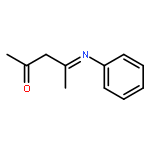
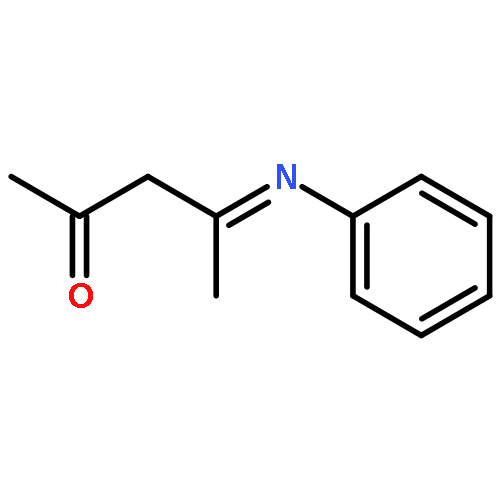
Co-reporter:Ku Sun Choung, Mohammad Din Islam, Richard W. Guo, and Thomas S. Teets
Inorganic Chemistry November 20, 2017 Volume 56(Issue 22) pp:14326-14326
Publication Date(Web):November 7, 2017
DOI:10.1021/acs.inorgchem.7b02438
Presented here are complexes of two different fluorinated β-diketiminate (NacNac) ligands with cyclometalated platinum. Reaction of the cyclometalated platinum dimers [Pt(C^N)(μ-X)]2 [C^N = 2-phenylpyridine (ppy), 2-(2,4-difluorophenyl)pyridine (F2ppy); X = Cl, Br] with lithium salts of backbone-fluorinated β-diketiminate ligands produces two structure types, depending on the temperature of the reaction. At milder temperatures (<80 °C), the major product is an unusual halide-bridged diplatinum complex, demonstrating a unique NacNac binding mode bridging the two platinum centers. At higher temperatures (>100 °C), the major species is a monoplatinum complex of the type Pt(C^N)(NacNac). The complexes display reduction waves in their cyclic voltammograms at mild potentials, as well as intense visible absorption bands (λ > 500 nm), that depend minimally on the identity of the C^N ligand or, in the case of the bimetallic complexes, the identity of the bridging halide. In addition, the monoplatinum complexes exhibit structured luminescence in the red and near-infrared regions deriving a NacNac-centered triplet state. All of these observations suggest that the NacNac π system contributes substantially to the frontier orbitals and motivates continued exploration of fluorinated β-diketiminate ligands in the design of complexes with desirable ligand-based redox and optical properties.
Co-reporter:Jong-Hwa Shon and Thomas S. Teets
Inorganic Chemistry December 18, 2017 Volume 56(Issue 24) pp:15295-15295
Publication Date(Web):November 27, 2017
DOI:10.1021/acs.inorgchem.7b02859
In this work, we outline a strategy to prepare a class of improved visible-light photosensitizers. Bis-cyclometalated iridium complexes with electron-rich β-diketiminate (NacNac) ancillary ligands are demonstrated to be potent excited-state electron donors. Evaluation of the photophysical and electrochemical properties establishes the excited-state redox potentials of the complexes, and Stern–Volmer quenching experiments inform on the kinetics of photoinduced electron transfer to the model substrates methyl viologen (MV2+) and benzophenone (BP). Compared to fac-Ir(ppy)3 (ppy = 2-phenylpyridine), widely regarded as a state-of-the-art photoreductant, the complexes we describe have excited-state redox potentials that are more potent by 300–400 mV and rates for photoinduced electron transfer that are accelerated by as much as a factor of 3. These complexes emerge as promising targets for application in photocatalytic reactions and other photochemical processes.
