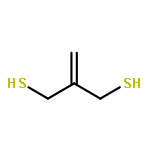
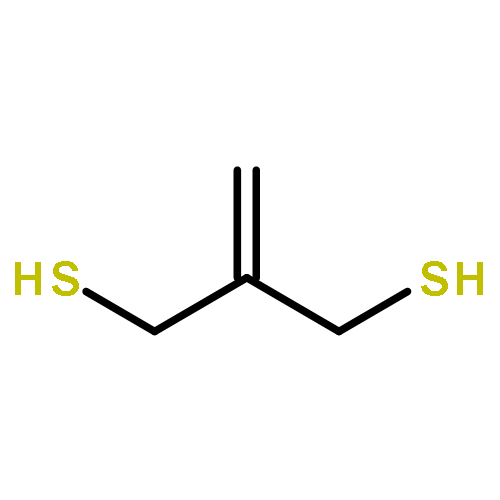
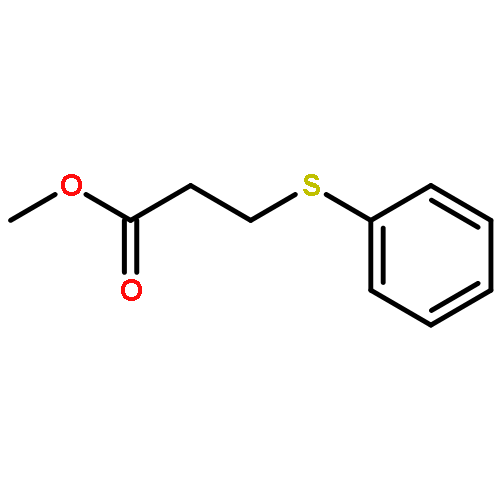
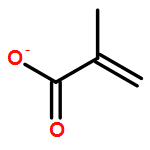
Co-reporter:Xinpeng Zhang, Weixian Xi, Sijia Huang, Katelyn Long, and Christopher N. Bowman
Macromolecules August 8, 2017 Volume 50(Issue 15) pp:5652-5652
Publication Date(Web):July 17, 2017
DOI:10.1021/acs.macromol.7b01117
We report a wavelength-selective polymerization process controlled by visible/UV light, whereby a base is generated for anion-mediated thiol–Michael polymerization reaction upon exposure at one wavelength (400–500 nm), while radicals are subsequently generated for a second stage radical polymerization at a second, independent wavelength (365 nm). Dual wavelength, light controlled sequential polymerization not only provides a relatively soft intermediate polymer that facilitates optimum processing and modification under visible light exposure but also enables a highly cross-linked, rigid final material after the UV-induced second stage radical polymerization. A photobase generator, NPPOC-TMG, and a photo-radical initiator, Irgacure 2959, were selected as the appropriate initiator pair for sequential thiol–Michael polymerization and acrylate homopolymerization. FT-IR and rheological tests were utilized to monitor the dual cure photopolymerization process, and mechanical performance of the polymer was characterized at each distinct stage by dynamic mechanical analysis (DMA). By demonstrating complete light control in another sequential polymerization system (thiol–Michael and thiol–ene hybrid polymerization), this initiator pair exhibits great potential to regulate many other coupled anion and radical hybrid polymerizations in both a sequential and controllable manner.
Co-reporter:Han Byul Song, Xiance Wang, James R. Patton, Jeffrey W. Stansbury, Christopher N. Bowman
Dental Materials 2017 Volume 33, Issue 6(Volume 33, Issue 6) pp:
Publication Date(Web):1 June 2017
DOI:10.1016/j.dental.2017.03.010
ObjectiveSeveral features necessary for polymer composite materials in practical applications such as dental restorative materials were investigated in photo-curable CuAAC (copper(I)-catalyzed azide-alkyne cycloaddition) thermosetting resin-based composites with varying filler loadings and compared to a conventional BisGMA/TEGDMA based composite.MethodsTri-functional alkyne and di-functional azide monomers were synthesized for CuAAC resins and incorporated with alkyne-functionalized glass microfillers for CuAAC composites. Polymerization kinetics, in situ temperature change, and shrinkage stress were monitored simultaneously with a tensometer coupled with FTIR spectroscopy and a data-logging thermocouple. The glass transition temperature was analyzed by dynamic mechanical analysis. Flexural modulus/strength and flexural toughness were characterized in three-point bending on a universal testing machine.ResultsThe photo-CuAAC polymerization of composites containing between 0 and 60 wt% microfiller achieved ∼99% conversion with a dramatic reduction in the maximum heat of reaction (∼20 °C decrease) for the 60 wt% filled CuAAC composites as compared with the unfilled CuAAC resin. CuAAC composites with 60 wt% microfiller generated more than twice lower shrinkage stress of 0.43 ± 0.01 MPa, equivalent flexural modulus of 6.1 ± 0.7 GPa, equivalent flexural strength of 107 ± 9 MPa, and more than 10 times higher energy absorption of 10 ± 1 MJ m−3 when strained to 11% relative to BisGMA-based composites at equivalent filler loadings.SignificanceMechanically robust and highly tough, photo-polymerized CuAAC composites with reduced shrinkage stress and a modest reaction exotherm were generated and resulted in essentially complete conversion.
Co-reporter:Zhenzhen Liu;Benjamin Fairbanks;Liangcan He;Tao Liu;Parag Shah;Jennifer N. Cha;Jeffrey W. Stansbury
Chemical Communications 2017 vol. 53(Issue 73) pp:10156-10159
Publication Date(Web):2017/09/12
DOI:10.1039/C7CC05542K
Synthetic biomacromolecules that mimic natural polymeric structures are of significant interest. For most applications of these materials, however, aqueous solubility is a necessity. Here, we present the synthesis of an intrinsically water soluble single stranded DNA analog formed by the synthesis of a Clickable Nucleic Acid (CNA). These molecules are formed with pendant hydroxyl groups present on the main polymer backbone, and subsequent modification of those hydroxyls with sulfonate moieties further enhances the hydrophilicity of these molecules.
Co-reporter:Matthew K. McBride;Matthew Hendrikx;Danqing Liu;Brady T. Worrell;Dirk J. Broer
Advanced Materials 2017 Volume 29(Issue 17) pp:
Publication Date(Web):2017/05/01
DOI:10.1002/adma.201606509
Photoactivated reversible addition fragmentation chain transfer (RAFT)-based dynamic covalent chemistry is incorporated into liquid crystalline networks (LCNs) to facilitate spatiotemporal control of alignment, domain structure, and birefringence. The RAFT-based bond exchange process, which leads to stress relaxation, is used in a variety of conditions, to enable the LCN to achieve a near-equilibrium structure and orientation upon irradiation. Once formed, and in the absence of subsequent triggering of the RAFT process, the (dis)order in the LCN and its associated birefringence are evidenced at all temperatures. Using this approach, the birefringence, including the formation of spatially patterned birefringent elements and surface-active topographical features, is selectively tuned by adjusting the light dose, temperature, and cross-linking density.
Co-reporter:Parag K. Shah;Jeffrey W. Stansbury
Polymer Chemistry (2010-Present) 2017 vol. 8(Issue 30) pp:4339-4351
Publication Date(Web):2017/08/01
DOI:10.1039/C7PY00702G
A new addition–fragmentation chain transfer (AFT) capable moiety was incorporated into a dimethacrylate monomer that participated readily in network formation by copolymerizing with multifunctional methacrylates or acrylates. The process of AFT occurred simultaneously with photopolymerization of the AFT monomer (AFM) and other (meth)acrylate monomers leading to polymer stress relaxation via network reconfiguration. At low loading levels of the AFM, a significant reduction in shrinkage stress, especially for acrylate monomers, was observed with nominal effects on conversion. At higher loading levels of the AFM, the photopolymerization reaction kinetics and final double bond conversion were significantly lowered along with a delay in the gel-point conversion. Electron paramagnetic resonance studies during polymerization revealed the presence of a distinct radical species that was present in proportional quantities to the AFM content in the system. The lifetime and the character of the persistent radicals were altered due to the presence of the distinctive radical, in turn affecting the polymerization kinetics. With polymerization conducted at higher irradiance, the differential conversion between the control resin and samples with moderate AFM content was minimal, especially for the methacrylate-based formulations.
Co-reporter:Gayla Berg Lyon;Austin Baranek
Advanced Functional Materials 2016 Volume 26( Issue 9) pp:1477-1485
Publication Date(Web):
DOI:10.1002/adfm.201505368
Step-growth Diels–Alder (DA) networks using furan and maleimide groups are particularly useful in forming thermally remendable crosslinked polymers, due to the dramatic shift in equilibrium over a relatively low temperature range as compared with other diene-dienophile pairs. However, the efficient healing observed in these materials at high temperature is directly tied to their ability to depolymerize and flow, and thermal treatment often results in deformation of the original shape. To overcome this limitation, a hybrid network material is developed, which consists of orthogonal Diels–Alder and polyurethane networks. Both step-growth networks form simultaneously at elevated temperature without the presence of a catalyst. At high temperatures, the Diels–Alder network depolymerizes and flows into fractures through capillary action, while the polyurethane serves as a scaffold to maintain the overall shape of the sample. The DA network then repolymerizes at lower temperatures, creating a crosslinked, scar-like “patch” throughout the crack. This healing process is repeatable without concern of monomer depletion. During heating through the glass transition, a shape memory “assist” is observed, which reverses some of the localized damage by bringing broken edges closer together. Samples are repeatedly damaged and then healed through temperature cycling, as evidenced through tensile fracture tests and electrochemical conductivity tests.
Co-reporter:Maciej Podgórski, Chen Wang, Ye Yuan, Danielle Konetski, Ivan Smalyukh, and Christopher N. Bowman
Chemistry of Materials 2016 Volume 28(Issue 14) pp:5102
Publication Date(Web):June 23, 2016
DOI:10.1021/acs.chemmater.6b02026
Polysulfide network oxidative modification is presented. Fundamental differences between the properties of sufide-based and sulfone-based networks are discussed, and a method for producing the sulfone-based materials from thioether-based materials is developed. Oxidation enables significant mechanical property enhancements of polysulfide materials without any deleterious effects that typically accompany cross-linking polymerizations. Various application examples such as sulfide-containing particle modification and hardening of soft lithography or thiol–ene 3D microprinted objects are also shown.
Co-reporter:Xinpeng Zhang, Weixian Xi, Chen Wang, Maciej Podgórski, and Christopher N. Bowman
ACS Macro Letters 2016 Volume 5(Issue 2) pp:229
Publication Date(Web):January 22, 2016
DOI:10.1021/acsmacrolett.5b00923
An efficient visible-light-sensitive photobase generator for thiol-Michael addition reactions was synthesized and evaluated. This highly reactive catalyst was designed by protecting a strong base (tetramethyl guanidine, TMG) with a visible-light-responsive group which was a coumarin derivative. The coumarin-coupled TMG was shown to exhibit extraordinary catalytic activity toward initiation of the thiol-Michael reaction, including thiol-Michael addition-based polymerization, upon visible-light irradiation, leading to a stoichiometric reaction of both thiol and vinyl functional groups. Owing to its features, this visible-light photobase generator enables homogeneous network formation in thiol-Michael polymerizations and also has the potential to be exploited in other visible-light-induced, base-catalyzed thiol-click processes such as thiol-isocynate and thiol-epoxy network-forming reactions.
Co-reporter:Han Byul Song, Austin Baranek and Christopher N. Bowman
Polymer Chemistry 2016 vol. 7(Issue 3) pp:603-612
Publication Date(Web):18 Nov 2015
DOI:10.1039/C5PY01655J
Photoinitiation of polymerizations based on the copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction enables spatio-temporal control and the formation of mechanically robust, highly glassy photopolymers. Here, we investigated several critical factors influencing photo-CuAAC polymerization kinetics via systematic variation of reaction conditions such as the physicochemical nature of the monomers; the copper salt and photoinitiator types and concentrations; light intensity; exposure time and solvent content. Real time Fourier transform infrared spectroscopy (FTIR) was used to monitor the polymerization kinetics in situ. Six different di-functional azide monomers and four different tri-functional alkyne monomers containing either aliphatic, aromatic, ether and/or carbamate substituents were synthesized and polymerized. Replacing carbamate structures with ether moieties in the monomers enabled an increase in conversion from 65% to 90% under similar irradiation conditions. The carbamate results in stiffer monomers and higher viscosity mixtures indicating that chain mobility and diffusion are key factors that determine the CuAAC network formation kinetics. Photoinitiation rates were manipulated by altering various aspects of the photo-reduction step; ultimately, a loading above 3 mol% per functional group for both the copper catalyst and the photoinitiator showed little or no rate dependence on concentration while a loading below 3 mol% exhibited 1st order rate dependence. Furthermore, a photoinitiating system consisting of camphorquinone resulted in 60% conversion in the dark after only 1 minute of 75 mW cm−2 light exposure at 400–500 nm, highlighting a unique characteristic of the CuAAC photopolymerization enabled by the combination of the copper(I)'s catalytic lifetime and the nature of the step-growth polymerization.
