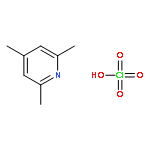
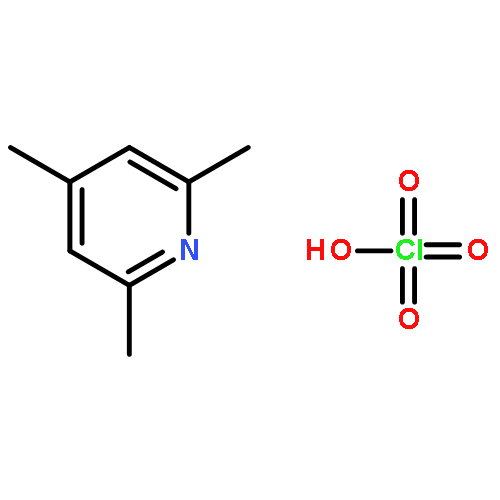
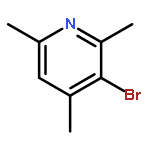
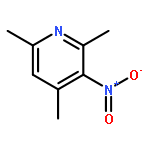
Co-reporter:Dr. Ilya G. Shenderovich;Dr. Stepan B. Lesnichin;Dr. Chingkuang Tu;Dr. David N. Silverman;Dr. Peter M. Tolstoy;Dr. Gleb S. Denisov;Dr. Hans-Heinrich Limbach
Chemistry - A European Journal 2015 Volume 21( Issue 7) pp:2915-2929
Publication Date(Web):
DOI:10.1002/chem.201404083
Abstract
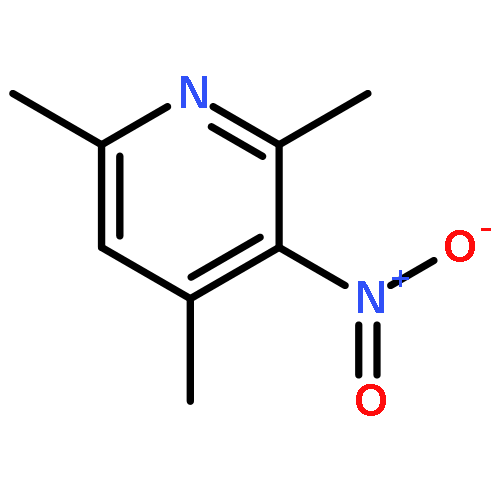
By using a combination of liquid and solid-state NMR spectroscopy, 15N-labeled 4-methylimidazole (4-MI) as a local probe of the environment has been studied: 1) in the polar, wet Freon CDF3/CDF2Cl down to 130 K, 2) in water at pH 12, and 3) in solid samples of the mutant H64A of human carbonic anhydrase II (HCA II). In the latter, the active-site His64 residue is replaced by alanine; the catalytic activity is, however, rescued by the presence of 4-MI. For the Freon solution, it is demonstrated that addition of water molecules not only catalyzes proton tautomerism but also lifts its quasidegeneracy. The possible hydrogen-bond clusters formed and the mechanism of the tautomerism are discussed. Information about the imidazole hydrogen-bond geometries is obtained by establishing a correlation between published 1H and 15N chemical shifts of the imidazole rings of histidines in proteins. This correlation is useful to distinguish histidines embedded in the interior of proteins and those at the surface, embedded in water. Moreover, evidence is obtained that the hydrogen-bond geometries of His64 in the active site of HCA II and of 4-MI in H64A HCA II are similar. Finally, the degeneracy of the rapid tautomerism of the neutral imidazole ring His64 reported by Shimahara et al. (J. Biol. Chem. 2007, 282, 9646) can be explained with a wet, polar, nonaqueous active-site conformation in the inward conformation, similar to the properties of 4-MI in the Freon solution. The biological implications for the enzyme mechanism are discussed.
Co-reporter:Monique Chan-Huot ; Alexandra Dos ; Reinhard Zander ; Shasad Sharif ; Peter M. Tolstoy ; Shara Compton ; Emily Fogle ; Michael D. Toney ; Ilya Shenderovich +; Gleb S. Denisov
Journal of the American Chemical Society 2013 Volume 135(Issue 48) pp:18160-18175
Publication Date(Web):October 22, 2013
DOI:10.1021/ja408988z
Using 15N solid-state NMR, we have studied protonation and H-bonded states of the cofactor pyridoxal 5′-phosphate (PLP) linked as an internal aldimine in alanine racemase (AlaR), aspartate aminotransferase (AspAT), and poly-l-lysine. Protonation of the pyridine nitrogen of PLP and the coupled proton transfer from the phenolic oxygen (enolimine form) to the aldimine nitrogen (ketoenamine form) is often considered to be a prerequisite to the initial step (transimination) of the enzyme-catalyzed reaction. Indeed, using 15N NMR and H-bond correlations in AspAT, we observe a strong aspartate-pyridine nitrogen H-bond with H located on nitrogen. After hydration, this hydrogen bond is maintained. By contrast, in the case of solid lyophilized AlaR, we find that the pyridine nitrogen is neither protonated nor hydrogen bonded to the proximal arginine side chain. However, hydration establishes a weak hydrogen bond to pyridine. To clarify how AlaR is activated, we performed 13C and 15N solid-state NMR experiments on isotopically labeled PLP aldimines formed by lyophilization with poly-l-lysine. In the dry solid, only the enolimine tautomer is observed. However, a fast reversible proton transfer involving the ketoenamine tautomer is observed after treatment with either gaseous water or gaseous dry HCl. Hydrolysis requires the action of both water and HCl. The formation of an external aldimine with aspartic acid at pH 9 also produces the ketoenamine form stabilized by interaction with a second aspartic acid, probably via a H-bond to the phenolic oxygen. We postulate that O-protonation is an effectual mechanism for the activation of PLP, as is N-protonation, and that enzymes that are incapable of N-protonation employ this mechanism.
Co-reporter:Brenda C. K. Ip, Ilya G. Shenderovich, Peter M. Tolstoy, Jaroslaw Frydel, Gleb S. Denisov, Gerd Buntkowsky, and Hans-Heinrich Limbach
The Journal of Physical Chemistry A 2012 Volume 116(Issue 46) pp:11370-11387
Publication Date(Web):August 3, 2012
DOI:10.1021/jp305863n
We have studied the hydrogen bond interactions of 15N labeled 4-methylpyridine (4-MP) with pentachlorophenol (PCP) in the solid state and in polar solution using various NMR techniques. Previous spectroscopic, X-ray, and neutron crystallographic studies showed that the triclinic 1:1 complex (4-MPPCP) exhibits the strongest known intermolecular OHN hydrogen bond in the solid state. By contrast, deuteration of the hydrogen bond gives rise to the formation of a monoclinic structure exhibiting a weaker hydrogen bond. By performing NMR experiments at different deuterium fractions and taking advantage of dipolar 1H–15N recoupling under combined fast MAS and 1H decoupling, we provide an explanation of the origin of the isotopic polymorphism of 4-MPPCP and improve previous chemical shift correlations for OHN hydrogen bonds. Because of anharmonic ground state vibrations, an ODN hydrogen bond in the triclinic form exhibits a shorter oxygen–hydron and a longer oxygen–nitrogen distance as compared to surrounding OHN hydrogen bonds, which also implies a reduction of the local dipole moment. The dipole–dipole interaction between adjacent coupled OHN hydrogen bonds which determines the structure of triclinic 4-MPPCP is then reduced by deuteration, and other interactions become dominant, leading to the monoclinic form. Finally, the observation of stronger OHN hydrogen bonds by 1H NMR in polar solution as compared to the solid state is discussed.