Co-reporter:Hanah Na, Ayan Maity, Rakib Morshed, and Thomas S. Teets
Organometallics August 14, 2017 Volume 36(Issue 15) pp:2965-2965
Publication Date(Web):July 19, 2017
DOI:10.1021/acs.organomet.7b00428
In this work, we report that covalent postsynthetic modification can be used for the preparation of a class of bis-cyclometalated iridium complexes featuring Chugaev-type chelating dicarbene ligands. Bis-cyclometalated iridium complexes with electron-deficient aryl isocyanide ancillary ligands react with hydrazine to form neutral dicarbene complexes. The neutral iridium carbene complexes have a basic site that can be protonated by strong acid, permitting access to complexes in two protonation states and allowing an additional layer of control over the key properties. These new Chugaev-type iridium complexes exhibit blue phosphorescence at both room temperature and 77 K. Compared to their bis-isocyanide precursors, the electrochemical and photophysical properties of these new complexes are substantially perturbed, demonstrating the concept that the electronic structure and excited state dynamics can be controlled by ancillary ligand modification. Furthermore, the emission spectra and excited-state dynamics are dependent on the protonation state of the dicarbene ancillary ligand, and we note an ∼2-fold increase in emission quantum yield when the ancillary ligand is protonated. This study demonstrates that ligand-based reactivity can be an alternative method for elaborating the structures of bis-cyclometalated iridium complexes and gives access to structures not readily obtainable by other means.
Co-reporter:Ayan Maity;Jonas C. Kölsch;Hanah Na
Dalton Transactions 2017 vol. 46(Issue 35) pp:11757-11767
Publication Date(Web):2017/09/11
DOI:10.1039/C7DT02540H
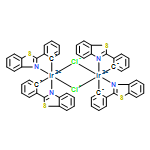
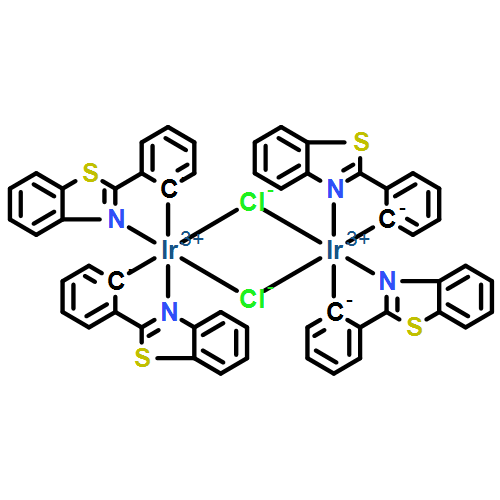
Transmetallation reactions involving organoboron reagents and transition metals are legion in synthetic organometallic chemistry and homogeneous catalysis. Triarylboranes (BAr3) have been observed to participate in transmetallation reactions with many transition metals, typically following abstraction of an alkyl (R−) or hydride ligand by the Lewis acidic borane. Here, an unusual transmetallation strategy is described where an aryl group from a borane replaces a weakly coordinated PF6− ligand. The precursors Ir(C^N)2(CNAr)(FPF5) (C^N = cyclometallating ligand, CNAr = aryl isocyanide) react smoothly with B(C6F5)3 to give complexes of the type Ir(C^N)2(CNAr)(C6F5), a previously unobserved structure type featuring an unchelated aryl ligand. The reaction tolerates a variety of C^N ligands and a range of electronically and sterically varied aryl isocyanide ancillary ligands. A total of six complexes of this type are described, two of which are characterized by single-crystal X-ray diffraction. All but one of the complexes luminesces at room temperature, with the emission wavelength dependent on the C^N ligand.
Co-reporter:Hanah Na;Ayan Maity
Dalton Transactions 2017 vol. 46(Issue 15) pp:5008-5016
Publication Date(Web):2017/04/10
DOI:10.1039/C7DT00694B
In this work we report a study on the effect of systematic ancillary ligand modifications on electrochemical and photophysical properties of cationic biscyclometalated bis(arylisocyanide)iridium(III) complexes. Nine new Ir(III) complexes were synthesized using three different cyclometalating (C^N) ligands (2,4-difluorophenylpyridine (F2ppy), 2-benzothienylpyridine (btp), and 2-phenylbenzothiazole (bt)) with three aryl isocyanide ancillary ligands (2,4-dimethoxyphenyl isocyanide (CNArOMe), 3,5-bis(trifluoromethyl)phenyl isocyanide (CNArCF3) and 4-nitrophenyl isocyanide (CNArNO2)). Systematic modifications of ancillary ligands with electron-donating or electron-withdrawing groups have a very minor influence on the positions of the absorption and emission bands, suggesting that aryl isocyanide ancillary ligands minimally perturb the primarily ligand-centered emissive states, but still can control the dynamics of the excited state. Replacing electron-donating groups with electron-withdrawing group influences kr and/or knr, resulting in changes in the lifetimes and quantum yields. In addition, we reveal that electronic structures can be substantially altered by incorporating electron-donating or electron-withdrawing groups on the aryl isocyanide ancillary ligand, with different magnitudes of the perturbation depending on the cyclometalating C^N ligand. Particularly, the formally IrIV/IrIII oxidation couple can be perturbed by over 200 mV when electron-donating substituents are replaced with electron-withdrawing groups.