Co-reporter:Gayla Berg Lyon, Lewis M. Cox, J. Taylor Goodrich, Austin D. Baranek, Yifu Ding, and Christopher N. Bowman
Macromolecules 2016 Volume 49(Issue 23) pp:8905-8913
Publication Date(Web):November 16, 2016
DOI:10.1021/acs.macromol.6b01281
Here, we introduce photocuring as a tool for the spatiotemporal control of vitrimer network synthesis via a photoinitiated thiol–ene polymerization. A difunctional norbornene monomer was synthesized containing ester linkages and pendant alcohol groups to participate in transesterification bond reshuffling reactions. The transesterification catalyst 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) was shown to be highly effective in promoting transesterification in these networks at high temperatures, without interfering with external spatiotemporal, photoinitiated control over the thiol–ene polymerization and associated network formation. A strong Arrhenius dependence of the stress relaxation time with inverse temperature was observed from 145 to 175 °C, which suggests a relaxation controlled by the transesterification reaction rate, similar to previously studied thermally cured vitrimers. These thiol–ene vitrimers are implemented in nanoimprint lithography (NIL) for creating surface features, where imprinting may be performed repeatedly on the same sample due to the reversible nature of the bond exchange reactions. Because the networks are photocurable, hierarchical structures were generated by photopatterning and developing a microscale pattern and then performing NIL on the surface of this pattern.
Co-reporter:Farbod Alimohammadi, Chen Wang, Olivia Z. Durham, Hannah R. Norton, Christopher N. Bowman, Devon A. Shipp
Polymer 2016 Volume 105() pp:180-186
Publication Date(Web):22 November 2016
DOI:10.1016/j.polymer.2016.10.016
•First report of radical mediated thiol-ene and thiol-yne dispersion polymerizations.•Polymer particles several hundred nanometers can be made.•Thermal, photo- or redox initiation methods were all shown to be effective.•Effects of monomer, stabilizer, and initiator on particle size were studied.We report the synthesis of polymeric particles using radical mediated step-growth thiol-ene and thiol-yne ‘click’ dispersion polymerizations in alcoholic solvents. Various alkene, alkyne and thiol monomers were used, and thermal, photo- or redox initiation methods were all shown to be effective means to initiate polymerization. Polymer particles typically were formed with diameters in the range of several hundred nanometers, with narrow size distributions though broader than typical free-radical chain-growth dispersion polymerizations. Photoinitiation yielded the smallest sizes due to the rapid nucleation of particles as compared to redox and thermal initiation methods. Reaction kinetics were monitored by FT-IR spectroscopy for aliquot samples taken at various reaction durations. The reaction achieved full conversion in photoinitiated systems within five minutes, while it took three hours for both thermal and redox initiation to be completed. The effects of polymerization conditions on particle size particularly the impact of monomer, stabilizer, and initiator concentrations were studied. Generally, average particle size increased with higher monomer concentration and decreased with additional stabilizer or initiator. Combinations of monomers with varying number of functional groups were investigated to form particles of various mechanical and physical behavior, including both linear and crosslinked systems.
Co-reporter:Danielle Konetski, Tao Gong, and Christopher N. Bowman
Langmuir 2016 Volume 32(Issue 32) pp:8195-8201
Publication Date(Web):July 22, 2016
DOI:10.1021/acs.langmuir.6b02043
Synthetic vesicles have a wide range of applications from drug and cosmetic delivery to artificial cell and membrane studies, making simple and controlled formation of vesicles a large focus of the field today. Here, we report the use of the photoinitiated copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction using visible light to introduce spatiotemporal control into the formation of vesicles. Upon the establishment of the spatiotemporal control over vesicle formation, it became possible to adjust initiation conditions to modulate vesicle sizes resulting in the formation of controllably small or large vesicles based on light intensity or giant vesicles when the formation was initiated in flow-free conditions. Additionally, this photoinitiated method enables vesicle formation at a density 400-fold higher than initiation using sodium ascorbate as the catalyst. Together, these advances enable the formation of high-density, controlled size vesicles using low-energy wavelengths while producing enhanced control over the formation characteristics of the vesicle.
Co-reporter:Mauro Claudino, Xinpeng Zhang, Marvin D. Alim, Maciej Podgórski, and Christopher N. Bowman
Macromolecules 2016 Volume 49(Issue 21) pp:8061-8074
Publication Date(Web):October 18, 2016
DOI:10.1021/acs.macromol.6b01605
A kinetic mechanism and the accompanying mathematical framework are presented for base-mediated thiol–Michael photopolymerization kinetics involving a photobase generator. Here, model kinetic predictions demonstrate excellent agreement with a representative experimental system composed of 2-(2-nitrophenyl)propyloxycarbonyl-1,1,3,3-tetramethylguanidine (NPPOC-TMG) as a photobase generator that is used to initiate thiol–vinyl sulfone Michael addition reactions and polymerizations. Modeling equations derived from a basic mechanistic scheme indicate overall polymerization rates that follow a pseudo-first-order kinetic process in the base and coreactant concentrations, controlled by the ratio of the propagation to chain-transfer kinetic parameters (kp/kCT) which is dictated by the rate-limiting step and controls the time necessary to reach gelation. Gelation occurs earlier as the kp/kCT ratio reaches a critical value, wherefrom gel times become nearly independent of kp/kCT. The theoretical approach allowed determining the effect of induction time on the reaction kinetics due to initial acid–base neutralization for the photogenerated base caused by the presence of protic contaminants. Such inhibition kinetics may be challenging for reaction systems that require high curing rates but are relevant for chemical systems that need to remain kinetically dormant until activated although at the ultimate cost of lower polymerization rates. The pure step-growth character of this living polymerization and the exhibited kinetics provide unique potential for extended dark-cure reactions and uniform material properties. The general kinetic model is applicable to photobase initiators where photolysis follows a unimolecular cleavage process releasing a strong base catalyst without cogeneration of intermediate radical species.
Co-reporter:Austin Baranek, Han Byul Song, Mathew McBride, Patricia Finnegan, and Christopher N. Bowman
Macromolecules 2016 Volume 49(Issue 4) pp:1191-1200
Publication Date(Web):February 2, 2016
DOI:10.1021/acs.macromol.6b00137
Bulk photopolymerization of a library of synthesized multifunctional azides and alkynes was carried out toward developing structure–property relationships for CuAAC-based polymer networks. Multifunctional azides and alkynes were formulated with a copper catalyst and a photoinitiator, cured, and analyzed for their mechanical properties. Material properties such as the glass transition temperatures (Tg) show a strong dependence on monomer structure with Tg values ranging from 41 to 90 °C for the series of CuAAC monomers synthesized in this study. Compared to the triazoles, analogous thioether-based polymer networks exhibit a 45–49 °C lower Tg whereas analogous monomers composed of ethers in place of carbamates exhibit a 40 °C lower Tg. Here, the formation of the triazole moiety during the polymerization represents a critical component in dictating the material properties of the ultimate polymer network where material properties such as the rubbery modulus, cross-link density, and Tg all exhibit strong dependence on polymerization conversion, monomer composition, and structure postgelation.
Co-reporter:Chen Wang, Shunsuke Chatani, Maciej Podgórski and Christopher N. Bowman
Polymer Chemistry 2015 vol. 6(Issue 20) pp:3758-3763
Publication Date(Web):13 Apr 2015
DOI:10.1039/C5PY00326A
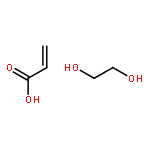
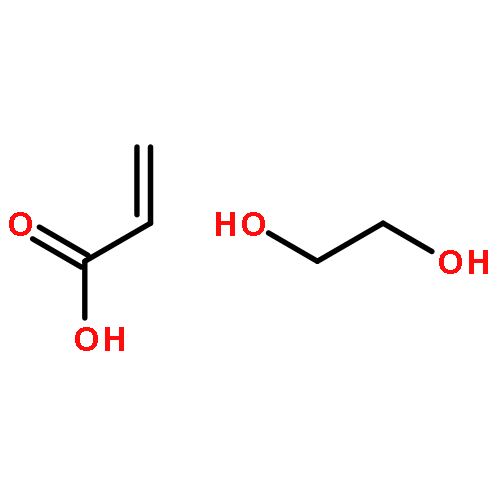
Thiol-Michael addition polymerization is successfully implemented in a miniemulsion polymerization system. By off-stoichiometric polymerization between thiols and acrylates, inherently functionalized particles are facilely prepared in a single step. We demonstrate that the latex films from such particles are readily available for further modification and second-stage photo-curing.
Co-reporter:Maciej Podgórski, Eftalda Becka, Shunsuke Chatani, Mauro Claudino and Christopher N. Bowman
Polymer Chemistry 2015 vol. 6(Issue 12) pp:2234-2240
Publication Date(Web):19 Jan 2015
DOI:10.1039/C4PY01552E
A series of thiol-Michael and radical thiol–ene network polymers were successfully prepared from ester-free as well as ester-containing monomer formulations. Polymerization reaction rates, dynamic mechanical analysis, and solvent resistance experiments were performed and compared between compositions with varied ester loading. The incorporation of ester-free alkyl thiol, vinyl sulfone and allylic monomers significantly improved the mechanical properties when compared with commercial, mercaptopropionate-based thiol–ene or thiol-Michael networks. For polymers with no hydrolytically degradable esters, glass transition temperatures (Tg's) as high as 100 °C were achieved. Importantly, solvent resistance tests demonstrated enhanced stability of ester-free formulations over PETMP-based polymers, especially in concentrated basic solutions. Kinetic analysis showed that glassy step-growth polymers are readily formed at ambient conditions with conversions reaching 80% and higher.
Co-reporter:M. Podgórski, C. Wang and C. N. Bowman
Soft Matter 2015 vol. 11(Issue 34) pp:6852-6858
Publication Date(Web):27 Jul 2015
DOI:10.1039/C5SM01260K
This investigation details the formation of polymer network trilayer laminates formed by thiol-X click chemistries, and their subsequent implementation and evaluation for quadruple shape memory behavior. Thiol-Michael addition and thiol-isocyanate-based crosslinking reactions were employed to fabricate each of the laminate's layers with independent control of the chemistry and properties of each layer and outstanding interlayer adhesion and stability. The characteristic features of step-growth thiol-X reactions, such as excellent network uniformity and narrow thermal transitions as well as their stoichiometric nature, enabled fabrication of trilayer laminates with three distinctly different glass transition temperatures grouped within a narrow range of 100 °C. Through variations in the layer thicknesses, a step-wise modulus drop as a function of temperature was achieved. This behavior allowed multi-step programming and the demonstration and quantification of quadruple shape memory performance. As is critical for this performance, the interface connecting the layers was evaluated in stoichiometric as well as off-stoichiometric systems. It was shown that the laminated structures exhibit strong interfacial binding and hardly suffer any delamination during cyclic material testing and deformation.
Co-reporter:Weixian Xi;Dr. Sankha Pattanayak;Chen Wang;Dr. Benjamin Fairbanks;Dr. Tao Gong;Justine Wagner;Dr. Christopher J. Kloxin;Dr. Christopher N. Bowman
Angewandte Chemie International Edition 2015 Volume 54( Issue 48) pp:
Publication Date(Web):
DOI:10.1002/anie.201509369
Co-reporter:Weixian Xi;Dr. Sankha Pattanayak;Chen Wang;Dr. Benjamin Fairbanks;Dr. Tao Gong;Justine Wagner;Dr. Christopher J. Kloxin;Dr. Christopher N. Bowman
Angewandte Chemie 2015 Volume 127( Issue 48) pp:
Publication Date(Web):
DOI:10.1002/ange.201509369
Co-reporter:Weixian Xi;Dr. Sankha Pattanayak;Chen Wang;Dr. Benjamin Fairbanks;Dr. Tao Gong;Justine Wagner;Dr. Christopher J. Kloxin;Dr. Christopher N. Bowman
Angewandte Chemie 2015 Volume 127( Issue 48) pp:14670-14675
Publication Date(Web):
DOI:10.1002/ange.201506711
Abstract
Synthetic polymer approaches generally lack the ability to control the primary sequence, with sequence control referred to as the holy grail. Two click chemistry reactions were now combined to form nucleobase-containing sequence-controlled polymers in simple polymerization reactions. Two distinct approaches are used to form these click nucleic acid (CNA) polymers. These approaches employ thiol–ene and thiol-Michael reactions to form homopolymers of a single nucleobase (e.g., poly(A)n) or homopolymers of specific repeating nucleobase sequences (e.g., poly(ATC)n). Furthermore, the incorporation of monofunctional thiol-terminated polymers into the polymerization system enables the preparation of multiblock copolymers in a single reaction vessel; the length of the diblock copolymer can be tuned by the stoichiometric ratio and/or the monomer functionality. These polymers are also used for organogel formation where complementary CNA-based polymers form reversible crosslinks.
