Co-reporter:Ruixue Wan;Gaoxingyu Huang;Chuangye Yan;Rui Bai
Science 2017 Volume 355(Issue 6321) pp:
Publication Date(Web):
DOI:10.1126/science.aak9979
Poised for the second step of splicing
In eukaryotes, noncoding sequences in transcribed precursor mRNA are cut out by a dynamic macromolecular machine, the spliceosome. This involves two sequential reactions. The first cuts one end of the noncoding intron and loops it back on itself to form an intron lariat, and the next excises the intron and ligates the coding mRNA. Insights into the first step of splicing have come from the structures of two intermediates: the Bact complex, which is primed for catalysis, and the C complex, which is formed after the first splicing reaction. Yan et al. now report a high-resolution structure of the step II catalytically activated spliceosome (the C* complex). This structure shows conformational changes that position catalytic motifs to accomplish the second splicing reaction.
Science, this issue p. 149
Co-reporter:Rui Bai, Chuangye Yan, Ruixue Wan, Jianlin Lei, Yigong Shi
Cell 2017 Volume 171, Issue 7(Volume 171, Issue 7) pp:
Publication Date(Web):14 December 2017
DOI:10.1016/j.cell.2017.10.038
•Cryo-EM structure of the yeast post-catalytic spliceosome (the P complex) at 3.6 Å•The ligated exon is anchored to the loop I of U5 snRNA•The 3′-splice site is recognized by conserved RNA nucleotides and the Prp8 1585-loop•Structural comparison reveals the active site changes in the C∗-P-ILS transitionRemoval of an intron from a pre-mRNA by the spliceosome results in the ligation of two exons in the post-catalytic spliceosome (known as the P complex). Here, we present a cryo-EM structure of the P complex from Saccharomyces cerevisiae at an average resolution of 3.6 Å. The ligated exon is held in the active site through RNA-RNA contacts. Three bases at the 3′ end of the 5′ exon remain anchored to loop I of U5 small nuclear RNA, and the conserved AG nucleotides of the 3′-splice site (3′SS) are specifically recognized by the invariant adenine of the branch point sequence, the guanine base at the 5′ end of the 5′SS, and an adenine base of U6 snRNA. The 3′SS is stabilized through an interaction with the 1585-loop of Prp8. The P complex structure provides a view on splice junction formation critical for understanding the complete splicing cycle.Download high-res image (281KB)Download full-size image
Co-reporter:Ruixue Wan, Chuangye Yan, Rui Bai, Jianlin Lei, Yigong Shi
Cell 2017 Volume 171, Issue 1(Volume 171, Issue 1) pp:
Publication Date(Web):21 September 2017
DOI:10.1016/j.cell.2017.08.029
•Cryo-EM structure of the S. cerevisiae intron lariat spliceosome (ILS) at 3.5 Å•The ATPase/helicase Prp43 binds Syf1 and is located near the 3′ end of U6 snRNA•The Ntr complex proteins Ntr1 and Ntr2 are anchored on Snu114 and Prp8, respectively•Prp43 may pull the intron or the 3′ end of U6 snRNA to disassemble the ILS complexThe disassembly of the intron lariat spliceosome (ILS) marks the end of a splicing cycle. Here we report a cryoelectron microscopy structure of the ILS complex from Saccharomyces cerevisiae at an average resolution of 3.5 Å. The intron lariat remains bound in the spliceosome whereas the ligated exon is already dissociated. The step II splicing factors Prp17 and Prp18, along with Cwc21 and Cwc22 that stabilize the 5′ exon binding to loop I of U5 small nuclear RNA (snRNA), have been released from the active site assembly. The DEAH family ATPase/helicase Prp43 binds Syf1 at the periphery of the spliceosome, with its RNA-binding site close to the 3′ end of U6 snRNA. The C-terminal domain of Ntr1/Spp382 associates with the GTPase Snu114, and Ntr2 is anchored to Prp8 while interacting with the superhelical domain of Ntr1. These structural features suggest a plausible mechanism for the disassembly of the ILS complex.Download high-res image (306KB)Download full-size image
Co-reporter:Linfeng Sun;Rui Zhou;Guanghui Yang
PNAS 2017 Volume 114 (Issue 4 ) pp:E476-E485
Publication Date(Web):2017-01-24
DOI:10.1073/pnas.1618657114
A hallmark of Alzheimer’s disease (AD) is the aggregation of β-amyloid peptides (Aβ) into amyloid plaques in patient brain.
Cleavage of amyloid precursor protein (APP) by the intramembrane protease γ-secretase produces Aβ of varying lengths, of which
longer peptides such as Aβ42 are thought to be more harmful. Increased ratios of longer Aβs over shorter ones, exemplified
by the ratio of Aβ42 over Aβ40, may lead to formation of amyloid plaques and consequent development of AD. In this study,
we analyzed 138 reported mutations in human presenilin-1 (PS1) by individually reconstituting the mutant PS1 proteins into
anterior-pharynx–defective protein 1 (APH-1)aL–containing γ-secretases and examining their abilities to produce Aβ42 and Aβ40
in vitro. About 90% of these mutations lead to reduced production of Aβ42 and Aβ40. Notably, 10% of these mutations result
in decreased Aβ42/Aβ40 ratios. There is no statistically significant correlation between the Aβ42/Aβ40 ratio produced by a
γ-secretase variant containing a specific PS1 mutation and the mean age at onset of patients from whom the mutation was isolated.
Co-reporter:Yini Li;Mengying Zhou;Qi Hu;Xiao-chen Bai;Weiyun Huang;Sjors H. W. Scheres
PNAS 2017 Volume 114 (Issue 7 ) pp:1542-1547
Publication Date(Web):2017-02-14
DOI:10.1073/pnas.1620626114
Mammalian intrinsic apoptosis requires activation of the initiator caspase-9, which then cleaves and activates the effector
caspases to execute cell killing. The heptameric Apaf-1 apoptosome is indispensable for caspase-9 activation by together forming
a holoenzyme. The molecular mechanism of caspase-9 activation remains largely enigmatic. Here, we report the cryoelectron
microscopy (cryo-EM) structure of an apoptotic holoenzyme and structure-guided biochemical analyses. The caspase recruitment
domains (CARDs) of Apaf-1 and caspase-9 assemble in two different ways: a 4:4 complex docks onto the central hub of the apoptosome,
and a 2:1 complex binds the periphery of the central hub. The interface between the CARD complex and the central hub is required
for caspase-9 activation within the holoenzyme. Unexpectedly, the CARD of free caspase-9 strongly inhibits its proteolytic
activity. These structural and biochemical findings demonstrate that the apoptosome activates caspase-9 at least in part through
sequestration of the inhibitory CARD domain.
Co-reporter:Xiaofeng Zhang, Chuangye Yan, Jing Hang, Lorenzo I. Finci, ... Yigong Shi
Cell 2017 Volume 169, Issue 5(Volume 169, Issue 5) pp:
Publication Date(Web):18 May 2017
DOI:10.1016/j.cell.2017.04.033
•The first atomic structure of a human spliceosome•The conformation of the spliceosome just prior to exon ligation•Mechanistic insights on Prp17, Slu7, PRKRIP1, and the EJC•Intron interlocked with RBM22 suggests mechanisms of intron recruitment and releaseMechanistic understanding of pre-mRNA splicing requires detailed structural information on various states of the spliceosome. Here we report the cryo electron microscopy (cryo-EM) structure of the human spliceosome just before exon ligation (the C∗ complex) at an average resolution of 3.76 Å. The splicing factor Prp17 stabilizes the active site conformation. The step II factor Slu7 adopts an extended conformation, binds Prp8 and Cwc22, and is poised for selection of the 3′-splice site. Remarkably, the intron lariat traverses through a positively charged central channel of RBM22; this unusual organization suggests mechanisms of intron recruitment, confinement, and release. The protein PRKRIP1 forms a 100-Å α helix linking the distant U2 snRNP to the catalytic center. A 35-residue fragment of the ATPase/helicase Prp22 latches onto Prp8, and the quaternary exon junction complex (EJC) recognizes upstream 5′-exon sequences and associates with Cwc22 and the GTPase Snu114. These structural features reveal important mechanistic insights into exon ligation.Download high-res image (376KB)Download full-size image
Co-reporter:Yigong Shi
Journal of Molecular Biology 2017 Volume 429, Issue 17(Volume 429, Issue 17) pp:
Publication Date(Web):18 August 2017
DOI:10.1016/j.jmb.2017.07.010
Pre-mRNA splicing is executed by the ribonucleoprotein machinery spliceosome. Nearly 40 years after the discovery of pre-mRNA splicing, the atomic structure of the spliceosome has finally come to light. Four distinct conformational states of the yeast spliceosome have been captured at atomic or near-atomic resolutions. Two catalytic metal ions at the active site are specifically coordinated by the U6 small nuclear RNA (snRNA) and catalyze both the branching reaction and the exon ligation. Of the three snRNAs in the fully assembled spliceosome, U5 and U6, along with 30 contiguous nucleotides of U2 at its 5′-end, remain structurally rigid throughout the splicing reaction. The rigidity of these RNA elements is safeguarded by Prp8 and 16 core protein components, which maintain the same overall conformation in all structurally characterized spliceosomes during the splicing reaction. Only the sequences downstream of nucleotide 30 of U2 snRNA are mobile; their movement, directed by the protein components, delivers the intron branch site into the close proximity of the 5′-splice site for the branching reaction. A set of additional structural rearrangement is required for exon ligation, and the lariat junction is moved out of the active site for recruitment of the 3′-splice site and 3′-exon. The spliceosome is proven to be a protein-directed metalloribozyme.
