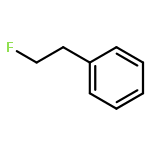
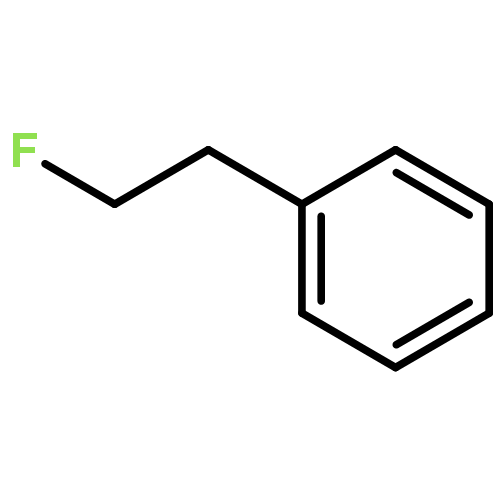
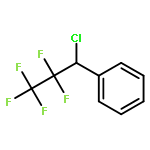
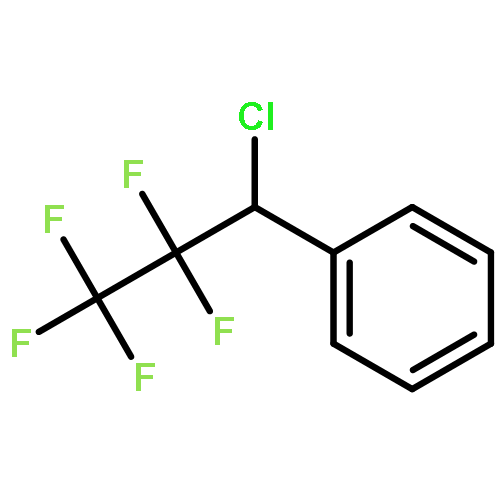
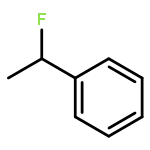
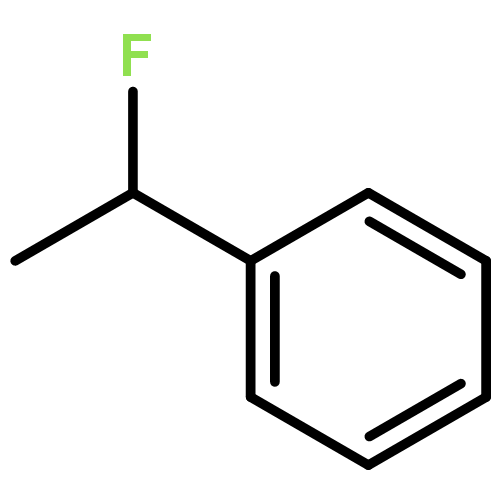
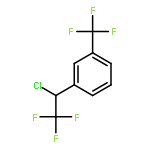
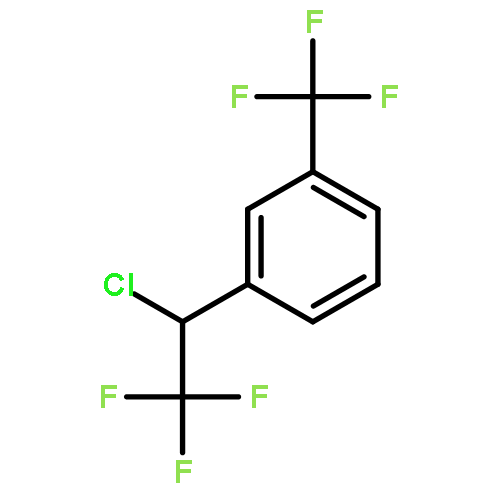
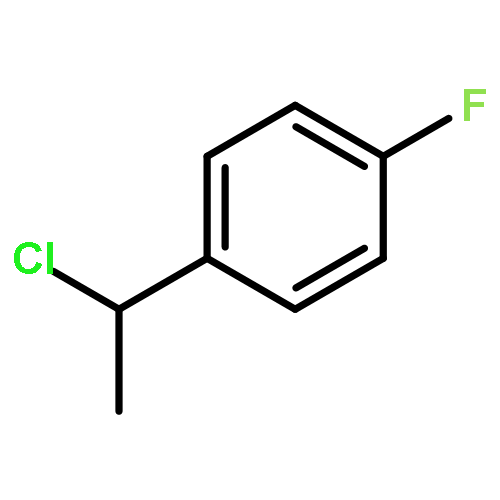
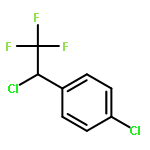
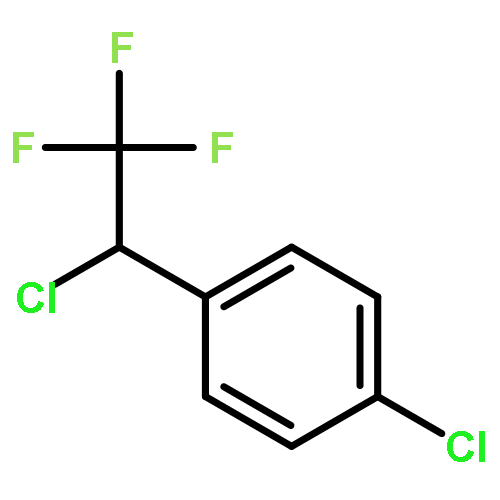
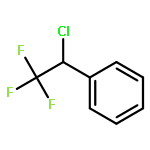
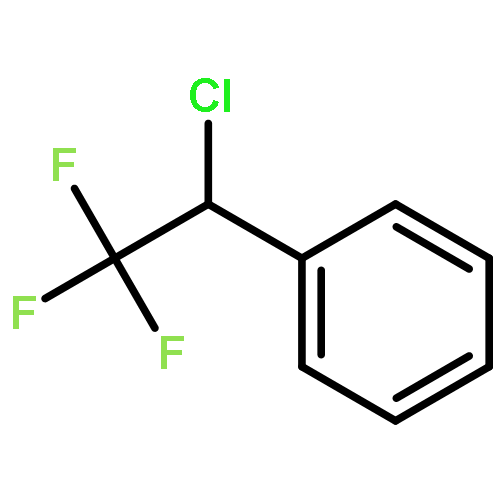
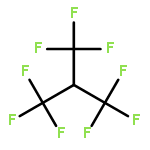
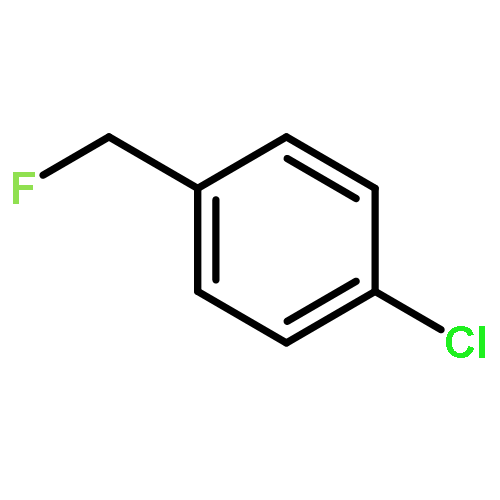
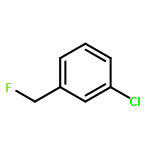
The gas-phase acidities (GA) of various aryl-substituted fluoroalkanes, XC6H4CH(R1)R2, were calculated at the B3LYP/6-311 + G(d,p)//B3LYP/6-311 + G(d,p). The acidity values of alkanes having a common substituent X varied significantly with the change of R1 and R2. Their changes in acidity of 1 and 2 having two strong electron-withdrawing groups (CF3 or C2F5) at the deprotonation site and 8, 9, 10, 11 having no fluorine atom at β-position were linearly correlated with the corrected number of fluorine atoms contained in the fluorinated alkyl group (R2 > 0.999). On the other hand, the GA values of β-fluorine substituted alkanes (3, 4, 5, 6, 7) deviated in a stronger acid direction from the line. The enhanced acidity was attributed to the additional stabilization of the conjugate anion caused by the β-fluorine negative hyperconjugation. The magnitude of β-fluorine negative hyperconjugation of the fluorinated alkyl group (ΔGoβ-F) given by the deviations from the line decreased with increasing electron-withdrawing ability of substituent X on the benzene ring, indicating that β-fluorine negative hyperconjugation competes with the electronic effect of the substituent X. The GAel values obtained by subtraction ΔGoβ-F from the apparent GA value were successfully correlated in terms of the Yukawa–Tsuno equation. The obtained ρel and r−el values were linearly related to the GAel value of the respective phenyl-substituted fluoroalkanes, supporting our previous conclusion that the ρ and r− values for the substituent effect caused by the electronic effects of the substituent on the acidity are determined by the thermodynamic stability of the parent ion (ring substituent = H). Copyright © 2015 John Wiley & Sons, Ltd.