Co-reporter:Weixian Xi;Dr. Sankha Pattanayak;Chen Wang;Dr. Benjamin Fairbanks;Dr. Tao Gong;Justine Wagner;Dr. Christopher J. Kloxin;Dr. Christopher N. Bowman
Angewandte Chemie International Edition 2015 Volume 54( Issue 48) pp:14462-14467
Publication Date(Web):
DOI:10.1002/anie.201506711
Abstract
Synthetic polymer approaches generally lack the ability to control the primary sequence, with sequence control referred to as the holy grail. Two click chemistry reactions were now combined to form nucleobase-containing sequence-controlled polymers in simple polymerization reactions. Two distinct approaches are used to form these click nucleic acid (CNA) polymers. These approaches employ thiol–ene and thiol-Michael reactions to form homopolymers of a single nucleobase (e.g., poly(A)n) or homopolymers of specific repeating nucleobase sequences (e.g., poly(ATC)n). Furthermore, the incorporation of monofunctional thiol-terminated polymers into the polymerization system enables the preparation of multiblock copolymers in a single reaction vessel; the length of the diblock copolymer can be tuned by the stoichiometric ratio and/or the monomer functionality. These polymers are also used for organogel formation where complementary CNA-based polymers form reversible crosslinks.
Co-reporter:Chen Wang, Xinpeng Zhang, Maciej Podgórski, Weixian Xi, Parag Shah, Jeffery Stansbury, and Christopher N. Bowman
Macromolecules 2015 Volume 48(Issue 23) pp:8461-8470
Publication Date(Web):November 19, 2015
DOI:10.1021/acs.macromol.5b02146
We report a dispersion polymerization method based on thiol–Michael addition reactions for the preparation of cross-linked, narrow dispersity microparticles with well-defined, tunable physicochemical properties. Polymerization between pentaerythritol tetra(3-mercaptopropionate) (PETMP) and trimethylolpropane triacrylate in methanol was chosen as a model system, with the addition of triethylamine as a catalyst and polyvinylpyrrolidone as a stabilizer. The formation of microparticles took place within seconds at ambient conditions, as a result of a polymerization driven phase transition from dissolved monomers to precipitated polymers. The particle size was found to be affected by the amount of catalyst, the monomer concentration, and the monomer/polymer solubility in the reaction media. Monodispersity was achieved within a range of particle diameters from 1.6 to 4.3 μm, as determined both by scanning electron microscopy and dynamic light scattering. The reaction kinetics were studied by Fourier transform infrared spectroscopy by analyzing aliquots withdrawn from the reaction system at various reaction time points. Nearly quantitative conversions were achieved within 6 h for stoichiometric systems and 1 h for off-stoichiometric systems, both initiated with triethylamine. By utilizing photolabile bases as the reaction catalyst, phototriggered formation of the microparticles was demonstrated with ultraviolet irradiation. Monodisperse particles were formed with hexylamine and 1,1,3,3-tetramethylguanidine, both with 2-(2-nitrophenyl)propyloxycarbonyl as the UV-labile photocage. Furthermore, as a demonstration of the versatility of this method, microparticles were prepared from copolymerizations between PETMP and four types of diacrylates with varied backbone structures. With increased backbone rigidity, the microparticle glass transition temperature increased from −36 to 8 °C. This method provides a platform for the realization of the nearly ideal step-growth networks in microscale, with highly tunable backbone structures, robust thermal transitions, and intrinsic functionalization capacity.
Co-reporter:Weixian Xi;Timothy F. Scott;Christopher J. Kloxin
Advanced Functional Materials 2014 Volume 24( Issue 18) pp:2572-2590
Publication Date(Web):
DOI:10.1002/adfm.201302847
Despite originating only a little more than a decade ago, click chemistry has become one of the most powerful paradigms in materials science, synthesis, and modification. By developing and implementing simple, robust chemistries that do not require difficult separations or harsh conditions, the ability to form, modify, and control the structure of materials on various length scales has become more broadly available to those in the materials science community. As such, click chemistry has seen broad implementation in polymer functionalization, surface modification, block copolymer and dendrimer synthesis, biomaterials fabrication, biofunctionalization, and in many other areas of materials science. Here, the basic reactions, approaches, and applications of click chemistry in materials science are highlighted, and a brief look is taken into the future enabling developments in this field.
Co-reporter:Devatha P. Nair, Maciej Podgórski, Shunsuke Chatani, Tao Gong, Weixian Xi, Christopher R. Fenoli, and Christopher N. Bowman
Chemistry of Materials 2014 Volume 26(Issue 1) pp:724
Publication Date(Web):August 19, 2013
DOI:10.1021/cm402180t
The key attribute of the thiol-Michael addition reaction that makes it a prized tool in materials science is its modular “click” nature, which allows for the implementation of this highly efficient, “green” reaction in applications that vary from small molecule synthesis to in situ polymer modifications in biological systems to the surface functionalization of material coatings. Over the past few decades, interest in the thiol-Michael addition reaction has increased dramatically, as is evidenced by the number of studies that have been dedicated to elucidating different aspects of the reaction that range from an in-depth analysis aimed at understanding the mechanistic pathways of the reaction to synthetic studies that have examined modifying molecular structures with the aim of yielding highly efficient thiol-Michael reaction monomers. This review examines the reaction mechanisms, the substrates and catalysts used in the reaction, and the subsequent implementation of the thiol-Michael reaction in materials science over the years, with particular emphasis on the recent developments in the arena over the past decade.Keywords: thiol-click chemistry; thiol-Michael addition reaction; thiol-Michael addition reaction mechanism;
Co-reporter:Haiyan Peng, Chen Wang, Weixian Xi, Benjamin A Kowalski, Tao Gong, Xiaolin Xie, Wentao Wang, Devatha P. Nair, Robert R. McLeod, and Christopher N. Bowman
Chemistry of Materials 2014 Volume 26(Issue 23) pp:6819
Publication Date(Web):November 20, 2014
DOI:10.1021/cm5034436
Freestanding substrates with high refractive index modulation, good oxygen resistance, and low volume shrinkage are critical in photolithography for the purpose of high density data storage, image patterning and anticounterfeiting. Herein, we demonstrate a novel paradigm of direct holographic image patterning via the radical-mediated thiol–yne click reaction subsequent to the base-catalyzed thiol-Michael addition reaction. With the benefit of a newly synthesized alkyne monomer, 9-(2-((2-(prop-2-yn-1-yloxy)ethyl)thio)ethyl)-9H-carbazole (POETEC), holograms with as high as 96% diffraction efficiency, refractive index modulation of 0.0036, dynamic range of 5.6 per 200 μm and volume shrinkage of 1.1%, are successfully patterned in an aerobic environment. Uniquely and distinctly, an inhibitor is unnecessary to prevent the initiation of the sequential reaction in this framework.
Co-reporter:Chen Wang, Maciej Podgórski and Christopher N. Bowman
Materials Horizons 2014 vol. 1(Issue 5) pp:535-539
Publication Date(Web):18 Jun 2014
DOI:10.1039/C4MH00082J
Herein, we demonstrate that, monodisperse, cross-linked clickable microspheres are made via step-growth thiol–acrylate Michael addition polymerization. The diameter of the microsphere is varied from 1–10 μm, depending on the cross-link density and the reaction conditions. Implementations of these microspheres including functionalized microspheres for “click” chemistry, polymeric composites, fluorescent labeling and polymer degradation are discussed and/or demonstrated.
Co-reporter:Abeer A. Alzahrani, Devatha P. Nair, David J. Smits, Mohand Saed, Christopher M. Yakacki, and Christopher N. Bowman
Chemistry of Materials 2014 Volume 26(Issue 18) pp:5303-5309
Publication Date(Web):August 26, 2014
DOI:10.1021/cm502237b
Two sequential click reactions are implemented in a facile methodology used to generate well-defined spatiotemporally controlled and persistent wrinkles on the surface of an elastomer. The click thiol-Michael addition reaction was utilized to form a cross-linked polymer with residual, reactive alkyne sites that remained tethered throughout the network. The latent, unreacted alkyne sites are subsequently reacted with diazide monomers via a photoinduced Cu(I)-catalyzed alkyne–azide cycloaddition (CuAAC) reaction to increase the cross-link density. Increased cross-linking raised the modulus and glass transition temperature from 1.6 MPa and 2 °C after the thiol–acrylate reaction to 4.4 MPa and 22 °C after the CuAAC reaction. However, the second-stage photopolymerization of the CuAAC reaction is significantly spatially restricted via limited Cu(II) ion diffusion into the thiol–acrylate elastomer, thereby creating the desired cross-linking gradient throughout the depth of the initial network and leading to the formation of a highly cross-linked skin layer. This approach leads to the formation of well-defined, persistent, reproducible wrinkles on the surface of the material with wavelength and amplitude of 8.50 ± 1.60 and 1.41 μm, respectively, for a polymer with a 1280 μm total film thickness. Control over the wavelength and amplitude of these wrinkles using the resin film thickness is further demonstrated by studying the surface profiles using scanning electron microscopy (SEM) and atomic force microscopy (AFM). Additionally, this novel technique allows the spatial selectivity of wrinkle formation with a wrinkled area that is only 8 μm wider than the photomasked area. This strategy represents a unique approach to generate photodirected wrinkling on the entire surface of the elastomer in one single step.
Co-reporter:Maciej Podgórski, Devatha P. Nair, Shunsuke Chatani, Gayla Berg, and Christopher N. Bowman
ACS Applied Materials & Interfaces 2014 Volume 6(Issue 9) pp:6111
Publication Date(Web):January 10, 2014
DOI:10.1021/am405371r
Thiol-isocyanate-methacrylate two-stage reactive network polymers were developed and used for fabrication of well-defined surface patterns as well as functional geometric shapes to demonstrate a new methodology for processing of “smart materials”. The dynamic stage I networks were synthesized in base-catalyzed thiol-isocyanate cross-linking reactions to yield tough, glassy materials at ambient conditions. Methacrylate-rich stage I networks, incorporating photoinitiator and photoabsorber, were irradiated with UV light to generate stage II networks with intricate property gradients. Upon directional straining and subsequent temperature-dependent stress relief of the predefined gradient regions, the desired surface or bulk geometric transformations were achieved. Depending on the gradient extent in conjunction with photoorthogonal initiators, the introduced deformations were shown to be easily erasable by heat or permanently fixable by bulk polymerization.Keywords: photopatterning; shape memory; stimuli-responsive materials; thiol isocyanate reaction; thiol-Michael addition;
Co-reporter:Abeer A. Alzahrani, Annette H. Erbse and Christopher N. Bowman
Polymer Chemistry 2014 vol. 5(Issue 6) pp:1874-1882
Publication Date(Web):13 Dec 2013
DOI:10.1039/C3PY01064C
Here, several distinct approaches for photoinitiation and subsequent utilization of the Copper catalyzed azide–alkyne cycloaddition (CuAAC) reaction are developed. In particular, Cu(II)–ligand complexes were synthesized that enabled direct photoreduction of the Cu(II). The sequential and orthogonal nature of the photo-CuAAC reaction and a chain-growth acrylate homopolymerization were demonstrated and used to form branched polymer structures. The efficiency of the photo-initiated Cu(II) complexes in regard to their ability to initiate the CuAAC reaction was examined by reacting a variety of amino-functional ligands with Cu(II) halides to form complexes capable of forming Cu(I) upon light irradiation. When irradiated with 365 nm light, the ligand donates an electron to Cu(II) to reduce it to Cu(I) which subsequently initiates the azide–alkyne cycloaddition (i.e., photo-CuAAC) reaction with exquisite spatiotemporal control. Aliphatic amine ligands were found to be the most efficient ligands in promoting photoreduction of Cu(II) and stabilizing Cu(I), once formed. Among the aliphatic amines studied, tertiary amines such as triethylamine (TEA), tetramethylethylenediamine (TMDA), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDTA), and hexamethylenetetramine (HMTETA) were found to be the most effective. In addition, the Cu(II)–amine complexes were insensitive to oxygen, indicating that the catalytic Cu(I) is largely prevented from re-oxidation by complexation with the amine ligand and/or the triazole. The reaction kinetics were accelerated by increasing the PMDETA:Cu(II) ratio with a ratio of ligand to Cu(II) of 4:1 yielding the maximum conversion in the shortest time.