Co-reporter:Jianping Wu;Tianyu Jiang;Minhao Liu
PNAS 2016 Volume 113 (Issue 8 ) pp:2074-2079
Publication Date(Web):2016-02-23
DOI:10.1073/pnas.1525616113
HBx is a hepatitis B virus protein that is required for viral infectivity and replication. Anti-apoptotic Bcl-2 family members
are thought to be among the important host targets of HBx. However, the structure and function of HBx are poorly understood
and the molecular mechanism of HBx-induced carcinogenesis remains unknown. In this study, we report biochemical and structural
characterization of HBx. The recombinant HBx protein contains metal ions, in particular iron and zinc. A BH3-like motif in
HBx (residues 110–135) binds Bcl-2 with a dissociation constant of ∼193 μM, which is drastically lower than that for a canonical
BH3 motif from Bim or Bad. Structural analysis reveals that, similar to other BH3 motifs, the BH3-like motif of HBx adopts
an amphipathic α-helix and binds the conserved BH3-binding groove on Bcl-2. Unlike the helical Bim or Bad BH3 motif, the C-terminal
portion of the bound HBx BH3-like motif has an extended conformation and makes considerably fewer interactions with Bcl-2.
These observations suggest that HBx may modulate Bcl-2 function in a way that is different from that of the classical BH3-only
proteins.
Co-reporter:Chuangye Yan;Ruixue Wan;Rui Bai;Gaoxingyu Huang
Science 2016 Vol 353(6302) pp:904-911
Publication Date(Web):26 Aug 2016
DOI:10.1126/science.aag0291
How spliceosomes make the first cut
In eukaryotes, transcribed precursor mRNA includes noncoding sequences that must be spliced out. This is done by the spliceosome, a dynamic complex in which five small nuclear RNAs and several proteins go through a series of ordered interactions and conformational rearrangements to achieve splicing. Two protein structures provide a look at the first catalytic step in the pathway. Yan et al. report the structure of the activated spliceosome (the Bact complex) at 3.5 Å resolution, revealing how latency is maintained even though the complex is mostly primed for catalysis. Wan et al. report the structure of the catalytic step 1 spliceosome (the C complex) at 3.4 Å resolution; this complex forms after the first step of the splicing reaction.
Science, this issue pp. 904 and 895
Co-reporter:Ruixue Wan;Chuangye Yan;Rui Bai;Lin Wang;Min Huang;Catherine C. L. Wong
Science 2016 Vol 351(6272) pp:466-475
Publication Date(Web):29 Jan 2016
DOI:10.1126/science.aad6466
Structure of a key spliceosomal complex
In eukaryotes, when DNA is transcribed into RNA, the primary transcript has protein-coding sequences interrupted by noncoding sequences called introns. Introns are removed by a complex molecular machine, the spliceosome. Wan et al. determined the structure of a key subcomplex, the tri-SNP, that comprises three small nuclear RNAs and more than 30 proteins. The structure, determined by electron microscopy at 3.8 Å resolution, unexpectedly shows a primary RNA transcript bound in the tri-SNP. Further analysis revealed how the spliceosome assembles to achieve its complex functions.
Science, this issue p. 466
Co-reporter:Ruixue Wan;Chuangye Yan;Rui Bai;Gaoxingyu Huang
Science 2016 Vol 353(6302) pp:895-904
Publication Date(Web):26 Aug 2016
DOI:10.1126/science.aag2235
How spliceosomes make the first cut
In eukaryotes, transcribed precursor mRNA includes noncoding sequences that must be spliced out. This is done by the spliceosome, a dynamic complex in which five small nuclear RNAs and several proteins go through a series of ordered interactions and conformational rearrangements to achieve splicing. Two protein structures provide a look at the first catalytic step in the pathway. Yan et al. report the structure of the activated spliceosome (the Bact complex) at 3.5 Å resolution, revealing how latency is maintained even though the complex is mostly primed for catalysis. Wan et al. report the structure of the catalytic step 1 spliceosome (the C complex) at 3.4 Å resolution; this complex forms after the first step of the splicing reaction.
Science, this issue pp. 904 and 895
Co-reporter:Jing Hang;Ruixue Wan;Chuangye Yan
Science 2015 Volume 349(Issue 6253) pp:1191-1198
Publication Date(Web):11 Sep 2015
DOI:10.1126/science.aac8159
Structure and function of the spliceosome
When RNA is transcribed from DNA in the eukaryotic cell nucleus, the initial transcript includes noncoding introns that must be spliced out. This splicing is done by a complex macromolecular machine, the spliceosome, which comprises five small nuclear RNAs and more than 100 associated proteins. Now, two papers reveal insights into the structure and function of the yeast spliceosome. Yan et al. describe a high-resolution structure determined by electron microscopy of a spliceosome complex comprising four RNAs and 37 proteins. Hang et al. focus on the catalytic site and show how protein components anchor the transcribed RNA and allow sufficient flexibility to deliver RNA components involved in catalyzing the splicing reaction.
Science, this issue pp. 1182 and 1191
Co-reporter:Chuangye Yan;Jing Hang;Ruixue Wan;Min Huang;Catherine C. L. Wong
Science 2015 Volume 349(Issue 6253) pp:1182-1191
Publication Date(Web):11 Sep 2015
DOI:10.1126/science.aac7629
Structure and function of the spliceosome
When RNA is transcribed from DNA in the eukaryotic cell nucleus, the initial transcript includes noncoding introns that must be spliced out. This splicing is done by a complex macromolecular machine, the spliceosome, which comprises five small nuclear RNAs and more than 100 associated proteins. Now, two papers reveal insights into the structure and function of the yeast spliceosome. Yan et al. describe a high-resolution structure determined by electron microscopy of a spliceosome complex comprising four RNAs and 37 proteins. Hang et al. focus on the catalytic site and show how protein components anchor the transcribed RNA and allow sufficient flexibility to deliver RNA components involved in catalyzing the splicing reaction.
Science, this issue pp. 1182 and 1191
Co-reporter:Linfeng Sun;Chuangye Yan;Xiaoyuan Zhou;Lingyun Zhao;Rui Zhou;Guanghui Yang;Tian Xie;Xueming Li;Yanyu Zhao;Shenjie Wu
PNAS 2015 Volume 112 (Issue 19 ) pp:6003-6008
Publication Date(Web):2015-05-12
DOI:10.1073/pnas.1506242112
The four-component intramembrane protease γ-secretase is intricately linked to the development of Alzheimer’s disease. Despite
recent structural advances, the transmembrane segments (TMs) of γ-secretase remain to be specifically assigned. Here we report
a 3D structure of human γ-secretase at 4.32-Å resolution, determined by single-particle, electron cryomicroscopy in the presence
of digitonin and with a T4 lysozyme fused to the amino terminus of presenilin 1 (PS1). The overall structure of this human
γ-secretase is very similar to that of wild-type γ-secretase determined in the presence of amphipols. The 20 TMs are unambiguously
assigned to the four components, revealing principles of subunit assembly. Within the transmembrane region, PS1 is centrally
located, with its amino-terminal fragment (NTF) packing against Pen-2 and its carboxyl-terminal fragment (CTF) interacting
with Aph-1. The only TM of nicastrin associates with Aph-1 at the thick end of the TM horseshoe, and the extracellular domain
of nicastrin directly binds Pen-2 at the thin end. TM6 and TM7 in PS1, which harbor the catalytic aspartate residues, are
located on the convex side of the TM horseshoe. This structure serves as an important framework for understanding the function
and mechanism of γ-secretase.
