Co-reporter:Kenji Sugisaki;Kazuo Toyota;Kazunobu Sato;Daisuke Shiomi
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 44) pp:30128-30138
Publication Date(Web):2017/11/15
DOI:10.1039/C7CP05533A
Spin–orbit contributions to the zero-field splitting (ZFS) tensor (DSO tensor) of MIII(acac)3 complexes (M = V, Cr, Mn, Fe and Mo; acac = acetylacetonate anion) are evaluated by means of ab initio (a hybrid CASSCF/MRMP2) and DFT (Pederson–Khanna (PK) and natural orbital-based Pederson–Khanna (NOB-PK)) methods, focusing on the behaviour of DFT-based approaches to the DSO tensors against the valence d-electron configurations of the transition metal ions in octahedral coordination. Both the DFT-based approaches reproduce trends in the D tensors. Significantly, the differences between the theoretical and experimental D (D = DZZ − (DXX + DYY)/2) values are smaller in NOB-PK than in PK, emphasising the usefulness of the natural orbital-based approach to the D tensor calculations of transition metal ion complexes. In the case of d2 and d4 electronic configurations, the DSO(NOB-PK) values are considerably underestimated in the absolute magnitude, compared with the experimental ones. The DSO tensor analysis based on the orbital region partitioning technique (ORPT) revealed that the DSO contributions attributed to excitations from the singly occupied region (SOR) to the unoccupied region (UOR) are significantly underestimated in the DFT-based approaches to all the complexes under study. In the case of d3 and d5 configurations, the (SOR → UOR) excitations contribute in a nearly isotropic manner, which causes fortuitous error cancellations in the DFT-based DSO values. These results indicate that more efforts to develop DFT frameworks should be directed towards the reproduction of quantitative DSO tensors of transition metal complexes with various electronic configurations and local symmetries around metal ions.
Co-reporter:Takeshi Yamane;Kenji Sugisaki;Tomoki Nakagawa;Hideto Matsuoka;Takahisa Nishio;Shigemori Kinjyo;Nobuyuki Mori;Satoshi Yokoyama;Chika Kawashima;Naoki Yokokura;Kazunobu Sato;Yuki Kanzaki;Daisuke Shiomi;Kazuo Toyota;David H. Dolphin;Wei-Ching Lin;Charles A. McDowell;Makoto Tadokoro
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 36) pp:24769-24791
Publication Date(Web):2017/09/20
DOI:10.1039/C7CP03850J
The fictitious spin-1/2 Hamiltonian approach is the putative method to analyze the fine-structure/hyperfine ESR spectra of high spin metallocomplexes having sizable zerofield splitting (ZFS), thus giving salient principal g-values far from around g = 2 without explicitly providing their ZFS parameters in most cases. Indeed, the significant departure of the g-values from g = 2 is indicative of the occurrence of their high spin states, but naturally they never agree with true g-values acquired by quantum chemical calculations such as sophisticated DFT or ab initio MO calculations. In this work, we propose facile approaches to determine the magnetic tensors of high spin metallocomplexes having sizable ZFS, instead of performing advanced high-field/high-frequency ESR spectroscopy. We have revisited analytical expressions for the relationship between effective g-values and true principal g-values for high spins. The useful analytical formulas for the geff–gtrue relationships are given for S's up to 7/2. The genuine Zeeman perturbation formalism gives the exact solutions for S = 3/2, and for higher S's it is much more accurate than the pseudo-Zeeman perturbation approach documented so far (A. Abragam and B. Bleaney, Electron Paramagnetic Resonance of Transition Metal Ions, 1970; J. R. Pilbrow, J. Magn. Reson., 1978, 31, 479; F. Trandafir et al., Appl. Magn. Reson., 2007, 31, 553; M. Fittipaldi et al., J. Phys. Chem. B, 2008, 112, 3859), in which the E(Sx2 − Sy2) term is putatively treated to the second order. To show the usefulness of the present approach, we exploit FeIII(Cl)OEP (S = 5/2) (OEP: 2,3,7,8,12,13,17,18-octaethylporphyrin) and CoIIOEP (S = 3/2) well magnetically diluted in the diamagnetic host crystal lattice of NiIIOEP. The advantage of single-crystal ESR spectroscopy lies in the fact that the molecular information on the principal axes of the magnetic tensors is crucial in comparing with reliable theoretical results. In high spin states of metallocomplexes with sizable ZFS in pseudo-octahedral symmetry, their fine-structure ESR transitions for the principal z-axis orientation appear in the lower field far from g = 2 at the X-band, disagreeing with the putative intuitive picture obtained using relevant ESR spectroscopy. A ReIII,IV dinuclear complex in a mixed valence state exemplifies the cases, whose fine-structure/hyperfine ESR spectra of the neat crystals have been analyzed in their principal-axis system. The DFT-based/ab initio MO calculations of the magnetic tensors for all the high spin entities in this work were carried out.
Co-reporter:Kenji Sugisaki, Satoru Yamamoto, Shigeaki Nakazawa, Kazuo Toyota, Kazunobu Sato, Daisuke Shiomi, and Takeji Takui
The Journal of Physical Chemistry A 2016 Volume 120(Issue 32) pp:6459-6466
Publication Date(Web):August 8, 2016
DOI:10.1021/acs.jpca.6b04932
Quantum computers are capable to efficiently perform full configuration interaction (FCI) calculations of atoms and molecules by using the quantum phase estimation (QPE) algorithm. Because the success probability of the QPE depends on the overlap between approximate and exact wave functions, efficient methods to prepare accurate initial guess wave functions enough to have sufficiently large overlap with the exact ones are highly desired. Here, we propose a quantum algorithm to construct the wave function consisting of one configuration state function, which is suitable for the initial guess wave function in QPE-based FCI calculations of open-shell molecules, based on the addition theorem of angular momentum. The proposed quantum algorithm enables us to prepare the wave function consisting of an exponential number of Slater determinants only by a polynomial number of quantum operations.
Co-reporter:Kenji SugisakiKazuo Toyota, Kazunobu Sato, Daisuke Shiomi, Takeji Takui
The Journal of Physical Chemistry A 2016 Volume 120(Issue 49) pp:9857-9866
Publication Date(Web):November 15, 2016
DOI:10.1021/acs.jpca.6b10253
A quasi-restricted orbital (QRO) approach for the calculation of the spin–orbit term of zero-field splitting tensors (DSO tensors) by means of density functional theory (DFT) importantly features in the fact that it is free from spin contamination problems because it uses spin eigenfunctions for the zeroth order wave functions. In 2011, however, Schmitt and co-workers pointed out that in the originally proposed QRO working equation some possible excitations were not included in their sum-over-states procedure, which causes spurious DSO contributions from closed-shell subsystems located far from the magnetic molecule under study. We have revisited the derivation of the QRO working equation and modified it, making it include all possible types of excitations in the sum-over-states procedure. We have found that the spurious DSO contribution can be eliminated by taking into account contributions from all possible types of singly excited configuration state functions. We have also found that only the SOMO(α) → SOMO(β) excited configurations have nonzero contributions to the DSO tensors as long as α and β spin orbitals have the same spatial distributions and orbital energies. For the DSO tensor calculations, by using a ground state wave function free from spin contamination, we propose a natural orbital-based Pederson–Khanna (NOB-PK) method, which utilizes the single determinant wave function consisting of natural orbitals in conjunction with the Pederson–Khanna (PK) type perturbation treatment. Some relevant calculations revealed that the NOB-PK method can afford more accurate DSO tensors than the conventional PK method as well as the QRO approach in MnII complexes and ReIV-based single molecule magnets.