Co-reporter:Stepan B. Lesnichin ; Ilya G. Shenderovich ; Titin Muljati ; David Silverman
Journal of the American Chemical Society 2011 Volume 133(Issue 29) pp:11331-11338
Publication Date(Web):June 20, 2011
DOI:10.1021/ja203478j
Using liquid-state NMR spectroscopy we have estimated the proton-donating ability of Zn-bound water in organometallic complexes designed as models for the active site of the metalloenzyme carbonic anhydrase (CA). This ability is important for the understanding of the enzyme reaction mechanism. The desired information was obtained by 1H and 15N NMR at 180 K of solutions of [TpPh,MeZnOH] [1, TpPh,Me = tris(2-methyl-4-phenylpyrazolyl)hydroborate] in CD2Cl2, in the absence and presence of the proton donors (C6F5)3BOH2 [aquatris(pentafluorophenyl)boron] and Col-H+ (2,4,6-trimethylpyridine-H+). Col-H+ forms a strong OHN hydrogen bond with 1, where the proton is located closer to nitrogen than to oxygen. (C6F5)3BOH2, which exhibits a pKa value of 1 in water, also forms a strong hydrogen bond with 1, where the proton is shifted slightly across the hydrogen-bond center toward the Zn-bound oxygen. Finally, a complex between Col and (C6F5)3BOH2 was identified, exhibiting a zwitterionic OHN hydrogen bond, where H is entirely shifted to nitrogen. The comparison with complexes of Col with carboxylic acids studied previously suggests that, surprisingly, the Zn-bound water exhibits in an aprotic environment a similar proton-donating ability as a carboxylic acid characterized in water by a pKa of 2.2 ± 0.6. This value is much smaller than the value of 9 found for [Zn(OH2)6]2+ in water and those between 5 and 8 reported for different forms of CA. Implications for the biological function of CA are discussed.
Co-reporter:Natalia Pérez-Hernández ; Martín Febles ; Cirilo Pérez ; Johann Spandl ; Julio D. Martín
The Journal of Physical Chemistry C 2011 Volume 115(Issue 19) pp:9393-9402
Publication Date(Web):April 22, 2011
DOI:10.1021/jp112105j
We have studied the properties of a series of solid hydrated organic porous networks with pore diameters ranging from approximately 0.4 to 0.9 nm using the attenuated total reflection infrared (ATR-IR) technique. These subnanometer organic pores are composed of water and of organic racemic bicyclic monomers containing carboxylic, alcoholic, ether functions and different appendices. In particular, the doubly hydrated hydroxyl acids 1 2H2O and 2 2H2O form cylindrical pores in which half of the water molecules are part of the walls and the other half are located inside the pores. In a first step, by a comparison of the spectra of a family of related compounds, the COOH, COH as well as the wall and pore water stretches were assigned. The COOH bands are broad and red-shifted as compared to carboxylic acid dimers, and exhibit a substructure assigned to Fermi resonances. The OH stretches fulfill well Novak’s correlation with the corresponding crystallographic O...O distances. In a second step, we have followed the deuteration of the different functional groups of solid 2 2H2O by ATR-IR by heavy water vapor. Surprisingly, we observe that the rates of deuteration are the same for all functional groups although exhibiting biexponential time dependence, in contrast to the liquid state where COOH groups exchange protons with water much faster than with alcohols. This result is rationalized in terms of slowly diffusing lattice defects resembling a local liquid or glass in the subnano scale in which the different exchange reactions take place. The nonexponential deuteration is explained in terms of a faster deuteration of crystal surface layers.
Co-reporter:Dr. Dariush Ajami;Dr. Peter M. Tolstoy;Dr. Henry Dube;Dr. Severin Odermatt;Benjamin Koeppe;Jing Guo;Dr. Hans-Heinrich Limbach; Dr. Julius Rebek, Jr.
Angewandte Chemie International Edition 2011 Volume 50( Issue 2) pp:528-531
Publication Date(Web):
DOI:10.1002/anie.201002182
Co-reporter:Dr. Dariush Ajami;Dr. Peter M. Tolstoy;Dr. Henry Dube;Dr. Severin Odermatt;Benjamin Koeppe;Jing Guo;Dr. Hans-Heinrich Limbach; Dr. Julius Rebek, Jr.
Angewandte Chemie 2011 Volume 123( Issue 2) pp:548-552
Publication Date(Web):
DOI:10.1002/ange.201002182
Co-reporter:Hans-Heinrich Limbach, Monique Chan-Huot, Shasad Sharif, Peter M. Tolstoy, Ilya G. Shenderovich, Gleb S. Denisov, Michael D. Toney
Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2011 Volume 1814(Issue 11) pp:1426-1437
Publication Date(Web):November 2011
DOI:10.1016/j.bbapap.2011.06.004
In this contribution we review recent NMR studies of protonation and hydrogen bond states of pyridoxal 5′-phosphate (PLP) and PLP model Schiff bases in different environments, starting from aqueous solution, the organic solid state to polar organic solution and finally to enzyme environments. We have established hydrogen bond correlations that allow one to estimate hydrogen bond geometries from 15N chemical shifts. It is shown that protonation of the pyridine ring of PLP in aspartate aminotransferase (AspAT) is achieved by (i) an intermolecular OHN hydrogen bond with an aspartate residue, assisted by the imidazole group of a histidine side chain and (ii) a local polarity as found for related model systems in a polar organic solvent exhibiting a dielectric constant of about 30. Model studies indicate that protonation of the pyridine ring of PLP leads to a dominance of the ketoenamine form, where the intramolecular OHN hydrogen bond of PLP exhibits a zwitterionic state. Thus, the PLP moiety in AspAT carries a net positive charge considered as a pre-requisite to initiate the enzyme reaction. However, it is shown that the ketoenamine form dominates in the absence of ring protonation when PLP is solvated by polar groups such as water. Finally, the differences between acid–base interactions in aqueous solution and in the interior of proteins are discussed. This article is part of a special issue entitled: Pyridoxal Phosphate Enzymology.Highlights► The OHN hydrogen bond geometries of pyridoxal 5'-phosphate can be determined by NMR. ► Protonation of the pyridine ring also polarizes the intramolecular OHN hydrogen bond. ► A net positive charge is required to activate the cofactor. ► Hydrogen bond and protonation states strongly depend on the environment. ► Pyridoxal 5'-phosphate behaves in enzymes similar as in polar organic solvents.