Co-reporter:Lijun Zhou, Yulin Zhou, Jing Hang, Ruixue Wan, Guifeng Lu, Chuangye Yan and Yigong Shi
Cell Research 2014 24(4) pp:497-500
Publication Date(Web):February 11, 2014
DOI:10.1038/cr.2014.18
The Lsm (Sm-like) family of proteins, characterized by the Sm fold1 and conserved among eukaryotes, plays an important role in RNA biogenesis. The Sm and Lsm complexes play an essential role in pre-mRNA splicing2. Of the five small ribonucleoproteins (snRNPs), four (U1, U2, U4, U5) contain the Sm heptamer ring, whereas the U6 snRNP contains a specific Lsm2-8 heptamer that comprises Lsm2, Lsm3, Lsm4, Lsm5, Lsm6, Lsm7, and Lsm83. Another Lsm heptameric complex, Lsm1-7, which differs from Lsm2-8 by one Lsm protein, is thought to function in mRNA decapping, a crucial step in the mRNA degradation pathway4.In yeast, mRNA degradation begins with deadenylation catalyzed mainly by the deadenylase Ccr4-Not complex to shorten the polyA tail5. Subsequently, whether the 3′-end of mRNA is bound by the Lsm1-7 complexes is an essential determinant for the directionality of mRNA degradation. In the absence of Lsm1-7, the exposed 3′-end of mRNA is captured by exosome and degradation begins from the 3′-end6. In the presence of Lsm1-7, the 3′-end of mRNA is recognized by the Pat1-Lsm1-7 complex6. Then the decapping enzymes Dcp1 and Dcp2 are recruited to decap mRNA, followed by Xrn1-mediated degradation from the 5′-end7.The Sm and Lsm proteins exhibit contrasting functions and distinct biochemical properties. First, the Sm proteins are located in the nucleus and only function in the spliceosome pathway of mRNA splicing. The Lsm proteins appear both in the nucleus and cytoplasm and are involved in various pathways of RNA metabolism. Second, the Sm proteins only assemble into a heptamer ring in the presence of snRNA, whereas the Lsm proteins can form two stable heptamer rings by themselves, the Lsm1-7 and Lsm2-8 complexes. The Sm ring binds the central part of snRNA whereas the Lsm2-8 ring recognizes the 3′-end of U6 snRNA8.Unlike the Sm proteins, whose heptameric ring can be readily assembled from stable heteromeric subcomplexes, the individually purified Lsm proteins, either alone or in heteromeric forms, failed to form a correctly assembled heptamer in the absence of denaturation/refolding. Lsm3 is known to form a stable homo-octamer by itself9; co-expression of Lsm5, Lsm6, and Lsm7 led to formation of a stable, but non-functional, hetero-hexamer10. In fact, every combination of hetero-dimeric or hetero-trimeric Lsm subcomplex would result in the formation of a stable oligomer, which can be reconstituted into a heptameric ring only by denaturation and refolding11. After numerous trials, we succeeded in co-expression of all seven Lsm proteins from S. cerevisiae using the pQLink plasmid12. The Lsm1-7 complex was biochemically purified to homogeneity (Supplementary information, Figure S1). Analysis by mass spectrometry confirmed the presence of all seven Lsm proteins.Despite rigorous trials, we were unable to crystallize the Lsm1-7 complex. To solve this problem, we launched a systematic protein engineering effort that involved removal of flexible sequences and mutation of cysteine to serine. Eventually we crystallized the Lsm1-7 complex in the P21 space group (Supplementary information, Figure S2). To facilitate structure determination, we also generated and crystallized the selenomethionine-labeled Lsm1-7 complex in the P1 space group. The structure of Lsm1-7 was determined by a combination of selenium-based single anomalous dispersion (SAD) and molecular replacement using the atomic coordinates of an archael Sm-like homo-heptamer (PDB code 1I8113). The final atomic model was refined at 3.0 Å resolution (Supplementary information, Table S1 and Figure S3).The overall appearance of the heptameric Lsm1-7 complex from S. cerevisiae resembles a thick donut, with an outer diameter of approximately 70 Å, an inner diameter of 5 Å, and a thickness of 45 Å (Figure 1A). The seven Lsm components sequentially interact with each other to form a closed ring, with the order of Lsm1-Lsm2-Lsm3-Lsm6-Lsm5-Lsm7-Lsm4. Except for Lsm1, each component within the ring only interacts with two neighboring Lsm proteins. For example, Lsm7 is in contact only with Lsm5 and Lsm4. The C-terminal sequence of Lsm1 forms an extended α-helix that crosses over the ring, with the ensuing loop associating with Lsm3 and Lsm6. The central hole of the Lsm1-7 ring is partially blocked by this α-helix from Lsm1, leaving a very small opening at the center of the ring. This structural feature contrasts that in the Lsm2-8 complex, where the central hole has a diameter of 15 Å8.Unlike the other six Lsm components, the N- and C-terminal sequences of Lsm1 have well-defined structures that extend out from the conserved Sm core fold (Figure 1B) and interact with neighboring Lsm subunits in the heptameric Lsm1-7 complex. Notably, a few amino acids from the C-terminal sequence of Lsm1 interact with Lsm3 and Lsm6 through hydrogen bonds (H-bonds) and van der Waals contacts (Figure 1C and 1D). In addition, Asp36 at the N-terminal loop of Lsm1 interacts with Lys8 of Lsm2 through a salt bridge (Figure 1E). These specific interactions likely constitute the basis why Lsm1, but not any other of the Lsm proteins, can replace Lsm8 to form a heptameric complex with Lsm2/3/4/5/6/7.The Lsm1-7 complex shares six common components with the heptameric Lsm2-8 complex, with Lsm1 in the Lsm1-7 complex substituted by Lsm8 in the Lsm2-8 complex. Consequently, the overall structures of these two complexes are very similar, with a root-mean-squared deviation (RMSD) of 0.89 Å over 461 aligned Cα atoms (Figure 1F). The main difference is between Lsm1 and Lsm8, which can be aligned to each other with an RMSD of 0.97 Å over 54 aligned Cα atoms in the core Sm fold (Figure 1G). Notably, these 54 aligned Cα atoms come from residues 32-115 of Lsm1 and residues 1-67 of Lsm8.Although the N- and C-terminal sequences of Lsm1 mediate interactions that are unique to the Lsm1-7 complex, the core Sm fold of Lsm1 appears to make a conserved set of interactions compared to Lsm8. Specifically, interactions between Lsm1 and Lsm2, including both main chain H-bonds and side chain van der Waals contacts, are similar to those between Lsm8 and Lsm2. The majority of the Lsm1-Lsm4 interactions are also preserved in the Lsm8-Lsm4 interface (Supplementary information, Figure S4).The Lsm1-7 complex plays an important role in RNA metabolism by facilitating degradation of mRNA. In yeast, the Lsm1-7 complex binds the 3′-end of mRNA and localizes to the P-bodies, ultimately resulting in mRNA degradation from the 5′-end4. We examined the RNA binding affinities of the Lsm1-7 complex using biolayer interferometry (BLI) on a Fortebio Octet system. The Lsm1-7 complex exhibits binding affinities of approximately 6 μM and 2 μM for the octa-nucleotides 5′-AAAAAAAA-3′ and 5′-UUUUUUUU-3′, respectively (Figure 1H). The binding affinity difference between the oligo-A and oligo-U sequences is only about 3-fold, suggesting that the Lsm1-7 complex may not recognize RNA sequences with the same level of specificity as the Lsm2-8 complex8. By contrast, the Lsm2-8 complex exhibits a binding affinity of about 20 nM for the oligo-U sequence, approximately 200-fold tighter than that for the oligo-A sequence (Figure 1H). These results suggest that the RNA binding mode of the Lsm1-7 complex may be similar to that of the Lsm2-8 complex towards the oligo-A, but not the oligo-U, sequence.Although Lsm1 and Lsm8 share limited sequence similarity, the RNA-binding residues in Lsm8 are largely preserved in Lsm1. For example, the D31xXxN35 and R57GX motifs in Lsm8 correspond to D72xXxN76 and R105GX in Lsm1, respectively. In addition, modeling studies suggest that the Lsm1-7 complex can bind to the 3′-end of oligo-U sequence similarly as the Lsm2-8 complex. However, the oligo-U binding affinity of Lsm1-7 is approximately 100-fold lower than that of Lsm2-8 (Figure 1H). Obviously, the sequence variation between Lsm1 and Lsm8 helps determine their RNA binding affinity as well as specificity. The molecular basis for this observation is likely to be revealed by the crystal structure of Lsm1-7 bound to RNA.An invariant Arg residue in Lsm6/3/2/8/4 plays an important role in RNA recognition by the Lsm2-8 complex8 (Figure 1I). We generated seven variants of the Lsm1-7 complex, each containing replacement of the conserved Arg (Ser in Lsm5) by Ala in one Lsm component, and examined their interactions with the oligo-A and oligo-U RNA elements (Figure 1J). As recently reported8, mutation of the conserved Arg to Ala in any of the Lsm6/3/2/8/4 subunits results in drastic reduction of binding affinity between the 3′-end U-rich sequence of U6 snRNA and the Lsm2-8 complex. In the Lsm1-7 complex, such point mutation in Lsm2 or Lsm3 caused the most pronounced reduction of RNA binding affinity to the oligo-U sequence (Figure 1J). By sharp contrast, such point mutation only caused modest changes (less than 2-fold) in binding affinities for the oligo-A sequence. This result is consistent with the observation that the Lsm1-7 complex exhibited a weak specificity for the oligo-U sequences (Figure 1H).In summary, we report the crystal structure of the heptameric Lsm1-7 complex and preliminary characterization of its RNA-binding properties. Our structural and biochemical characterization serves as a framework for mechanistic understanding of the function of Lsm1-7 complex in RNA metabolism. In the final phase of manuscript preparation, we noted publication of two related manuscripts14,15, of which Sharif et al.14 reported the crystal structure of Lsm1-7 without RNA binding studies.This work was supported by the National Natural Science Foundation of China (31130002 and 31021002). The atomic coordinates have been deposited in the Protein Data Bank with the accession code 4M75.(Supplementary information is linked to the online version of the paper on the Cell Research website.)