Co-reporter:Satoru Yamamoto, Shigeaki Nakazawa, Kenji Sugisaki, Kazunobu Sato, Kazuo Toyota, Daisuke Shiomi and Takeji Takui
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 4) pp:2742-2749
Publication Date(Web):27 Nov 2014
DOI:10.1039/C4CP04744C
A molecular spin quantum computer (MSQC) requires electron spin qubits, which pulse-based electron spin/magnetic resonance (ESR/MR) techniques can afford to manipulate for implementing quantum gate operations in open shell molecular entities. Importantly, nuclear spins, which are topologically connected, particularly in organic molecular spin systems, are client qubits, while electron spins play a role of bus qubits. Here, we introduce the implementation for an adiabatic quantum algorithm, suggesting the possible utilization of molecular spins with optimized spin structures for MSQCs. We exemplify the utilization of an adiabatic factorization problem of 21, compared with the corresponding nuclear magnetic resonance (NMR) case. Two molecular spins are selected: one is a molecular spin composed of three exchange-coupled electrons as electron-only qubits and the other an electron-bus qubit with two client nuclear spin qubits. Their electronic spin structures are well characterized in terms of the quantum mechanical behaviour in the spin Hamiltonian. The implementation of adiabatic quantum computing/computation (AQC) has, for the first time, been achieved by establishing ESR/MR pulse sequences for effective spin Hamiltonians in a fully controlled manner of spin manipulation. The conquered pulse sequences have been compared with the NMR experiments and shown much faster CPU times corresponding to the interaction strength between the spins. Significant differences are shown in rotational operations and pulse intervals for ESR/MR operations. As a result, we suggest the advantages and possible utilization of the time-evolution based AQC approach for molecular spin quantum computers and molecular spin quantum simulators underlain by sophisticated ESR/MR pulsed spin technology.
Co-reporter:Kenji Sugisaki, Kazuo Toyota, Kazunobu Sato, Daisuke Shiomi, Masahiro Kitagawa and Takeji Takui
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 19) pp:9171-9181
Publication Date(Web):21 Mar 2014
DOI:10.1039/C4CP00822G
The CASSCF and the hybrid CASSCF–MRMP2 methods are applied to the calculations of spin–spin and spin–orbit contributions to the zero-field splitting tensors (D tensors) of the halogen-substituted spin-septet 2,4,6-trinitrenopyridines, focusing on the heavy atom effects on the spin–orbit term of the D tensors (DSO tensors). The calculations reproduced experimentally determined |D| values within an error of 15%. Halogen substitutions at the 3,5-positions are less influential in the spin–spin dipolar (DSS) term of 2,4,6-trinitrenopyridines, although the DSO terms are strongly affected by the introduction of heavier halogens. The absolute sign of the DSO value (D = DZZ − (DXX + DYY)/2) of 3,5-dibromo derivative 3 is predicted to be negative, which contradicts the Pederson–Khanna (PK) DFT result previously reported. The large negative contributions to the DSO value of 3 arise from the excited spin-septet states ascribed mainly to the excitations of in-plane lone pair of bromine atoms → SOMO of π nature. The importance of the excited states involving electron transitions from the lone pair orbital of the halogen atom is also confirmed in the DSO tensors of halogen-substituted para-phenylnitrenes. A new scheme based on the orbital region partitioning is proposed for the analysis of the DSO tensors as calculated by means of the PK-DFT approach.
Co-reporter:Dr. Shinsuke Nishida;Yosuke Yamamoto;Dr. Takeji Takui;Dr. Yasushi Morita
ChemSusChem 2013 Volume 6( Issue 5) pp:794-797
Publication Date(Web):
DOI:10.1002/cssc.201300010
Co-reporter:Dr. Akira Ueda;Dr. Shuichi Suzuki;Kenta Yoshida;Dr. Kozo Fukui;Dr. Kazunobu Sato;Dr. Takeji Takui;Dr. Kazuhiro Nakasuji;Dr. Yasushi Morita
Angewandte Chemie 2013 Volume 125( Issue 18) pp:4895-4899
Publication Date(Web):
DOI:10.1002/ange.201301435
Co-reporter:Dr. Akira Ueda;Hideki Wasa;Dr. Shinsuke Nishida;Dr. Yuki Kanzaki;Dr. Kazunobu Sato;Dr. Takeji Takui;Dr. Yasushi Morita
Chemistry – An Asian Journal 2013 Volume 8( Issue 9) pp:2057-2063
Publication Date(Web):
DOI:10.1002/asia.201300471
Abstract
A triangulene-based C2-symmetric 33 π-conjugated stable neutral π-radical, 2., which possesses two dicyanomethylene groups and one oxo group, has been designed, synthesized, and isolated as an analogue of tris(dicyanomethylene) derivative 1. and trioxo derivative TOT. with C3 symmetry. Effects of molecular-symmetry reduction and electron-accepting substituents on this fused polycyclic neutral π-radical system were studied in terms of their molecular structure, electronic-spin structure, and electrochemical and optical properties with the help of theoretical calculations. Interestingly, this system (2.) has a four-stage redox ability, like TOT., as well as low frontier energy levels and a small SOMO–LUMO gap, similar to 1., in spite of the loss of the degenerate LUMOs in symmetry-lowered 2., which is associated with the attachment of the weaker electron-accepting oxo group instead of the dicyanomethylene group in 1.. These prominent results are attributable to the structural and electronic properties in the triangulene-based highly delocalized fused polycyclic neutral π-radical system.
Co-reporter:Dr. Akira Ueda;Dr. Shuichi Suzuki;Kenta Yoshida;Dr. Kozo Fukui;Dr. Kazunobu Sato;Dr. Takeji Takui;Dr. Kazuhiro Nakasuji;Dr. Yasushi Morita
Angewandte Chemie International Edition 2013 Volume 52( Issue 18) pp:4795-4799
Publication Date(Web):
DOI:10.1002/anie.201301435
Co-reporter:Dr. Shinsuke Nishida;Dr. Junya Kawai;Miki Moriguchi;Tomohiro Ohba;Naoki Haneda;Dr. Kozo Fukui; Akira Fuyuhiro; Daisuke Shiomi; Kazunobu Sato; Takeji Takui; Kazuhiro Nakasuji; Yasushi Morita
Chemistry - A European Journal 2013 Volume 19( Issue 36) pp:11904-11915
Publication Date(Web):
DOI:10.1002/chem.201301783
Abstract
The tri-tert-butylphenalenyl (TBPLY) radical exists as a π dimer in the crystal form with perfect overlapping of the singly occupied molecular orbitals (SOMOs) causing strong antiferromagnetic exchange interactions. 2,5-Di-tert-butyl-6-oxophenalenoxyl (6OPO) is a phenalenyl-based air-stable neutral π radical with extensive spin delocalization and is a counter analogue of phenalenyl in terms of the topological symmetry of the spin density distribution. X-ray crystal structure analyses showed that 8-tert-butyl- and 8-(p-XC6H4)-6OPOs (X=I, Br) also form π dimers in the crystalline state. The π-dimeric structure of 8-tert-butyl-6OPO is seemingly similar to that of TBPLY even though its SOMO–SOMO overlap is small compared with that of TBPLY. The 8-(p-XC6H4) derivatives form slipped stacking π dimers in which the SOMO–SOMO overlaps are greater than in 8-tert-butyl-6OPO, but still smaller than in TBPLY. The solid-state electronic spectra of the 6OPO derivatives show much weaker intradimer charge-transfer bands, and SQUID measurements for 8-(p-BrC6H4)-6OPO show a weak antiferromagnetic exchange interaction in the π dimer. These results demonstrate that the control of the spin distribution patterns of the phenalenyl skeleton switches the mode of exchange interaction within the phenalenyl-based π dimer. The formation of the relevant multicenter–two-electron bonds is discussed.