Co-reporter:Shunsuke Chatani, Christopher J. Kloxin and Christopher N. Bowman
Polymer Chemistry 2014 vol. 5(Issue 7) pp:2187-2201
Publication Date(Web):24 Dec 2013
DOI:10.1039/C3PY01334K
As the demand for polymeric materials transitions towards the need for customizable, high value, specialty polymeric materials, the ability to use light to initiate various physicochemical changes in polymers represents one of the most powerful and rapidly evolving approaches. Whether for polymer formation, polymer modification, shape change, or inducing smart material responses, light has the unique capacity for enabling 4D manipulation of each of those processes. Given the simple, 3D ability to focus light on a targeted voxel and the even simpler ability to turn a light on and off to facilitate temporal control, light has been used widely in various polymer modifications. Further, in addition to the ability to enhance the control of various reactive processes, due to the much greater energy available in a photon as compared to the thermal energy available, light enables chemical processes to occur at ambient conditions that are otherwise inaccessible without heating. In particular, within the polymer chemistry field, light has been used to cause bond formation, bond degradation, and isomerization, with subsequent reactions including polymerization, polymer degradation, polymer functionalization, and responsive changes in properties of smart materials. Here, this article attempts to provide a fundamental basis for the various photochemical processes implemented in polymer systems, followed by selected examples of that implementation in various polymerization, functionalization, degradation, and other reactions.
Co-reporter:Shunsuke Chatani, Tao Gong, Brittany A. Earle, Maciej Podgórski, and Christopher N. Bowman
ACS Macro Letters 2014 Volume 3(Issue 4) pp:315
Publication Date(Web):March 19, 2014
DOI:10.1021/mz500132j
A visible-light base generating system was successfully employed in catalyzing the thiol-Michael addition reaction to yield cross-linked polymers from a stoichiometric mixture of model thiol and vinyl monomers. Implementation of the radical inhibitor TEMPO with a combination of a photosensitizer (isopropylthioxanthone, ITX) and a photobase generator (triazabicyclodecene tetraphenylborate, TBD·HBPh4) resulted in suppression of radical mediated side reactions and provided stoichiometric and complete conversion of both thiol and vinyl functional groups. The new initiating system acts as an efficient visible-light photobase generator that improves the orthogonality of the thiol-Michael addition with respect to off-stoichiometric radical thiol-vinyl addition/vinyl chain reactions. This approach opens up a variety of possibilities for base-catalyzed reactions in multiple applications such as coatings and biomaterials that require biocompatible, environmentally friendly, and low-energy visible-light initiation.
Co-reporter:Christopher R. Fenoli and Christopher N. Bowman
Polymer Chemistry 2014 vol. 5(Issue 1) pp:62-68
Publication Date(Web):14 Aug 2013
DOI:10.1039/C3PY00709J
In this study, we present a mild, efficient, and high yield synthesis of a variety of trithiocarbonates and allyl sulphide-containing AFT (addition–fragmentation termination) monomers containing terminal (meth)acrylate functional groups. The trithiocarbonate moiety core was synthesized with reaction yields often above 95%. Synthesis of monomers with allyl sulphide cores gave yields ranging from 48–92%. These monomers, designated as CRAFT (Controllable Reversible Addition Chain Transfer) monomers, offer the ability to control the mechanical and polymerization properties of the resulting thiol-Michael and chain-growth polymer networks in a previously unattainable manner. The triothiocarbonates reached 70–81% acrylate conversion in 10–200 s of light exposure while the allyl sulphide monomers reached 74–85% conversion in 10–50 s.
Co-reporter:Maciej Podgórski;Shunsuke Chatani
Macromolecular Rapid Communications 2014 Volume 35( Issue 17) pp:1497-1502
Publication Date(Web):
DOI:10.1002/marc.201400260
Co-reporter:James W. Wydra;Christopher R. Fenoli;Neil B. Cramer;Jeffrey W. Stansbury
Journal of Polymer Science Part A: Polymer Chemistry 2014 Volume 52( Issue 9) pp:1315-1321
Publication Date(Web):
DOI:10.1002/pola.27120
ABSTRACT
The effects of the addition of small amounts of multifunctional monomers that contain functional groups capable of undergoing addition-fragmentation during radical polymerizations are investigated. Specifically, up to 6 wt % of phenyl trithiocarbonate (TTC)-containing diacrylate was added to conventional thiol-multiacrylate photopolymerizations where its addition led to up to 60% reduction in polymerization-induced shrinkage stress. The higher levels of TTC achieve the lowest stress though they also significantly depress the polymerization rate. Using up to 0.5 wt % phenyl TTC successfully reduces the stress by nearly 20%, demonstrating the effectiveness of the phenyl TTC, while minimizing the influence that the RAFT activity of the TTC unit has on the polymerization rate. When the polymerization rates of the TTC-containing resins are increased by changing the incident light intensity, complete acrylate conversion is achieved and the stress remains up to 40% lower in the TTC-containing resins. © 2014 Wiley Periodicals, Inc. J. Polym. Sci., Part A: Polym. Chem. 2014, 52, 1315–1321
Co-reporter:Christopher R. Fenoli, James W. Wydra, and Christopher N. Bowman
Macromolecules 2014 Volume 47(Issue 3) pp:907-915
Publication Date(Web):January 31, 2014
DOI:10.1021/ma402548e
With the advent of systematically designed controllable reversible addition–fragmentation termination (CRAFT) compounds, we have identified structure–property relationships related to the RAFT compositional structure as it impacts photoplasticity in covalent adaptable networks (CANs). In this study, we have expanded the range and functional capabilities of addition–fragmentation capable network forming monomers by synthesizing and evaluating systematically varying CRAFT monomers with the general formula ABCBA. Subsequent assessment of the impact of these monomers on photoplasticity and stress relaxation was performed. Structural variation of the A and B segments, in particular, imparts increased efficiency and efficacy in stress relaxation and photoplasticity. The CRAFT monomers employed have highly efficient stress relaxation properties demonstrating stress reduction of up to 54% and 75%, respectively, in postpolymerization network photoplasticity experiments. Furthermore, polymerization stress reduction in purely acrylate and acrylate–thiol networks with CRAFT monomers shows a remarkably enhanced efficacy with the inclusion of relatively small amounts of the monomers. With a loading of only 1.5 wt % of the alkene trithiocarbonate monomer in each system more than 75% stress reduction was achieved.
Co-reporter:Gayla J. Berg, Tao Gong, Christopher R. Fenoli, and Christopher N. Bowman
Macromolecules 2014 Volume 47(Issue 10) pp:3473-3482
Publication Date(Web):May 12, 2014
DOI:10.1021/ma500244r
A Diels–Alder (DA) network containing dissolved multiacrylate monomers is demonstrated as a novel two-stage reactive polymer network, with a potential application in self-supporting stereolithography. Initially, a thermoreversible Diels–Alder “scaffold” network is formed, containing unreacted acrylate monomers and photoinitiator. During photopatterning with light at 15 mW/cm2 from a 365 nm source for 16 s of exposure at either ambient temperature or 70 °C, both acrylates and unreacted maleimides polymerize to form a permanent, covalently cross-linked network structure that simultaneously maintains the thermoreversible characteristics afforded by the underlying DA network. Light exposure of a DA network containing between 25 and 50 wt % acrylate monomer resulted in a sharp increase in cross-link density and a 60 °C jump in glass transition temperature of the material. As a result of the temperature-dependent DA equilibrium, the temperature of the film during light exposure has dramatic effects on the resulting acrylate conversion (as measured by FT-IR) and mechanical behavior (as measured by DMA) of the complex dynamic network structure. For example, despite the irreversible acrylate network, the rubbery modulus of the material decreases above the glass transition temperature due to the presence of the dynamic thermosensitive DA network. The shape of the modulus curve was also affected by the ratio of DA monomers to acrylate monomers; higher DA monomer content resulted in greater temperature sensitivity of the rubbery modulus in light-exposed films. 3D structures with feature sizes ranging from 50 to 500 μm were produced in geometries such as stacked rectangles and “logpile” structures. In the unexposed regions, free acrylate and maleimide groups were shown to tolerate temperatures as high as 120 °C with no premature gel formation observed. Removal of unexposed material during the development step was achieved at 120 °C, where the Diels–Alder equilibrium shifts toward the furan and maleimide reactants and the network depolymerizes. Finally, a process was developed for the fabrication of 3D microstructures via layer-by-layer photopatterning. The process is highly repeatable and results in complete elimination of unexposed regions. Additionally, excess quantities of the unexposed mixture may be stored at 4 °C for at least several weeks and then reused by heating to 120 °C to fully depolymerize the DA network, subsequently using the liquid mixture to make films.
Co-reporter:Shunsuke Chatani, Maciej Podgórski, Chen Wang, and Christopher N. Bowman
Macromolecules 2014 Volume 47(Issue 15) pp:4894-4900
Publication Date(Web):July 22, 2014
DOI:10.1021/ma501418r
A kinetically selective thiol–Michael addition “click” reaction was employed for facile and efficient synthesis of dendrimers as a first example of using solely a single chemistry without protection/deprotection reactions in dendrimer synthesis. First, a wide range of thiols and vinyls were assessed for their reaction selectivity, and several combinations demonstrated superior selectivity. This result led to a design of new monomers A*A2 (vinyl) and B*B2 (thiol) that both have three functional groups with one much more reactive and two much less reactive moieties. Starting from a multifunctional core thiol, a fifth-generation dendrimer with 96 peripheral functional groups was synthesized in less than a half day by sequentially reacting A*A2 and B*B2 monomers all under thiol–Michael addition reaction conditions. Furthermore, a one-pot dendritic–linear polymer conjugation was demonstrated by a convergent synthesis approach starting from an alkyne-terminated dendron synthesis that was subsequently coupled with azide-terminated poly(ethylene glycol), all in one pot with just a single, final purification step needed for the entire procedure. The results herein will provide a new, robust, and efficient methodology for synthesis of dendrimers as well as other complex polymer architectures such as dendritic–linear polymer conjugates, heterofunctional dendrimers, and dendronized surfaces.
Co-reporter:Gayla J. Berg, Matthew K. McBride, Chen Wang, Christopher N. Bowman
Polymer 2014 Volume 55(Issue 23) pp:5849-5872
Publication Date(Web):5 November 2014
DOI:10.1016/j.polymer.2014.07.052
The rapidly expanding field of shape memory polymers (SMPs) is driven by a growing number of potential applications, such as biomaterials, optics, and electronics. The basic concept involves polymers that can be trapped in a thermodynamically-unfavorable shape, then triggered by an external stimulus to return to their original shape, doing useful work in the process. Part of the attraction of using SMPs is that the energy released during actuation is stored in the polymer itself, rather than requiring an external force to change shape. This approach is beneficial for applications where external actuation is impossible or inconvenient. Polymers are also advantageous over shape memory metal alloys or ceramics in that there are endless combinations of functional groups and material properties to suit a variety of purposes, based on the monomers and polymerization conditions chosen. This advantage of SMPs is of particular interest in the development of materials with additional, desirable physicochemical attributes that are not necessarily coupled to the shape memory (SM) behavior itself. The SM behavior is quantitatively measured to facilitate comparison of various polymer systems, and researchers have used a number of defining parameters to guide the development and characterization of materials with extremely precise and reliable SM responses. In this review, recent trends in the structural or chemical characteristics of SMPs are explored, with an emphasis on how the molecular structure and functionality of each polymer affects its mechanical response.
Co-reporter:Christopher N. Bowman
Polymer 2014 Volume 55(Issue 23) pp:5847-5848
Publication Date(Web):5 November 2014
DOI:10.1016/j.polymer.2014.09.058
Co-reporter:Matthew K. McBride, Tao Gong, Devatha P. Nair, Christopher N. Bowman
Polymer 2014 Volume 55(Issue 23) pp:5880-5884
Publication Date(Web):5 November 2014
DOI:10.1016/j.polymer.2014.08.001
•Shape memory attributes of CuAAC polymer networks are studied.•Polymers are developed with Tg's ranging from 55 to 114 °C using the CuAAC.•Polymerization with the CuAAC reaction offers control over the final properties.The formation of polymer networks polymerized with the Copper (I) – catalyzed azide – alkyne cycloaddition (CuAAC) click reaction is described along with their accompanying utilization as shape memory polymers. Due to the click nature of the reaction and the synthetic accessibility of azide and alkyne functional-monomers, the polymer architecture was readily controlled through monomer design to manipulate crosslink density, ability for further functionalization, and the glass transition temperature (55–114 °C). Free strain recovery is used to quantify the shape memory properties of a model CuAAC network resulting in excellent shape fixity and recovery of 99%. The step growth nature of this polymerization results in homogenous network formation with narrow glass transitions ranges having half widths of the transition close to 15 °C for these materials resulting in shape recovery sharpness of 3.9%/°C in a model system comparable to similarly crosslinked chain growth polymers. Utilization of the CuAAC reaction to form shape memory materials opens a range of possibilities and behaviors that are not readily achieved in other shape memory materials such as (meth) acrylates, thiol-ene, thiol-Michael, and poly(caprolactone) based shape memory materials.