Co-reporter:Alexandra Dos, Volkmar Schimming, Monique Chan-Huot and Hans-Heinrich Limbach
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 35) pp:10235-10245
Publication Date(Web):20 Jul 2010
DOI:10.1039/C002730H
Using high resolution solid state 15N and 13C NMR spectroscopy we have studied the effects of successive hydration on the 15N labeled side chain amino groups of solid poly-L-lysine (PLL) in the presence of acids. Generally, hydration leads to the formation of local “ionic fluid” phases composed by flexible side chain ammonium groups, acid anions and small amounts of water. The associated local dynamics reduces the widths of the inhomogeneously broadened 15N amino signals found for the dry states. The hydration of free base PLL—which consists of mixtures of α-helices and β-pleated sheets—is monitored by a small low-field shift of the amino group signal arising from hydrogen bonding with water, reaching eventually the value of PLL in water at pH 13. No difference for the two conformations is observed. PLL×HF adopts a similar secondary structure with isolated NHF hydrogen bonds; hydration leads only to small low-field shifts which are nevertheless compatible with the formation of ammonium groups in aqueous solution. PLL doped with small amounts of HCl contains ammonium groups which are internally solvated by neighboring free amino groups. Both nitrogen environments are characterized by different chemical shifts. Hydration with less than one water molecule per amino group leads already to a chemical shift averaging arising from fast proton motions along NHN-hydrogen bonds and fast side chain and anion motions.
By contrast, the hydration of fully doped PLL×HBr and PLL×HCl is more complex. These systems exist only in β-pleated sheet conformations forming alkyl ammonium salt structures. Separate 15N signal components are observed for (i) the dry states, for (ii) wet β-pleated sheets and for (iii) wet α-helices which are successively formed upon hydration. Exchange between these environments is slow, but water motions lead to averaged amino group signals within each of the two wet environments. These results indicate that the different environments form domains. As the replacement of NHBr or of NHCl hydrogen bonds by NHO hydrogen bonds leads to high-field shifts the observation of separated signals is the result of different water content in the three domains. In agreement with previous X-ray powder diffraction studies we observe a dominance of the α-helical regions at above 3 water molecules per amino group in the case of PLL×HBr and at about 5 water molecules in the case of PLL×HCl, an effect arising from the limited space between β-pleated sheets and the larger volume of bromide as compared to chloride.
Co-reporter:Monique Chan-Huot, Shasad Sharif, Peter M. Tolstoy, Michael D. Toney, and Hans-Heinrich Limbach
Biochemistry 2010 Volume 49(Issue 51) pp:
Publication Date(Web):November 10, 2010
DOI:10.1021/bi101061m
We have measured the pH-dependent 1H, 13C, and 15N NMR spectra of pyridoxal 5′-phosphate (13C2-PLP) mixed with equal amounts of either doubly 15N-labeled diaminopropane, 15Nα-labeled l-lysine, or 15Nε-labeled l-lysine as model systems for various intermediates of the transimination reaction in PLP-dependent enzymes. At low pH, only the hydrate and aldehyde forms of PLP and the free protonated diamines are present. Above pH 4, the formation of single- and double-headed aldimines (Schiff bases) with the added diamines is observed, and their 13C and 15N NMR parameters have been characterized. For 1:1 mixtures the single-headed aldimines dominate. In a similar way, the NMR parameters of the geminal diamine formed with diaminopropane at high pH are measured. However, no geminal diamine is formed with l-lysine. In contrast to the aldimine formed with the ε-amino group of lysine, the aldimine formed with the α-amino group is unstable at moderately high pH but dominates slightly below pH 10. By analyzing the NMR data, both the mole fractions of the different PLP species and up to 6 different protonation states including their pKa values were obtained. Furthermore, the data show that all Schiff bases are subject to a proton tautomerism along the intramolecular OHN hydrogen bond, where the zwitterionic form is favored before deprotonation occurs at high pH. This observation, as well as the observation that around pH 7 the different PLP species are present in comparable amounts, sheds new light on the mechanism of the transimination reaction.
Co-reporter:Hans-Heinrich Limbach;K. Barbara Schowen;Richard L. Schowen
Journal of Physical Organic Chemistry 2010 Volume 23( Issue 7) pp:586-605
Publication Date(Web):
DOI:10.1002/poc.1663
Abstract
Arrhenius curves of selected hydrogen transfer reactions in organic molecules and enzymes are reviewed with the focus on systems exhibiting temperature-independent kinetic isotope effects. The latter can be rationalized in terms of a ‘pre-tunneling state’ which is formed from the reactants by heavy atom motions and which represents a suitable molecular configuration for tunneling to occur. Within the Bell–Limbach tunneling model, formation of the pre-tunneling state dominates the Arrhenius curves of the H and the D transfer even at higher temperatures if a large energy Em is required to reach the pre-tunneling state. Tunneling from higher vibrational levels and the over-barrier reaction via the transition state which lead to temperature-dependent kinetic isotope effects dominate the Arrhenius curves only if Em is small compared to the energy of the transition state. Using published data on several hydrogen transfer systems, the type of motions leading to the pre-tunneling state is explored. Among the phenomena which lead to large energies of the pre-tunneling state are (i) cleavage of hydrogen bonds or coordination bonds of the donor or acceptor atoms to molecules or molecular groups in order to allow the formation of the pre-tunneling state, (ii) the occurrence of an energetic intermediate on the reaction pathway within which tunneling takes place, and (iii) major reorganization of a molecular skeleton, requiring the excitation of specific vibrations in order to reach the pre-tunneling state. This model suggests a solution to the puzzle of Kwart's findings of temperature-independent kinetic isotope effects for hydrogen transfer in small organic molecules. Copyright © 2010 John Wiley & Sons, Ltd.
Co-reporter:Monique Chan-Huot, Christiane Niether, Shasad Sharif, Peter M. Tolstoy, Michael D. Toney, Hans-Heinrich Limbach
Journal of Molecular Structure 2010 Volume 976(1–3) pp:282-289
Publication Date(Web):15 July 2010
DOI:10.1016/j.molstruc.2010.03.032
We have measured the 13C NMR spectra of the cofactor pyridoxal-5′-phosphate (vitamin B6, PLP) at 278 K in aqueous solution as a function of pH. By 13C enrichment of PLP in the C-4′ and C-5′ positions we were able to measure spectra down to pH 1. From the dependence of the 13C chemical shifts on pH, the pKa values of PLP could be determined. In particular, the heretofore uncharacterized protonation state of PLP, in which the phosphate group as well as the pyridine ring and the phenolic groups are fully protonated, has been analyzed. The corresponding pKa value of 2.4 indicates that the phosphate group is solely involved in the first deprotonation step. The 15N chemical shifts of the pyridine ring of PLP published previously are in good agreement with the new results. These shifts contain information about the tautomerism of the different protonation states of PLP. The implications of these findings for the biological function of PLP are discussed.
Co-reporter:Mariusz Pietrzak Dr.;JensP. Wehling;Shushu Kong;PeterM. Tolstoy Dr.;IlyaG. Shenderovich Dr.;Concepción López Dr.;Rosa María Claramunt Dr.;José Elguero Dr.;GlebS. Denisov Dr. Dr.