Co-reporter:Kazuki Ayabe, Kazunobu Sato, Shinsuke Nishida, Tomoaki Ise, Shigeaki Nakazawa, Kenji Sugisaki, Yasushi Morita, Kazuo Toyota, Daisuke Shiomi, Masahiro Kitagawa and Takeji Takui
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 25) pp:9137-9148
Publication Date(Web):23 Apr 2012
DOI:10.1039/C2CP40778G
Weakly exchange-coupled biradicals have attracted much attention in terms of their DNP application in NMR spectroscopy for biological systems or the use of synthetic electron-spin qubits. Pulse-ESR based electron spin nutation (ESN) spectroscopy applied to biradicals is generally treated as transition moment spectroscopy from the theoretical side, illustrating that it is a powerful and facile tool to determine relatively short distances between weakly exchange-coupled electron spins. The nutation frequency as a function of the microwave irradiation strength ω1 (angular frequency) for any cases of weakly exchange-coupled systems can be classified into three categories; D12 (spin dipolar interaction)-driven, Δg-driven and ω1-driven nutation behaviour with the increasing strength of ω1. For hetero-spin biradicals, Δg effects can be a dominating characteristic in the biradical nutation spectroscopy. Two-dimensional pulse-based electron spin nutation (2D-ESN) spectroscopy operating at the X-band can afford to determine small values of D12 in weakly exchange-coupled biradicals in rigid glasses. The analytical expressions derived here for ω1-dependent nutation frequencies are based on only four electronic spin states relevant to the biradicals, while real biradical systems often have sizable hyperfine interactions. Thus, we have evaluated nuclear hyperfine effects on the nutation frequencies to check the validity of the present theoretical treatment. The experimental spin dipolar coupling of a typical TEMPO-based biradical 1, (2,2,6,6-tetra[(2H3)methyl]-[3,3-2H2,4-2H1,5,5-2H2]piperidin-N-oxyl-4-yl)(2,2,6,6-tetra[(2H3)methyl]-[3,3-2H2,4-2H1,5,5-2H2,15N]piperidin-15N-oxyl-4-yl) terephthalate in a toluene glass, with a distance of 1.69 nm between the two spin sites is D12 = −32 MHz (the effect of the exchange coupling J12 is vanishing due to the homo-spin sites of 1, i.e. Δg = 0), while 0 < |J12| ≦ 1.0 MHz as determined by simulating the random-orientation CW ESR spectra of 1. In addition, we have carried out Q-band pulsed ELDOR (ELectron–electron DOuble Resonance) experiments to confirm whether the obtained values for D12 and J12 are accurate. The distance is in a fuzzy region for the distance-measurements capability of the conventional, powerful ELDOR spectroscopy. The strong and weak points of the ESN spectroscopy with a single microwave frequency applicable to weakly exchange-coupled multi-electron systems are discussed in comparison with conventional ELDOR spectroscopy. The theoretical spin dipolar tensor and exchange interaction of the TEMPO biradical, as obtained by sophisticated quantum chemical calculations, agree with the experimental ones.
Co-reporter:Dr. Akira Ueda;Hideki Wasa;Dr. Shuichi Suzuki;Dr. Keiji Okada;Dr. Kazunobu Sato;Dr. Takeji Takui;Dr. Yasushi Morita
Angewandte Chemie 2012 Volume 124( Issue 27) pp:6795-6799
Publication Date(Web):
DOI:10.1002/ange.201202654
Co-reporter:Yuki Kanzaki, Daisuke Shiomi, Kazunobu Sato, and Takeji Takui
The Journal of Physical Chemistry B 2012 Volume 116(Issue 3) pp:1053-1059
Publication Date(Web):December 20, 2011
DOI:10.1021/jp211391x
An ESR hyperfine splitting pattern of a biradical in solution depends on the magnitude of the intramolecular exchange interaction Jintra compared with the hyperfine coupling constant A. Some biradicals exhibit their hyperfine splitting patterns characteristic of a monoradical, even though their exchange interaction is strong enough, |Jintra| ≫ |A|. The contradiction in ESR spectroscopy is known as “biradical paradox”, puzzling scientists for a long time. In this study, it is shown from ESR spectral simulations underlain by a theoretical model of a series of spin Hamiltonians that noncovalent aggregation of biradical molecules in solution leads to the appearance of paradoxical ESR spectra. Most of the spins in an aggregate of one dimension lose their contribution to the ESR spectra owing to intermolecular antiferromagnetic interactions Jinter, leaving two outermost spins ESR-active in the aggregate of one dimension. Paradoxical ESR spectra appear only when Jintra and Jinter fall within a particular range of the magnitudes which depends on the number of molecules in the aggregate.
Co-reporter:Dr. Akira Ueda;Hideki Wasa;Dr. Shinsuke Nishida;Dr. Yuki Kanzaki;Dr. Kazunobu Sato;Dr. Daisuke Shiomi;Dr. Takeji Takui;Dr. Yasushi Morita
Chemistry - A European Journal 2012 Volume 18( Issue 51) pp:16272-16276
Publication Date(Web):
DOI:10.1002/chem.201203755
Co-reporter:Dr. Shigeaki Nakazawa;Dr. Shinsuke Nishida;Dr. Tomoaki Ise;Dr. Tomohiro Yoshino;Dr. Nobuyuki Mori;Dr. Robabeh D. Rahimi;Dr. Kazunobu Sato;Dr. Yasushi Morita;Dr. Kazuo Toyota;Dr. Daisuke Shiomi;Dr. Masahiro Kitagawa;Dr. Hideyuki Hara;Dr. Patrick Carl;Dr. Peter Höfer;Dr. Takeji Takui
Angewandte Chemie International Edition 2012 Volume 51( Issue 39) pp:9860-9864
Publication Date(Web):
DOI:10.1002/anie.201204489
Co-reporter:Dr. Akira Ueda;Hideki Wasa;Dr. Shuichi Suzuki;Dr. Keiji Okada;Dr. Kazunobu Sato;Dr. Takeji Takui;Dr. Yasushi Morita
Angewandte Chemie International Edition 2012 Volume 51( Issue 27) pp:6691-6695
Publication Date(Web):
DOI:10.1002/anie.201202654
Co-reporter:Kenji Sugisaki, Kazuo Toyota, Kazunobu Sato, Daisuke Shiomi, Masahiro Kitagawa and Takeji Takui
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 15) pp:6970-6980
Publication Date(Web):10 Mar 2011
DOI:10.1039/C0CP02809F
Spin–orbit and spin–spin contributions to the zero-field splitting (ZFS) tensors (D tensors) of spin-triplet phenyl-, naphthyl-, and anthryl-nitrenes in their ground state are investigated by quantum chemical calculations, focusing on the effects of the ring size and substituted position of nitrene on the D tensor. A hybrid CASSCF/MRMP2 approach to the spin–orbit term of the D tensor (DSO tensor), which was recently proposed by us, has shown that the spin–orbit contribution to the entire D value, termed the ZFS parameter or fine-structure constant, is about 10% in all the arylnitrenes under study and less depends on the size and connectivity of the aryl groups. Order of the absolute values for DSO can be explained by the perturbation on the energy level and spatial distributions of π-SOMO through the orbital interaction between SOMO of the nitrene moiety and frontier orbitals of the aryl scaffolds. Spin–spin contribution to the D tensor (DSS tensor) has been calculated in terms of the McWeeny–Mizuno equation with the DFT/EPR-II spin densities. The DSS value calculated with the RO-B3LYP spin density agrees well with the D(Exptl) − DSO reference value in phenylnitrene, but agreement with the reference value gradually becomes worse as the D value decreases. Exchange–correlation functional dependence on the DSS tensor has been explored with standard 23 exchange–correlation functionals in both RO- and U-DFT methodologies, and the RO-HCTH/407 method gives the best agreement with the D(Exptl) − DSO reference value. Significant exchange–correlation functional dependence is observed in spin-delocalized systems such as 9-anthrylnitrene (6). By employing the hybrid CASSCF/MRMP2 approach and the McWeeny–Mizuno equation combined with the RO-HCTH/407/EPR-II//U-HCTH/407/6-31G* spin densities for DSO and DSS, respectively, a quantitative agreement with the experiment is achieved with errors less than 10% in all the arylnitrenes under study. Guidelines to the putative approaches to DSS tensor calculations are given.
Co-reporter:Shigeaki Nakazawa, Kazunobu Sato, Daisuke Shiomi, Masafumi Yano, Takamasa Kinoshita, Maria Luisa T. M. B. Franco, Maria Celina R. L. R. Lazana, Maria Candida B. L. Shohoji, The late Koichi Itoh and Takeji Takui
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 4) pp:1424-1433
Publication Date(Web):22 Nov 2010
DOI:10.1039/C0CP00730G
Trianionic spin-quartet and tetraanionic spin-quintet molecular clusters derived from m-dibenzoylbenzene in solution were identified by CW-ESR/pulse-ESR based two-dimensional electron spin transient nutation spectroscopy, and their spin and clustering structures in the ground state were determined in terms of a D-tensor based phenomenological approach and DFT calculations. The molecular structures obtained semiempirically are supported by DFT-based quantum chemical calculations. The DFT calculations have been tested for a sodium ion bridged fluorenone-based cluster, [fluorenone−˙ {Na+(dme)2}]2, whose crystal structure was reported in the literature [H. Bock, H.-F. Herrmann, D. Fenske and H. Goesmann, Angew. Chem., Int. Ed. Engl., 1988, 27, 1067], reproducing the experimentally determined moelcular structure of the dimer cluster. It is suggested that both the quartet and quintet clusters in the 2-MTHF glass and solution form the cross-typed structures with the two m-dibenzoylbenzene moieties in cis-configuration. A dianionic spin-triplet m-dibenzoylbenzene derivative was detected for the first time and its charge and spin densities were studied by the quantum chemical calculations. The high-spin states of the open-shell entities under study were confirmed by X-band pulse-ESR based electron spin nutation spectroscopy in organic frozen glasses. The D values and other spin Hamiltonian parameters of all the polyanionic high-spin species were determined by the hybrid eigenfield spectral simulation for fine-structure ESR spectra. m-Dibenzoylbenzene provides pseudo-degenerate π-LUMOs arising from its topological symmetry of the π-electron network and its dianion in the triplet ground state is a prototypical model for topologically-controlled genuinely organic ferromagnetic metals.