Co-reporter:Weixian Xi, Haiyan Peng, Alan Aguirre-Soto, Christopher J. Kloxin, Jeffery W. Stansbury, and Christopher N. Bowman
Macromolecules 2014 Volume 47(Issue 18) pp:6159-6165
Publication Date(Web):September 8, 2014
DOI:10.1021/ma501366f
Photochemical processes enable spatial and temporal control of reactions, which can be implemented as an accurate external control approach in both polymer synthesis and materials applications. “Click” reactions have also been employed as efficient tools in the same field. Herein, we combined photochemical processes and thiol-Michael “click” reactions to achieve a “photo-click” reaction that can be used in surface patterning and controlled polymer network formation, owing to the ease of spatial and temporal control through use of photolabile amines as appropriate catalysts.
Co-reporter:Shunsuke Chatani, Chen Wang, Maciej Podgórski, and Christopher N. Bowman
Macromolecules 2014 Volume 47(Issue 15) pp:4949-4954
Publication Date(Web):July 23, 2014
DOI:10.1021/ma501028a
We present a composite material composed of dual polymer networks uniquely formed from a single reaction type and catalyst but involving monomers with dramatically different reactivities. This powerful new approach to creating polymer networks produces two narrow glass transition, homogeneous networks sequentially from a single reaction but with all monomers present and uniformly mixed prior to any polymerization. These materials exhibit a triple shape memory effect based on the dual polymer networks, which were both formed using the thiol–Michael addition reaction. Two multifunctional thiol monomers (i.e., mercaptoacetate (MA) and mercaptopropionate (MP)) and two multifunctional vinyls (i.e., vinyl sulfone (V) and acrylate (A)) were polymerized in situ using a nucleophilic initiator. The MA-V polymer network (Tg = 55 °C) was generated first associated with the higher functional group reactivities followed by the formation of the MP-A network (Tg = 10 °C) which was confirmed by FT-IR, SEM, DMA, and a separately prepared composite polymer consisting of MA-V particles embedded in an MP-A matrix. The triple shape memory effect was characterized using DMA, and it was demonstrated that the shapes could be programmed either by a one-step (single temperature) or a two-step method (two different temperatures). This material was able to hold its transitional shape for an extended time period (>1 h) at intermediate temperature (20 °C) between its two Tgs, mainly due to narrow transitions of two separate networks. This new approach to obtain dual polymer networks with distinct transitions and characteristics is simple and robust, thus enabling applications in areas such as triple shape memory polymers, biomedical materials, and composites.
Co-reporter:Haiyan Peng, Devatha P. Nair, Benjamin A. Kowalski, Weixian Xi, Tao Gong, Chen Wang, Michael Cole, Neil B. Cramer, Xiaolin Xie, Robert R. McLeod, and Christopher N. Bowman
Macromolecules 2014 Volume 47(Issue 7) pp:2306-2315
Publication Date(Web):March 28, 2014
DOI:10.1021/ma500167x
Orthogonal, sequential “click” reactions were implemented to yield novel polymeric substrates with the ability to record holographic data. The base-catalyzed thiol–acrylate Michael “click” reaction was implemented to yield a writable, stage 1 polymeric substrate with glass transition temperatures (Tg) ranging from 0 to −26 °C and rubbery storage moduli (E′) from 11.1 to 0.3 MPa. The loosely cross-linked matrix also contained a novel high refractive index monomer 9-(2,3-bis(allyloxy)propyl)-9H-carbazole (BAPC) that did not participate in the thiol–Michael reaction but allowed for large index gradients to be developed within the network upon subsequent exposure to coherent laser beams and initiation of the radical-mediated thiol–ene reaction. The holographic gratings were recorded with 96% diffraction efficiency and ca. 2.4 cm/mJ of light sensitivity in 2 s under a 405 nm exposure with an intensity of 20 mW/cm2. Subsequent to pattern formation, via a thiol–allyl radical “click” photopolymerization initiated by flood illumination of the sample, holographic materials with high Tg, high modulus, diffraction efficiency as high as 82%, and refractive index modulation of 0.004 were obtained. Graded rainbow holograms that displayed colors from blue to red at a single viewing angle were readily formed through this new technique.
Co-reporter:Christopher J. Kloxin and Christopher N. Bowman
Chemical Society Reviews 2013 vol. 42(Issue 17) pp:7161-7173
Publication Date(Web):12 Apr 2013
DOI:10.1039/C3CS60046G
Covalently crosslinked materials, classically referred to as thermosets, represent a broad class of elastic materials that readily retain their shape and molecular architecture through covalent bonds that are ubiquitous throughout the network structure. These materials, in particular in their swollen gel state, have been widely used as stimuli responsive materials with their ability to change volume in response to changes in temperature, pH, or other solvent conditions and have also been used in shape memory applications. However, the existence of a permanent, unalterable shape and structure dictated by the covalently crosslinked structure has dramatically limited their abilities in this and many other areas. These materials are not generally reconfigurable, recyclable, reprocessable, and have limited ability to alter permanently their stress state, topography, topology, or structure. Recently, a new paradigm has been explored in crosslinked polymers – that of covalent adaptable networks (CANs) in which covalently crosslinked networks are formed such that triggerable, reversible chemical structures persist throughout the network. These reversible covalent bonds can be triggered through molecular triggers, light or other incident radiation, or temperature changes. Upon application of this stimulus, rather than causing a temporary shape change, the CAN structure responds by permanently adjusting its structure through either reversible addition/condensation or through reversible bond exchange mechanisms, either of which allow the material to essentially reequilibrate to its new state and condition. Here, we provide a tutorial review on these materials and their responsiveness to applied stimuli. In particular, we review the broad classification of these materials, the nature of the chemical bonds that enable the adaptable structure, how the properties of these materials depend on the reversible structure, and how the application of a stimulus causes these materials to alter their shape, topography, and properties.
Co-reporter:Tao Gong;Brian J. Adzima;Noah H. Baker
Advanced Materials 2013 Volume 25( Issue 14) pp:2024-2028
Publication Date(Web):
DOI:10.1002/adma.201203815
Co-reporter:Tao Gong;Brian J. Adzima;Noah H. Baker
Advanced Materials 2013 Volume 25( Issue 14) pp:
Publication Date(Web):
DOI:10.1002/adma.201370086
Co-reporter:Raveesh Shenoy, Mark W. Tibbitt, Kristi S. Anseth, and Christopher N. Bowman
Chemistry of Materials 2013 Volume 25(Issue 5) pp:761
Publication Date(Web):February 12, 2013
DOI:10.1021/cm303913f
A unique design paradigm to form core–shell particles based on interfacial radical polymerization is described. The interfacial initiation system is comprised of an enzymatic reaction between glucose and glucose oxidase (GOx) to generate hydrogen peroxide, which, in the presence of iron (Fe2+), generates hydroxyl radicals that initiate polymerization. Shell formation on prefabricated polymeric cores is achieved by localizing the initiation reaction to the interface of the core and a surrounding aqueous monomer formulation into which it is immersed. The interfacially confined initiation reaction is accomplished by incorporating one or more of the initiating species in the particle core and the remainder of the complementary initiating components in the surrounding media such that interactions and the resulting initiation reaction occur at the interface. This work is focused on engineering the reaction behavior and mass transport processes to promote interfacially confined polymerization, controlling the rate of shell formation, and manipulating the structure of the core–shell particle. Specifically, incorporating GOx in the precursor solution used to fabricate cores ranging from 100 to 200 μm, and the remainder of the complementary initiating components and monomer in the bulk solution prior to interfacial polymerization yielded shells whose average thickness was 20 μm after 4 min of immersion and at a bulk iron concentration of 12.5 mM. When the locations of glucose and GOx are interchanged, the average thickness of the shell was 15 or 100 μm for bulk iron concentrations of 45 and 12.5 mM, respectively. The initial locations of glucose and GOx also determine the degree of interpenetration of the core and the shell. Specifically, for a bulk iron concentration of 45 mM, the thickness of the interpenetrating layer averaged 12 μm when GOx was initially within the core, whereas no interpenetrating layer was observed when glucose was incorporated in the core. The polymeric shell formed by this technique is also demonstrated to be self-supporting following core degradation. This behavior is accomplished by fabricating the particle core hydrogel from monomers possessing degradable groups that can be irreversibly cleaved by light exposure following shell formation. When the coated particle was exposed to light, the shell remained intact while the core degraded as evidenced by a dramatic change in diffusion coefficient of fluorescent beads immobilized within the core.Keywords: core−shell particles; interfacial radical polymerization; stimuli-responsive microparticles;
Co-reporter:Shunsuke Chatani, Richard J. Sheridan, Maciej Podgórski, Devatha P. Nair, and Christopher N. Bowman
Chemistry of Materials 2013 Volume 25(Issue 19) pp:3897
Publication Date(Web):September 20, 2013
DOI:10.1021/cm402229j
A chemical clock protocol that enables enhanced temporal control over the onset of two base-catalyzed ‘click’ reactions, the thiol-Michael addition reaction, and the thiol-isocyanate reaction, is described and used in polymerization reactions. Initiating protocols with predictable induction times for both click reactions are developed and characterized using a pair consisting of an electron deficient vinylic species and a nucleophile with an acid. The approach was successfully demonstrated such that the reaction onset is effectively and predictably delayed by up to 20 min, with rapid complete reaction following the controllable induction period. By implementing initiation systems with varying relative concentrations of the electron deficient vinyl, nucleophile, and acid, this approach to formulating a comprehensive initiator system affords a previously unavailable degree of temporal control that is extremely useful for designing and processing cross-linked polymers and other thiol-Michael and thiol-isocyanate polymerizations.Keywords: chemical clock; click chemistry; thiol isocyanate reaction; thiol-Michael addition reaction;
Co-reporter:Tao Gong, Brian J. Adzima and Christopher N. Bowman
Chemical Communications 2013 vol. 49(Issue 72) pp:7950-7952
Publication Date(Web):25 Jul 2013
DOI:10.1039/C3CC43637C
A novel copper(II) complex has been developed in which the counter anion, acylphosphinate, serves as a visible light photoinitiator. This molecule is inactive in the dark but, upon visible light exposure, both CuAAC and ATRP reactions are readily and rapidly initiated.
Co-reporter:Weixian Xi, Matthias Krieger, Christopher J. Kloxin and Christopher N. Bowman
Chemical Communications 2013 vol. 49(Issue 40) pp:4504-4506
Publication Date(Web):08 Mar 2013
DOI:10.1039/C3CC41123K
The utilization of 2-(2-nitrophenyl)propyloxycarbonyl (NPPOC) as a photolabile primary amine cage enables the thiol-Michael ‘click’ reaction to be photo-triggered. The photolabile amine exhibits efficient catalytic activity upon UV irradiation and is shown to initiate the photopolymerization of tetrathiol and diacrylate comonomers viaMichael addition.
Co-reporter:Richard J. Sheridan and Christopher N. Bowman
Polymer Chemistry 2013 vol. 4(Issue 18) pp:4974-4979
Publication Date(Web):05 Dec 2012
DOI:10.1039/C2PY20960H
When Diels–Alder-based thermoreversible covalent adaptable networks (TR-CANs), are applied in fracture healing applications, the contributions of network structure tend to take a back seat to explanations based solely on the chemical behaviour of the reversible bonds binding the network. However, for TR-CANs near the gel point, rheological experiments have shown that accounting for network structure via scaling relationships is necessary to understand their viscoelastic behaviour. By extension, the structure of the network should have a substantial effect on fracture healing performance. In this work we demonstrate this effect in a model hysteresis heated Diels–Alder network material. The effective functionality of the monomers was varied from 3.0 to 3.5, changing the gel temperature from 106 °C to 122 °C. By subjecting these materials to identical healing conditions, we observed the change due to network structure while holding e.g. bond conversion and bond lifetime constant. We showed with statistical confidence that both healing time, and the interaction between healing time and composition (p = 0.016 and p = 0.014, respectively) are necessary to explain the observed differences in healing performance. A single-parameter model of healing was developed based on the scaling relationship that determined mechanical relaxation, and the model was interpreted to understand how network structure and fracture healing interact in TR-CANs.