Co-reporter:Ayan Maity and Thomas S. Teets
Chemical Reviews 2016 Volume 116(Issue 15) pp:8873-8911
Publication Date(Web):May 10, 2016
DOI:10.1021/acs.chemrev.6b00034
This Review highlights stoichiometric reactions and elementary steps of catalytic reactions involving cooperative participation of transition-metal hydrides and main group Lewis acids. Included are reactions where the transition-metal hydride acts as a reactant as well as transformations that form the metal hydride as a product. This Review is divided by reaction type, illustrating the diverse roles that Lewis acids can play in mediating transformations involving transition-metal hydrides as either reactants or products. We begin with a discussion of reactions where metal hydrides form direct adducts with Lewis acids, elaborating the structure and dynamics of the products of these reactions. The bulk of this Review focuses on reactions where the transition metal and Lewis acid act in cooperation, and includes sections on carbonyl reduction, H2 activation, and hydride elimination reactions, all of which can be promoted by Lewis acids. Also included is a section on Lewis acid–base secondary coordination sphere interactions, which can influence the reactivity of hydrides. Work from the past 50 years is included, but the majority of this Review focuses on research from the past decade, with the intent of showcasing the rapid emergence of this field and the potential for further development into the future.
Co-reporter:Evanta Kabir; Chia-Hua Wu; Judy I-Chia Wu
Inorganic Chemistry 2016 Volume 55(Issue 2) pp:956-963
Publication Date(Web):December 24, 2015
DOI:10.1021/acs.inorgchem.5b02595
Formazanates are a ligand class featuring a 1,2,4,5-tetraazapentadienyl core, with variable substitution at the 1, 3, and 5 positions. Here we describe a set of four heteroleptic cylcometalated platinum complexes containing triarylformazanate ligands. The complexes are prepared by metathesis reactions of chloro-bridged dimers [Pt(C∧N)(μ-Cl)]2 (C∧N = 2-phenylpyridine or 2-(2,4-difluorophenyl)pyridine) with triarylformazans in the presence of base. X-ray diffraction studies reveal the molecular structures of three such complexes. Cyclic voltammograms and UV–vis absorption spectra of the complexes show features characteristic of both the cyclometalated platinum fragment and the formazanate, with the latter giving rise to two reversible one-electron reductions in the CV and an intense visible π → π* absorption which is red-shifted by >100 nm relative to the free formazan. The electronic structures and redox properties of the complexes were further investigated by UV–vis spectroelectrochemistry and density functional theory calculations. All of the experimental and theoretical work points to a frontier molecular orbital manifold where the formazanate π and π* orbitals are substantially mixed with d-orbitals derived from the platinum center.