Co-reporter:YiGong Shi
Science China Life Sciences 2014 Volume 57( Issue 1) pp:1-3
Publication Date(Web):2014 January
DOI:10.1007/s11427-013-4594-x
Co-reporter:Sheng Wang;Renhong Yan;Xi Zhang;Qi Chu;
Proceedings of the National Academy of Sciences 2014 111(35) pp:12734-12739
Publication Date(Web):August 18, 2014
DOI:10.1073/pnas.1414093111
Enteropathogenic bacteria, exemplified by Escherichia coli, rely on acid-resistance systems (ARs) to survive the acidic environment of the stomach. AR3 consumes intracellular protons
through decarboxylation of arginine (Arg) in the cytoplasm and exchange of the reaction product agmatine (Agm) with extracellular
Arg. The latter process is mediated by the Arg:Agm antiporter AdiC, which is activated in response to acidic pH and remains
fully active at pH 6.0 and below. Despite our knowledge of structural information, the molecular mechanism by which AdiC senses
acidic pH remains completely unknown. Relying on alanine-scanning mutagenesis and an in vitro proteoliposome-based transport
assay, we have identified Tyr74 as a critical pH sensor in AdiC. The AdiC variant Y74A exhibited robust transport activity
at all pH values examined while maintaining stringent substrate specificity for Arg:Agm. Replacement of Tyr74 by Phe, but
not by any other amino acid, led to the maintenance of pH-dependent substrate transport. These observations, in conjunction
with structural information, identify a working model for pH-induced activation of AdiC in which a closed conformation is
disrupted by cation–π interactions between proton and the aromatic side chain of Tyr74.
Co-reporter:Qi Hu;Di Wu;Tianxi He;Zhen Yan;Chuangye Yan;Qionglin Liang;Wen Chen
PNAS 2014 Volume 111 (Issue 46 ) pp:16254-16261
Publication Date(Web):2014-11-18
DOI:10.1073/pnas.1418000111
Autocatalytic activation of an initiator caspase triggers the onset of apoptosis. In dying cells, caspase-9 activation is
mediated by a multimeric adaptor complex known as the Apaf-1 apoptosome. The molecular mechanism by which caspase-9 is activated
by the Apaf-1 apoptosome remains largely unknown. Here we demonstrate that the previously reported 1:1 interaction between
Apaf-1 caspase recruitment domain (CARD) and caspase-9 CARD is insufficient for the activation of caspase-9. Rather, formation
of a multimeric CARD:CARD assembly between Apaf-1 and caspase-9, which requires three types of distinct interfaces, underlies
caspase-9 activation. Importantly, an additional surface area on the multimeric CARD assembly is essential for caspase-9 activation.
Together, these findings reveal mechanistic insights into the activation of caspase-9 by the Apaf-1 apoptosome and support
the induced conformation model for initiator caspase activation by adaptor complexes.
Co-reporter:Tian Xie;Rui Zhou;Yanyu Zhao;Linfeng Sun;Chuangye Yan;Peilong Lu;Dan Ma;Guanghui Yang
PNAS 2014 Volume 111 (Issue 37 ) pp:13349-13354
Publication Date(Web):2014-09-16
DOI:10.1073/pnas.1414837111
γ-Secretase is an intramembrane protease responsible for the generation of amyloid-β (Aβ) peptides. Aberrant accumulation
of Aβ leads to the formation of amyloid plaques in the brain of patients with Alzheimer's disease. Nicastrin is the putative
substrate-recruiting component of the γ-secretase complex. No atomic-resolution structure had been identified on γ-secretase
or any of its four components, hindering mechanistic understanding of γ-secretase function. Here we report the crystal structure
of nicastrin from Dictyostelium purpureum at 1.95-Å resolution. The extracellular domain of nicastrin contains a large lobe and a small lobe. The large lobe of nicastrin,
thought to be responsible for substrate recognition, associates with the small lobe through a hydrophobic pivot at the center.
The putative substrate-binding pocket is shielded from the small lobe by a lid, which blocks substrate entry. These structural
features suggest a working model of nicastrin function. Analysis of nicastrin structure provides insights into the assembly
and architecture of the γ-secretase complex.
Co-reporter:Peilong Lu;Dan Ma;Chuangye Yan;Xinqi Gong;Mingjian Du
PNAS 2014 Volume 111 (Issue 5 ) pp:1813-1818
Publication Date(Web):2014-02-04
DOI:10.1073/pnas.1323931111
Vitamin C, also known as ascorbate, is required in numerous essential metabolic reactions in eukaryotes. The eukaryotic ascorbate-dependent
oxidoreductase cytochrome b561 (Cyt b561), a family of highly conserved transmembrane enzymes, plays an important role in ascorbate recycling and iron absorption.
Although Cyt b561 was identified four decades ago, its atomic structure and functional mechanism remain largely unknown. Here, we report the
high-resolution crystal structures of cytochrome b561 from Arabidopsis thaliana in both substrate-free and substrate-bound states. Cyt b561 forms a homodimer, with each protomer consisting of six transmembrane helices and two heme groups. The negatively charged
substrate ascorbate, or monodehydroascorbate, is enclosed in a positively charged pocket on either side of the membrane. Two
highly conserved amino acids, Lys81 and His106, play an essential role in substrate recognition and catalysis. Our structural and biochemical analyses allow the proposition
of a general electron transfer mechanism for members of the Cyt b561 family.
Co-reporter:Weijiao Huang, Wooyoung Choi, Yuling Chen, Qi Zhang, Haiteng Deng, Wei He and Yigong Shi
Cell Research 2013 23(5) pp:724-727
Publication Date(Web):January 29, 2013
DOI:10.1038/cr.2013.15
Cancer cells exhibit a greatly increased level of aerobic glycolysis with accumulation of lactic acid, a phenomenon known as the Warburg effect. Apparently, survival of cancer cells requires an elaborate system for acid resistance. L-glutamine (Gln) has long been known to be essential for cancer cell growth, which is generally thought to relate to the nutritional value of Gln as carbon and nitrogen source. On the basis of our recent finding that Gln provides acid resistance for E. coli through release of ammonia, we hypothesized that the primary role of Gln in cancer cells is to fight acid, rather than provide nutrition, through enzymatic deamidation. In this letter, we provide preliminary experimental evidence that supports this hypothesis. We demonstrate that Gln helps cancer cells survive acidic stress, which is compromised by inhibition of specific glutaminase activity. Our data suggests that glutaminase inhibitors, currently under clinical trials as an anti-cancer drug, may work by countering the ability of cancer cells to survive under acidic environment. We further speculate that the general requirement of Gln in cell culture is also due to its crucial role in acid resistance.A hallmark of cancer cell is described as the Warburg Effect, with increased aerobic glycolysis and reduced oxidative phosphorylation1. Even in the presence of oxygen, cancer cells convert glucose mainly into lactic acid, which, with a pK of 3.7, contributes to an acidic environment in tumor2. The extracellular pH value of human cancer tissues can be as low as 5.63. As numerous biological reactions are strictly pH dependent, lowered pH value may be detrimental to cancer cell growth. Consequently, cancer cells must be able to efficiently neutralize lactic acid to ensure normal growth. Supporting this analysis, the intracellular pH in tumors was reported to remain largely unchanged even when the extracellular pH value precipitously dropped as a consequence of cell growth4. This observation suggests presence of a robust acid resistance system within cancer cells. Much to our surprise, there is little description or research into the acid resistance system in cancer cells.L-glutamine (Gln) is the most abundant free amino acid in human body5, with its concentration higher than that of all other 19 amino acids combined. Gln is also the most abundant free amino acid in a large variety of food sources, including meat and vegetables6. Remarkably, cancer cells heavily depend on Gln for growth and proliferation5, and glutamine deprivation in cell culture leads to rapid loss of cell viability7. Strikingly, most tumor cells consume Gln at a much higher rate compared to normal cells8. The prevailing view is that Gln provides carbon and nitrogen source for cell growth, as Gln is converted into L-glutamate (Glu) and ammonia through glutaminolysis9. Unfortunately, this common belief is contradicted by the experimental observation that, despite efficient transport systems for Glu and ammonium, Glu at any concentration, even if supplemented by NH4+ and ATP, failed to permit the growth of L-fibroblast7. Thus, we believe that the question of why Gln is required for cancer cell growth and more generally cell culture remains largely enigmatic.In the accompanying paper6, we report a novel bacterial acid resistance system, which relies on the release of ammonia from Gln via the enzymatic activity of glutaminases. The released ammonia neutralizes the acid in E. coli. This finding, together with the use of ammonia by Helicobactor polari to fight acid in the stomach10, motivated us to hypothesize an important role for Gln in cancer cell growth. In