The gas-phase acidity (GA) of a series of 1,1-bis(trifluoromethanesulfonyl)propane derivatives, Tf2CHCH2CH(R1)R2, and Tf2CHCH2Ar (Ar = phenol derivatives) was determined by measuring proton-transfer equilibria using a Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer and by computing the free energy of the deprotonated carbanions and the corresponding neutrals. The effects of R1 and R2 or Ar groups on the acidity were examined. In Tf2CHCH2CH(R1)R2, the GA values calculated using the conformers of neutral molecules that are free from the intramolecular hydrogen-bonding interaction between the acidic hydrogen atom of the Tf2CH moiety and R1 or R2 group and between the hydrogen atom of the CHR1R2 moiety and the SO2CF3 group were correlated in terms of an equation, GA = −17.0ΣσI + 3.4Σσα + 299.5. On the basis of this correlation, it was elucidated that the intramolecular hydrogen bonding or dipole–dipole interaction in the neutral molecule weakens significantly the acidity. In Tf2CHCH2Ar (5, 6 and 7), the GA was strengthened by the strong hydrogen-bonding interaction between the phenolic hydrogen in the aromatic moiety and the SO2CF3 group in the conjugate anion compared with that in the neutral molecule. Copyright © 2014 John Wiley & Sons, Ltd.
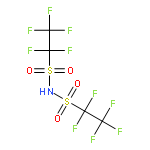
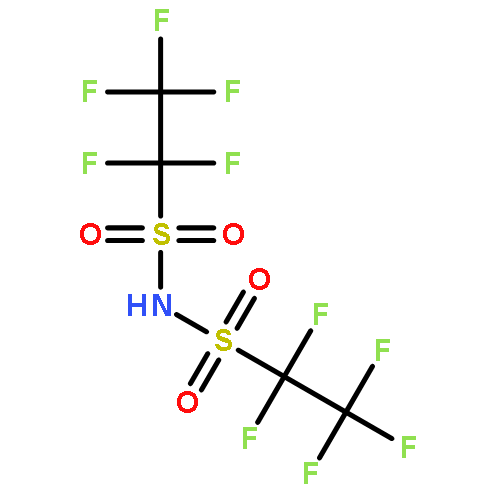
The gas-phase acidity (GA) values were determined for a number of perfluoroalkyl-substituted sulfonylimides by measuring proton-transfer equilibria using a Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer. The GA scale below 286.5 kcal mol−1 for (CF3SO2)2NH was extended and partially revised. The GA value of (C4F9SO2)2NH which is currently the strongest acid was revised from 284.1 to 278.6 kcal mol−1. The effect of fluorine atoms on the acidity of perfluoroalkyl-substituted sulfonylimides was described with the following model
where N(α), N(β), N(γ), and N(δ) are the numbers of fluorine atoms at α, β, γ, and δ position in RfSO2 (Rf = perfluoroalkyl group), respectively. This correlation indicates that the electron-withdrawing ability of the RfSO2 group can be described in terms of the number of fluorine atoms in the perfluoroalkyl group corrected by taking into account their positions. Copyright © 2014 John Wiley & Sons, Ltd.