Co-reporter:Megan A. Cole, Katherine C. Jankousky and Christopher N. Bowman
Polymer Chemistry 2013 vol. 4(Issue 4) pp:1167-1175
Publication Date(Web):26 Nov 2012
DOI:10.1039/C2PY20843A
The unique formation–structure–property attributes and reaction behavior of the thiol–ene “click” reaction have been explored extensively for photochemically and thermally initiated reactions but have been much less explored for redox initiation. Therefore, the objective of this work is to characterize fully the impact of the initiation system, monomer structure, degree of functionalization, and inhibitor level on the redox-mediated thiol–ene polymerization rate and behavior. Moreover, this study confirms the ability of redox initiation to achieve full conversion of desired thiol–ene “click” products for small molecules in solution. For the multifunctional thiol–ene systems, polymerization rate was shown to be comparable to photo- and thermally initiated systems, but with the additional advantages of unlimited depth of cure and mild reaction conditions. Additionally, the network properties of the redox-initiated thiol–ene systems were on par with a photocured material formulated with identical monomers and radical initiating potential. Lastly, control over the polymerization rate and preceding induction period was garnered from the concentration of inhibitor included in the reaction mixture. The mechanism of action of quinone inhibition in redox-mediated thiol–ene polymerizations is shown to depend on both the presence of an aniline reducing agent and the concentration of inhibitor, with quinone concentrations in great excess of oxidizing agent concentrations actually leading to heightened polymerization rates when aniline is present.
Co-reporter:Shunsuke Chatani, Devatha P. Nair and Christopher N. Bowman
Polymer Chemistry 2013 vol. 4(Issue 4) pp:1048-1055
Publication Date(Web):19 Nov 2012
DOI:10.1039/C2PY20826A
The reactivity, selectivity and kinetics of vinyl sulfones and acrylates in base and nucleophile-catalyzed thiol–Michael addition reactions were examined in detail in this study. The vinyl sulfones react selectively and more rapidly with thiols in the presence of acrylates, which was clearly indicated from reactions of hexanethiol (HT), ethyl vinyl sulfone (EVS) and hexyl acrylate (HA) at a molar ratio of 2:1:1. EVS reaches 100% conversion with minimal consumption (<10%) of HA, which demonstrates the high selectivity of vinyl sulfones over acrylates. The reaction rate of EVS with HT was approximately 7 times higher than that of HA. A detailed study of the kinetics of the nucleophile-catalyzed thiol–Michael addition reaction was carried out, and it was shown that the delay observed in the initial stages of the nucleophile-catalyzed thiol–Michael addition reaction is due to the relatively slow attack of the nucleophiles on the vinyl. The presence of protic species other than thiols in the reaction mixture has also been shown to significantly impede the reaction rate, and in extreme cases, has been shown to inhibit the Michael addition reaction. These results provided a better understanding of the conditions under which the thiol–Michael addition reaction can or cannot be considered as a click reaction. Finally, the high reaction selectivity of vinyl sulfones over acrylates via thiol–Michael addition reaction in ternary systems is used to control gelation behavior in crosslinked polymer networks formed by thiol–Michael addition reactions.
Co-reporter:Raveesh Shenoy
Macromolecular Theory and Simulations 2013 Volume 22( Issue 2) pp:115-126
Publication Date(Web):
DOI:10.1002/mats.201200062
Co-reporter:Devatha P. Nair;Neil B. Cramer;Mathew K. McBride;John C. Gaipa;Nathan C. Lee;Robin Shas
Macromolecular Symposia 2013 Volume 329( Issue 1) pp:101-107
Publication Date(Web):
DOI:10.1002/masy.201200105
Summary
High modulus two-stage reactive polymer systems are fabricated and characterized in regards to their thermomechanical properties and behavior. The polymer networks comprise thiol-acrylate formulations in which a polymer matrix is initially formed via an amine catalyzed thiol-Michael addition ‘click’ reaction, eventually followed by photoinitiated, free radical polymerization of the excess acrylic functional groups to result in formation of a highly crosslinked, high modulus polymer material. Composites were formed and evaluated using two distinct polymerizable thiol-acrylate formulations, each with three different filler types. Here, the fillers were used primarily to improve the mechanical performance of the polymer material following the initial Michael addition reaction though improvements were also observed in some materials following the photopolymerization as well. The fillers used were 0.7 µm methacrylated silica particles, translucent Kevlar veil and PET mesh. Thermomechanical analysis showed that the fillers resulted in a significant increase in the modulus in both the polymer networks formed at the end of each of the orthogonal reactions without a significant change in the glass transition temperatures (Tg). The two-stage matrix formed with a hexa-acrylate matrix and 20 volume % silica particles showed a 125% increase in the modulus at the end of the Michael-addition reaction and a 100% increase in the modulus after photopolymerization of the acrylates, when compared with the modulus of the unfilled polymer.
Co-reporter:Kenneth Christopher Koehler, Kristi S. Anseth, and Christopher N. Bowman
Biomacromolecules 2013 Volume 14(Issue 2) pp:
Publication Date(Web):January 14, 2013
DOI:10.1021/bm301789d
A synthetic amino acid bearing a furan functionality was developed and incorporated into peptide sequences using solid phase synthesis. Peptides expressing the furan moiety were attached to poly(ethylene glycol) (PEG) hydrogels through a thermally reversible covalent bond formed by a Diels–Alder reaction. Reactions of thiol and maleimide PEG macromers in an off-stoichiometric Michael addition were performed, such that the maleimide moiety was in excess, to create hydrogel networks with pendant Diels–Alder compatible tethering sites, that is, the maleimide. By making use of the Diels–Alder reaction, it was possible to control the release rate of reversibly bound moieties from the hydrogel by changing the temperature; higher temperatures favor a faster retro-Diels–Alder reaction and, therefore, a faster release from the polymer network. This concept was demonstrated by incorporating a fluorescently labeled furan-RGDS sequence into a hydrogel possessing excess maleimide functionalities and monitoring the subsequent liberation of RGDS at various temperatures, illustrating a Diels–Alder mediated release mechanism. The release profile was quantified at temperatures ranging from physiological (37 °C) to 80 °C. By changing the temperature, varying extents of release were attained over the time course of several days, ranging from 40% release for lower temperatures to complete release for the highest temperature considered. Further confirmation of a reaction-diffusion controlled release mechanism was obtained through comparison of experimental release data to a reaction-diffusion model of the release. In addition to thermal modulation, tuning of the release rate was accomplished by altering the number of possible Diels–Alder tethering sites present in the hydrogel. Increasing the amount of free maleimide and, therefore, the number of potential Diels–Alder reaction sites, effectively slowed the release of peptide from the polymer. For instance, doubling the amount of maleimide sites present in the hydrogel system decreased the amount of peptide released from approximately 60% to about 40% in the same span of time.
Co-reporter:Megan A. Cole
Journal of Polymer Science Part A: Polymer Chemistry 2013 Volume 51( Issue 8) pp:1749-1757
Publication Date(Web):
DOI:10.1002/pola.26551
Abstract
Polysiloxanes are commonly used in a myriad of applications, and the “click” nature of the thiol-ene reaction is well suited for introducing alternative functionalities or for crosslinking these ubiquitous polymers. As such, understanding of the thiol-ene reaction in the presence of silicones is valuable and would lead to enhanced methodologies for modification and crosslinking. Here, the thiol-ene reaction kinetics were investigated in functionalized oligosiloxanes having varying degrees of thiol functionalization (SH), π–π interactions (from diphenyls, DP), and ene types (CC). In the ene-functionalized oligomers, π–π interactions were controlled through the use of dioctyl repeats (DO). The polymerization rate and rate-limiting steps were determined for all systems containing an allyl-functionalized oligomer, and rates ranging from 0.10 to 0.54 mol L−1 min−1 were seen. The rate-limiting step varied with the oligomer composition; examples of rate-limited propagation (5:3:2 CC:DP:DO/1:1 SH:DP) or chain transfer (5:3:2 CC:DP:DO/3:1 SH:DP) were found in addition to cases with similar reaction rate constants (5:2:3 CC:DP:DO/1:1 SH:DP). None of the siloxanes were found to exhibit autoacceleration despite their relatively high viscosities. Instead, the allyl-, vinyl-, and acrylate-functionalized siloxanes were all found to undergo unimolecular termination based on their high α scaling values (0.98, 0.95, and 0.82, respectively) in the relation Rp ∝ Riα. © 2013 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2013
Co-reporter:Kenneth C. Koehler, Daniel L. Alge, Kristi S. Anseth, Christopher N. Bowman
Biomaterials 2013 34(16) pp: 4150-4158
Publication Date(Web):
DOI:10.1016/j.biomaterials.2013.02.020
Co-reporter:Kenneth Christopher Koehler;Daniel L. Alge
International Journal of Peptide Research and Therapeutics 2013 Volume 19( Issue 3) pp:265-274
Publication Date(Web):2013 September
DOI:10.1007/s10989-013-9347-y
An amino acid possessing a maleimide side chain was developed and synthesized in good yield. With a propensity to undergo the Michael addition reaction, the creation of a maleimide amino acid derivative was targeted for use as a highly functional tool for enabling peptide conjugation and structural modifications. After addressing the inherent potential side reactions of maleimides during solid phase peptide synthesis, the ability to incorporate the maleimide amino acid in an RGDS peptide sequence was demonstrated. 1H NMR and mass spectroscopic techniques enabled thorough characterization of the peptide sequence, confirming the presence of the maleimide functionality. Once characterized, the ability to use the maleimide moiety as a peptide modification tool was investigated. Specifically, it was shown that the maleimide functional group could be exploited, given the proper reaction conditions, to anchor a peptide to a surface and create a cyclic conformation from a linear sequence. Furthermore, bioactivity of the peptide containing maleimide amino acid was evaluated by studying cellular interactions with surfaces functionalized with an integrin binding sequence.
Co-reporter:Devatha P. Nair;Neil B. Cramer;John C. Gaipa;Matthew K. McBride;Emily M. Matherly;Robert R. McLeod;Robin Shas
Advanced Functional Materials 2012 Volume 22( Issue 7) pp:1502-1510
Publication Date(Web):
DOI:10.1002/adfm.201102742
Abstract
There are distinct advantages to designing polymer systems that afford two distinct sets of material properties– an intermediate polymer that would enable optimum handling and processing of the material, while maintaining the ability to tune in different, final polymer properties that enable the optimal functioning of the material. In this study, by designing a series of non-stoichiometric thiol-acrylate systems, a polymer network is initially formed via a base catalyzed Michael addition reaction that proceeds stoichiometrically via the thiol-acrylate “click” reaction. This self-limiting reaction results in a polymer with excess acrylic functional groups within the network. At a later point in time, the photoinitiated, free radical polymerization of the excess acrylic functional groups results in a highly crosslinked, robust material system. These two stage reactive thiol-acrylate networks that have intermediate stage rubbery moduli and glass transition temperatures that range from 0.5 MPa and -10 °C to 22 MPa and 22 °C, respectively, are formulated and characterized. The same polymer networks can then attain glass transition temperatures that range from 5 °C to 195 °C and rubbery moduli of up to 200 MPa after the subsequent photocuring stage. The two stage reactive networks formed by varying the stoichiometric ratios of the thiol and acrylate monomers were shown to perform as substrates for three specific applications: shape memory polymers, impression materials, and as optical materials for writing refractive index patterns.
Co-reporter:Weixian Xi, Chen Wang, Christopher J. Kloxin, and Christopher N. Bowman
ACS Macro Letters 2012 Volume 1(Issue 7) pp:811
Publication Date(Web):June 12, 2012
DOI:10.1021/mz3001918
A new group of nitrogen-centered nucleophilic catalysts for the thiol-Michael addition “click” reactions is examined. These nucleophiles showed efficient catalytic activities as compared with traditional base catalysts, such as triethylamine, and are demonstrated to be a viable strategy for cross-linking polymerization reactions. Additionally, an experimental and computational mechanistic study was performed, suggesting a pathway for the nitrogen-centered catalyst to undergo the nucleophilic addition mechanism.
Co-reporter:Brad J. Berron, Allison M. May, Zheng Zheng, Vivek Balasubramaniam and Christopher N. Bowman
Lab on a Chip 2012 vol. 12(Issue 4) pp:708-710
Publication Date(Web):20 Dec 2011
DOI:10.1039/C2LC21101G
The growing need for medical diagnostics in resource limited settings is driving the development of simple, standalone immunoassay devices. A capillary flow device using polymerization based amplification is capable of blocking a microfluidic channel in response to target biomaterials, enabling multiple modes of detection that require little or no supplemental instrumentation.
Co-reporter:Brian J. Adzima;Christopher J. Kloxin;Cole A. DeForest;Kristi S. Anseth
Macromolecular Rapid Communications 2012 Volume 33( Issue 24) pp:2092-2096
Publication Date(Web):
DOI:10.1002/marc.201200599
Abstract
3D structures are written and developed in a crosslinked polymer initially formed by a Diels–Alder reaction. Unlike conventional liquid resists, small features cannot sediment, as the reversible crosslinks function as a support, and the modulus of the material is in the MPa range at room temperature. The support structure, however, can be easily removed by heating the material, and depolymerizing the polymer into a mixture of low-viscosity monomers. Complex shapes are written into the polymer network using two-photon techniques to spatially control the photoinitiation and subsequent thiol–ene reaction to selectively convert the Diels–Alder adducts into irreversible crosslinks.
