Co-reporter:Francisco Velazquez Escobar, Christina Lang, Aref Takiden, Constantin Schneider, Jens Balke, Jon Hughes, Ulrike Alexiev, Peter HildebrandtMaria Andrea Mroginski
The Journal of Physical Chemistry B 2017 Volume 121(Issue 1) pp:
Publication Date(Web):December 14, 2016
DOI:10.1021/acs.jpcb.6b09600
Phytochromes are biological red/far-red light sensors found in many organisms. Photoisomerization of the linear methine-bridged tetrapyrrole triggers transient proton translocation events in the chromophore binding pocket (CBP) leading to major conformational changes of the protein matrix that are in turn associated with signaling. By combining pH-dependent resonance Raman and UV–visible absorption spectroscopy, we analyzed protonation-dependent equilibria in the CBP of Cph1 involving the proposed Pr-I and Pr-II substates that prevail below and above pH 7.5, respectively. The protonation pattern and vibrational properties of these states were further characterized by means of hybrid quantum mechanics/molecular mechanics calculations. From this combined experimental–theoretical study, we were able to identify His260 as the key residue controlling pH-dependent equilibria. This residue is not only responsible for the conformational heterogeneity of CBP in the Pr state of prokaryotic phytochromes, discussed extensively in the past, but it constitutes the sink and source of protons in the proton release/uptake mechanism involving the tetrapyrrole chromophore which finally leads to the formation of the Pfr state. Thus, this work provides valuable information that may guide further experiments toward the understanding of the specific role of protons in controlling structure and function of phytochromes in general.
Co-reporter:Alexandre Ciaccafava, Daria Tombolelli, Lilith Domnik, Jochen Fesseler, Jae-Hun Jeoung, Holger Dobbek, Maria Andrea Mroginski, Ingo Zebger and Peter Hildebrandt
Chemical Science 2016 vol. 7(Issue 5) pp:3162-3171
Publication Date(Web):27 Jan 2016
DOI:10.1039/C5SC04554A
Carbon monoxide dehydrogenase (CODH) is a key enzyme for reversible CO interconversion. To elucidate structural and mechanistic details of CO binding at the CODH active site (C-cluster), cyanide is frequently used as an iso-electronic substitute and inhibitor. However, previous studies revealed conflicting results on the structure of the cyanide-bound complex and the mechanism of cyanide-inhibition. To address this issue in this work, we have employed IR spectroscopy, crystallography, site directed mutagenesis, and theoretical methods to analyse the cyanide complex of the CODH from Carboxydothermus hydrogenoformans (CODHIICh). IR spectroscopy demonstrates that a single cyanide binds to the Ni ion. Whereas the inhibitor could be partially removed at elevated temperature, irreversible degradation of the C-cluster occurred in the presence of an excess of cyanide on the long-minute time scale, eventually leading to the formation of [Fe(CN)6]4− and [Ni(CN)4]2− complexes. Theoretical calculations based on a new high-resolution structure of the cyanide-bound CODHIICh indicated that cyanide binding to the Ni ion occurs upon dissociation of the hydroxyl ligand from the Fe1 subsite of the C-cluster. The hydroxyl group is presumably protonated by Lys563 which, unlike to His93, does not form a hydrogen bond with the cyanide ligand. A stable deprotonated ε-amino group of Lys563 in the cyanide complex is consistent with the nearly unchanged CN stretching in the Lys563Ala variant of CODHIICh. These findings support the view that the proton channel connecting the solution phase with the active site displays a strict directionality, controlled by the oxidation state of the C-cluster.
Co-reporter:Tillmann Utesch, Diego Millo, Maria Ana Castro, Peter Hildebrandt, Ingo Zebger, and Maria Andrea Mroginski
Langmuir 2013 Volume 29(Issue 2) pp:673-682
Publication Date(Web):December 7, 2012
DOI:10.1021/la303635q
Understanding the interaction and immobilization of [NiFe] hydrogenases on functionalized surfaces is important in the field of biotechnology and, in particular, for the development of biofuel cells. In this study, we investigated the adsorption behavior of the standard [NiFe] hydrogenase of Desulfovibrio gigas on amino-terminated alkanethiol self-assembled monolayers (SAMs) with different levels of protonation. Classical all-atom molecular dynamics (MD) simulations revealed a strong correlation between the adsorption behavior and the level of ionization of the chemically modified electrode surface. While the hydrogenase undergoes a weak but stable initial adsorption process on SAMs with a low degree of protonation, a stronger immobilization is observable on highly ionized SAMs, affecting protein reorientation and conformation. These results were validated by complementary surface-enhanced infrared absorption (SEIRA) measurements on the comparable [NiFe] standard hydrogenases from Desulfovibrio vulgaris Miyazaki F and allowed in this way for a detailed insight into the adsorption mechanism at the atomic level.