Basicities of a number of ring-substituted N,N-dimethylanilines (DMA) and some related bases in water, acetonitrile, and THF and in the gas phase have been experimentally determined. Gas-phase basicities of DMAs and related bases were calculated at DFT B3LYP 6-311+G** and G3(MP2) levels. Structure–Basicity relationships in these four media were discussed. By comparison of gas-phase basicity shifts induced by stepwise substitution, starting from ammonia and ending at triphenylamine, it was observed that solvent effects of water and acetonitrile exceed the structural effects on intrinsic basicity. It was shown that the influence of substituents in the phenyl ring on DMA basicity is reduced by two to three times when going from the gas phase into the abovementioned condensed media. In the gas phase, 4-NO, 4-NO2, 4-CN, 4-COMe, and 4-CHO substituted DMAs protonate on substituent, whereas in solvents, only 4-NO-DMA probably protonates on substituent. The sensitivity of DMA basicity toward substitution in the phenyl ring was compared with the related family of phenylphosphazene bases, and it was found that the substituent effect is stronger in DMAs by 1.35 times in acetonitrile, 1.45 times in the gas phase, 2.0 times in THF, and 2.6 times in water. Copyright © 2012 John Wiley & Sons, Ltd.
The transient intermediates with infrared bands at 1676–1680 cm−1 observed for reaction of substituted phenylketenes with diethylamine in acetonitrile were suggested to be the amide enols rather than the zwitterions on the basis of the theoretical calculations. A single broad band at 1674 cm−1 observed for reaction with the primary amines was attributed to overlap of two bands of the intermediate (amide enol) and the final product (amide). The substituent effect for the second-order rate constants of diethylamine-catalyzed tautomerization of the amide enol intermediates to give the amides was analyzed successfully by the Yukawa–Tsuno equation, giving a ρ value of 0.63 and an r− value of 1.31. The r− value larger than unity for pKa of phenols indicates that the negative charge formed at an oxygen atom of the amide enol at the transition state is significantly delocalized into the aromatic π-system through the ethenyl group. This r− value was considered to reflect an intrinsic property of β-phenylenolate skeleton. A remarkably small ρ value attributes to the cyclic transition structure where the negative charge disperses in a six-member ring. Copyright © 2013 John Wiley & Sons, Ltd.
Pseudo-first-order rate constants (kobsd) for reactions of 4-nitrophenyl salicylate (7) with alkali metal ethoxides (EtOM, M=K, Na, and Li) in anhydrous ethanol have been measured spectrophotometrically. Interestingly, the kobsd value decreases significantly as the concentration of EtOM increases. Because the phenolic moiety of substrate 7 would be deprotonated and exist as an anionic form (i.e., 7−) under kinetic conditions, the ground-state stabilization of 7− through formation of a six-membered cyclic complex with M+ (i.e., 8) is proposed to be responsible for the decreasing kobsd trend. The kobsd value at a given concentration of EtOK increases steeply upon addition of [18]crown-6 ether (18C6) up to [18C6]/[EtOK]=1 in the reaction mixture and then remains relatively constant thereafter. In contrast, kobsd decreases upon addition of salts (e.g., LiClO4 or KSCN) to the reaction mixture, which indicates that M+ ions inhibit the reaction. However, in the presence of 18C6, the kobsd value is independent of the concentration of EtOK but remains constant, which indicates that the reaction proceeds through a unimolecular mechanism in the presence of the complexing agent. Although two conceivable unimolecular pathways (formation of ketene 9 and lactone 10) can account for the kinetic results, the reaction has been concluded to proceed via formation of ketene 9 as the reactive intermediate on the basis of theoretical calculations.