Co-reporter:Ayan Maity, Linh Q. Le, Zhuan Zhu, Jiming Bao, and Thomas S. Teets
Inorganic Chemistry 2016 Volume 55(Issue 5) pp:2299-2308
Publication Date(Web):February 24, 2016
DOI:10.1021/acs.inorgchem.5b02691
Cyclometalated iridium complexes with efficient phosphorescence and good electrochemical stability are important candidates for optoelectronic devices. Isocyanide ligands are strong-field ligands: when attached to transition metals, they impart larger HOMO–LUMO energy gaps, engender higher oxidative stability at the metal center, and support rugged organometallic complexes. Aryl isocyanides offer more versatile steric and electronic control by selective substitution at the aryl ring periphery. Despite a few reports of alkyl isocyanide of cyclometalated iridium(III), detailed studies on analogous aryl isocyanide complexes are scant. We report the synthesis, photophysical properties, and electrochemical properties of 11 new luminescent cationic biscyclometalated bis(aryl isocyanide)iridium(III) complexes. Three different aryl isocyanides—2,6-dimethylphenyl isocyanide (CNArdmp), 2,6-diisopropylphenyl isocyanide (CNArdipp), and 2-naphthyl isocyanide (CNArnap)—were combined with four cyclometalating ligands with differential π–π* energies—2-phenylpyridine (ppy), 2,4-difluorophenylpyridine (F2ppy), 2-benzothienylpyridine (btp), and 2-phenylbenzothiazole (bt). Five of them were crystallographically characterized. All new complexes show wide redox windows, with reduction potentials falling in a narrow range of −2.02 to −2.37 V and oxidation potentials spanning a wider range of 0.97–1.48 V. Efficient structured emission spans from the blue region for [(F2ppy)2Ir(CNAr)2]PF6 to the orange region for [(btp)2Ir(CNAr)2]PF6, demonstrating that isocyanide ligands can support redox-stable luminescent complexes with a range of emission colors. Emission quantum yields were generally high, reaching a maximum of 0.37 for two complexes, whereas btp-ligated complexes had quantum yields below 1%. The structure of the CNAr ligand has a minimal effect on the photophysical properties, which are shown to arise from ligand-centered excited states with very little contribution from metal-to-ligand charge transfer in most cases.
Co-reporter:Rosa A. Maya, Ayan Maity, and Thomas S. Teets
Organometallics 2016 Volume 35(Issue 17) pp:2890-2899
Publication Date(Web):August 16, 2016
DOI:10.1021/acs.organomet.6b00453
In this work we describe a series of bis-cyclometalated iridium complexes with ancillary β-ketoiminate (acNac) and β-diketiminate (NacNac) ligands, prepared by a general synthetic route. Fluorination of these ligands by introducing CF3 substituents onto the ligand backbone and/or N-aryl substituent(s) leads to pronounced changes in the redox properties and photophysical properties. All of the complexes show a reversible IrIV/IrIII redox couple that is sensitive to the degree of fluorination on the ancillary ligand. Introduction of CF3 groups at the 3- and 5-positions of the N-aryl substituent shifts the potential positive by ca. 50–70 mV per CF3 group, whereas fluorination of the acNac or NacNac backbone induces larger shifts of ca. 200–300 mV per CF3 group. Fluorination of the NacNac backbone gives rise to substantially altered excited-state properties. Complexes with backbone CF3 groups luminesce in the red and near-infrared regions out of an excited state that is predominately a π → π* NacNac-centered triplet state. A preponderance of evidence supports the assignment of this low-energy feature, including minimal dependence of this emission feature on the identity of the cyclometalating ligand, pronounced vibronic structure, and microsecond lifetimes. These results demonstrate that acNac and NacNac ancillary ligands can engender cyclometalated iridium complexes with desirable and readily tailorable redox and optical properties, motivating continued application of this important ligand class to the design of phosphorescent organometallic molecules.
Co-reporter:Hanah Na, Ayan Maity, and Thomas S. Teets
Organometallics 2016 Volume 35(Issue 13) pp:2267-2274
Publication Date(Web):June 17, 2016
DOI:10.1021/acs.organomet.6b00332
In this work we show that postsynthetic addition of borane Lewis acids to Lewis base decorated organoplatinum photosensitizers induces significant changes in the optical and electrochemical properties. In particular, the charge transfer (CT) energies of these chromophores are significantly modified by these outer-sphere interactions. The direction of the CT shift depends on the site of Lewis acid binding, which occurs either at the diimine ligand in bipyrazine-linked molecules or at an ancillary acetylide ligand in pyridyl-substituted bis(acetylide) molecules. The magnitude of the shift depends on the Lewis acidity of the borane and the number of equivalents added and is comparable to the perturbation brought on by covalent substituent modification of supporting ligands in related complexes. This approach offers a new means of tuning the properties of organometallic phosphors that complements the traditional approach of covalent modification and other postsynthetic modification strategies.