Co-reporter:Yumi Yakiyama;Tsuyoshi Murata;Tomoaki Ise;Daisuke Shiomi;Kazunobu Sato;Kazuhiro Nakasuji;Yasushi Morita
European Journal of Inorganic Chemistry 2011 Volume 2011( Issue 23) pp:3438-3445
Publication Date(Web):
DOI:10.1002/ejic.201100488
Abstract
Complex formation of quaterimidazole with FeII, CoII, and NiII yielded dinuclear triple helicates in the solution state by the self-assembling ability of the ligand based on the strong chelating coordination bonds. Crystallization of the CoII complex from aqueous solution afforded the CoIII complex with a triple-helical structure. X-ray crystal structure analyses revealed that the helicates are linked through N–H···X hydrogen bonds with counteranions and solvent molecules to form three-dimensional networks. The spectrophotometric titration experiment showed that quaterimidazole forms triple helicates with various d-block transition metal ions with high selectivity. The outward intermolecular interactions and high stability of the helicates from the rigid molecular structures combined with the intramolecular π–π interactions of imidazole moieties and their strong chelating coordination enabled the successful optical resolution of right- and left-handed helicates by chiral-column HPLC.
Co-reporter:Dr. Shinsuke Nishida ;Kazuki Kariyazono;Azusa Yamanaka;Dr. Kozo Fukui; Kazunobu Sato; Takeji Takui; Kazuhiro Nakasuji; Yasushi Morita
Chemistry – An Asian Journal 2011 Volume 6( Issue 5) pp:1188-1196
Publication Date(Web):
DOI:10.1002/asia.201000793
Abstract
A new 2,5-di-tert-butyl-6-oxophenalenoxyl (6OPO) derivative with a cyano group at the 8-position, where a large spin density resides, has been synthesized. This neutral radical exhibits high stability in the solid state in air despite the low steric protection on the 8-position; the stability is comparable to that of a corresponding 8-tert-butylated 6OPO derivative. EPR/1H-ENDOR/TRIPLE (electron paramagnetic resonance/1H-electron-nuclear double resonance/TRIPLE) spectroscopy and cyclic voltammetry showed an extended spin delocalization on the cyano group and a significant increase in electron-accepting ability relative to that of the 8-tert-butylated 6OPO derivative. DFT calculations indicated the extension of a singly occupied molecular orbital (SOMO) onto the cyano group and the lower-lying SOMO and LUMO in comparison with those of the 8-tert-butylated 6OPO derivative, which was consistent with experimental results. Furthermore, the extended nature of π conjugation onto the cyano group was quantitatively evaluated by calculating the contributing weights of resonance structures in terms of a molecular orbital (MO)-based valence-bond (VB) method. Herein, the synthesis and physical properties of the 8-cyano-6OPO derivative are described, emphasizing that the high stability arises from the electronic effect of the cyano group. Also, the usefulness of the quantitative resonance structure analysis is shown.
Co-reporter:Tomohiro Yoshino, Shinsuke Nishida, Kazunobu Sato, Shigeaki Nakazawa, Robabeh D. Rahimi, Kazuo Toyota, Daisuke Shiomi, Yasushi Morita, Masahiro Kitagawa, and Takeji Takui
The Journal of Physical Chemistry Letters 2011 Volume 2(Issue 5) pp:449-453
Publication Date(Web):February 11, 2011
DOI:10.1021/jz101650z
Electron and nuclear spins as quantum bits (qubits) have been the focus of current issues in quantum information science/technology and related fields. From the viewpoint of chemistry, synthetic spin qubits are emerging. Diphenylnitroxide (DPNO) and its novel fluorine-substituted radicals are characterized as synthetic electron bus spin-qubits by continuous-wave ESR and 1H-,19F-ENDOR/TRIPLE spectroscopy in solution and by DFT calculations. The partially fluorinated DPNOs have been synthesized to illustrate that they are candidates for the synthetic bus spin-qubits with well-defined client qubits. The fluorinated DPNOs undergo spin delocalization, dominating the robust spin polarization in the π-conjugation of phenyl rings, serving to increase the number of distinguishable client qubits from three to six.Keywords: client qubit; fluorinated diphenylnitroxide; solution ENDOR; spin delocalization; spin polarization; synthetic bus spin-qubit radical;
Co-reporter:Daisuke Shiomi, Yuki Kanzaki, Sho Okada, Ryosuke Arima, Yuji Miyazaki, Akira Inaba, Rika Tanaka, Kazunobu Sato, and Takeji Takui
The Journal of Physical Chemistry Letters 2011 Volume 2(Issue 24) pp:3036-3039
Publication Date(Web):November 15, 2011
DOI:10.1021/jz2014105
We report the ferromagnetic ordering phenomena occurring in organic molecular crystals with structural chirality. Achiral radical 1 has been found to crystallize in two enantiomorphs with chiral space groups of P43 and P41. The P43 form (1L) has left-handed stacking of the molecules, giving the helical chirality in a crystalline solid. In the other form of P41 (1R), the right-handed stacking corresponds to a mirror image of 1L. Magnetic susceptibility measurements show that both the crystals undergo a ferromagnetic phase transition at TC = 1.1 K. The ferromagnetic ordering has been confirmed by heat capacity measurements. The magnetic heat capacity exhibits a λ-shaped peak at TC = 1.1 K with an entropy change of R ln 2, as expected for S = 1/2 spins. This is the first example of genuinely organic molecule-based ferromagnetism associated with the structural chirality based on the helical molecular packing in the crystalline solid.Keywords: chirality; ferromagnet; heat capacity; helical structure; magnetic susceptibility; nitronyl nitroxide; organic radical;
Co-reporter:Yasushi Morita ; Yumi Yakiyama ; Shigeaki Nakazawa ; Tsuyoshi Murata ; Tomoaki Ise ; Daisuke Hashizume ; Daisuke Shiomi ; Kazunobu Sato ; Masahiro Kitagawa ; Kazuhiro Nakasuji
Journal of the American Chemical Society 2010 Volume 132(Issue 20) pp:6944-6946
Publication Date(Web):May 3, 2010
DOI:10.1021/ja102030w
We have first achieved the synthesis of triple-stranded metallo-helicates composed of 4,4′:2′,2′′:4′′,4′′′-quaterimidazole (Qim) and Mn(II) or Zn(II) ions, which serve as synthetic electron spin qubits (quantum bits). In the crystal structure, a hydrogen-bonding network through counteranions and/or crystal solvents was constructed by the outward N−H hydrogen-bonding functional groups intrinsic to the imidazole skeleton. Importantly, these helicates showed high stability even in a solution state at room temperature. These salient features of triple helicates of Qim are different from those of reported metallo-helicates. These chemical properties of the Qim-based triple helicates allow us to synthesize magnetically diluted single crystals composed of Mn(II) (S = 5/2) and diamagnetic Zn(II) complexes of Qim in an appropriate Mn(II)/Zn(II) ratio. The magnetically diluted crystals can afford to build up the prototype of electron-spin qubits of Lloyd’s one-dimensional periodic system, which gives a practical approach to scalable quantum computers/quantum information processing systems (QCs/QIPSs). The experiments have proven the practical capability of oligo(imidazole)s as a component of Lloyd’s system which has nonequivalent g-tensors within the helicate (g-engineering). The helical symmetry plays an important role in giving a prototype of the synthetic spin qubits of the formidable Lloyd model. This result links supramolecular chemistry to the field of QCs/QIPSs.