Co-reporter:Diana Leung
Macromolecular Chemistry and Physics 2012 Volume 213( Issue 2) pp:198-204
Publication Date(Web):
DOI:10.1002/macp.201100402
Abstract
The trithiocarbonate 1a reduces the volumetric shrinkage stress in crosslinked multi(meth)acrylate networks by promoting a reversible, free-radical-mediated network rearrangement that enables network adaptation and stress relaxation. Addition of 1a or 1b into a dimethacrylate system is examined by FT-IR. The polymerization rate is reduced in samples containing 1a, indicating a changed polymerization mechanism. The volumetric shrinkage stress is reduced by more than 50% compared to a pure dimethacrylate system with the addition of as little as 2 wt% of 1a, while maintaining the favorable mechanical properties of the methacrylate-crosslinked networks. No synthetic modification of the methacrylate monomer units is needed, allowing for adaptation to almost any radically polymerized system.
Co-reporter:Megan A. Cole
Journal of Polymer Science Part A: Polymer Chemistry 2012 Volume 50( Issue 20) pp:4325-4333
Publication Date(Web):
DOI:10.1002/pola.26245
Abstract
Three types of linear thiol-functionalized siloxane oligomers and three types of ene-functionalized oligomers were synthesized and subsequently photopolymerized. Within each type of thiol-functionalized oligomer, the ratio of mercaptan repeat units to nonreactive phenyl repeat units was varied to manipulate both the crosslink density and the degree of secondary interactions through π–π stacking. Similarly, the repeat units of the three ene-functionalized oligomers are composed of allyl-functional monomers, benzene-functional monomers, and octyl-functional monomers in varying ratios of benzene:octyl but with a constant fraction of allyl moieties. The structural composition of the siloxane oligomers plays a pivotal role in the observed material properties of networks formed through thiol–ene photopolymerization. Networks with a high concentration of thiol functionalities exhibit higher rubbery moduli, ultimate strengths, and Young's moduli than networks with lower thiol concentrations. Moreover, the concentration of functionalities capable of participating in secondary interactions via hydrogen bonding or π–π stacking directly impacts the network glass transition temperature and elasticity. The combination of low crosslink density and high secondary interactions produces networks with the greatest toughness. Finally, the fraction of octyl repeats correlates with the hydrophobic nature of the network. © 2012 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2012
Co-reporter:Richard J. Sheridan and Christopher N. Bowman
Macromolecules 2012 Volume 45(Issue 18) pp:7634-7641
Publication Date(Web):September 14, 2012
DOI:10.1021/ma301329u
Although the gel point conversion of a thermoreversible polymer network is certainly a key parameter in determining the material properties, it is not a conventional liquid–solid transition as in common, irreversible networks. Rather, the material’s viscosity is time-dependent and finite at the gel point and beyond, as bond breakage works in concert with diffusion to relax stresses imposed on the forming transient network of the material. For example, in a model Diels–Alder network with functionality 3.8 and a stoichiometric ratio of 10:6 furan:maleimide used here, a crossover frequency (0.52, 0.32, and 0.12 rad/s) was measured below the temperature corresponding to gelation (3, 5, and 10 °C below, respectively). In this work, we describe this complex process occurring in model thermoreversible networks with a simple relationship from the work of Semenov and Rubinstein on associative transient networks. This relationship provides a toolkit for the prediction of the important engineering and rheological properties of the material in the postgel regime, such as viscosity, plateau modulus, and relaxation time, based upon the straightforward estimation of two material-dependent parameters: the gel point conversion pgel and a proportionality constant C. We show key agreement between theory and experiment as the gel point conversion estimated from network dynamics matches the classical prediction of the gel point within 4% conversion. We discuss the applicability criteria of the Semenov–Rubinstein scaling relationship and compare it to time–temperature superposition methods of describing transient network relaxation.
Co-reporter:Hee Young Park, Christopher J. Kloxin, Mark F. Fordney, and Christopher N. Bowman
Macromolecules 2012 Volume 45(Issue 14) pp:
Publication Date(Web):July 3, 2012
DOI:10.1021/ma300225q
Since polymerization-induced shrinkage stress is detrimental in many applications, addition–fragmentation chain transfer (AFCT) was employed to induce network relaxation and adaptation that mitigate the shrinkage stress. Here, to form high glass transition temperature, high modulus polymers while still minimizing stress, multifunctional methacrylate monomers were incorporated into allyl sulfide-containing thiol–yne resins to provide simultaneously high glass transition temperatures and a facile mechanism for AFCT throughout the network. As a negative control, in an attempt to isolate just the effects of AFCT in the polymerization, a propyl sulfide-based diyne, which has a nearly identical chemical structure though absent any AFCT-capable functional group, was synthesized and implemented in place of the allyl sulfide-based diyne. The glass transition temperature of the ternary systems increased from 39 to 79 °C as the methacrylate content increased while the shrinkage stress of the optimal ternary resin was lower than either the binary thiol–yne resin or the pure methacrylate resin. The stress relaxation benefit associated with AFCT increased with increasing allyl sulfide concentration as shown by a decrease in the relative stress from 0.98 to 0.53. The allyl sulfide-based thiol–yne–methacrylate system exhibits stress relaxation up to 55% and increased Tg up to 40 °C compared with the control, AFCT-incapable thiol–yne. This ternary system has less than 1/3 of the stress of conventional dimethacrylate monomer resins while possessing similarly outstanding mechanical behavior.
Co-reporter: Christopher N. Bowman; Christopher J. Kloxin
Angewandte Chemie International Edition 2012 Volume 51( Issue 18) pp:4272-4274
Publication Date(Web):
DOI:10.1002/anie.201200708
Co-reporter:Raveesh Shenoy, Christopher N. Bowman
Biomaterials 2012 33(29) pp: 6909-6914
Publication Date(Web):
DOI:10.1016/j.biomaterials.2012.06.014
Co-reporter: Christopher N. Bowman; Christopher J. Kloxin
Angewandte Chemie 2012 Volume 124( Issue 18) pp:4346-4348
Publication Date(Web):
DOI:10.1002/ange.201200708
Co-reporter:Hee Young Park, Christopher J. Kloxin, Ahmed S. Abuelyaman, Joe D. Oxman, and Christopher N. Bowman
Macromolecules 2012 Volume 45(Issue 14) pp:
Publication Date(Web):July 3, 2012
DOI:10.1021/ma300228z
To reduce shrinkage stress which arises during the polymerization of cross-linked polymers, allyl sulfide functional groups were incorporated into methacrylate polymerizations to determine their effect on stress relaxation via addition–fragmentation chain transfer (AFCT). Additionally, stoichiometrically balanced thiol and allyl sulfide-containing norbornene monomers were incorporated into the methacrylate resin to maximize the overall functional group conversion and promote AFCT while also enhancing the polymer’s mechanical properties. Shrinkage stress and reaction kinetics for each of the various functional groups were measured by tensometry and Fourier-transform infrared (FTIR) spectroscopy, respectively. The glass transition temperature (Tg) and elastic moduli (E′) were measured using dynamic mechanical analysis. When the allyl sulfide functional group was incorporated into dimethacrylates, the polymerization-induced shrinkage stress was not relieved as compared with analogous propyl sulfide-containing resins. These analogous propyl sulfide-containing monomers are incapable of undergoing AFCT while having similar chemical structure and cross-link density to the allyl sulfide-containing methacrylates. Here, a monomethacrylate monomer that also contains a cyclic allyl sulfide (PAS) was found to increase the cross-linking density nearly 20 times as compared to an analogous monomethacrylate in which the allyl sulfide was replaced with an ethyl sulfide. Despite the much higher cross-link density, the PAS formulation exhibited no concomitant increase in stress. Thiol–norbornene resins were copolymerized in PAS to promote AFCT as well as to synergistically combine the ring-opening benefits associated with the thiol–ene reaction. AFCT resulted in a 63% reduction of polymerization stress and a 45 °C enhancement of the glass transition temperature in the allyl sulfide-containing thiol–norbornene–methacrylate system compared with rubbery dimethacrylates. When compared with conventional glassy dimethacrylates, this combined system has less than 10% of the typical shrinkage stress level while having similarly excellent mechanical properties.
Co-reporter:Devatha P. Nair, Neil B. Cramer, Matthew K. McBride, John C. Gaipa, Robin Shandas, Christopher N. Bowman
Polymer 2012 Volume 53(Issue 12) pp:2429-2434
Publication Date(Web):25 May 2012
DOI:10.1016/j.polymer.2012.04.007
In this study, we develop thiol/acrylate two-stage reactive network forming polymer systems that exhibit two distinct and orthogonal stages of curing. Using a thiol-acrylate system with excess acrylate functional groups, a first stage polymer network is formed via a 1 to 1 stoichiometric thiol-acrylate Michael addition reaction (stage 1). At a later point in time, the excess acrylate functional groups are homopolymerized via a photoinitiated free radical polymerization to form a second stage polymer network (stage 2). By varying the monomers within the system as well as the stoichiometery of the thiol to acrylate functional groups, we demonstrate the ability of the two-stage polymer network forming systems to encompass a wide range of properties at the end of both the stage 1 and stage 2 polymerizations. Using urethane di- and hexa-acrylates within the formulations led to two-stage reactive polymeric systems with stage 1 Tgs that ranged from −12 to 30° C. The systems were then photocured, upon which the Tg of the systems increases by up to 90°C while also achieving a nearly 20 fold modulus increase.Graphical abstract
Co-reporter:Christopher J. Kloxin;Timothy F. Scott;Hee Young Park
Advanced Materials 2011 Volume 23( Issue 17) pp:1977-1981
Publication Date(Web):
DOI:10.1002/adma.201100323
Co-reporter:Christopher J. Kloxin;Timothy F. Scott;Hee Young Park
Advanced Materials 2011 Volume 23( Issue 17) pp:
Publication Date(Web):
DOI:10.1002/adma.201190062
Co-reporter:Timothy F. Scott, Christopher J. Kloxin, Darren L. Forman, Robert R. McLeod and Christopher N. Bowman
Journal of Materials Chemistry A 2011 vol. 21(Issue 37) pp:14150-14155
Publication Date(Web):08 Jul 2011
DOI:10.1039/C1JM11915J
Optical direct write lithography (ODWL) has the capacity for generating three dimensional arbitrary patterns. Here we examine principles for voxel refinement and relate several techniques for achieving nanoscale resolution. The influence of optics, gelation, and polymerization scaling behavior are expounded, demonstrating the necessity for adopting a multidisciplinary mindset to control both voxel dimensions and minimize out-of-focus reactions. Aspects of two-photon ODWL are reviewed and recent multi-beam ODWL approaches that draw inspiration from STED microscopy are examined.
Co-reporter:Sheng Ye, Neil B. Cramer, and Christopher N. Bowman
Macromolecules 2011 Volume 44(Issue 3) pp:490-494
Publication Date(Web):January 10, 2011
DOI:10.1021/ma101296j
Monomeric resins are frequently polymerized at a temperature that is far below the desired glass transition temperature for the final polymer network. This work quantitatively investigates the relationship between the glass transition temperature (Tg), the cure temperature (Tcure), the maximum achievable glass transition temperature for a particular monomer formulation (Tgmax), and the network heterogeneity as indicated by the half-width of the glass transition region (Tg1/2width) for both chain growth and step growth polymerization systems. Various monomer resins were systematically photopolymerized at 25, 50, 75, and 100 °C to the maximum achievable double bond conversion with living radical photopolymerizations employed as necessary to eliminate issues associated with postcuring during heating of the samples for dynamic mechanical analysis. For polymer samples that have the potential for much higher glass transition temperatures than the curing temperature (i.e., where Tcure < Tgmax − Tg1/2width), mass transfer limitations and the network heterogeneity combine to control Tg such that Tg ≈ Tcure + Tg1/2width. For systems polymerized at temperatures closer to their maximum glass transition temperature, i.e., where Tcure ≥ Tgmax − Tg1/2width, the polymer structure becomes the dominant factor and Tg ≈ Tgmax. These relationships were broadly and successfully applied to cross-linked networks formed from both chain growth and step growth polymerizations, incorporating networks of vastly different heterogeneity, with Tg’s ranging from 35 to 205 °C with approximately 10% error found between predicted and experimental glass transition temperatures.