Co-reporter:Yousf K. Radwan; Ayan Maity
Inorganic Chemistry 2015 Volume 54(Issue 14) pp:7122-7131
Publication Date(Web):July 9, 2015
DOI:10.1021/acs.inorgchem.5b01401
A series of cyclometalated iridium complexes with β-ketoiminate and β-diketiminate ligands are described. Two different cyclometalating (C^N) ligands—2-phenylpridine (ppy) and 2-phenylbenzothiazole (bt)—are used in concert with three different ancillary (LX) ligands—a phenyl-substituted β-ketoiminate (acNacMe), a phenyl-substituted β-diketiminate (NacNacMe), and a fluorinated version of the β-diketiminate (NacNacCF3)—to furnish a suite of six complexes. The complexes are prepared by metathesis reactions of chloro-bridged dimers [Ir(C^N)2(μ-Cl)]2 with potassium or lithium salts of the ancillary LX ligand. Four of the complexes are characterized by X-ray crystallography, and all six were subjected to in-depth optical and electrochemical interrogation. Cyclic voltammetry shows both reduction and oxidation waves, with the latter strongly dependent on the identity of the LX ligand. The complexes are all luminescent, with the nature of the emissive excited state and the quantum yield (Φ) dependent on the identity of both the C^N and LX ligands. Whereas the complexes Ir(ppy)2(NacNacMe) and Ir(ppy)2(acNacMe) are weakly luminescent (Φ ≈ 0.01), the complexes Ir(bt)2(NacNacMe) and Ir(bt)2(acNacMe) are strongly luminescent, with the latter’s quantum efficiency (Φ = 0.82) among the highest ever observed for cyclometalated iridium complexes. Fluorination of the NacNac ligand gives rise to completely disparate emission behavior suggestive of a NacNac-centered emissive state. The results described here, in comparison with previous groups’ studies on acetylacetonate (acac) analogues, suggest that the weaker-field NacNac and acNac ligands raise the energy of the metal-centered HOMO, with energy of the HOMO increasing in the order NacNacCF3 < acNacMe < NacNacMe.
Co-reporter:Hanah Na, Ayan Maity and Thomas S. Teets
Dalton Transactions 2017 - vol. 46(Issue 15) pp:NaN5016-5016
Publication Date(Web):2017/03/22
DOI:10.1039/C7DT00694B
In this work we report a study on the effect of systematic ancillary ligand modifications on electrochemical and photophysical properties of cationic biscyclometalated bis(arylisocyanide)iridium(III) complexes. Nine new Ir(III) complexes were synthesized using three different cyclometalating (C^N) ligands (2,4-difluorophenylpyridine (F2ppy), 2-benzothienylpyridine (btp), and 2-phenylbenzothiazole (bt)) with three aryl isocyanide ancillary ligands (2,4-dimethoxyphenyl isocyanide (CNArOMe), 3,5-bis(trifluoromethyl)phenyl isocyanide (CNArCF3) and 4-nitrophenyl isocyanide (CNArNO2)). Systematic modifications of ancillary ligands with electron-donating or electron-withdrawing groups have a very minor influence on the positions of the absorption and emission bands, suggesting that aryl isocyanide ancillary ligands minimally perturb the primarily ligand-centered emissive states, but still can control the dynamics of the excited state. Replacing electron-donating groups with electron-withdrawing group influences kr and/or knr, resulting in changes in the lifetimes and quantum yields. In addition, we reveal that electronic structures can be substantially altered by incorporating electron-donating or electron-withdrawing groups on the aryl isocyanide ancillary ligand, with different magnitudes of the perturbation depending on the cyclometalating C^N ligand. Particularly, the formally IrIV/IrIII oxidation couple can be perturbed by over 200 mV when electron-donating substituents are replaced with electron-withdrawing groups.
.jpg)










![N-[(e)-4-phenyliminopent-2-en-2-yl]aniline](http://img.cochemist.com/ccimg/19200/19164-92-2.png)
![N-[(e)-4-phenyliminopent-2-en-2-yl]aniline](http://img.cochemist.com/ccimg/19200/19164-92-2_b.png)