Co-reporter:Hiroyuki Tanaka, Daisuke Shiomi, Shuichi Suzuki, Masatoshi Kozaki, Keiji Okada, Kazunobu Sato and Takeji Takui
CrystEngComm 2010 vol. 12(Issue 2) pp:526-531
Publication Date(Web):11 Sep 2009
DOI:10.1039/B909757K
As a novel radical building block possessing multiple hydrogen bond functionality, we have designed and synthesised diaminotriazine-substituted nitronyl nitroxide (DAT-NN; 1). The diaminotriazine moiety of 1 forms two kinds of hydrogen bonding and molecule 1 forms an S = 1/2 uniform antiferromagnetic Heisenberg chain with the exchange interaction of J/kB = −15.3 K in the crystalline solid, which is attributable to close contacts between the nitronyl nitroxide oxygen atoms. 1H-ENDOR spectroscopy has shown that the triazine moiety has little significant spin density. Thus, the diaminotriazine moiety plays a role primarily in determining the molecular packing instead of propagating intermolecular magnetic interactions in the crystalline solid.
Co-reporter:Akira Ueda Dr.;Kanako Ogasawara;Shinsuke Nishida Dr.;Tomoaki Ise Dr.;Tomohiro Yoshino;Shigeaki Nakazawa Dr.;Kazunobu Sato Dr. Dr.;Kazuhiro Nakasuji Dr.;Yasushi Morita Dr.
Angewandte Chemie 2010 Volume 122( Issue 36) pp:6477-6481
Publication Date(Web):
DOI:10.1002/ange.201002626
Co-reporter:Akira Ueda;Shinsuke Nishida Dr.;Kozo Fukui Dr.;Tomoaki Ise Dr.;Daisuke Shiomi Dr.;Kazunobu Sato Dr. Dr.;Kazuhiro Nakasuji Dr.;Yasushi Morita Dr.
Angewandte Chemie 2010 Volume 122( Issue 9) pp:1722-1726
Publication Date(Web):
DOI:10.1002/ange.200906666
Co-reporter:Dr. Kenji Sugisaki;Dr. Kazuo Toyota; Dr. Kazunobu Sato; Dr. Daisuke Shiomi; Dr. Masahiro Kitagawa; Dr. Takeji Takui
ChemPhysChem 2010 Volume 11( Issue 14) pp:3146-3151
Publication Date(Web):
DOI:10.1002/cphc.201000492
Abstract
Zero-field splitting (ZFS) tensors (D tensors) of organic high-spin oligonitrenes/oligocarbenes up to spin-septet are quantitatively determined on the basis of quantum chemical calculations. The spin–orbit contributions, DSO tensors are calculated in terms of a hybrid CASSCF/MRMP2 approach, which was recently proposed by us. The spin–spin counterparts, DSS tensors are computed based on McWeeny–Mizuno’s equation in conjunction with the RODFT spin densities. The present calculations show that more than 10 % of ZFS arises from spin–orbit interactions in the high-spin nitrenes under study. Contributions of spin-bearing site–site interactions are estimated with the aid of a semi-empirical model for the D tensors and found to be ca. 5 % of the DSO tensor. The analysis of intermediate states reveal that the largest contributions to the calculated DSO tensors are attributed to intra-site spin flip excitations and delocalized π and π* orbitals play an important role in the inter-site spin–orbit interactions.
Co-reporter:Akira Ueda;Shinsuke Nishida Dr.;Kozo Fukui Dr.;Tomoaki Ise Dr.;Daisuke Shiomi Dr.;Kazunobu Sato Dr. Dr.;Kazuhiro Nakasuji Dr.;Yasushi Morita Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 9) pp:1678-1682
Publication Date(Web):
DOI:10.1002/anie.200906666
Co-reporter:Akira Ueda Dr.;Kanako Ogasawara;Shinsuke Nishida Dr.;Tomoaki Ise Dr.;Tomohiro Yoshino;Shigeaki Nakazawa Dr.;Kazunobu Sato Dr. Dr.;Kazuhiro Nakasuji Dr.;Yasushi Morita Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 36) pp:6333-6337
Publication Date(Web):
DOI:10.1002/anie.201002626
Co-reporter:Kenji Sugisaki, Kazuo Toyota, Kazunobu Sato, Daisuke Shiomi, Masahiro Kitagawa, Takeji Takui
Chemical Physics Letters 2009 Volume 477(4–6) pp:369-373
Publication Date(Web):6 August 2009
DOI:10.1016/j.cplett.2009.07.007
Abstract
The spin–orbit contributions (DSO tensors) to the zero-field splitting tensors (D tensors) in the excited triplet states of CS2 and diazabenzenes are investigated by using the sum-over-states formula with the CASSCF spin–orbit coupling matrices and the CASSCF or MRMP2 energies. The DSO tensor calculated using the CASSCF energies is in some cases far from the experiment due to inaccurate energy differences, but the discrepancy can be circumvented by replacing the CASSCF energies with the MRMP2 ones. Effects of the geometry relaxation in the excited states on the D tensors are also discussed.
Co-reporter:Teruaki Koto, Kazunobu Sato, Daisuke Shiomi, Kazuo Toyota, Koichi Itoh, Edel Wasserman and Takeji Takui
The Journal of Physical Chemistry A 2009 Volume 113(Issue 34) pp:9521-9526
Publication Date(Web):August 5, 2009
DOI:10.1021/jp9042717
In high-spin chemistry, random-orientation fine-structure (FS) electron spin resonance (ESR) spectroscopy entertains advantages as the most facile and convenient method to identify high-spin systems, as frequently reported in the literature. Random-orientation ESR spectroscopy applicable to organic high-spin entities can date back to the Wasserman and co-workers’ attempt on the first spin-quintet dicarbene, m-phenylenebis(phenylmethlene) (m-PBPM), in the 2-MTHF glass in 1963 and 1967, following their successful work on randomly oriented triplet-state ESR spectroscopy. The FS ESR spectrum of m-PBPM in the 2-MTHF glass, however, has never fully been analyzed due to a peculiar line-broadening appearing at many canonical peaks. Organic high-spin spectra from most quintet dinitrenes also suffer from similar phenomena. Seemingly intrinsic line-diffusing or -broadening phenomena adversely affect the reliable determination of FS parameters for organic high-spin entities in rigid glasses. In high-spin chemistry, the line-broadening has been an obstacle that masks key FS transitions in many cases. Thus, both the origin of the broadening and the comprehensive spectral analysis have been a long-standing issue. In this report, we examine the origin of the line-broadening appearing in the FS ESR spectra, illustrated by a comprehensive spectral analysis for m-PBPM in the quintet ground state and the first-documented quintet-state dinitrene, m-phenylenebis(nitrene) (m-PBN) in the 2-MTHF glass. A complete analysis of the random-orientation FS spectra from m-PBPM diluted in the benzophenone crystal has shown that the g-anisotropy of m-PBPM is not prominent. Also the higher-order FS terms such as Si2Sj2 group-theoretically allowed for S = 2 are not necessary in spite of the argument for a hydrocarbon-based tetraradical (S = 2) in the ground state. Our new approach to the line-broadening analysis invokes both exact analytical solutions for the resonance fields of canonical peaks and the magnetic-parameters gradient method. The D- and E-values of m-PBPM acquired by the spectral simulation in this study give different molecular structures of the quintet dicarbene in the benzophenone crystal lattice (D = +0.07030 cm−1, E = +0.02120 cm−1) and in the 2-MTHF glass (D = +0.07800 cm−1, E = +0.02210 cm−1). Microscopic origins of the line-broadening observed for high-spin oligocarbenes or oligonitrenes generated by photolysis in organic glasses have been proposed.