this hypothesis, Gln is thought to facilitate cancer cell growth not just by providing carbon and nitrogen source, but more importantly, by fighting acidic stress through enzymatic release of ammonia. Our proposed hypothesis is consistent with a body of existing literatures and knowledge. Two different glutaminases (GLS) have been identified in mammalian cells: GLS1 (with two splice forms KGA and GAC11,12), which is mainly expressed in the kidney, and GLS2 (also called LGA13), which exists mainly in the liver. The mRNA levels of GAC have been found to be elevated in various tumors12. In fact, GAC was shown to be essential for the growth of non-small cell lung cancer14. Glutaminase inhibitors have been under clinical trials in the United States as a potential anti-cancer therapy15. It should be noted, however, these previous studies did not link the important role of glutaminase to the requirement of Gln in acid resistance for cancer cell growth.To help validate this hypothesis, we studied the impact of Gln in cell growth using HeLa and MCF-7 cells, derived from cervical and breast cancers, respectively. In this letter, we report that, as cells grow, the extracellular environment becomes increasingly acidic, and continued cell growth requires Gln. In culture medium of lower pH, cancer cells consumed more Gln, with more Glu accumulated in the extracellular environment. The pro-survival role of Gln under acidic pH is sabotaged by specific glutaminase inhibitor. These data collectively support the hypothesis of an essential role for Gln in cancer cell growth under acidic stress. Our preliminary experimental evidence is briefly outlined below.First, we show that cancer cell lines appear to depend on Gln to maintain normal growth. HeLa and MCF-7 cells were cultured following manufacturer's protocol but with provision of varying concentrations of Gln (0, 5, 10, and 25 mM); cell density was measured at 0, 8, 16, 24, 48, 72, and 96 h. There is a strong correlation between cell growth and the presence of Gln (Supplementary information, Figure S1). In the presence of 5 mM or higher concentrations of Gln, the growth curves appear qualitatively the same for HeLa or MCF-7 cells. By contrast, the HeLa or MCF7 cell density was considerably lower in the absence of Gln (Supplementary information, Figure S1). Reduced levels of Gln also gave rise to reduced rate of growth for HeLa and MCF-7 cells (Supplementary information, Figure S2).Next, we measured the pH values of the culture medium at different time points. Consistent with the notion that cell growth results in acidification of the culture medium, the pH decreased over time for both cell lines (Supplementary information, Figure S3). Reflecting a reciprocal relationship to their growth curves, the culture medium for HeLa or MCF-7 cells exhibited a similar temporal decrease of pH values in the presence of 5 mM or higher Gln. By contrast, the pH value of the HeLa or MCF-7 culture medium showed much less decrease in the absence of Gln, presumably due to lack of cell growth. In the absence of cells, the culture medium pH remained largely unchanged over time (Supplementary information, Figure S3).If acidic pH caused by cell growth is detrimental to continued cell growth, then continued supply of buffered medium might alleviate this problem. To examine this scenario, we cultured HeLa or MCF-7 cells at relatively constant pH, with medium change every 8 h. At pH 7.3, HeLa and MCF-7 cells exhibited robust growth even in the absence of Gln (Figure 1A). By contrast, cell growth was considerably less at pH 6.3 in the absence of Gln (Figure 1A). Consistent with this analysis, the presence of 10 mM Gln allowed robust cell growth at both pH 6.3 and pH 7.3 (Supplementary information, Figure S4). Together, our data suggest that, for HeLa or MCF-7 cancer cells, Gln might provide an efficient means of countering the acidic stress.If Gln is used for acid resistance, then we would expect to see consumption of Gln from, and release of Glu into, the culture medium. In addition, there might be more consumption of Gln at lower pH. To investigate this scenario, we cultured HeLa or MCF-7 cells in the presence of 0.5 mM L-[U-13C5, -U-15N2] Gln and 10 mM unlabelled Gln at pH 6.5 or 7.0. After 8 h, the remaining Gln in the culture medium was quantified using mass spectrometry. For both HeLa and MCF-7 cells, there is more cell growth at pH 7.0 than at pH 6.5 (Supplementary information, Figure S5); yet consumption of Gln at pH 6.5, normalized for cell density, is considerably more than that at pH 7.0 (Figure 1B, left panel). Specifically, compared to pH 7.0, HeLa and MCF-7 cells at pH 6.5 exhibit 61% and 37% more consumption of Gln, respectively. Consistent with intracellular conversion of Gln to Glu, the concentrations of Glu in the culture medium increased by 39% and 33%, respectively, for HeLa and MCF-7 cells (Figure 1B, right panel).If conversion of Gln to Glu is required for acid resistance, then inhibition of the glutaminase activity is expected to preferentially inhibit cell growth at lower pH. To examine this scenario, we cultured HeLa and MCF-7 cells in medium with or without glutaminase inhibitor at pH 6.0 and 6.5. At both pH values, despite the presence of 10 mM Gln, there were much less cells in the presence of the glutaminase GLS1-specific inhibitor 968 compared to its absence (Figure 1C). Serving as a control in these experiments, cell growth in the absence of Gln was considerably worse than that in the presence of Gln. These results also suggest that GLS1 is likely a crucial enzyme for glutaminolysis in HeLa and MCF-7 cells under acidic stress.Many mammalian enzymes are active only within a narrow pH window. To examine the pH dependence of glutaminase activity, we expressed and purified the two GLS1 isoforms (KGA and GAC) to homogeneity and measured their enzymatic activity in vitro. The result clearly shows that, even at pH 6.0, both GAC and KGA retained significant glutaminase activity (Figure 1D). GAC exhibited a higher level of glutaminase activity than KGA at pH 6.0, 7.0, and 8.0. In particular, GAC exhibits approximately 108% higher activity compared to KGA at pH 6.0. This result suggests that the GLS1 splice variant GAC may be more important for cancer cell growth under acidic pH.These experimental observations serve as preliminary evidence to support our hypothesis that Gln may play an important role in cancer cell survival and growth through enzymatic release of ammonia for acid resistance (Figure 1E). In other words, the reliance of cancer cells on Gln is not just for nutrition but more importantly also for the purpose of fighting acid stress as a result of Warburg Effect. As the first step for metabolic utilization of Gln involves the glutaminase activity, it is inherently difficult to separate the two potential functions of Gln — carbon/nitrogen source versus acid resistance. Any molecular biology manipulation of glutaminases simultaneously affects both metabolic utilization of Gln and its role in acid resistance. Nonetheless, our finding that Gln is preferentially converted to Glu in the culture medium of lower pH strongly supports the acid resistance hypothesis. To the best of our knowledge, this may be the first time that an important role for Gln in cancer cell growth through acid resistance is proposed. Such a conceptual advance has important ramifications for understanding cancer cell homeostasis and for designing potential therapeutic treatment. Detailed methods are described in the Supplementary information, Data S1.This work was supported by the Ministry of Science and Technology of China (2009CB918801), the National Natural Science Foundation of China (31130002, 31021002, and 30888001), and Beijing Municipal Commissions of Education and Science and Technology. We apologize to those colleagues whose papers are not cited due to strict reference limit.(Supplementary information is linked to the online version of the paper on the Cell Research website.)
Co-reporter:Peilong Lu, Dan Ma, Yuling Chen, Yingying Guo, Guo-Qiang Chen, Haiteng Deng and Yigong Shi
Cell Research 2013 23(5) pp:635-644
Publication Date(Web):January 22, 2013
DOI:10.1038/cr.2013.13
Bacteria, exemplified by enteropathogenic Escherichia coli (E. coli), rely on elaborate acid resistance systems to survive acidic environment (such as the stomach). Comprehensive understanding of bacterial acid resistance is important for prevention and clinical treatment. In this study, we report a previously uncharacterized type of acid resistance system in E. coli that relies on L-glutamine (Gln), one of the most abundant food-borne free amino acids. Upon uptake into E. coli, Gln is converted to L-glutamate (Glu) by the acid-activated glutaminase YbaS, with concomitant release of gaseous ammonia. The free ammonia neutralizes proton, resulting in elevated intracellular pH under acidic environment. We show that YbaS and the amino acid antiporter GadC, which exchanges extracellular Gln with intracellular Glu, together constitute an acid resistance system that is sufficient for E. coli survival under extremely acidic environment.