Chemistry - A European Journal 2010 Volume 16( Issue 5) pp:1679-1690
Publication Date(Web):
DOI:10.1002/chem.200902259
Abstract
The properties of the intramolecular hydrogen bonds of doubly 15N-labeled protonated sponges of the 1,8-bis(dimethylamino)naphthalene (DMANH+) type have been studied as a function of the solvent, counteranion, and temperature using low-temperature NMR spectroscopy. Information about the hydrogen-bond symmetries was obtained by the analysis of the chemical shifts δH and δN and the scalar coupling constants J(N,N), J(N,H), J(H,N) of the 15NH15N hydrogen bonds. Whereas the individual couplings J(N,H) and J(H,N) were averaged by a fast intramolecular proton tautomerism between two forms, it is shown that the sum |J(N,H)+J(H,N)| generally represents a measure of the hydrogen-bond strength in a similar way to δH and J(N,N). The NMR spectroscopic parameters of DMANH+ and of 4-nitro-DMANH+ are independent of the anion in the case of CD3CN, which indicates ion-pair dissociation in this solvent. By contrast, studies using CD2Cl2, [D8]toluene as well as the freon mixture CDF3/CDF2Cl, which is liquid down to 100 K, revealed an influence of temperature and of the counteranions. Whereas a small counteranion such as trifluoroacetate perturbed the hydrogen bond, the large noncoordinating anion tetrakis[3,5-bis(trifluoromethyl)phenyl]borate B[{C6H3(CF3)2}4]− (BARF−), which exhibits a delocalized charge, made the hydrogen bond more symmetric. Lowering the temperature led to a similar symmetrization, an effect that is discussed in terms of solvent ordering at low temperature and differential solvent order/disorder at high temperatures. By contrast, toluene molecules that are ordered around the cation led to typical high-field shifts of the hydrogen-bonded proton as well as of those bound to carbon, an effect that is absent in the case of neutral NHN chelates.
Co-reporter:Alexandra Dos ; Volkmar Schimming ; Monique Chan Huot
Journal of the American Chemical Society 2009 Volume 131(Issue 22) pp:7641-7653
Publication Date(Web):May 15, 2009
DOI:10.1021/ja901082a
The acid−base and base−base interactions of the 15N-labeled side-chain amino groups of dry solid poly-l-lysine (PLL) and the consequences for the secondary structure have been studied using high-resolution solid state 15N and 13C CPMAS NMR spectroscopy. In a previous study we had shown that at acid/base ratios of 1 per amino group the halogen acids HI, HCl and HBr form PLL salts in the β-pleated sheet but not in the α-helical structure, whereas HF and various oxygen acids form 1:1 acid−base hydrogen-bonded complexes in both secondary structures. In the present study we performed NMR experiments at reduced acid/base ratios in order to elucidate whether also 1:2 and 1:3 acid−base complexes are formed under these conditions. Generally, the PLL samples containing HF, HBr, HCl, HI, CH3COOH, H3PO4, H2SO4, or HNO3 were obtained by lyophilization at different pH. For comparison, samples were also obtained by letting dry acid-free PLL interact with gaseous HCl. In a theoretical section we first study the probability of the different acid−base complexes as a function of the acid/base ratio and the equilibrium constants of the complex formation. Using this information, the 15N NMR spectra of acid doped PLL obtained were analyzed and assigned. Indeed, evidence for the formation of 1:2 and 1:3 acid−base complexes at lower acid/base ratios could be obtained. Moreover, the salt structures of the halides of PLL are already destroyed at acid/base ratios of about 0.8. By contrast, when acid-free poly-l-lysine is exposed to HCl gas, a biexponential conversion of amino groups into ammonium groups is observed without formation of 1:2 and 1:3 complexes. 13C NMR reveals that the β-pleated sheet environments of acid-free PLL react rapidly with HCl, whereas the α-helices first have to be converted in a slow reaction to β-pleated sheets before they can react. Interestingly, after partial doping with HCl, exposure to gaseous H2O catalyzes the interconversion of the ammonium and amino groups into a mixture of 1:1, 1:2 and 1:3 complexes. Finally, the 15N NMR assignments were assisted by DFT calculations on methylamine−acid model complexes.
Co-reporter:Sven Macholl, Jochen Matthes, Hans-Heinrich Limbach, Sylviane Sabo-Etienne, Bruno Chaudret, Gerd Buntkowsky
Solid State Nuclear Magnetic Resonance 2009 Volume 36(Issue 3) pp:137-143
Publication Date(Web):November 2009
DOI:10.1016/j.ssnmr.2009.08.001
2H solid-state, variable temperature magic angle spinning (MAS) NMR spectra of precipitated samples of the deutero dideuterium complexes Ru(D)2(η2-D2)2(PCy3)2 and RuD(η2-D2)I(PCy3)2 [Cy=cyclohexyl] are presented. They show that even at moderate MAS speed, high resolution is achieved at 7 and 14 T allowing 2H chemical shifts and quadrupole couplings to be obtained and assigned to different solid and gaseous 2H species. These two parameters allow identifying chemically different hydrogen species in the material. The analysis of these parameters in this study reveals the presence of three different species in the sample, namely the complexes RuD(η2-D2)I(PCy3)2 and RuD(η2-D2)2I(PCy3)2, and highly mobile HD/D2. These assignments are supported by 2H T1 relaxation times and 31P MAS NMR spectra. Moreover, variable temperature MAS NMR spectra reveal temperature-dependent line-shape changes, which are clear indications of intramolecular hydrogen exchange of the deutero and the dideuterium ligands and which give an estimate for the activation energy of this process.
Co-reporter:Hans-Heinrich Limbach;Peter M. Tolstoy;Natalia Pérez-Hernández;Jing Guo;Ilya G. Shenderovich;Gleb S. Denisov
Israel Journal of Chemistry 2009 Volume 49( Issue 2) pp:199-216
Publication Date(Web):
DOI:10.1560/IJC.49.2.199
Abstract
Hydrogen bond geometries and 1H NMR chemical shifts of OHO hydrogen-bonded systems have been analyzed using an improved valence bond order model. This model predicts that the heavy atom hydrogen bond coordinate q2 = r1 + r2 is a function of the proton coordinate q1 = (r1 - r2), where r1 and r2 represent the OH and the HO distances.
In the first part, it is shown that this correlation reproduces published equilibrium geometries of the Zundel cation H5O2+ as well as those of water clusters in the gas phase and embedded in the fullerene C180. Using the example of the water hexamer, it is shown that changing the level of calculation shifts the calculated geometries along the correlation curve, but not away from the curve. In order to take quantum zero-point vibrational effects (QZPVE) into account, an empirical correction is proposed. It is shown that this correction properly describes the calculated classical and quantum hydrogen bond geometries of compressed ice as well as calculated geometric H/D isotope effects. The improved valence bond order model is used to analyze a large number of OHO hydrogen bond geometries contained in the Cambridge Structural Database.
In the second part, a relation between the geometries and the 1H NMR chemical shieldings of OHO hydrogen bonded systems is established using the valence bond order model. GIAO calculations of the isolated symmetric Zundel cation where H is located in the hydrogen bond center show only a small dependence of the chemical shifts on the O…O distance. This result is rationalized in terms of neighbor group effects and deshielding in the naked proton. The consequence is that the 1H NMR chemical shifts are not much affected by QZPVE. Calculations on water clusters indicate that the influence of the chemical environment of the OHO hydrogen bonds on their 1H NMR chemical shifts is smaller for the strong hydrogen bond regime but large for the weak hydrogen bond regime. A simple chemical shift vs. q1 relation is then used to calculate the average chemical shifts of water clusters in the regime of fast hydrogen bond exchange between hydrogen bonded and free OH groups. It is shown that average chemical shifts of about 6 ppm are possible as the clusters considered exhibit a broad distribution of stronger and weaker hydrogen bonds. The implications for water in organic solvents and for liquid water are discussed, based on published data on the 1H chemical shift distribution in the latter.