Co-reporter:Yvonne Rippers;Marius Horch; Peter Hildebrt;Dr. Ingo Zebger ; Maria Andrea Mroginski
ChemPhysChem 2012 Volume 13( Issue 17) pp:3852-3856
Publication Date(Web):
DOI:10.1002/cphc.201200562
Abstract
Combined molecular dynamics (MD) and quantum mechanical/molecular mechanical (QM/MM) calculations were performed on the crystal structure of the reduced membrane-bound [NiFe] hydrogenase (MBH) from Ralstonia eutropha to determine the absolute configuration of the CO and the two CN− ligands bound to the active-site iron of the enzyme. For three models that include the CO ligand at different positions, often indistinguishable on the basis of the crystallographic data, we optimized the structures and calculated the ligand stretching frequencies. Comparison with the experimental IR data reveals that the CO ligand is in trans position to the substrate-binding site of the bimetallic [NiFe] cluster.
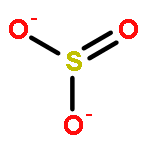
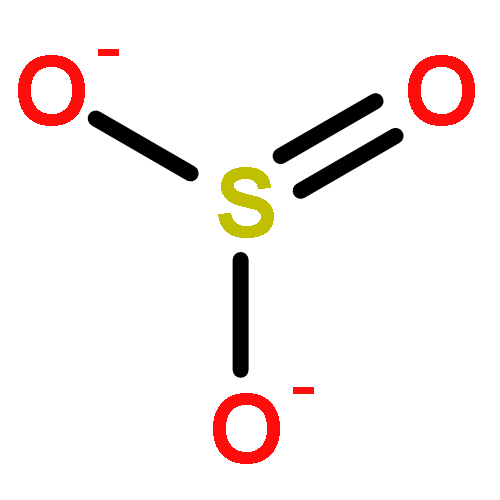
Co-reporter:Tillmann Utesch, Murat Sezer, Inez M. Weidinger, and Maria Andrea Mroginski
Langmuir 2012 Volume 28(Issue 13) pp:5761-5769
Publication Date(Web):March 2, 2012
DOI:10.1021/la205055g
Sulfite oxidase (SO) is an enzyme catalyzing the terminal step of the metabolism of sulfur-containing amino acids that is essential for almost all living organisms. The catalytic activity of SO in vertebrates strongly depends on the efficiency of the intramolecular electron transfer (IET) between the catalytic Moco domain and the cytochrome b5 (cyt b5) domain. The IET process is assumed to be mediated by large domain motions of the cyt b5 domains within the enzyme. Thus, the interaction of SO with charged surfaces may affect the mobility of the cyt b5 domain required for IET and consequently hinder SO activation. In this study, we present a molecular dynamics approach to investigating the ionic strength dependence of the initial surface adsorption of SO in two different conformations—the crystallographic structure and the model structure for an activated SO—onto mixed amino- and hydroxyl-terminated SAMs. The results show for both conformations at low ionic strengths a strong adsorption of the cyt b5 units onto the SAM, which inhibits the domain motion event required for IET. Under higher ion concentrations, however, the interaction with the surface is weakened by the negatively charged ions acting as a buffer and competing in adsorption with the cathodic cyt b5 domains. This competition prevents the immobilization of the cytochrome b5 units onto the surface, allowing the intramolecular domain motions favoring IET. Our predictions support the interpretation of recent experimental spectroelectrochemical studies on SO.