Co-reporter:Dong Deng, Ping Yin, Chuangye Yan, Xiaojing Pan, Xinqi Gong, Shiqian Qi, Tian Xie, Magdy Mahfouz, Jian-Kang Zhu, Nieng Yan and Yigong Shi
Cell Research 2012 22(10) pp:1502-1504
Publication Date(Web):September 4, 2012
DOI:10.1038/cr.2012.127
TALE (transcription activator-like effector) DNA-binding repeats, which represent a modular assembly for specific target DNA of almost any sequences, provide a powerful tool for genetic editing. All the codes are for the four fundamental bases. There was no report on the recognition of modified DNA by TALE up to date. In this study, we report two crystal structures of engineered TALE repeats in complex with methylated DNA elements at 1.85 Å and 1.95 Å resolutions, respectively. Biochemical analysis shows that the TALE code NG, but not HD, binds to 5-methylcytosine (mC). Our findings will extend the application of TALE in epigenetic modification and cancer research, but also reveal the previously unconsidered limits for the applications of TALEs.DNA methylation is a major epigenetic mark, and plays a pivotal role in diverse biological processes in a wide range of organisms. In mammals, DNA methylation usually occurs to the C5 position of cytosine in the CG context. Hypermethylation of the CpG islands may lead to gene silencing1,2.The TALEs are a family of DNA-binding proteins3,4,5. A TALE contains a number of sequence repeats, each recognizing one DNA base. Each TALE repeat consists of 33-35 highly conserved amino acids except for those at positions 12 and 13, which are named RVD (repeat variable diresidue) and determine DNA-binding specificity. The recognition codes between the RVDs and the DNA bases have been established through experimental and computational approaches6,7. For example, the bases A, G, C and T can be recognized by the RVDs NI (Asn and Ile), NN (Asn and Asn), HD (His and Asp) and NG (Asn and Gly), respectively4. The modular nature of TALE repeats provides an important tool for genetic manipulation8,9,10.We recently determined the high-resolution structure of DNA-bound TALE dHax3, which provided the molecular basis for base-specific DNA recognition11. The 34-residue TALE repeat comprises two α-helices connected by a short loop where RVD resides. The RVD loop tracks along the major groove of DNA (Figure 1A). Only the second residue of RVD, namely the one at position 13, is in direct contact with the base in the sense DNA strand, whereas the first residue helps maintain the RVD loop conformation through hydrogen bond.Notably, the DNA base T is recognized by Gly13 in most cases. The lack of side chain in Gly not only provides sufficient space to accommodate the 5-methyl group of thymine but also allows optimal van der Waals interactions between the Cα atom of Gly13 and the 5-methyl group11 (Figure 1B). This observation immediately suggests the possibility that mC might be recognized by Gly13 in RVD, because the only difference between the bases T and mC is at position 4, which is not involved in binding to TALE repeats. To examine this possibility, we replaced three T bases in the sense DNA strand by three mC bases and performed DNA-binding studies using the electrophoretic mobility shift assay (EMSA).Confirming our prediction, the dHax3 protein binds to the triply modified DNA, with the forward strand 5′-TCCCT(mC)TA(mC)CTC(mC)-3′ (Figure 1C). This binding is very similar to that for the unmodified dsDNA, with the forward strand 5′-TCCCTTTATCTCT-3′ (Figure 1C). This result is rather striking, considering the fact that three T-A base pairs have been replaced by three mC-G base pairs in the dsDNA.Next, we crystallized the binary complex between dHax3 and the triply modified DNA-binding sequence, and determined its structure at 1.85 Å (Figure 1D and Supplementary information, Figures S1A, S2A and Table S1). As anticipated, the 5-methyl group of mC points to the Cα of Gly13 with a distance of 3.4-4.0 Å for the three mC bases (Figure 1D and Supplementary information, Figure S2B). As DNA methylation mostly occurs to cytosine in the CG context, we constructed a dHax3 variant that is expected to recognize the DNA elements 5′-TCCCTT(mC)G(mC)GTCT-3′, where the RVDs NG and NN are designed for bases mC and G, respectively (Supplementary information, Figure S1A). The crystal structure of this dHax3 variant, which we name dHax3-mCG, in complex with its target dsDNA was also obtained and refined at 1.95 Å resolution (Figure 1E and Supplementary information, Figure S2C and Table S1). The coordination of mC bases by Gly13 residues is identical to that in the first structure (Figure 1D). In fact, the two structures of dHax3 variants in complex with triply and doubly methylated DNA elements are nearly identical to that of dHax3 bound to the unmodified DNA11, with root-mean-squared deviation values of less than 0.3 Å over more than 900 Cα atoms (Supplementary information, Figure S2D).Encouraged by the structural findings, we replaced all six T bases by mC in the sense DNA strand. Subsequent EMSA study revealed that dHax3 retained similar binding to this DNA element as to the unmodified DNA (Figure 1F, lanes 1-10; Supplementary information, Figure S1B). In contrast, there was no detectable binding between dHax3 and the DNA element in which the six T bases were replaced by the base C (Figure 1F, lanes 11-20). This is rather striking, because this result suggests a qualitative and reliable method for differentiating the bases mC and C. We next examined whether the RVD code HD, which favors the base C, may also recognize mC. Substitution of the five C bases with mC or T in the sense DNA strand led to complete abrogation of DNA binding by dHax3 (Figure 1F, lanes 21-30). Substitution of the five C bases with A or G resulted in significant impairment of DNA binding (Figure 1F, lanes 31-40). These results illustrate the specific nature of mC recognition by TALE repeats involving the RVD NG, but not HD.Our experimental characterization provides a molecular basis for distinguishing methylated and unmethylated cytosine. Binding of mC by TALE repeat through the RVD NG extends the DNA recognition code and has potential application in epigenetics and cancer research. For example, specific TALE repeats may be designed to recognize the hypermethylated DNA region; detection can be facilitated by fusing TALEs with fluorescence proteins.Our study also strongly argues that the in vivo methylation status of the target DNA sequence must be considered for the design of specific DNA-binding TALEs. Methylation of the base C in vivo might render the DNA sequence unfit for binding by the designed TALEs. Because the methylation status of DNA sequences is frequently under dynamic control, one would have to design at least two TALEs for one DNA sequence (i.e., one for methylated and one for unmethylated). In fact, assessment of methylation status of specific DNA sequences in vivo can be greatly facilitated through quantification of fluorescence signal of designed GFP-TALEs. Alternatively, the CpG sequences may be avoided for the application of TALEs, although this practice will somehow limit the potential application. Despite these complexities, the discovery of mC binding by TALEs with RVD NG opens a number of exciting opportunities.This work was supported by funds from the Ministry of Science and Technology of China (2011CB910501, 2009CB918801, and 2009CB918802), the National Natural Science Foundation of China (30888001, 91017011 and 31070644) and Tsinghua University. The atomic coordinates have been deposited in the Protein Data Bank (PDB; accession code: 4GJP and 4GJR).(Supplementary information is linked to the online version of the paper on the Cell Research website.)
Co-reporter:Weijiao Huang, Wooyoung Choi, Wanqiu Hu, Na Mi, Qiang Guo, Meisheng Ma, Mei Liu, Yuan Tian, Peilong Lu, Feng-Liang Wang, Haiteng Deng, Lei Liu, Ning Gao, Li Yu and Yigong Shi
Cell Research 2012 22(3) pp:473-489
Publication Date(Web):February 7, 2012
DOI:10.1038/cr.2012.24
The Beclin 1 gene is a haplo-insufficient tumor suppressor and plays an essential role in autophagy. However, the molecular mechanism by which Beclin 1 functions remains largely unknown. Here we report the crystal structure of the evolutionarily conserved domain (ECD) of Beclin 1 at 1.6 Å resolution. Beclin 1 ECD exhibits a previously unreported fold, with three structural repeats arranged symmetrically around a central axis. Beclin 1 ECD defines a novel class of membrane-binding domain, with a strong preference for lipid membrane enriched with cardiolipin. The tip of a surface loop in Beclin 1 ECD, comprising three aromatic amino acids, acts as a hydrophobic finger to associate with lipid membrane, consequently resulting in the deformation of membrane and liposomes. Mutation of these aromatic residues rendered Beclin 1 unable to stably associate with lipid membrane in vitro and unable to fully rescue autophagy in Beclin 1-knockdown cells in vivo. These observations form an important framework for deciphering the biological functions of Beclin 1.
Co-reporter:Jong W. Yu;Philip D. Jeffrey;Jun Yong Ha;Xiaolu Yang
PNAS 2011 108 (52 ) pp:
Publication Date(Web):2011-12-27
DOI:10.1073/pnas.1111708108
The mucosa-associated lymphoid tissue lymphoma translocation 1 (MALT1) paracaspase, a key component of the Carma1/Bcl10/MALT1
signalosome, is critical for NF-κB signaling in multiple contexts. MALT1 is thought to function as a scaffold and protease
to promote signaling; however, the biochemical and structural basis of paracaspase action remains largely unknown. Here we
report the 1.75-Å resolution crystal structure of the MALT1 paracaspase region, which contains the paracaspase domain and
an ensuing Ig-like domain. The paracaspase and the Ig domains appear as a single folding unit and interact with each other
through extensive van der Waals contacts and hydrogen bonds. The paracaspase domain adopts a fold that is nearly identical
to that of classic caspases and homodimerizes similarly to form an active protease. Unlike caspases, the active and mature
form of the paracaspase domain remains a single uncleaved polypeptide and specifically recognizes the bound peptide inhibitor
Val-Arg-Pro-Arg. In particular, the carboxyl-terminal amino acid Arg of the inhibitor is coordinated by three highly conserved
acidic residues. This structure serves as an important framework for deciphering the function and mechanism of paracaspases
exemplified by MALT1.