Co-reporter:Hans-Heinrich Limbach;Peter M. Tolstoy;Natalia Pérez-Hernández;Jing Guo;Ilya G. Shenderovich;Gleb S. Denisov
Israel Journal of Chemistry 2009 Volume 49( Issue 2) pp:s1-s36
Publication Date(Web):
DOI:10.1560/IJC.49.2.S1
Co-reporter:Juan Miguel Lopez del Amo, Uwe Langer, Verónica Torres, Mariusz Pietrzak, Gerd Buntkowsky, Hans-Martin Vieth, Mohamed F. Shibl, Oliver Kühn, Martin Bröring and Hans-Heinrich Limbach
The Journal of Physical Chemistry A 2009 Volume 113(Issue 10) pp:2193-2206
Publication Date(Web):December 18, 2008
DOI:10.1021/jp8079414
Using high resolution solid state 15N and 2H spectroscopy and longitudinal relaxometry we have studied the tautomerism of porphycene in the solid state, corresponding to a double proton transfer in two cooperative hydrogen bonds. The tautomerism is degenerate above 225 K but the degeneracy is lifted below this temperature, indicating a phase transition. Thus, the high-temperature phase is characterized by a dynamic proton disorder and the low-temperature phase by a dynamic proton order. 15N magnetization transfer experiments obtained under cross polarization (CP) and magic angle spinning (MAS) conditions reveal the presence of two nonequivalent molecules A and B in the unit cell of phase II, exhibiting slightly different equilibrium constants of the tautomerism. Rate constants of the tautomerism in phase I could be obtained by the analysis of the longitudinal 15N and 2H relaxation times. The former, obtained at 9.12 MHz, exhibit a T1 minimum around 270 K and are consistent with proton transfer induced dipolar 1H−15N relaxation mechanism. The latter, obtained at 46.03 MHz, exhibit a minimum around 330 K and arise from quadrupole relaxation. Within the margin of error, the rate constants of the HH and of the HD/DD tautomerism are the same, exhibiting a barrier of about 30 kJ mol−1, as expected for an overbarrier reaction in a configuration with two compressed hydrogen bonds. By contrast, in the low-temperature phase a switch of the DD transfer kinetics into the nanosecond time scale is observed, exhibiting a non-Arrhenius temperature dependence which is typical for tunneling. This increase of the rate constants by lowering the temperature is discussed in terms of a switch from a concerted HH transfer in phase I to a stepwise transfer in phase II, where intermolecular interactions lower the energy of one of the cis-intermediates.
Co-reporter:Mariusz Pietrzak Dr.;AndrewC. Try Dr.;Bruno Andrioletti Dr.;JonathanL. Sessler ;Pavel Anzenbacher Jr. Dr. Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 6) pp:1123-1126
Publication Date(Web):
DOI:10.1002/anie.200704411
Co-reporter:Mariusz Pietrzak Dr.;AndrewC. Try Dr.;Bruno Andrioletti Dr.;JonathanL. Sessler ;Pavel Anzenbacher Jr. Dr. Dr.
Angewandte Chemie 2008 Volume 120( Issue 6) pp:1139-1142
Publication Date(Web):
DOI:10.1002/ange.200704411
Co-reporter:Alexandra Dos, Volkmar Schimming, Sergio Tosoni and Hans-Heinrich Limbach
The Journal of Physical Chemistry B 2008 Volume 112(Issue 49) pp:15604-15615
Publication Date(Web):November 14, 2008
DOI:10.1021/jp806551u
The interactions of the 15N-labeled amino groups of dry solid poly-l-lysine (PLL) with various halogen and oxygen acids HX and the relation to the secondary structure have been studied using solid-state 15N and 13C CPMAS NMR spectroscopy (CP = cross polarization and MAS = magic angle spinning). For comparison, 15N NMR spectra of an aqueous solution of PLL were measured as a function of pH. In order to understand the effects of protonation and hydration on the 15N chemical shifts of the amino groups, DFT and chemical shielding calculations were performed on isolated methylamine−acid complexes and on periodic halide clusters of the type (CH3NH3+X−)n. The combined experimental and computational results reveal low-field shifts of the amino nitrogens upon interaction with the oxygen acids HX = HF, H2SO4, CH3COOH, (CH3)2POOH, H3PO4, HNO3, and internal carbamic acid formed by reaction of the amino groups with gaseous CO2. Evidence is obtained that only hydrogen-bonded species of the type (Lys−NH2···H−X)n are formed in the absence of water. 15N chemical shifts are maximum when H is located in the hydrogen bond center and then decrease again upon full protonation, as found for aqueous solution at low pH. By contrast, halogen acids interact in a different way. They form internal salts of the type (Lys−NH3+X−)n via the interaction of many acid−base pairs. This salt formation is possible only in the β-sheet conformation. By contrast, the formation of hydrogen-bonded complexes can occur both in β-sheet domains as well as in α-helical domains. The 15N chemical shifts of the protonated ammonium groups increase when the size of the interacting halogen anions is increased from chloride to iodide and when the number of the interacting anions is increased. Thus, the observed high-field 15N shift of ammonium groups upon hydration is the consequence of replacing interacting halogen atoms by oxygen atoms.
Co-reporter:Juan Miguel Lopez, Ferdinand Männle, Iwona Wawer, Gerd Buntkowsky and Hans-Heinrich Limbach
Physical Chemistry Chemical Physics 2007 vol. 9(Issue 32) pp:4498-4513
Publication Date(Web):25 Jun 2007
DOI:10.1039/B704384H
Using dynamic NMR spectroscopy, the kinetics of the degenerate double proton transfer in cyclic dimers of polycrystalline 15N,15N′-di-(4-bromophenyl)-formamidine (DBrFA) have been studied including the kinetic HH/HD/DD isotope effects in a wide temperature range. This transfer is controlled by intermolecular interactions, which in turn are controlled by the molecular conformation and hence the molecular structure. At low temperatures, rate constants were determined by line shape analysis of 15N NMR spectra obtained using cross-polarization (CP) and magic angle spinning (MAS). At higher temperatures, in the microsecond time scale, rate constants and kinetic isotope effects were obtained by a combination of longitudinal 15N and 2H relaxation measurements. 15N CPMAS line shape analysis was also employed to study the non-degenerate double proton transfer of polycrystalline 15N,15N′-diphenyl-formamidine (DPFA). The kinetic results are in excellent agreement with the kinetics of DPFA and 15N,15N′-di-(4-fluorophenyl)-formamidine (DFFA) studied previously for solutions in tetrahydrofuran. Two large HH/HD and HD/DD isotope effects are observed in the whole temperature range which indicates a concerted double proton transfer mechanism in the domain of the reaction energy surface. The Arrhenius curves are non-linear indicating a tunneling mechanism. Arrhenius curve simulations were performed using the Bell–Limbach tunneling model. The role of the phenyl group conformation and hydrogen bond compression on the barrier of the proton transfer is discussed.
