-reporter:Dr Neal M. Davies;Jody K. Takemoto;Dion R. Brocks
Clinical Pharmacokinetics 2010 Volume 49( Issue 6) pp:351-377
Publication Date(Web):2010 June
DOI:10.2165/11319320-000000000-00000
multiple peaking in the blood fluid concentration-time curve is a phenomenon occasionally encountered in pharmacokinetics. When it occurs, it can create difficulties in the determination and interpretation of pharmacokinetic parameters. Multiple peaking can occur as a consequence of a number of different mechanisms. These include, in addition to others, factors related to the formulation, be it the drug chemical entity itself or other formulation-related factors such as the excipients incorporated into the product design. Another contributing factor that can work in concert with the formulation is the physiological makeup of the gastrointestinal tract itself. This includes the pH and components of bile such as bile salts and phospholipids, the secretion of which is regulated by hormonal and dietary factors. In some cases, biochemical differences in the regional areas of the gastrointestinal tract, such as regiospecificity in bile concentrations and/or transport proteins, could contribute to windows for absorption that result in multiple peaking of xenobiotics. One of the most common sources of multiple peaking is contributed by biliary secretion followed by intestinal reabsorption of a drug, a process for which the term ‘enterohepatic recycling’ has been coined. This cause of multiple peaking is associated with special consideration in the calculation and interpretation of the drug clearance and volume of distribution. In this review, each of these various causes of multiple peaking is discussed, with incorporation of relevant examples for illustrative purposes.
Co-reporter:Jaime A. Yáñez;M. Laird Forrest;Yusuke Ohgami;Glen S. Kwon;Neal M. Davies
Cancer Chemotherapy and Pharmacology 2008 Volume 61( Issue 1) pp:133-144
Publication Date(Web):2008/01/01
DOI:10.1007/s00280-007-0458-z
To determine the pharmacokinetics, tissue, and blood distribution of rapamycin PEG-block-poly(ε-caprolactone) (PEG-b-PCL) micelle formulations with and without the addition of α-tocopherol compared to control rapamycin in Tween 80/PEG 400/N,N-dimethylacetamide (DMA) (7:64:29).Rapamycin was incorporated at 10% w/w into PEG-b-PCL micelles (5:10 kDa) using a solvent extraction technique. The co-incorporation of 2:1 α-tocopherol:PEG-b-PCL was also studied. Rapamycin was quantified utilizing LC/MS in a Waters XTerra MS C18 column with 32-desmethoxyrapamycin as the internal standard. Male Sprague Dawley rats (N = 4 per group; ∼200 g) were cannulated via the left jugular and dosed intravenously (IV) with the rapamycin control and micelle formulations (10 mg/kg, 1:9 ratio for rapamycin to PEG-b-PCL). For tissue distribution 24 h after IV dosing, whole blood, plasma, red blood cells, and all the representative tissues were collected. The tissues were rapidly frozen under liquid nitrogen and ground to a fine powder. The rapamycin concentrations in plasma and red blood cells were utilized to determine the blood distribution (partition coefficient between plasma and red blood cells). For the determination of the pharmacokinetic parameters, blood, plasma, and urine samples were collected over 48 h. The pharmacokinetic parameters were calculated using WinNonlin® (Version 5.1) software.Rapamycin concentrations were considerably less in brain after administration of both micelle formulations compared to a rapamycin in the Tween 80/PEG 400/DMA control group. There was a 2-fold and 1.6-fold increase in the plasma fraction for rapamycin micelles with and without α-tocopherol. There was a decrease in volume of distribution for both formulations, an increase in AUC, a decrease in clearance, and increase in half life respectively for rapamycin in PEG-b-PCL + α-tocopherol micelles and in PEG-b-PCL micelles. There was no mortality with the micelle formulations compared to 60% mortality with rapamycin in Tween 80/PEG 400/DMA.The decreased distribution into the brain of rapamycin in PEG-b-PCL micelles may ameliorate rapamycin neurotoxicity. Both micelle formulations increase rapamycin distribution in plasma, which could facilitate access into solid tumors. The micellar delivery systems of rapamycin impart in vivo controlled release, resulting in altered disposition, and dramatically reduced mortality.