Co-reporter:Maria Andrea Mroginski, Steve Kaminski, David von Stetten, Simone Ringsdorf, Wolfgang Gärtner, Lars-Oliver Essen, and Peter Hildebrandt
The Journal of Physical Chemistry B 2011 Volume 115(Issue 5) pp:1220-1231
Publication Date(Web):December 30, 2010
DOI:10.1021/jp108265h
A homology structural model was generated for plant phytochrome phyA utilizing the crystal structure of the sensory module of cyanobacterial phytochrome Cph1 (Cph1Δ2). As chromophores, either the native phytochromobilin cofactor (PΦB) or phycocyanobilin (PCB), the natural cofactor in Cph1, was incorporated. These homology models were further optimized by molecular dynamics (MD) simulations revealing a satisfying overall agreement with the crystal structure of Cph1Δ2. Notable differences in the PΦB adduct of phyA result from a restructuring of the small helical segment α7 that leads to displacements of a few amino acids away from the cofactor. This repositioning of residues also include aspartate 218 such that, instead of its carbonyl function as in Cph1Δ2, an additional water molecule forms hydrogen bonds with the ring B and C NH groups. To validate the phyA structural model in the chromophore binding pocket, Raman spectra of the cofactor were calculated by means of the quantum mechanics/molecular mechanics (QM/MM) hybrid methodology and compared with the experimental resonance Raman (RR) spectra. The satisfactory overall agreement between calculated and experimental spectra is taken as an indication for the good quality of the structural model. Moreover, the methine bridge stretching modes and the effects of isotopic labeling at selected positions of the chromophore are very well reproduced to allow confirming even details of the methine bridge geometry as predicted by the homology model. Specifically, it is demonstrated that the experimental RR spectra are consistent with a torsional angle of ring D with respect to ring C that is distinctly higher for phyA−PCB (45°) and phyA−PΦB (42°) than for Cph1Δ2 (30°). Raman spectra calculated from different points of the MD trajectory display variations of the mode frequencies and intensities reflecting the structural fluctuations from snapshot to snapshot. The snapshot spectrum of the lowest energy structure and the sum of all snapshot spectra afford an equally good description of the experimental data. Particularly large variations between the snapshots are noted for the N−H in-plane bending mode of the pyrrole rings B and C, which reflect alterations of the hydrogen bond interactions brought about by fluctuations of water molecules in the cofactor cavity. This overestimation of the water molecule mobility is a consequence of the deficiency of the current QM/MM methodology that, due to the lack of appropriate protein force fields, cannot adequately account for the electrostatics in the cofactor pocket.
Co-reporter:Tillmann Utesch, Grazia Daminelli, and Maria Andrea Mroginski
Langmuir 2011 Volume 27(Issue 21) pp:13144-13153
Publication Date(Web):September 29, 2011
DOI:10.1021/la202489w
Bone morphogenetic protein-2 (BMP-2) plays a crucial role in osteoblast differentiation and proliferation. Its effective therapeutic use for ectopic bone and cartilage regeneration depends, among other factors, on the interaction with the carrier at the implant site. In this study, we used classical molecular dynamics (MD) and a hybrid approach of steered molecular dynamics (SMD) combined with MD simulations to investigate the initial stages of the adsorption of BMP-2 when approaching two implant surfaces, hydrophobic graphite and hydrophilic titanium dioxide rutile. Surface adsorption was evaluated for six different orientations of the protein, two end-on and four side-on, in explicit water environment. On graphite, we observed a weak but stable adsorption. Depending on the initial orientation, hydrophobic patches as well as flexible loops of the protein were involved in the interaction with graphite. On the contrary, BMP-2 adsorbed only loosely to hydrophilic titanium dioxide. Despite a favorable interaction energy between protein and the TiO2 surface, the rapid formation of a two-layer water structure prevented the direct interaction between protein and titanium dioxide. The first water adlayer had a strong repulsive effect on the protein, while the second attracted the protein toward the surface. For both surfaces, hydrophobic graphite and hydrophilic titanium dioxide, denaturation of BMP-2 induced by adsorption was not observed on the nanosecond time scale.