Co-reporter:Mariusz Pietrzak, Claudia Benedict, Holger Gehring, Ewald Daltrozzo, Hans-Heinrich Limbach
Journal of Molecular Structure 2007 Volumes 844–845() pp:222-231
Publication Date(Web):12 November 2007
DOI:10.1016/j.molstruc.2007.04.023
The 1H, 15N, and 13C NMR spectra of partially 15N labeled bis-(2-pyridyl)-acetonitrile (1) dissolved in CDCl3 and CD2Cl2 have been measured in order to characterize its intramolecular NHN hydrogen bond. A fast proton tautomerism renders the molecule symmetric within the NMR timescale which complicates the determination of the scalar coupling constant 2hJ(15N,15N) ≡ J(N,N) across the intramolecular NHN-hydrogen bond. It is shown that an isotopic labeling scheme where experiments are performed on a mixture of 1-14N14N, 1-14N15N, and 1-15N15N facilitates the direct determination of J(N,N) from the non-decoupled 15N NMR spectra as well as the indirect detection via 13C NMR. Thus, a value of J(N,N) = 10.3 ± 0.5 Hz is obtained, which is similar to the corresponding value of 10.6 ± 0.5 Hz found previously for the seven-membered H-chelate N,N′-diphenyl-6-aminopentafulvene-1-aldimine-15N2 (2). By contrast, the crystallographic N…N distances and hydrogen bond angles of both compounds are very different, i.e. 2.65 Å and about 140° in the case of 1 and 2.79 Å and about 160° in the case of 2. However, the sum of the calculated NH and H…N distances is the same for both compounds, i.e. 2.75 Å. This finding supports the previous proposition that the values of J(A,B) of a hydrogen bond AHB are correlated with the sum of the two hydrogen – heavy atom distances rather than with the heavy atom distance.
Co-reporter:Gerd Buntkowsky Dr.;Jochen Matthes Dr. ;Tal Pery;Stefan Gründemann Dr.;Bernadeta Walaszek;Bruno Chaudret Dr.
ChemPhysChem 2006 Volume 7(Issue 3) pp:551-554
Publication Date(Web):22 FEB 2006
DOI:10.1002/cphc.200500559
The phenomenon of exchange coupling is taken into account in the description of the magnetic nuclear spin conversion between bound ortho- and para-dihydrogen. This conversion occurs without bond breaking, in contrast to the chemical spin conversion. It is shown that the exchange coupling needs to be reduced so that the corresponding exchange barrier can increase and the given magnetic interaction can effectively induce a spin conversion. The implications for related molecules such as water are discussed. For ice, a dipolar magnetic conversion and for liquid water a chemical conversion are predicted to occur within the millisecond timescale. It follows that a separation of water into its spin isomers, as proposed by Tikhonov and Volkov (Science2002, 296, 2363), is not feasible. Nuclear spin temperatures of water vapor in comets, which are smaller than the gas-phase equilibrium temperatures, are proposed to be diagnostic for the temperature of the ice or the dust surface from which the water was released.
Co-reporter:Hans-Heinrich Limbach, Ferdinand Männle, Carsten Detering, Gleb S. Denisov
Chemical Physics 2005 Volume 319(1–3) pp:69-92
Publication Date(Web):7 December 2005
DOI:10.1016/j.chemphys.2005.05.021
Abstract
In this paper, we explore the mechanisms of degenerate base-catalyzed intra- and intermolecular proton transfer using dynamic liquid state NMR. For this purpose, the model compound 1,3-bis(4-fluorophenyl)[1,3-15N2]triazene (1) was studied with and without the presence of dimethylamine (2), trimethylamine (3) and water, using tetrahydrofuran-d8 and methylethylether-d8 as solvents, down to 130 K. Compound 1 represents an analog of carboxylic acids and of diarylamidines forming cyclic dimers in which a fast double proton transfer takes place. By contrast, the structure of 1 was chosen in such a way that this double proton transfer is suppressed, thus revealing the base catalyzed transfer by dynamic 1H and 19F NMR. Surprisingly, both 2 and 3 can pick up the mobile proton of 1 at one nitrogen atom and carry it to the other nitrogen atom of 1, resulting in an intramolecular transfer process catalyzed each time by a different base molecule. Even more surprising is that the intramolecular transfer catalyzed by 2 is faster than the superimposed intermolecular double proton transfer. In the absence of added bases, a 1 is subject to a slow proton exchange with 2-amino-5,4′-difluoro-diphenyl-diazene (4) which is formed in small quantities from 1 in the presence of acid impurities. This process can be minimized by a proper sample preparation technique.
The kinetic H/D isotope effects are small, especially in the catalysis by 2, indicating a major heavy atom rearrangement and absence of tunneling. Semi-empirical PM3 and ab initio DFT calculations indicate a reaction pathway via a hydrogen bond switch of the protonated amine representing the transition state. The Arrhenius curves of all processes exhibit strong convex curvatures. This phenomenon is explained in terms of the hydrogen bond association of 1 with the added bases, preceding the proton transfer. At low temperatures, all catalysts are in a hydrogen bonded reactive complex with 1, and the rate constants observed equal to those of the reacting complex. However, at high temperatures, dissociation of the complex occurs, and the temperature dependence of the observed rate constants is affected also by the enthalpy of the hydrogen bond association. Finally, implications of this study for the mechanisms of enzyme proton transfers are discussed.
Co-reporter:Tal Pery;Katrin Pelzer;Gerd Buntkowsky Dr.;Karine Philippot Dr. Dr.;Bruno Chaudret Dr.
ChemPhysChem 2005 Volume 6(Issue 4) pp:
Publication Date(Web):12 APR 2005
DOI:10.1002/cphc.200400621
Surface-reactive metal colloids: Ruthenium nanoparticles are able to accommodate hydrogen atoms at, or immediately below, their surface, as shown by a combination of liquid, gas-phase and solid-state NMR techniques. The hydrogen atoms are mobile on the ruthenium particles—and reactive—which leads to fast hydrogen–deuterium exchange with the ligands (see graphic) even in the solid state.
Co-reporter:Hans-Heinrich Limbach, Mariusz Pietrzak, Hans Benedict, Peter M. Tolstoy, Nikolai S. Golubev, Gleb S. Denisov
Journal of Molecular Structure 2004 Volume 706(1–3) pp:115-119
Publication Date(Web):12 November 2004
DOI:10.1016/j.molstruc.2004.03.006
In this paper, empirical corrections for anharmonic ground-state vibrations of hydrogen and deuterium in the hydrogen bridges A–L⋯B, L=H, D are introduced into the geometric hydrogen bond correlation analysis based on the empirical Pauling valence bond orders. The method is verified using the examples of the hydrogen bonded anions in [(CO)5Cr–CN⋯H⋯NC–Cr(CO)5]− As(Ph)4+ (1h), in [(CO)5Cr–CN⋯H⋯NC–Cr(CO)5]− N(n-propyl)4+ (2h), in the model system [CN⋯H⋯NC]− Li+ (3h), and their deuterated isotopologs (1d, 2d and 3d) studied previously by dipolar NMR and theoretical methods by H. Benedict et al. [J. Am. Chem. Soc. 120 (1998) 2939]. The new corrections are able to describe isotope effects on hydrogen bond geometries from the weak to the strong hydrogen bond regime, taking into account single and double-well situations.