Co-reporter:Connie M. Remsberg;Jonathan K. Reynolds;Jody K. Takemoto;Karina R. Vega-Villa;Neal M. Davies
Clinical Pharmacokinetics 2008 Volume 47( Issue 11) pp:703-720
Publication Date(Web):2008/11/01
DOI:10.2165/00003088-200847110-00002
The NSAID etoricoxib is a selective inhibitor of cyclo-oxygenase 2 (COX-2), approved for treatment of patients with chronic arthropathies and musculoskeletal and dental pain. The rate of absorption of etoricoxib is moderate when given orally (the maximum plasma drug concentration occurs after ∼1 hour), and the extent of absorption is similar with oral and intravenous doses. Etoricoxib is extensively protein bound, primarily to plasma albumin, and has an apparent volume of distribution of 120 L in humans. The area under the plasma concentration-time curve (AUC) of etoricoxib increases in proportion to increasing oral doses between 5 and 120 mg. The elimination half-life of ∼20 hours in healthy subjects enables once-daily dosing. Etoricoxib is eliminated following biotransformation to carboxylic acid and glucuronide metabolites that are excreted in urine and faeces, with little of the drug (<1%) being eliminated unchanged in the urine. Etoricoxib is metabolized primarily by the cytochrome P450 (CYP) 3A4 isoenzyme. Plasma concentrations (AUC) of etoricoxib appear not to be different in patients with chronic renal insufficiency compared with individuals who have normal renal function. Compared with healthy subjects, it has been reported that the AUC is increased by approximately 40% in patients with moderate hepatic impairment. No inhibitory effects on CYP2C9, 2C19, 2D6, 2E1 or 3A4 are expected to occur with etoricoxib. Coadministration of etoricoxib with other drugs has been examined only to a limited extent, thus further assessment is necessary. Etoricoxib has been assessed for the management of several specific disease states, including pain, osteoarthritis, and rheumatoid arthritis, and has shown similar efficacy in comparison with traditional NSAIDs (including naproxen, diclofenac and ibuprofen) in these conditions. Etoricoxib has demonstrated a significant reduction in gastrointestinal toxicity compared with many traditional NSAIDs. The renal adverse effects of etoricoxib appear to be similar to those of other NSAIDs, and the cardiovascular adverse effects of this selective COX-2 inhibitor require further clinical scrutiny. Further study is necessary to delineate the relevance of the pharmacokinetic disposition in terms of the clinical benefits and risks of etoricoxib compared with other options in the clinical arsenal.
Co-reporter:Jaime A. Yáñez, Preston K. Andrews, Neal M. Davies
Journal of Chromatography B 2007 Volume 848(Issue 2) pp:159-181
Publication Date(Web):1 April 2007
DOI:10.1016/j.jchromb.2006.10.052
Although the analysis of the enantiomers and epimers of chiral flavanones has been carried out for over 20 years, there often remains a deficit within the pharmaceutical, agricultural, and medical sciences to address this issue. Hence, despite increased interest in the potential therapeutic uses, plant physiology roles, and health-benefits of chiral flavanones, the importance of stereoselectivity in agricultural, nutrition, pharmacokinetic, pharmacodynamic, pharmacological activity and disposition has often been ignored. This review presents both the general principles that allow separation of chiral flavanones, and discusses both the advantages and disadvantages of the available chromatographic assay methods and procedures used to separately quantify flavanone enantiomers and epimers in biological matrices.
Co-reporter:Renee L. Good, Kathryn A. Roupe, Chie Fukuda, G. Dennis Clifton, Marc W. Fariss, Neal M. Davies
Journal of Pharmaceutical and Biomedical Analysis 2005 Volume 39(1–2) pp:33-38
Publication Date(Web):1 September 2005
DOI:10.1016/j.jpba.2005.02.029
A method of analysis of a Vitamin E derivative d-tocopheryl acid succinate (TS) in biological fluids and commercially available products is necessary to study the kinetics of in vitro and in vivo metabolism, tissue distribution, and content uniformity. A simple and inexpensive high-performance liquid chromatographic method was developed for the direct determination of d-tocopheryl acid succinate in commercially available products, rat serum, and rat tissues. This method can also be applied to the determination of 15 Vitamin E derivatives. Rat serum (0.1 ml) was extracted with sodium dodecyl sulfate, ethanol, hexane, and then dried under nitrogen gas after addition of the internal standard, dl-α-tocopherol acetate. Separation was achieved on a C18 column with UV detection at 205 nm. The calibration curve for d-tocopheryl acid succinate was linear ranging from 0.025 to 100 μg/ml. The mean extraction efficiency was >92%. Precision of the assay was <5% (CV), and was within 5% at the limit of quantitation (0.025 μg/ml). Bias of the assay was lower than 5%, and was within 5% at the limit of quantitation. The assay was applied successfully to the serum and tissue distribution of d-tocopheryl acid succinate in rats, various Vitamin E derivatives, and content uniformity in commercially available products containing d-tocopheryl acid succinate.