Co-reporter:Steve Kaminski, Michael Gaus, Prasad Phatak, David von Stetten, Marcus Elstner and Maria Andrea Mroginski
Journal of Chemical Theory and Computation 2010 Volume 6(Issue 4) pp:1240-1255
Publication Date(Web):February 24, 2010
DOI:10.1021/ct900660x
The Self-Consistent Charge Density Functional Tight Binding (SCC-DFTB) method has been extended for the calculation of vibrational Raman spectra employing the Fourier Transform of Time-Correlation Function (FTTCF) formalism. As Witek and co-workers have already shown for a set of various organic molecules, the minimal basis SCC-DFTB approach performs surprisingly good in terms of polarizability calculations. Therefore, we were encouraged to use this electronic structure method for the purpose of Raman spectra calculations via FTTCF. The molecular polarizability was accessed via second order numeric derivatives of the SCC-DFTB energy with respect to the components of an external electric field “on-the-fly” during a molecular dynamics (MD) simulation. The finite electric field approach delivers Raman spectra that are in overall good agreement for most of 10 small organic model compounds examined in the gas phase compared to a standard Normal Mode Analysis (NMA) approach at the same (SCC-DFTB) and at a higher level of theory (BLYP aug-cc-pVTZ). With the use of reparametrized SCC-DFTB repulsive potentials, a distinct improvement of the Raman spectra from the SCC-DFTB/FTTCF protocol of conjugated hydrocarbons has been observed. Further QM/MM test calculations of l-phenylalanine in aqueous solution revealed larger deviations concerning vibrational frequencies and relative intensities for several stretching and bending modes in the benzene ring as compared to experimental results. Our SCC-DFTB/FTTCF approach was also tested against a hybrid method, in which polarizability calculations at the B3YLP 6-31G(d) level were performed on a trajectory at the SCC-DFTB level. We found that our SCC-DFTB/FTTCF protocol is not only much more efficient but in terms of the resulting Raman spectra also of similar accuracy compared to the hybrid approach. In our opinion, the more accurately calculated polarizabilities at the B3YLP 6-31G(d) level cannot compensate for the usually insufficient sampling of phase space when employing high level QM methods in a FTTCF framework.
Co-reporter:Maria A. Mroginski Dr.;Steve Kaminski ;Peter Hildebrt Dr.
ChemPhysChem 2010 Volume 11( Issue 6) pp:1265-1274
Publication Date(Web):
DOI:10.1002/cphc.200900895
Abstract
The Raman spectrum of the phycoviolobilin cofactor of the α-subunit of phycoerythrocyanin was computed using a hybrid quantum mechanical/molecular mechanics (QM/MM) method in order to evaluate the performance of the QM/MM approach for calculating the vibrational spectra of protein-bound tetrapyrroles as found in phytochrome photoreceptors. A good overall agreement between the experimental and the calculated spectra was achieved. In addition, calculation of the vibrational properties of several snapshots extracted from a molecular dynamics simulation allowed us to investigate in detail the effect of the protein environment on the vibrational spectra. Heterogeneous broadening of most of the experimental bands could be reproduced in a satisfactory manner as the sum of individual spectra obtained by normal-mode-analysis (NMA). An exception is the bandwidth of the peak at 1646 cm−1, which is underestimated by the NMA sum as well as by the instantaneous normal mode analysis (INMA) approach.