Co-reporter:Hans-Heinrich Limbach ;Mariusz Pietrzak;Shasad Sharif;Peter M. Tolstoy Dr.;Ilya G. Shenderovich Dr.;Sergei N. Smirnov;Nikolai S. Golubev Dr.;Gleb S. Denisov
Chemistry - A European Journal 2004 Volume 10(Issue 20) pp:
Publication Date(Web):9 SEP 2004
DOI:10.1002/chem.200400212
In this paper, equations are proposed which relate various NMR parameters of OHN hydrogen-bonded pyridine–acid complexes to their bond valences which are in turn correlated with their hydrogen-bond geometries. As the valence bond model is strictly valid only for weak hydrogen bonds appropriate empirical correction factors are proposed which take into account anharmonic zero-point energy vibrations. The correction factors are different for OHN and ODN hydrogen bonds and depend on whether a double or a single well potential is realized in the strong hydrogen-bond regime. One correction factor was determined from the known experimental structure of a very strong OHN hydrogen bond between pentachlorophenol and 4-methylpyridine, determined by the neutron diffraction method. The remaining correction factors which allow one also to describe H/D isotope effects on the NMR parameters and geometries of OHN hydrogen bond were determined by analysing the NMR parameters of the series of protonated and deuterated pyridine- and collidine–acid complexes. The method may be used in the future to establish hydrogen-bond geometries in biologically relevant functional OHN hydrogen bonds.
Co-reporter:Bob Grünberg;Thomas Emmler;Egbert Gedat Dr.;Ilja Shenderovich Dr.;Gerhard H. Findenegg Dr. Dr.;Gerd Buntkowsky Priv. Doz. Dr.
Chemistry - A European Journal 2004 Volume 10(Issue 22) pp:
Publication Date(Web):7 OCT 2004
DOI:10.1002/chem.200400351
The adsorption of water in two mesoporous silica materials with cylindrical pores of uniform diameter, MCM-41 and SBA-15, was studied by 1H MAS (MAS=magic angle spinning) and static solid-state NMR spectroscopy. All observed hydrogen atoms are either surface SiOH groups or hydrogen-bonded water molecules. Unlike MCM-41, some strongly bound water molecules exist at the inner surfaces of SBA-15 that are assigned to surface defects. At higher filling levels, a further difference between MCM-41 and SBA-15 is observed. Water molecules in MCM-41 exhibit a bimodal line distribution of chemical shifts, with one peak at the position of inner-bulk water, and the second peak at the position of water molecules in fast exchange with surface SiOH groups. In SBA-15, a single line is observed that shifts continuously as the pore filling is increased. This result is attributed to a different pore-filling mechanism for the two silica materials. In MCM-41, due to its small pore diameter (3.3 nm), pore filling by pore condensation (axial-pore-filling mode) occurs at a low relative pressure, corresponding roughly to a single adsorbed monolayer. For SBA-15, owing to its larger pore diameter (8 nm), a gradual increase in the thickness of the adsorbed layer (radial-pore-filling mode) prevails until pore condensation takes place at a higher level of pore filling.
Co-reporter:Natalia V. Belkova;Alexei V. Ionidis;Lina M. Epstein;Elena S. Shubina;Stephan Gruendemann;Nikolai S. Golubev
European Journal of Inorganic Chemistry 2001 Volume 2001(Issue 7) pp:
Publication Date(Web):28 MAY 2001
DOI:10.1002/1099-0682(200107)2001:7<1753::AID-EJIC1753>3.0.CO;2-H
The interaction of the ruthenium hydride complex RuH ≡ CpRuH(CO)(PCy3) (1) with various proton donors AH ≡ CF3CH2OH (2a), (CF3)2CHOH (2b), (CF3)3COH (2c), CF3COOH (2d), and HBF4 (2e) has been studied by variable-temperature IR spectroscopy using hexane and CH2Cl2 as solvents of different polarity. A low-temperature NMR study of the interaction of 1 with 2c was performed using [D8]methylcyclohexane, CD2Cl2, and a liquefied mixture of CDF2Cl/CDF3 (2:1). The first stage of the proton transfer process was found to be the formation of hydrogen-bonded complexes of the type RuH···HA. The hydrogen bonds in these complexes are of medium strength (−ΔH° = 5.3−7.6 kcal mol−1). The second stage is the slow conversion of the H-complex to a dihydrogen complex to which a hydrogen-bonded ion-pair structure [Ru(η2-H2)]+···A− was assigned. The kinetics of this unusually slow proton transfer reaction was monitored in the case of 2c at 200 K in CH2Cl2. Fast protonation of 1 by 2d leads additionally to a species assigned as the free cationic complex [Ru(η2-H2)]+, whose formation is driven by the formation of the homoconjugated anionic complex [AHA]−. At temperatures above 220 K both the hydrogen-bonded ion pair and the free cationic complex easily release dihydrogen, producing RuA.
Co-reporter:Ilja G. Shenderovich;Andrej P. Burtsev;Gleb S. Denisov;Nikolai S. Golubev
Magnetic Resonance in Chemistry 2001 Volume 39(Issue S1) pp:S91-S99
Publication Date(Web):5 NOV 2001
DOI:10.1002/mrc.938
The influence of solvent polarity on the properties of hydrogen-bonded 1 : 1 complexes of 2,4,6-trimethylpyridine-15N with HF and DF, labeled below as FHN and FDN, has been studied by multinuclear magnetic resonance spectroscopy in the slow hydrogen bond exchange regime reached below 190 K. Mixtures of CDF3/CDClF2 were employed as solvent, which is liquid down to 90 K. In order to evaluate their polarity, the static dielectric constants εo of the CHF3, CHClF2 and of the binary 1 : 1 mixture were measured from 160 K down to 90 K. A strong increase of εo from 14 at 190 K to 38 at 103 K is observed for the mixtures used in the NMR measurements. Upon cooling, i.e. increase of the dielectric constant, the NMR spectra indicate a gradual transformation of an asymmetric molecular complex FH···N to a quasi-symmetric complex Fδ−···H···Nδ+ and eventually to a more or less zwitterionic species F−···H N+. These changes are not only manifested in the scalar couplings J(1H,19F) and J(1H,15N) but also lead to characteristic primary and secondary H/D isotope effects on the chemical shifts of the hydrogen bonded nuclei. Whereas the primary isotope chemical shift effect pΔ(D/H) ≡ δ(F2HN) − δ(F1HN) = −0.2 ppm is negative at 190 K and in agreement with an asymmetric hydrogen bond in the molecular complex, it changes its sign when the temperature is lowered, goes through a maximum of +0.27 ppm at εo ≈ 22 and finally decreases again. The positive value of pΔ(D/H) is in agreement with D more confined to the hydrogen bond center compared with H, which constitutes a fingerprint of a quasi-symmetric hydrogen bond involving a single well potential for the proton motion. The quasi-symmetric complex is further characterized by the following NMR parameters, J(1H,19F) = 30 Hz, J(1H,15N) = −50 Hz, J(19F,15N) = −96 Hz, δ(F1HN) = 20.0 ppm, δ(19FHN) = −114.2 ppm, δ(FH15N) = −63.5 ppm, and the one-bond H/D-isotope effects δ(F2HN) − δ(F1HN) = +0.27 ppm, δ(19FDN) − δ(19FHN) = 1.4 ppm and δ(FD15N) − δ(FH15N) = −3.4 ppm. Copyright © 2001 John Wiley & Sons, Ltd.