The relative free energy changes (lanthanum cation basicity, LaCB[L2]) for the reaction [La(OMe)2]L ⇌ La(OMe) + 2L were determined in the gas phase for m- and p-substituted acetophenones based on the measurement of ligand exchange equilibria using an FT-ICR mass spectrometer. The substituent effect on ΔLaCB[L2] of acetophenone is described in terms of the Yukawa–Tsuno equation, ΔG = ρ(σ° + r+ Δ σ), with a ρ value of −11.2 and an r+ value of 0.49. From this result, a ρ value of −7.0 and an r+ value of 0.49 were estimated for the monomeric complex [LLa(OMe)] with the aid of theoretical calculations. This ρ value was found to be significantly smaller than that for protonation, and even smaller than Li+ basicity. Such a small ρ value has been attributed to the largely ionic (ion–dipole interaction) nature of the bonding interaction between La(OMe) and the carbonyl oxygen atom and, in part, to the long distance between La(OMe) and the substituent. Contrary to the ρ value, the r+ value is identical in both La(OMe) and Li+ basicities, suggesting that the r+ value of 0.49 can be regarded as a limiting one in a series of Lewis cation basicities of the acetophenone system, H+ (0.86) > Me3Si+ (0.75) > Me3Ge+ (0.71) > Cu+ (0.60) > Li+ = La(OMe) (0.49). Since the binding interaction between La(OMe) or Li+ and a neutral ligand is mostly electrostatic, the moderate r+ was interpreted to result from the redistribution of the induced positive charge within the acetophenone moiety upon binding with a metal ion rather than transfer of positive charge from a metal ion to the aromatic moiety. Copyright © 2010 John Wiley & Sons, Ltd.
The relative free energy changes for the reaction ML+ = M+ + L (M = Cu+ and Li+) were determined in the gas phase for a series of dimethoxyalkanes (MeO(CH2)nOMe, n = 2–9) by measuring the equilibrium constants of ligand-transfer reactions using a FT-ICR mass spectrometry. Stable 1:1 Cu+-complexes (CuL+) were observed when the chain is longer than n = 4 while the 1:2 complexes (CuL) were formed for smaller compounds as stable ions. The dissociation free energy for CuL+ significantly increases with increasing chain length, by 10 kcal mol−1 from n = 4 to 9. This increase is attributed to the release of constrain involved in the cyclic conformation of the Cu+-complexes. This is consistent with the geometrical and energetic features of the complexes obtained by the DFT calculations at B3LYP/6-311G level of theory. On the contrary, the corresponding dissociation free energy for LiL+ increases only 3 kcal mol−1 from n = 2 to 9, although the structures of the 1:1 Li+-complexes are also considered to be cyclic. From these results it is concluded that the Cu[MeO(CH2)nOMe]+ requires linear alignment for OCuO, indicating the importance of sdσ hybridization of Cu+ in the first two ligands binding energy, while the stability of the Li+ complex is less sensitive to binding geometries except for the system forming a small ring such as n = 1 and 2. Copyright © 2006 John Wiley & Sons, Ltd.