Co-reporter:Shigeaki Nakazawa, Kazunobu Sato, Daisuke Shiomi, M. Luisa T.M.B. Franco, M. Celina R.L.R. Lazana, M. Candida B.L. Shohoji, Koichi Itoh, Takeji Takui
Inorganica Chimica Acta 2008 Volume 361(14–15) pp:4031-4037
Publication Date(Web):1 October 2008
DOI:10.1016/j.ica.2008.03.048
C60-based polyanionic high-spin clusters (S = 1–3) in their ground state have been prepared by successive chemical reductions of pristine C60 fullerene with potassium in the presence of dicyclohexano-18-crown-6-ether in solution. Intermolecular spin-triplet, quintet, and septet states arising from the C60-based polyanionic molecular clusters have been generated at ambient temperature and identified by CW-ESR and pulse-ESR-based two-dimensional (2D) electron spin transient nutation (2D-ESTN) spectroscopy in organic rigid glasses, for the first time. Intermolecular exchange interactions between mono- and polyanionic C60 fullerenes are ferromagnetic via bridging potassium metal cations. The molecular structures of the polyanionic high-spin C60 clusters in solution have been proposed by a well-established phenomenological spin Hamiltonian approach in terms of the D-tensor-based calculations for the high-spin clusters. The findings of the C60-based high-spin molecular clusters evidence the occurrence of an intramolecular triplet C60 dianion in the ground state. Unequivocal spin identification for molecular high-spin entities by 2D-ESTN spectroscopy and its powerfulness have been illustrated, emphasizing that the 2D-ESTN spectroscopy is useful for mixtures of molecular species with different spin multiplicities, characterized by a small g-anisotropy, for which the powerfulness of advanced high-frequency ESR spectroscopy is hampered.C60-based polyanionic high-spin clusters (S = 1–3) in their ground state have been prepared by chemical reductions of pristine C60 fullerene in 2-methyltetrahydrofuran, being identified by CW-ESR and pulse-ESR-based two-dimensional electron spin transient nutation spectroscopy in the glass. The molecular structures of the clusters have been determined by a well-established semi-empirical D-tensor approach.
Co-reporter:Takatoshi Sawai;Kazunobu Sato Dr.;Tomoaki Ise Dr.;Daisuke Shiomi Dr.;Kazuo Toyota Dr.;Yasushi Morita Dr. Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 21) pp:3988-3990
Publication Date(Web):
DOI:10.1002/anie.200705583
Co-reporter:Yasushi Morita Dr.;Akira Ueda;Shinsuke Nishida Dr.;Kozo Fukui Dr.;Tomoaki Ise Dr.;Daisuke Shiomi Dr.;Kazunobu Sato Dr. Dr.;Kazuhiro Nakasuji Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 11) pp:2035-2038
Publication Date(Web):
DOI:10.1002/anie.200704752
Co-reporter:Yuki Kanzaki, Daisuke Shiomi, Chika Kaneda, Tomoaki Ise, Kazunobu Sato and Takeji Takui
Journal of Materials Chemistry A 2006 vol. 16(Issue 21) pp:2064-2073
Publication Date(Web):16 Mar 2006
DOI:10.1039/B518416A
We propose a strategy of crystal-engineering for heteromolecular crystals composed of organic open-shell molecules, in which two kinds of open-shell molecular entity are co-crystallized and, at the same time, distinct molecular entities with differing spin multiplicities are adjacent to each other in a crystalline solid state. Stable organic triradicals of nitronylnitroxide and iminonitroxide based on the benzene-1,3,5-triyl framework, trisNN (3) and trisIN (4), have been designed and synthesized. These molecules consist of a m-phenylene-substituted biradical and a monoradical moiety united by an ester bridge. From X-ray crystallography, these molecules have been found to possess an approximately isosceles triangular geometry and fully head-to-tail molecular packing in three dimensions has been achieved in the crystalline solid state. Thus, the co-crystallization is attained by binding the two π-conjugated radicals by σ-bonds, retaining the magnetic degree of freedom for the two distinct open-shell moieties. The alternating aggregation of the heteromolecular assembly is achieved by tuning the molecular geometry of the building-block molecules, leading to the close packing of isosceles triangular molecules in the head-to-tail fashion. Local contacts of the nitroxide groups with large spin densities between the nearest-neighbor molecules give a six-spin molecular cluster on a tetrad of the bi-, mono-, mono-, and biradical moieties in the head-to-tail stacking. Temperature dependence of magnetic susceptibility of the crystalline solids of 3 and 4 has been analyzed on the basis of their crystal structures. The six-spin cluster model has reasonably explained the susceptibility.
Co-reporter:Kenichi Hayakawa, Daisuke Shiomi, Tomoaki Ise, Kazunobu Sato and Takeji Takui
Journal of Materials Chemistry A 2006 vol. 16(Issue 42) pp:4146-4154
Publication Date(Web):04 Sep 2006
DOI:10.1039/B609760J
A 3,5-pyridine-substituted biradical of nitronyl nitroxide (1) has been designed, synthesized and the magnetic properties fully characterized. The ground-state spin multiplicity of 1 has been found to be triplet (S = 1) with a singlet–triplet energy gap of 2J/kB = 40 K from magnetic susceptibility measurements on a magnetically diluted system dispersed in organic polymer films. The 3,5-substituted pyridine 1 has a hydrogen-accepting site which is more accessible to hydrogen donors than previously known biradicals with sterically hindered 2,6-pyridine frameworks. N-Methylation of 1 has yielded a stable cationic species in a trifluoromethanesulfonate salt (2+·TfO−). The ground state of the cation 2+ has been found to be triplet as well with 2J/kB = 32 K from magnetic susceptibility measurements for magnetically diluted films. The magnetic susceptibility of neat crystalline solids of 1 and 2+·TfO− has been explained by Heisenberg exchange coupling models based on their X-ray crystal structures. It is well known that the energy preference of a high-spin ground state for m-phenylene, or m-xylylene, coupling units is disturbed in such cases as the π-conjugation is affected by heteroatomic substitution, an ionic charge, or molecular conformation. The present experimental results show that the high-spin preference in 1 and 2+ is little influenced by the heterocycle or the ionic charge. Intermolecular noncovalent bonds such as hydrogen bonding and electrostatic interactions are a driving force for crystallization of open-shell molecules in a controllable manner. The ground-state triplet biradicals serve as building blocks for molecule-based magnets of S > 1/2 based on intermolecular noncovalent bonding architecture.
Co-reporter:Tomoaki Ise, Daisuke Shiomi, Kazunobu Sato and Takeji Takui
Chemical Communications 2006 (Issue 46) pp:4832-4834
Publication Date(Web):03 Oct 2006
DOI:10.1039/B611236F
A Watson–Crick type molecular complex of adenine and thymine bases substituted with the stable radical of nitronylnitroxide has been synthesized, which forms a double-chain spin system in the crystal.
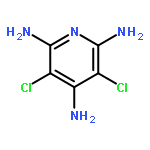
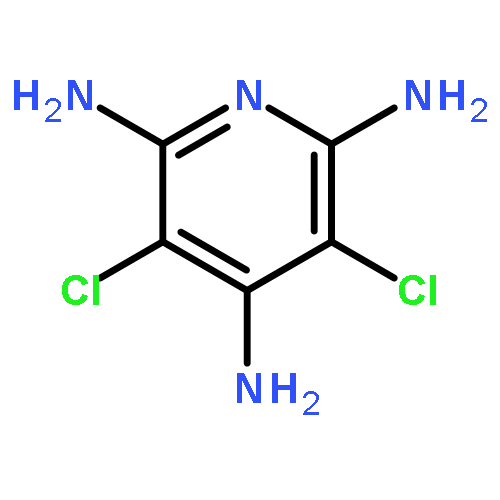
Co-reporter:Kenji Sugisaki;Kazuo Toyota;Kazunobu Sato;Daisuke Shiomi
Angewandte Chemie 2006 Volume 118(Issue 14) pp:
Publication Date(Web):6 MAR 2006
DOI:10.1002/ange.200502695
Das Konzept von Orbitalkorrelationen zwischen dem Benzolgerüst und den Nitreneinheiten von 2-Methylphenylen-1,3-dinitren wurde genutzt, um die Energieniveaus und Merkmale der angeregten elektronischen Zustände dieses typischen High-Spin-Quintettmoleküls mithilfe anspruchsvoller Ab-initio-MO-Rechnungen zu interpretieren. So konnte erstmals das UV/Vis-Absorptionsspektrum dieses Dinitrens theoretisch analysiert werden (siehe Balkenspektrum im Bild).