Co-reporter:Mariusz Pietrzak;Marta Pérez-Torralba;Dionísia Sanz;Rosa María Claramunt;José Elguero
Magnetic Resonance in Chemistry 2001 Volume 39(Issue S1) pp:S100-S108
Publication Date(Web):5 NOV 2001
DOI:10.1002/mrc.937
Multinuclear liquid state magnetic resonance experiments have been performed on two seven-membered 15N-labeled H-chelates of the 6-aminofulvene-1-aldimines type in order to characterize the strong intramolecular NHN hydrogen bonds as a function of the molecular symmetry. In particular, the symmetrically substituted N,N′-diphenyl-6-aminopentafulvene-1-aldimine-15N2 (1) and its asymmetric analog N-phenyl-N′-(1,3,4-triazol)- 6-aminopentafulvene-1-aldimine-15N5 (2) have been studied. For 1, an NN coupling constant across the hydrogen bridge of 2hJ(15N,15N) = 10.6 Hz was determined indirectly by 13C NMR at two different Larmor frequencies, 125.76 and 67.93 MHz; this coupling constant is characteristically enhanced compared with the value of 8.6 Hz obtained previously for 2. Because of a fast degenerate proton tautomerism the hydrogen bond proton in 1 is coupled with both nitrogen atoms with a coupling constant of −40.8 Hz. {15N} tickling experiments were performed on 2 in order to determine the relative signs of the coupling constants of the NHN hydrogen bridge. We find that 2hJ(15N,15N) and 1hJ(1H,15N) = +4.4 Hz exhibit the same sign, i.e. the opposite sign compared with 1J(15N,1H) = −88.6 Hz. This finding proves that 1hJ(1H,15N) corresponds to an intrinsic coupling, which is not induced by a tautomerism absent in 2 because of the large difference in basicities of the aniline and the amino-1,3,4-triazole substituents. Therefore, these observations indicate a sign change of J(15N,1H) when the proton is transferred successively from one nitrogen to the other, as observed previously for FHF hydrogen bonds. The relation between the values of the coupling constants and the hydrogen bond geometries is discussed in terms of the valence bond order model, as are the implications for obtaining equilibrium constants of tautomerism from coupling constant data. Copyright © 2001 John Wiley & Sons, Ltd.
Co-reporter:Nikolai S. Golubev;Ilja G. Shenderovich;Sergei N. Smirnov;Gleb S. Denisov
Chemistry - A European Journal 1999 Volume 5(Issue 2) pp:
Publication Date(Web):4 FEB 1999
DOI:10.1002/(SICI)1521-3765(19990201)5:2<492::AID-CHEM492>3.0.CO;2-I
Spin–spin coupling between all three nuclei of a hydrogen bridge is seen for the first time. The observation of these couplings in the hydrogen fluoride/collidine complex (see figure) reveals a covalent character of the hydrogen bridge. Its geometry is strongly affected by temperature dependent solvent electric field effects.
Co-reporter:Dr. Ferdin Männle and; Dr. Hans-Heinrich Limbach Dipl.-Chem.
Angewandte Chemie 1996 Volume 108(Issue 4) pp:
Publication Date(Web):31 JAN 2006
DOI:10.1002/ange.19961080419
Co-reporter:Juan Miguel Lopez, Ferdinand Männle, Iwona Wawer, Gerd Buntkowsky and Hans-Heinrich Limbach
Physical Chemistry Chemical Physics 2007 - vol. 9(Issue 32) pp:NaN4513-4513
Publication Date(Web):2007/06/25
DOI:10.1039/B704384H
Using dynamic NMR spectroscopy, the kinetics of the degenerate double proton transfer in cyclic dimers of polycrystalline 15N,15N′-di-(4-bromophenyl)-formamidine (DBrFA) have been studied including the kinetic HH/HD/DD isotope effects in a wide temperature range. This transfer is controlled by intermolecular interactions, which in turn are controlled by the molecular conformation and hence the molecular structure. At low temperatures, rate constants were determined by line shape analysis of 15N NMR spectra obtained using cross-polarization (CP) and magic angle spinning (MAS). At higher temperatures, in the microsecond time scale, rate constants and kinetic isotope effects were obtained by a combination of longitudinal 15N and 2H relaxation measurements. 15N CPMAS line shape analysis was also employed to study the non-degenerate double proton transfer of polycrystalline 15N,15N′-diphenyl-formamidine (DPFA). The kinetic results are in excellent agreement with the kinetics of DPFA and 15N,15N′-di-(4-fluorophenyl)-formamidine (DFFA) studied previously for solutions in tetrahydrofuran. Two large HH/HD and HD/DD isotope effects are observed in the whole temperature range which indicates a concerted double proton transfer mechanism in the domain of the reaction energy surface. The Arrhenius curves are non-linear indicating a tunneling mechanism. Arrhenius curve simulations were performed using the Bell–Limbach tunneling model. The role of the phenyl group conformation and hydrogen bond compression on the barrier of the proton transfer is discussed.
Co-reporter:Alexandra Dos, Volkmar Schimming, Monique Chan-Huot and Hans-Heinrich Limbach
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 35) pp:NaN10245-10245
Publication Date(Web):2010/07/20
DOI:10.1039/C002730H
Using high resolution solid state 15N and 13C NMR spectroscopy we have studied the effects of successive hydration on the 15N labeled side chain amino groups of solid poly-L-lysine (PLL) in the presence of acids. Generally, hydration leads to the formation of local “ionic fluid” phases composed by flexible side chain ammonium groups, acid anions and small amounts of water. The associated local dynamics reduces the widths of the inhomogeneously broadened 15N amino signals found for the dry states. The hydration of free base PLL—which consists of mixtures of α-helices and β-pleated sheets—is monitored by a small low-field shift of the amino group signal arising from hydrogen bonding with water, reaching eventually the value of PLL in water at pH 13. No difference for the two conformations is observed. PLL×HF adopts a similar secondary structure with isolated NHF hydrogen bonds; hydration leads only to small low-field shifts which are nevertheless compatible with the formation of ammonium groups in aqueous solution. PLL doped with small amounts of HCl contains ammonium groups which are internally solvated by neighboring free amino groups. Both nitrogen environments are characterized by different chemical shifts. Hydration with less than one water molecule per amino group leads already to a chemical shift averaging arising from fast proton motions along NHN-hydrogen bonds and fast side chain and anion motions.
By contrast, the hydration of fully doped PLL×HBr and PLL×HCl is more complex. These systems exist only in β-pleated sheet conformations forming alkyl ammonium salt structures. Separate 15N signal components are observed for (i) the dry states, for (ii) wet β-pleated sheets and for (iii) wet α-helices which are successively formed upon hydration. Exchange between these environments is slow, but water motions lead to averaged amino group signals within each of the two wet environments. These results indicate that the different environments form domains. As the replacement of NHBr or of NHCl hydrogen bonds by NHO hydrogen bonds leads to high-field shifts the observation of separated signals is the result of different water content in the three domains. In agreement with previous X-ray powder diffraction studies we observe a dominance of the α-helical regions at above 3 water molecules per amino group in the case of PLL×HBr and at about 5 water molecules in the case of PLL×HCl, an effect arising from the limited space between β-pleated sheets and the larger volume of bromide as compared to chloride.