Co-reporter:Yigong Shi;Yi Rao
Science 2010 Vol 329(5996) pp:1128
Publication Date(Web):03 Sep 2010
DOI:10.1126/science.1196916
Summary
Government research funds in China have been growing at an annual rate of more than 20%, exceeding even the expectations of China's most enthusiastic scientists. In theory, this could allow China to make truly outstanding progress in science and research, complementing the nation's economic success. In reality, however, rampant problems in research funding—some attributable to the system and others cultural—are slowing down China's potential pace of innovation.
Co-reporter:Xiang Gao,
Lijun Zhou,
Xuyao Jiao,
Feiran Lu,
Chuangye Yan,
Xin Zeng,
Jiawei Wang
&
Yigong Shi
Nature 2010 463(7282) pp:828
Publication Date(Web):2010-01-20
DOI:10.1038/nature08741
The amino acid antiporter AdiC is important for the survival of enteric bacteria such as Escherichia coli in extremely acid environments. Although the structure of substrate-free AdiC is known, how the substrate (arginine or agmatine) is recognized and transported by AdiC remains unclear. The crystal structure of an E. coli AdiC variant bound to arginine is now reported and analysed.
Co-reporter:Xiang Gao;Feiran Lu;Lijun Zhou;Shangyu Dang;Linfeng Sun;Xiaochun Li;Jiawei Wang
Science 2009 Vol 324(5934) pp:1565-1568
Publication Date(Web):19 Jun 2009
DOI:10.1126/science.1173654
Co-reporter:Jason Huhn, Philip D. Jeffrey, Kristofer Larsen, Thomas Rundberget, Frode Rise, Neil R. Cox, Vickery Arcus, Yigong Shi and Christopher O. Miles
Chemical Research in Toxicology 2009 Volume 22(Issue 11) pp:1782
Publication Date(Web):October 21, 2009
DOI:10.1021/tx9001622
Okadaic acid (OA), dinophysistoxin-1 (DTX-1), and dinophysistoxin-2 (DTX-2) are algal toxins that can accumulate in shellfish and cause diarrhetic shellfish poisoning. Recent studies indicate that DTX-2 is about half as toxic and has about half the affinity for protein phosphatase 2A (PP2A) as OA. NMR structural studies showed that DTX-1 possessed an equatorial 35-methyl group but that DTX-2 had an axial 35-methyl group. Molecular modeling studies indicated that an axial 35-methyl could exhibit unfavorable interactions in the PP2A binding site, and this has been proposed as the reason for the reduced toxicity of DTX-2. Statistical analyses of published data indicate that the affinity of PP2A for DTX-1 is 1.6-fold higher, and for DTX-2 is 2-fold lower, than for OA. We obtained X-ray crystal structures of DTX-1 and DTX-2 bound to PP2A. The crystal structures independently confirm the C-35 stereochemistries determined in the earlier NMR study. The structure for the DTX-1 complex was virtually identical to that of the OA−PP2A complex, except for the presence of the equatorial 35-methyl on the ligand. The favorable placement of the equatorial 35-methyl group of DTX-1 against the aromatic π-bonds of His191 may account for the increased affinity of PP2A toward DTX-1. In contrast, the axial 35-methyl of DTX-2 caused the side chain of His191 to rotate 140° so that it pointed toward the solvent, thereby opening one end of the hydrophobic binding cage. This rearrangement to accommodate the unfavorable interaction from the axial 35-methyl of DTX-2 reduces the binding energy and appears to be responsible for the reduced affinity of PP2A for DTX-2. These results highlight the potential of molecular modeling studies for understanding the relative toxicity of analogues once the binding site at the molecular target has been properly characterized.
Co-reporter:Xiaochun Li;Lihui Feng;Boyuan Wang;Hui Kang;Yang Qi;Jiawei Wang
PNAS 2009 Volume 106 (Issue 35 ) pp:14837-14842
Publication Date(Web):2009-09-01
DOI:10.1073/pnas.0903289106
Regulated intramembrane proteolysis (RIP) by the Site-2 protease (S2P) results in the release of a transmembrane signaling
protein. Curiously, however, S2P cleavage must be preceded by the action of the Site-1 protease (S1P). To decipher the underlying
mechanism, we reconstituted sequential, in vitro cleavages of the Escherichia coli transmembrane protein RseA by DegS (S1P) and RseP (S2P). After DegS cleavage, the newly exposed carboxyl-terminal residue
Val-148 of RseA plays an essential role for RseP cleavage, and its mutation to charged or dissimilar amino acids crippled
the Site-2 cleavage. By contrast, the identity of residues 146 and 147 of RseA has no impact on Site-2 cleavage. These results
explain why Site-1 cleavage must precede Site-2 cleavage. Structural analysis reveals that the putative peptide-binding groove
in the second, but not the first, PDZ domain of RseP is poised for binding to a single hydrophobic amino acid. These observations
suggest that after DegS cleavage, the newly exposed carboxyl terminus of RseA may facilitate Site-2 cleavage through direct
interaction with the PDZ domain.
Co-reporter:Yi Wang,
Yongjian Huang,
Jiawei Wang,
Chao Cheng,
Weijiao Huang,
Peilong Lu,
Ya-Nan Xu,
Pengye Wang,
Nieng Yan
&
Yigong Shi
Nature 2009 462(7272) pp:467
Publication Date(Web):2009-11-26
DOI:10.1038/nature08610
The formate–nitrite transporter family, of which FocA is a representative member, is known to transport short-chain acids in bacteria, archaea, fungi, algae and parasites; however, the structure and transport mechanism of these transporters remain unknown. Here, study of the crystal structure of Escherichia coli FocA reveals that the overall structure of FocA closely resembles that of aquaporin, suggesting that it is in fact a channel, rather than a transporter.
Co-reporter:Yigong Shi
Science China Life Sciences 2009 Volume 52( Issue 2) pp:130-132
Publication Date(Web):2009 February
DOI:10.1007/s11427-009-0024-5
Co-reporter:YiGong Shi
Science China Life Sciences 2009 Volume 52( Issue 2) pp:135-146
Publication Date(Web):2009 February
DOI:10.1007/s11427-009-0018-3
Protein phosphatase 2A (PP2A) represents a conserved family of important protein serine/threonine phosphatases in species ranging from yeast to human. The PP2A core enzyme comprises a scaffold subunit and a catalytic subunit. The heterotrimeric PP2A holoenzyme consists of the core enzyme and a variable regulatory subunit. The catalytic subunit of PP2A is subject to reversible methylation, mediated by two conserved enzymes. Both the PP2A core and holoenzymes are regulated through interaction with a large number of cellular cofactors. Recent biochemical and structural investigation reveals critical insights into the assembly and function of the PP2A core enzyme as well as two families of holoenzyme. This review focuses on the molecular mechanisms revealed by these latest advances.
Co-reporter:Jong W. Yu;Philip D. Jeffrey
PNAS 2009 Volume 106 (Issue 20 ) pp:8169-8174
Publication Date(Web):2009-05-19
DOI:10.1073/pnas.0812453106
Cellular FLICE-inhibitory protein (c-FLIPL) is a key regulator of the extrinsic cell death pathway. Although widely regarded as an inhibitor of initiator caspase activation
and cell death, c-FLIPL is also capable of enhancing procaspase-8 activation through heterodimerization of their respective protease domains. However,
the underlying mechanism of this activation process remains enigmatic. Here, we demonstrate that cleavage of the intersubunit
linker of c-FLIPL by procaspase-8 potentiates the activation process by enhancing heterodimerization between the two proteins and vastly improving
the proteolytic activity of unprocessed caspase-(C)8. The crystal structures of the protease-like domain of c-FLIPL alone and in complex with zymogen C8 identify the unique determinants that favor heterodimerization over procaspase-8 homodimerization,
and induce the latent active site of zymogen C8 into a productive conformation. Together, these findings provide molecular
insights into a key aspect of c-FLIPL function that modulates procaspase-8 activation to elicit diverse responses in different cellular contexts.