Co-reporter:Ryoko Santo;Riichi Miyamoto;Rika Tanaka Dr.;Takanori Nishioka Dr.;Kazunobu Sato Dr.;Kazuo Toyota Dr.;Makoto Obata Dr.;Shigenobu Yano Dr.;Isamu Kinoshita Dr.;Akio Ichimura Dr. Dr.
Angewandte Chemie International Edition 2006 Volume 45(Issue 45) pp:
Publication Date(Web):20 OCT 2006
DOI:10.1002/anie.200603127
All change: The stable, diamagnetic, trigonal-bipyramidal CuIII complex (see picture) arising from one-electron oxidation of a CuII complex undergoes conversion into a paramagnetic octahedral CuIII complex upon addition of Cl− ions. Characterization of the complexes and density functional calculations indicate that the initial one-electron oxidation is metal- rather than ligand-centered.
Co-reporter:Kenji Sugisaki, Kazuo Toyota, Kazunobu Sato, Daisuke Shiomi,Takeji Takui
Angewandte Chemie International Edition 2006 45(14) pp:2257-2260
Publication Date(Web):
DOI:10.1002/anie.200502695
Co-reporter:Ryoko Santo;Riichi Miyamoto;Rika Tanaka Dr.;Takanori Nishioka Dr.;Kazunobu Sato Dr.;Kazuo Toyota Dr.;Makoto Obata Dr.;Shigenobu Yano Dr.;Isamu Kinoshita Dr.;Akio Ichimura Dr. Dr.
Angewandte Chemie 2006 Volume 118(Issue 45) pp:
Publication Date(Web):20 OCT 2006
DOI:10.1002/ange.200603127
Alles ändert sich: Der im Bild gezeigte stabile, diamagnetische, trigonal-bipyramidale CuIII-Komplex, der bei der Einelektronenoxidation eines CuII-Komplexes erhalten wird, lagert sich bei der Zugabe von Cl−-Ionen in einen paramagnetischen, oktaedrischen CuIII-Komplex um. Die Charakterisierung der Komplexe und Dichtefunktionalrechnungen sprechen dafür, dass die urspüngliche Einelektronenoxidation metall- und nicht ligandenzentriert ist.
Co-reporter:Shinsuke Nishida Dr.;Yasushi Morita Dr.;Kozo Fukui Dr.;Kazunobu Sato Dr.;Daisuke Shiomi Dr. Dr.;Kazuhiro Nakasuji Dr.
Angewandte Chemie 2005 Volume 117(Issue 44) pp:
Publication Date(Web):13 OCT 2005
DOI:10.1002/ange.200502180
Die richtige Lösung für den Spin: Spintransfer und Solvatochromie/Thermochromie zeigt ein rein organisches offenschaliges System, in dem ein neutrales 6-Oxophenalenoxyl-Radikal als Elektronenacceptor direkt mit einer Tetrathiafulvalen-Einheit als Elektronendonor verbunden ist. Ein intramolekularer Elektronentransfer (IET, siehe Schema) bedingt die Umwandlung, die auf geringfügige Änderungen des Lösungsmittels und der Temperatur anspricht.
Co-reporter:Shinsuke Nishida, Yasushi Morita, Kozo Fukui, Kazunobu Sato, Daisuke Shiomi, Takeji Takui,Kazuhiro Nakasuji
Angewandte Chemie International Edition 2005 44(44) pp:7277-7280
Publication Date(Web):
DOI:10.1002/anie.200502180
Co-reporter:Yasushi Morita Dr.;Takashi Aoki;Kozo Fukui Dr.;Shigeaki Nakazawa Dr.;Koichi Tamaki Dr.;Shuichi Suzuki;Akira Fuyuhiro Dr.;Kagetoshi Yamamoto Dr.;Kazunobu Sato Dr.;Daisuke Shiomi Dr.;Akira Naito Dr. Dr.;Kazuhiro Nakasuji Dr.
Angewandte Chemie International Edition 2002 Volume 41(Issue 10) pp:
Publication Date(Web):15 MAY 2002
DOI:10.1002/1521-3773(20020517)41:10<1793::AID-ANIE1793>3.0.CO;2-G
Stable gable radical: 1,3-Diazaphenalenyl radical, a typical example of an isoelectronic mode of modification for phenalenyl, has been isolated for the first time as a crystalline solid by introducing bulky substituents (see picture, tert-butyl groups are omitted from the crystal structure). The gable syn dimer with a column motif shows an extremely strong antiferromagnetic exchange coupling of 2J/kB=−4.19(2)×103 K.
Co-reporter:Yasushi Morita Dr.;Takashi Aoki;Kozo Fukui Dr.;Shigeaki Nakazawa Dr.;Koichi Tamaki Dr.;Shuichi Suzuki;Akira Fuyuhiro Dr.;Kagetoshi Yamamoto Dr.;Kazunobu Sato Dr.;Daisuke Shiomi Dr.;Akira Naito Dr. Dr.;Kazuhiro Nakasuji Dr.
Angewandte Chemie 2002 Volume 114(Issue 10) pp:
Publication Date(Web):15 MAY 2002
DOI:10.1002/1521-3757(20020517)114:10<1871::AID-ANGE1871>3.0.CO;2-S
Stabiles Radikal: 1,3-Diazaphenalenyl, ein zum Phenalenylradikal isoelekronisches Radikal, wurde nach der Einführung sperriger Substituenten erstmals in kristalliner Form erhalten (siehe Bild, tert-Butylgruppen sind rechts weggelassen). Im dargestellten „giebelförmigen“ syn-Dimer tritt eine extrem starke, antiferromagnetische Kopplung von 2J/kB=−4.19(2)×103 K auf.
Co-reporter:Syuichiro Hase, Daisuke Shiomi, Kazunobu Sato and Takeji Takui
Journal of Materials Chemistry A 2001 vol. 11(Issue 3) pp:756-760
Publication Date(Web):06 Feb 2001
DOI:10.1039/B007316O
The crystal structures of two kinds of nitronyl nitroxide biradical
with a phenolic substituent, 2,4-bis(1-oxyl-3-oxido-4,4,5,5-tetramethyl-2-imidazolin-2-yl)phenol (1a) and 4-methyl-2,6-bis(1-oxyl-3-oxido-4,4,5,5-tetramethyl-2-imidazolin-2-yl)phenol (2a) have been solved. The biradical 1a belongs to the monoclinic system with space
group P21/n, a = 11.806(4) Å, b = 25.330(5) Å, c = 7.337(7) Å, β = 104.65(4)°,
and Z = 4, while the biradical 2a to the orthorhombic system with Pbca, a = 20.206(4) Å, b = 40.405(4) Å, c = 11.888(3) Å,
and Z = 8. From the magnetic susceptibility
in the crystalline solid state, both 1a
and 2a are found to have a triplet (S = 1)
ground state with the intramolecular ferromagnetic interaction of 2J/kB = 26.0 ± 0.5 K
for 1a and 12 ± 1 K
for 2a. The triplet ground states
are confirmed by the EPR measurements on the isolated molecules in diluted
glassy solutions. The singlet–triplet energy gaps of the biradicals
with a phenolic hydroxy substituent are found to be reduced as compared with
a parent non-substituted biradical.