Co-reporter:Kathryn Roupe, Steven Halls, Neal M. Davies
Journal of Pharmaceutical and Biomedical Analysis 2005 Volume 38(Issue 1) pp:148-154
Publication Date(Web):1 June 2005
DOI:10.1016/j.jpba.2004.12.015
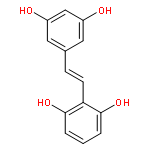
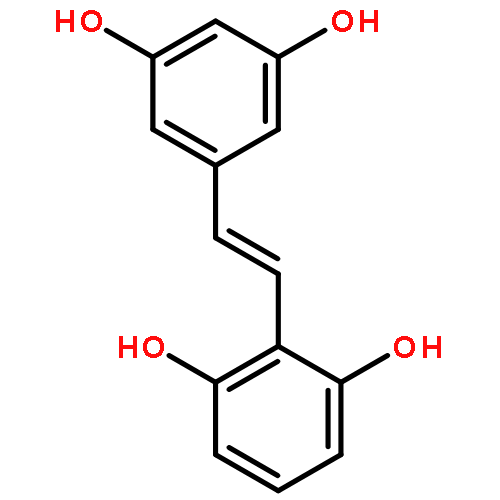
A method of analysis of pinosylvin in biological fluids is necessary to study the kinetics of in vitro and in vivo metabolism and determine its concentration in natural products. A novel and simple high-performance liquid chromatographic method was developed for simultaneous determination of pinosylvin and products of its metabolism in rat serum and liver microsomes. Serum, or microsomes (0.1 mL) were precipitated with acetonitrile after addition of the internal standard, 7-ethoxycoumarin. Separation was achieved on an amylose tris 3,5 dimethylphenylcarbamate column (150 mm × 4.6 mm, ID, 5 μm) with UV detection at 308 nm. The calibration curves were linear ranging from 0.5 to 100 μg/mL. The mean extraction efficiency was >99%. Precision of the assay (coefficient of variation) was <10%, including the limit of quantitation (0.5 μg/mL). Bias of the assay was lower than 15%. The limit of detection was 100 ng/mL for a 0.1 mL sample. The assay was successfully applied to both the in vitro and in vivo metabolic kinetic study of pinosylvin. Three metabolites of pinosylvin, two oxidative and one glucuronidated, have been identified. The two oxidative metabolites of pinosylvin have been identified as E- and Z-resveratrol.
Co-reporter:Xiao Wei Teng, Neal M. Davies
Journal of Pharmaceutical and Biomedical Analysis 2004 Volume 35(Issue 5) pp:1143-1147
Publication Date(Web):3 September 2004
DOI:10.1016/j.jpba.2004.03.013
Co-reporter:Neal M. Davies
Journal of Pharmaceutical Sciences 2004 Volume 93(Issue 12) pp:2877-2880
Publication Date(Web):27 SEP 2004
DOI:10.1002/jps.20189
Co-reporter:Xiao Wei Teng, Stephen W.J Wang, Neal M Davies
Journal of Chromatography B 2003 Volume 796(Issue 2) pp:225-231
Publication Date(Web):5 November 2003
DOI:10.1016/j.jchromb.2003.07.010
A simple, rapid and sensitive high-performance liquid chromatographic method was developed for determination of ibuprofen, (±)-(R, S)-2-(4-isobutylphenyl)-propionic acid, enantiomers in rat serum. Serum (0.1 ml) was extracted with 2,2,4-trimethylpentane/isopropanol (95:5, v/v) after addition of the internal standard, (S)-naproxen, and acidification with H2SO4. Enantiomeric resolution of ibuprofen was achieved on ChiralPak AD-RH column with ultraviolet (UV) detection at 220 nm without interference from endogenous co-extracted solutes. The calibration curve demonstrated excellent linearity between 0.1 and 50 μg/ml for each enantiomer. The mean extraction efficiency was >92%. Precision of the assay was within 11% (relative standard deviation (R.S.D.)) and bias of the assay was lower than 15% at the limit of quantitation (0.1 μg/ml). The assay was applied successfully to an oral pharmacokinetic study of ibuprofen in rats.
Co-reporter:Karina R. Vega-Villa, Jody K. Takemoto, Jaime A. Yáñez, Connie M. Remsberg, ... Neal M. Davies
Advanced Drug Delivery Reviews (22 May 2008) Volume 60(Issue 8) pp:929-938
Publication Date(Web):22 May 2008
DOI:10.1016/j.addr.2007.11.007
Toxicity of nanocarrier systems involves physiological, physicochemical, and molecular considerations. Nanoparticle exposures through the skin, the respiratory tract, the gastrointestinal tract and the lymphatics have been described. Nanocarrier systems may induce cytotoxicity and/or genotoxicity, whereas their antigenicity is still not well understood. Nanocarrier may alter the physicochemical properties of xenobiotics resulting in pharmaceutical changes in stability, solubility, and pharmacokinetic disposition. In particular, nanocarriers may reduce toxicity of hydrophobic cancer drugs that are solubilized. Nano regulation is still undergoing major changes to encompass environmental, health, and safety issues. The rapid commercialization of nanotechnology requires thoughtful environmental, health and safety research, meaningful, and an open discussion of broader societal impacts, and urgent toxicological oversight action.