Co-reporter:Guanghui Yang, Rui Zhou, Yigong Shi
Current Opinion in Structural Biology (October 2017) Volume 46() pp:55-64
Publication Date(Web):1 October 2017
DOI:10.1016/j.sbi.2017.05.013
•Methods on sample preparation and electron microscopy of human γ-secretase.•A summary of cryo-EM structures of human γ-secretase.•Functional insights revealed by the cryo-EM structures of γ-secretase.γ-secretase, a membrane-embedded aspartate protease, catalyzes peptide bond hydrolysis of a large variety of type I integral membrane proteins exemplified by amyloid precursor protein (APP). Cleavage of APP leads to formation of β-amyloid plaque, which is a hallmark of Alzheimer’s disease (AD). Over 200 AD-associated mutations are mapped to presenilin 1 (PS1), the catalytic component of γ-secretase. In the past three years, several cryo-electron microscopy (cryo-EM) structures of human γ-secretase have been determined at near atomic resolutions. Here we summarize the methods involved and discuss structural features of γ-secretase and the associated functional insights.
Co-reporter:Tian Xie, Wei Peng, Yexing Liu, Chuangye Yan, ... Yigong Shi
Structure (5 March 2013) Volume 21(Issue 3) pp:493-499
Publication Date(Web):5 March 2013
DOI:10.1016/j.str.2013.01.016
Necroptosis is a cellular mechanism that mediates necrotic cell death. The receptor-interacting serine/threonine protein kinase 1 (RIP1) is an essential upstream signaling molecule in tumor-necrosis-factor-α-induced necroptosis. Necrostatins, a series of small-molecule inhibitors, suppress necroptosis by specifically inhibiting RIP1 kinase activity. Both RIP1 structure and the mechanisms by which necrostatins inhibit RIP1 remain unknown. Here, we report the crystal structures of the RIP1 kinase domain individually bound to necrostatin-1 analog, necrostatin-3 analog, and necrostatin-4. Necrostatin, caged in a hydrophobic pocket between the N- and C-lobes of the kinase domain, stabilizes RIP1 in an inactive conformation through interactions with highly conserved amino acids in the activation loop and the surrounding structural elements. Structural comparison of RIP1 with the inhibitor-bound oncogenic kinase B-RAF reveals partially overlapping binding sites for necrostatin and for the anticancer compound PLX4032. Our study provides a structural basis for RIP1 inhibition by necrostatins and offers insights into potential structure-based drug design.Highlights► Three necrostatin-bound RIP1 kinase domain structures were reported ► Necrostatin is caged in a hydrophobic pocket between the N- and C-lobes of RIP1 ► Necrostatin-bound RIP1 is in the inactive conformation ► The binding site for necrostatin partially overlaps with that for PLX4032 in B-RAF
Co-reporter:Yanhui Xu, Yu Chen, Ping Zhang, Philip D. Jeffrey, Yigong Shi
Molecular Cell (26 September 2008) Volume 31(Issue 6) pp:873-885
Publication Date(Web):26 September 2008
DOI:10.1016/j.molcel.2008.08.006
Protein phosphatase 2A (PP2A) regulates many essential aspects of cellular physiology. Members of the regulatory B/B55/PR55 family are thought to play a key role in the dephosphorylation of Tau, whose hyperphosphorylation contributes to Alzheimer's disease. The underlying mechanisms of the PP2A-Tau connection remain largely enigmatic. Here, we report the complete reconstitution of a Tau dephosphorylation assay and the crystal structure of a heterotrimeric PP2A holoenzyme involving the regulatory subunit Bα. We show that Bα specifically and markedly facilitates dephosphorylation of the phosphorylated Tau in our reconstituted assay. The Bα subunit comprises a seven-bladed β propeller, with an acidic, substrate-binding groove located in the center of the propeller. The β propeller latches onto the ridge of the PP2A scaffold subunit with the help of a protruding β hairpin arm. Structure-guided mutagenesis studies revealed the underpinnings of PP2A-mediated dephosphorylation of Tau.
Co-reporter:Fan Zhang, Min Hu, Geng Tian, Ping Zhang, ... Yigong Shi
Molecular Cell (14 May 2009) Volume 34(Issue 4) pp:473-484
Publication Date(Web):14 May 2009
DOI:10.1016/j.molcel.2009.04.021
Eukaryotic proteasome consists of a core particle (CP), which degrades unfolded protein, and a regulatory particle (RP), which is responsible for recognition, ATP-dependent unfolding, and translocation of polyubiquitinated substrate protein. In the archaea Methanocaldococcus jannaschii, the RP is a homohexameric complex of proteasome-activating nucleotidase (PAN). Here, we report the crystal structures of essential elements of the archaeal proteasome: the CP, the ATPase domain of PAN, and a distal subcomplex that is likely the first to encounter substrate. The distal subcomplex contains a coiled-coil segment and an OB-fold domain, both of which appear to be conserved in the eukaryotic proteasome. The OB domains of PAN form a hexameric ring with a 13 Å pore, which likely constitutes the outermost constriction of the substrate translocation channel. These studies reveal structural codes and architecture of the complete proteasome, identify potential substrate-binding sites, and uncover unexpected asymmetry in the RP of archaea and eukaryotes.
Co-reporter:Fan Zhang, Zhuoru Wu, Ping Zhang, Geng Tian, ... Yigong Shi
Molecular Cell (14 May 2009) Volume 34(Issue 4) pp:485-496
Publication Date(Web):14 May 2009
DOI:10.1016/j.molcel.2009.04.022
In the archaebacterium Methanocaldococcus jannaschii (M. jannaschii), the proteasomal regulatory particle (RP), a homohexameric complex of proteasome-activating nucleotidase (PAN), is responsible for target protein recognition, followed by unfolding and translocation of the bound protein into the core particle (CP) for degradation. Guided by structure-based mutagenesis, we identify amino acids and structural motifs that are essential for PAN function. Key residues line the axial channel of PAN, defining the apparent pathway of substrate translocation. Subcomplex II of PAN, comprising the ATPase domain, associates with the CP and drives ATP-dependent unfolding of the substrate protein, whereas the distal subcomplex I forms the entry port of the substrate translocation channel. A linker segment between subcomplexes I and II is essential for PAN function, implying functional and perhaps mechanical coupling between these domains. Sequence conservation suggests that the principles of PAN function are likely to apply to the proteasomal RP of eukaryotes.

![4-Piperidineacetic acid,1-[(1R)-1-(4-chlorophenyl)-4-methylpentyl]-2-[4-(trifluoromethyl)phenyl]-,(2S,4R)-](http://img.cochemist.com/ccimg/884700/884600-68-4.png)
![4-Piperidineacetic acid,1-[(1R)-1-(4-chlorophenyl)-4-methylpentyl]-2-[4-(trifluoromethyl)phenyl]-,(2S,4R)-](http://img.cochemist.com/ccimg/884700/884600-68-4_b.png)
![(E)-1-[(1S)-1-(4-Fluorophenyl)ethyl]-3-[3-methoxy-4-(4-methyl-1H-imidazol-1-yl)benzylidene]piperidin-2-one](http://img.cochemist.com/ccimg/870900/870843-42-8.png)
![(E)-1-[(1S)-1-(4-Fluorophenyl)ethyl]-3-[3-methoxy-4-(4-methyl-1H-imidazol-1-yl)benzylidene]piperidin-2-one](http://img.cochemist.com/ccimg/870900/870843-42-8_b.png)
![Carbamic acid,N-[(1S)-3-(dimethoxyphosphinyl)-2-oxo-1-(phenylmethyl)propyl]-,1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/176600/176504-90-8.png)
![Carbamic acid,N-[(1S)-3-(dimethoxyphosphinyl)-2-oxo-1-(phenylmethyl)propyl]-,1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/176600/176504-90-8_b.png)
![Carbamic acid,[(1S)-2-phenyl-1-[(2R)-tetrahydro-5-oxo-2-furanyl]ethyl]-,1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/135200/135130-98-2.png)
![Carbamic acid,[(1S)-2-phenyl-1-[(2R)-tetrahydro-5-oxo-2-furanyl]ethyl]-,1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/135200/135130-98-2_b.png)
![TERT-BUTYL N-[(2S,3S,5R)-6-[[(2S)-1-[[(2S)-1-AMINO-1-OXO-3-PHENYLPROPAN-2-YL]AMINO]-4-METHYL-1-OXOPENTAN-2-YL]AMINO]-5-BENZYL-3-HYDROXY-6-OXO-1-PHENYLHEXAN-2-YL]CARBAMATE](http://img.cochemist.com/data/images/126409-24-3.png)
![TERT-BUTYL N-[(2S,3S,5R)-6-[[(2S)-1-[[(2S)-1-AMINO-1-OXO-3-PHENYLPROPAN-2-YL]AMINO]-4-METHYL-1-OXOPENTAN-2-YL]AMINO]-5-BENZYL-3-HYDROXY-6-OXO-1-PHENYLHEXAN-2-YL]CARBAMATE](http://img.cochemist.com/data/images/126409-24-3_b.png)








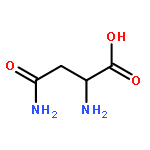
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)