Co-reporter:Kazuki Ayabe, Kazunobu Sato, Shinsuke Nishida, Tomoaki Ise, Shigeaki Nakazawa, Kenji Sugisaki, Yasushi Morita, Kazuo Toyota, Daisuke Shiomi, Masahiro Kitagawa and Takeji Takui
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 25) pp:NaN9148-9148
Publication Date(Web):2012/04/23
DOI:10.1039/C2CP40778G
Weakly exchange-coupled biradicals have attracted much attention in terms of their DNP application in NMR spectroscopy for biological systems or the use of synthetic electron-spin qubits. Pulse-ESR based electron spin nutation (ESN) spectroscopy applied to biradicals is generally treated as transition moment spectroscopy from the theoretical side, illustrating that it is a powerful and facile tool to determine relatively short distances between weakly exchange-coupled electron spins. The nutation frequency as a function of the microwave irradiation strength ω1 (angular frequency) for any cases of weakly exchange-coupled systems can be classified into three categories; D12 (spin dipolar interaction)-driven, Δg-driven and ω1-driven nutation behaviour with the increasing strength of ω1. For hetero-spin biradicals, Δg effects can be a dominating characteristic in the biradical nutation spectroscopy. Two-dimensional pulse-based electron spin nutation (2D-ESN) spectroscopy operating at the X-band can afford to determine small values of D12 in weakly exchange-coupled biradicals in rigid glasses. The analytical expressions derived here for ω1-dependent nutation frequencies are based on only four electronic spin states relevant to the biradicals, while real biradical systems often have sizable hyperfine interactions. Thus, we have evaluated nuclear hyperfine effects on the nutation frequencies to check the validity of the present theoretical treatment. The experimental spin dipolar coupling of a typical TEMPO-based biradical 1, (2,2,6,6-tetra[(2H3)methyl]-[3,3-2H2,4-2H1,5,5-2H2]piperidin-N-oxyl-4-yl)(2,2,6,6-tetra[(2H3)methyl]-[3,3-2H2,4-2H1,5,5-2H2,15N]piperidin-15N-oxyl-4-yl) terephthalate in a toluene glass, with a distance of 1.69 nm between the two spin sites is D12 = −32 MHz (the effect of the exchange coupling J12 is vanishing due to the homo-spin sites of 1, i.e. Δg = 0), while 0 < |J12| ≦ 1.0 MHz as determined by simulating the random-orientation CW ESR spectra of 1. In addition, we have carried out Q-band pulsed ELDOR (ELectron–electron DOuble Resonance) experiments to confirm whether the obtained values for D12 and J12 are accurate. The distance is in a fuzzy region for the distance-measurements capability of the conventional, powerful ELDOR spectroscopy. The strong and weak points of the ESN spectroscopy with a single microwave frequency applicable to weakly exchange-coupled multi-electron systems are discussed in comparison with conventional ELDOR spectroscopy. The theoretical spin dipolar tensor and exchange interaction of the TEMPO biradical, as obtained by sophisticated quantum chemical calculations, agree with the experimental ones.
Co-reporter:Shigeaki Nakazawa, Kazunobu Sato, Daisuke Shiomi, Masafumi Yano, Takamasa Kinoshita, Maria Luisa T. M. B. Franco, Maria Celina R. L. R. Lazana, Maria Candida B. L. Shohoji, The late Koichi Itoh and Takeji Takui
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 4) pp:NaN1433-1433
Publication Date(Web):2010/11/22
DOI:10.1039/C0CP00730G
Trianionic spin-quartet and tetraanionic spin-quintet molecular clusters derived from m-dibenzoylbenzene in solution were identified by CW-ESR/pulse-ESR based two-dimensional electron spin transient nutation spectroscopy, and their spin and clustering structures in the ground state were determined in terms of a D-tensor based phenomenological approach and DFT calculations. The molecular structures obtained semiempirically are supported by DFT-based quantum chemical calculations. The DFT calculations have been tested for a sodium ion bridged fluorenone-based cluster, [fluorenone−˙ {Na+(dme)2}]2, whose crystal structure was reported in the literature [H. Bock, H.-F. Herrmann, D. Fenske and H. Goesmann, Angew. Chem., Int. Ed. Engl., 1988, 27, 1067], reproducing the experimentally determined moelcular structure of the dimer cluster. It is suggested that both the quartet and quintet clusters in the 2-MTHF glass and solution form the cross-typed structures with the two m-dibenzoylbenzene moieties in cis-configuration. A dianionic spin-triplet m-dibenzoylbenzene derivative was detected for the first time and its charge and spin densities were studied by the quantum chemical calculations. The high-spin states of the open-shell entities under study were confirmed by X-band pulse-ESR based electron spin nutation spectroscopy in organic frozen glasses. The D values and other spin Hamiltonian parameters of all the polyanionic high-spin species were determined by the hybrid eigenfield spectral simulation for fine-structure ESR spectra. m-Dibenzoylbenzene provides pseudo-degenerate π-LUMOs arising from its topological symmetry of the π-electron network and its dianion in the triplet ground state is a prototypical model for topologically-controlled genuinely organic ferromagnetic metals.
Co-reporter:Kenji Sugisaki, Kazuo Toyota, Kazunobu Sato, Daisuke Shiomi, Masahiro Kitagawa and Takeji Takui
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 15) pp:NaN6980-6980
Publication Date(Web):2011/03/10
DOI:10.1039/C0CP02809F
Spin–orbit and spin–spin contributions to the zero-field splitting (ZFS) tensors (D tensors) of spin-triplet phenyl-, naphthyl-, and anthryl-nitrenes in their ground state are investigated by quantum chemical calculations, focusing on the effects of the ring size and substituted position of nitrene on the D tensor. A hybrid CASSCF/MRMP2 approach to the spin–orbit term of the D tensor (DSO tensor), which was recently proposed by us, has shown that the spin–orbit contribution to the entire D value, termed the ZFS parameter or fine-structure constant, is about 10% in all the arylnitrenes under study and less depends on the size and connectivity of the aryl groups. Order of the absolute values for DSO can be explained by the perturbation on the energy level and spatial distributions of π-SOMO through the orbital interaction between SOMO of the nitrene moiety and frontier orbitals of the aryl scaffolds. Spin–spin contribution to the D tensor (DSS tensor) has been calculated in terms of the McWeeny–Mizuno equation with the DFT/EPR-II spin densities. The DSS value calculated with the RO-B3LYP spin density agrees well with the D(Exptl) − DSO reference value in phenylnitrene, but agreement with the reference value gradually becomes worse as the D value decreases. Exchange–correlation functional dependence on the DSS tensor has been explored with standard 23 exchange–correlation functionals in both RO- and U-DFT methodologies, and the RO-HCTH/407 method gives the best agreement with the D(Exptl) − DSO reference value. Significant exchange–correlation functional dependence is observed in spin-delocalized systems such as 9-anthrylnitrene (6). By employing the hybrid CASSCF/MRMP2 approach and the McWeeny–Mizuno equation combined with the RO-HCTH/407/EPR-II//U-HCTH/407/6-31G* spin densities for DSO and DSS, respectively, a quantitative agreement with the experiment is achieved with errors less than 10% in all the arylnitrenes under study. Guidelines to the putative approaches to DSS tensor calculations are given.
Co-reporter:Kenji Sugisaki, Kazuo Toyota, Kazunobu Sato, Daisuke Shiomi, Masahiro Kitagawa and Takeji Takui
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 19) pp:
Publication Date(Web):
DOI:10.1039/C4CP00822G
Co-reporter:Satoru Yamamoto, Shigeaki Nakazawa, Kenji Sugisaki, Kazunobu Sato, Kazuo Toyota, Daisuke Shiomi and Takeji Takui
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 4) pp:NaN2749-2749
Publication Date(Web):2014/11/27
DOI:10.1039/C4CP04744C
A molecular spin quantum computer (MSQC) requires electron spin qubits, which pulse-based electron spin/magnetic resonance (ESR/MR) techniques can afford to manipulate for implementing quantum gate operations in open shell molecular entities. Importantly, nuclear spins, which are topologically connected, particularly in organic molecular spin systems, are client qubits, while electron spins play a role of bus qubits. Here, we introduce the implementation for an adiabatic quantum algorithm, suggesting the possible utilization of molecular spins with optimized spin structures for MSQCs. We exemplify the utilization of an adiabatic factorization problem of 21, compared with the corresponding nuclear magnetic resonance (NMR) case. Two molecular spins are selected: one is a molecular spin composed of three exchange-coupled electrons as electron-only qubits and the other an electron-bus qubit with two client nuclear spin qubits. Their electronic spin structures are well characterized in terms of the quantum mechanical behaviour in the spin Hamiltonian. The implementation of adiabatic quantum computing/computation (AQC) has, for the first time, been achieved by establishing ESR/MR pulse sequences for effective spin Hamiltonians in a fully controlled manner of spin manipulation. The conquered pulse sequences have been compared with the NMR experiments and shown much faster CPU times corresponding to the interaction strength between the spins. Significant differences are shown in rotational operations and pulse intervals for ESR/MR operations. As a result, we suggest the advantages and possible utilization of the time-evolution based AQC approach for molecular spin quantum computers and molecular spin quantum simulators underlain by sophisticated ESR/MR pulsed spin technology.









{kind=link}
{kind=link}