![Ethanesulfonamide, 1,1,2,2,2-pentafluoro-N-[(trifluoromethyl)sulfonyl]-](http://img.cochemist.com/ccimg/152900/152894-11-6.png)
![Ethanesulfonamide, 1,1,2,2,2-pentafluoro-N-[(trifluoromethyl)sulfonyl]-](http://img.cochemist.com/ccimg/152900/152894-11-6_b.png)

![1-Hexanesulfonamide, 1,1,2,2,3,3,4,4,5,5,6,6,6-tridecafluoro-N-[(trifluoromethyl)sulfonyl]-](http://img.cochemist.com/ccimg/110900/110874-76-5.png)
![1-Hexanesulfonamide, 1,1,2,2,3,3,4,4,5,5,6,6,6-tridecafluoro-N-[(trifluoromethyl)sulfonyl]-](http://img.cochemist.com/ccimg/110900/110874-76-5_b.png)








![Propane, 1,1,3,3-tetrakis[(trifluoromethyl)sulfonyl]-](http://img.cochemist.com/ccimg/60900/60805-11-0.png)
![Propane, 1,1,3,3-tetrakis[(trifluoromethyl)sulfonyl]-](http://img.cochemist.com/ccimg/60900/60805-11-0_b.png)

![8-METHOXYPYRIDO[2,3-D]PYRIDAZINE](http://img.cochemist.com/ccimg/39900/39847-39-7.png)
![8-METHOXYPYRIDO[2,3-D]PYRIDAZINE](http://img.cochemist.com/ccimg/39900/39847-39-7_b.png)










![BICYCLO[2.2.1]HEPTANE, 2,2,3,3,5,5,6,6,7,7-DECAFLUORO-](http://img.cochemist.com/ccimg/5000/4957-92-0.png)
![BICYCLO[2.2.1]HEPTANE, 2,2,3,3,5,5,6,6,7,7-DECAFLUORO-](http://img.cochemist.com/ccimg/5000/4957-92-0_b.png)
![2,2,3,3,4,5,5,6,6,7,7-UNDECAFLUOROBICYCLO[2.2.1]HEPTANE](http://img.cochemist.com/ccimg/5000/4934-61-6.png)
![2,2,3,3,4,5,5,6,6,7,7-UNDECAFLUOROBICYCLO[2.2.1]HEPTANE](http://img.cochemist.com/ccimg/5000/4934-61-6_b.png)




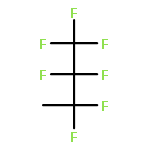
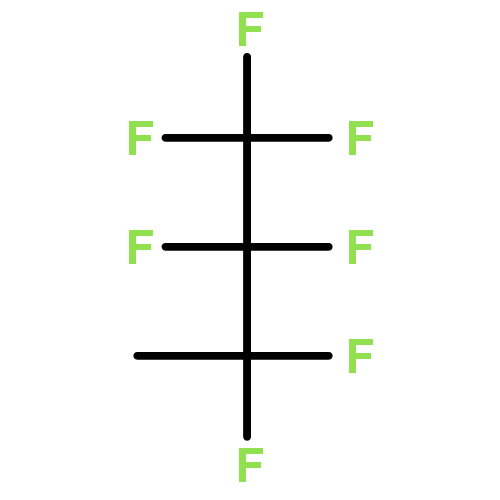



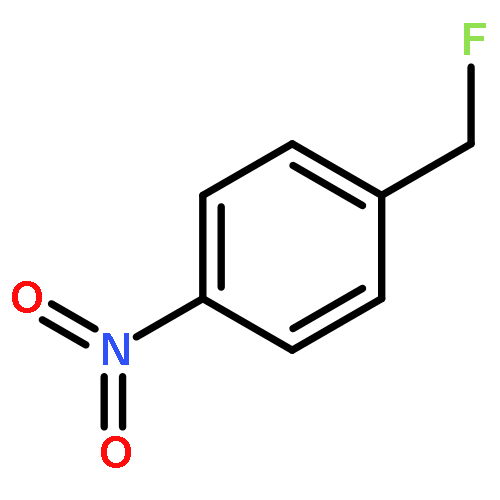




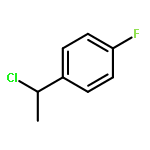








![Benzene, [3,3,3-trifluoro-2,2-bis(trifluoromethyl)propyl]-](http://img.cochemist.com/ccimg/36600/36553-72-7.png)
![Benzene, [3,3,3-trifluoro-2,2-bis(trifluoromethyl)propyl]-](http://img.cochemist.com/ccimg/36600/36553-72-7_b.png)