Co-reporter:Maria Andrea Mroginski, David von Stetten, Francisco Velazquez Escobar, Holger M. Strauss, Steve Kaminski, Patrick Scheerer, Mina Günther, Daniel H. Murgida, Peter Schmieder, Christian Bongards, Wolfgang Gärtner, Jo Mailliet, Jon Hughes, Lars-Oliver Essen, Peter Hildebrandt
Biophysical Journal (20 May 2009) Volume 96(Issue 10) pp:
Publication Date(Web):20 May 2009
DOI:10.1016/j.bpj.2009.02.029
A quantum mechanics (QM)/molecular mechanics (MM) hybrid method was applied to the Pr state of the cyanobacterial phytochrome Cph1 to calculate the Raman spectra of the bound PCB cofactor. Two QM/MM models were derived from the atomic coordinates of the crystal structure. The models differed in the protonation site of His260 in the chromophore-binding pocket such that either the δ-nitrogen (M-HSD) or the ɛ-nitrogen (M-HSE) carried a hydrogen. The optimized structures of the two models display small differences specifically in the orientation of His260 with respect to the PCB cofactor and the hydrogen bond network at the cofactor-binding site. For both models, the calculated Raman spectra of the cofactor reveal a good overall agreement with the experimental resonance Raman (RR) spectra obtained from Cph1 in the crystalline state and in solution, including Cph1 adducts with isotopically labeled PCB. However, a distinctly better reproduction of important details in the experimental spectra is provided by the M-HSD model, which therefore may represent an improved structure of the cofactor site. Thus, QM/MM calculations of chromoproteins may allow for refining crystal structure models in the chromophore-binding pocket guided by the comparison with experimental RR spectra. Analysis of the calculated and experimental spectra also allowed us to identify and assign the modes that sensitively respond to chromophore-protein interactions. The most pronounced effect was noted for the stretching mode of the methine bridge A-B adjacent to the covalent attachment site of PCB. Due a distinct narrowing of the A-B methine bridge bond angle, this mode undergoes a large frequency upshift as compared with the spectrum obtained by QM calculations for the chromophore in vacuo. This protein-induced distortion of the PCB geometry is the main origin of a previous erroneous interpretation of the RR spectra based on QM calculations of the isolated cofactor.
Co-reporter:Alexandre Ciaccafava, Daria Tombolelli, Lilith Domnik, Jochen Fesseler, Jae-Hun Jeoung, Holger Dobbek, Maria Andrea Mroginski, Ingo Zebger and Peter Hildebrandt
Chemical Science (2010-Present) 2016 - vol. 7(Issue 5) pp:
Publication Date(Web):
DOI:10.1039/C5SC04554A



























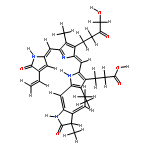
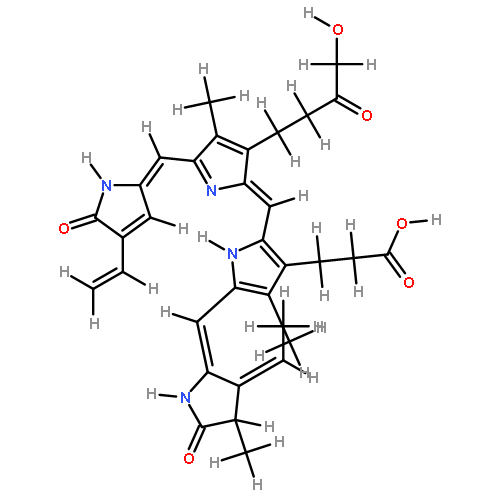
![3-[(2E,5Z)-2-[[4-(2-CARBOXYETHYL)-5-[(Z)-[(3Z,4R)-3-ETHYLIDENE-4-METHYL-5-OXOPYRROLIDIN-2-YLIDENE]METHYL]-3-METHYL-1H-PYRROL-2-YL]METHYLIDENE]-5-[(4-ETHENYL-3-METHYL-5-OXOPYRROL-2-YL)METHYLIDENE]-4-METHYLPYRROL-3-YL]PROPANOIC ACID](http://img.cochemist.com/data/images/143392-71-6.png)
![3-[(2E,5Z)-2-[[4-(2-CARBOXYETHYL)-5-[(Z)-[(3Z,4R)-3-ETHYLIDENE-4-METHYL-5-OXOPYRROLIDIN-2-YLIDENE]METHYL]-3-METHYL-1H-PYRROL-2-YL]METHYLIDENE]-5-[(4-ETHENYL-3-METHYL-5-OXOPYRROL-2-YL)METHYLIDENE]-4-METHYLPYRROL-3-YL]PROPANOIC ACID](http://img.cochemist.com/data/images/143392-71-6_b.png)


![1H-Pyrrole,2,3,4-trimethyl-5-[(Z)-(3,4,5-trimethyl-2H-pyrrol-2-ylidene)methyl]-](http://img.cochemist.com/ccimg/66600/66585-53-3.png)
![1H-Pyrrole,2,3,4-trimethyl-5-[(Z)-(3,4,5-trimethyl-2H-pyrrol-2-ylidene)methyl]-](http://img.cochemist.com/ccimg/66600/66585-53-3_b.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)