Co-reporter:Kathleen M. Schreck, Diana Leung, and Christopher N. Bowman
Macromolecules 2011 Volume 44(Issue 19) pp:7520-7529
Publication Date(Web):September 15, 2011
DOI:10.1021/ma201695x
The thiol–ene reaction serves as a more oxygen-tolerant alternative to traditional (meth)acrylate chemistry for forming photopolymerized networks with numerous desirable attributes including energy absorption, optical clarity, and reduced shrinkage stress. However, when utilizing commercially available monomers, many thiol–ene networks also exhibit decreases in properties such as glass transition temperature (Tg) and cross-link density. In this study, hybrid organic/inorganic thiol–ene resins incorporating silsesquioxane (SSQ) species into the photopolymerized networks were investigated as a route to improve these properties. Thiol- and ene-functionalized SSQs (SH-SSQ and allyl-SSQ, respectively) were synthesized via alkoxysilane hydrolysis/condensation chemistry, using a photopolymerizable monomer [either pentaerythriol tetrakis(3-mercaptopropionate) (PETMP) or 1,3,5-triallyl-1,3,5-triazine-2,4,6(1H,3H,5H)-trione (TATATO)] as the reaction solvent. The resulting SSQ-containing solutions (SSQ-PETMP and SSQ-TATATO) were characterized, and their incorporation into photopolymerized networks was evaluated.
Co-reporter:Sheng Ye, Neil B. Cramer, Blake E. Stevens, Robert L. Sani, and Christopher N. Bowman
Macromolecules 2011 Volume 44(Issue 12) pp:4988-4996
Publication Date(Web):June 1, 2011
DOI:10.1021/ma200098e
Induction curing is demonstrated as a novel type of in situ radiation curing that maintains most of the advantages of photocuring while eliminating the restriction of light accessibility. Induction curing is utilized to polymerize opaque composites comprised of thiol–acrylate and thiol–ene resins, nanoscale magnetic particles, and carbon nanotubes. Nanoscale magnetic particles are dispersed in the resin, and upon exposure to the magnetic field, these particles lead to induction heating that rapidly initiates the polymerization. Heat transfer profiles and reaction kinetics of the samples are modeled during the reactions with varying induction heater power, species concentration, species type, and sample thickness, and the model is compared with the experimental results. Thiol–ene polymerizations achieved full conversion between 1.5 min and 1 h, depending on the field intensity and the composition, with the maximum reaction temperature decreasing from 146 to 87 °C when the induction heater power was decreased from 8 to 3 kW. The polymerization reactions of the thiol–acrylate system were demonstrated to achieve full conversion between 0.6 and 30 min with maximum temperatures from 139 to 86 °C. The experimental behavior was characterized and the temperature profile modeled for the thiol–acrylate composite comprised of sub-100 nm nickel particles and induction heater power in the range of 32–20 kW. A 9 °C average deviation was observed between the modeling and experimental results for the maximum temperature rise. The model also was utilized to predict reaction temperatures and kinetics for systems with varying thermal initiator concentration, initiator half-life, monomer molecular weight, and temperature gradients in samples with varying thickness, thereby demonstrating that induction curing represents a designable and tunable polymerization method. Finally, induction curing was utilized to cure thiol–acrylate systems containing carbon nanotubes where 1 wt % carbon nanotubes resulted in systems where the storage modulus increased from 17.6 ± 0.2 to 21.6 ± 0.1 MPa and an electrical conductivity that increased from <10–7 to 0.33 ± 0.5 S/m.
Co-reporter:Charles E. Hoyle, Andrew B. Lowe and Christopher N. Bowman
Chemical Society Reviews 2010 vol. 39(Issue 4) pp:1355-1387
Publication Date(Web):09 Feb 2010
DOI:10.1039/B901979K
The merits of thiol-click chemistry and its potential for making new forays into chemical synthesis and materials applications are described. Since thiols react to high yields under benign conditions with a vast range of chemical species, their utility extends to a large number of applications in the chemical, biological, physical, materials and engineering fields. This critical review provides insight into emerging venues for application as well as new mechanistic understanding of this exceptional chemistry in its many forms (81 references).
Co-reporter:Brian J. Adzima;Christopher J. Kloxin
Advanced Materials 2010 Volume 22( Issue 25) pp:2784-2787
Publication Date(Web):
DOI:10.1002/adma.200904138
Co-reporter:Junghwan Shin, Hironori Matsushima, Christopher M. Comer, Christopher N. Bowman and Charles E. Hoyle
Chemistry of Materials 2010 Volume 22(Issue 8) pp:2616
Publication Date(Web):March 4, 2010
DOI:10.1021/cm903856n
Thiol−isocyanate−ene ternary networks with systematic variations (100/100/0, 100/80/20, 100/60/40, 100/40/60, 100/20/80, and 100/0/100) were prepared by sequential and simultaneous thiol−ene and thiol−isocyanate click reactions. The thiol−isocyanate coupling reaction was triggered thermally or photolytically to control the sequence with the thiol−ene photopolymerization. Triethyl amine (TEA) and 2,2-dimethoxy-2-phenyl acetophenone (DMPA) were used for the sequential thermally induced thiol−isocyanate coupling and photochemically initiated thiol−ene reaction, respectively. A thermally stable photolatent base catalyst (tributylamine·tetraphenylborate salt, TBA·HBPh4) capable of in situ generation of tributylamine by UV light was used with isopropylthioxanthone (ITX) for the simultaneous thiol−isocyanate/thiol−ene curing systems. The kinetics of the hybrid networks investigated using real-time IR indicate that both thiol−isocyanate and thiol−ene reactions were quantitatively rapid and efficient (>90% of conversion in a matter of minutes and seconds, respectively). The Tg of the thiourethane/thiol−ene hybrid networks progressively increases (−5 to 35 °C by DSC) as a function of the thiourethane content due to the higher extent of hydrogen bonding, also resulting in enhanced mechanical properties. Highly uniform and dense network structures exhibiting narrow full width at half-maximum (∼10 °C) were obtained for both the sequential and the simultaneous thiol click reactions, resulting in identical thermal properties that are independent of the sequence of the curing processes.
Co-reporter:Andrew B. Lowe, Charles E. Hoyle and Christopher N. Bowman
Journal of Materials Chemistry A 2010 vol. 20(Issue 23) pp:4745-4750
Publication Date(Web):19 Feb 2010
DOI:10.1039/B917102A
Radical mediated thiol-yne polymerization reactions complement the more well-known thiol-ene radical polymerization processes, with the added advantage of increased functionality. In one system studied, the rate constant for the addition of the thiol to the vinyl sulfide created by the initial reaction of the thiol with the alkyne is three times faster than the initial reaction. When hydrocarbon based dialkynes and dithiols were copolymerized, the resulting thiol-alkyne networks containing only hydrocarbon and sulfide linking groups exhibited refractive index values tunable above 1.65, with the refractive index directly related to the sulfur content. The thiol-yne reaction was also found to be useful in functionalizing thiol-terminated polymer chain ends via sequential Michael thiol-ene addition followed by the thiol-yne reaction: the result is the dual functionalization of the polymer chain end. A thermally responsive polymer hydrogel network was formed when an yne terminated water-soluble homopolymer was polymerized with a tetrafunctional thiol.
Co-reporter:Leah M. Johnson, Cole A. DeForest, Aishwarya Pendurti, Kristi S. Anseth and Christopher N. Bowman
ACS Applied Materials & Interfaces 2010 Volume 2(Issue 7) pp:1963
Publication Date(Web):June 29, 2010
DOI:10.1021/am100275n
Enzyme-mediated redox chain initiation involving glucose oxidase (GOX) was employed in an iterative solution dip-coating technique to polymerize multiple, three-dimensional hydrogel layers using mild aqueous conditions at ambient temperature and oxygen levels. To the best of our knowledge, sequential enzyme-mediated dip-coating resulting in an interfacial radical chain polymerization and subsequent formation of three-dimensional hydrogel layers has not been previously explored. Conformal, micrometer-scale, uniform poly(ethylene glycol) (PEG)-based hydrogel layers were polymerized within seconds and remained securely associated after incubation in water for 16 weeks. Incorporation of either small molecules (i.e., rhodamine-B acrylate, fluorescein acrylate) or fluorescent nanoparticles into crosslinked hydrogel layers during the polymerization reaction was also achieved. The encapsulation of 0.2 μm-diameter nanoparticles into hydrogels during polymerization of a 2-hydroxyethyl acrylate (HEA)/PEG575 diacrylate monomer formulation, using the GOX-mediated initiation, resulted in minimal effects on polymerization kinetics, with final acrylate conversions of 95% (± 1%) achieved within minutes. The temporal control and spatial localization afforded by this interfacial redox approach resulted in the polymerization of uniform secondary layers ranging between 150 (± 10) μm and 650 (± 10) μm for 15 and 120 s immersion times, respectively. Moreover, increasing the PEG575-fraction within the initial hydrogel substrate from 10 to 50% decreased the subsequent layer thicknesses from 690 (± 30) μm to 490 (± 10) μm because of lowered glucose concentration at the hydrogel interface. The ability to sequentially combine differing initiation mechanisms with this coating approach was achieved by using GOX-mediated interfacial polymerization on hydrogel substrates initially photopolymerized in the presence of glucose. The strict control of layer thicknesses combined with the rapid, water-soluble, and mild polymerization will readily benefit applications requiring formation of stratified, complex, and three-dimensional polymer structures.Keywords: coatings; enzyme-mediated initiation; glucose oxidase; hydrogels; nanoparticle hydrogel incorporation; poly(ethylene glycol) (PEG); redox chain initiation
Co-reporter:Heather J. Avens, Christopher N. Bowman
Acta Biomaterialia 2010 Volume 6(Issue 1) pp:83-89
Publication Date(Web):January 2010
DOI:10.1016/j.actbio.2009.06.008
Abstract
Antibody microarrays are a critical tool for proteomics, requiring broad, highly sensitive detection of numerous low abundance biomarkers. Fluorescent polymerization-based amplification (FPBA) is presented as a novel, non-enzymatic signal amplification method that takes advantage of the chain-reaction nature of radical polymerization to achieve a highly amplified fluorescent response. A streptavidin–eosin conjugate localizes eosin photoinitiators for polymerization on the chip where biotinylated target protein is bound. The chip is contacted with acrylamide as a monomer, N-methyldiethanolamine as a coinitiator and yellow/green fluorescent nanoparticles (NPs) which, upon initiation, combine to form a macroscopically visible and highly fluorescent film. The rapid polymerization kinetics and the presence of cross-linker favor entrapment of the fluorescent NPs in the polymer, enabling highly sensitive fluorescent biodetection. This method is demonstrated as being appropriate for antibody microarrays and is compared to detection approaches which utilize streptavidin–fluorescein isothiocyanate (SA–FITC) and streptavidin-labeled yellow/green NPs (SA–NPs). It is found that FPBA is able to detect 0.16 ± 0.01 biotin–antibody μm−2 (or 40 zmol surface-bound target molecules), while SA–FITC has a limit of detection of 31 ± 1 biotin–antibody μm−2 and SA–NPs fail to achieve any significant signal under the conditions evaluated here. Further, FPBA in conjunction with fluorescent stereomicroscopy yields equal or better sensitivity compared to fluorescent detection of SA–eosin using a much more costly microarray scanner. By facilitating highly sensitive detection, FPBA is expected to enable detection of low abundance antigens and also make possible a transition towards less expensive fluorescence detection instrumentation.
Co-reporter:Leah M. Johnson, Ryan R. Hansen, Milan Urban, Robert D. Kuchta and Christopher N. Bowman
Biomacromolecules 2010 Volume 11(Issue 4) pp:
Publication Date(Web):March 25, 2010
DOI:10.1021/bm901441v
Co-reporter:Benjamin D. Fairbanks, Evan A. Sims, Kristi S. Anseth and Christopher N. Bowman
Macromolecules 2010 Volume 43(Issue 9) pp:4113-4119
Publication Date(Web):April 6, 2010
DOI:10.1021/ma1002968
Because of its utility in network polymerization, dendrimer synthesis, and monomer development, the photoinitiated addition of thiols to alkynes has rapidly become an important tool for polymer scientists. Yet, because this chemistry has only recently been applied to cross-linked polymer development, understanding of the nature of how the yne structure affects the reactions and information on the relative reactivities of alkynes bearing various substituents is unavailable as is the relative addition rate of the thiol to the yne as compared to the vinyl sulfide. Herein, the photoinitiated addition of octanethiol to various alkynes is explored. The most rapid addition of thiols to alkynes is that to cyclooctyne, although the resulting vinyl sulfide does not permit subsequent thiol addition. Furthermore, in the absence of radical initiators and light, thiols add spontaneously to cyclooctynes, suggesting limitations to the orthogonality of the strain-promoted copper-less azide, alkyne cycloadditions. In order of decreasing reaction rates, the consecutive addition of two thiols occurs with the aliphatic 1-octyne > propargyl acetate > methyl propargyl ether > 2-octyne. Ethyl propiolate and methyl propargylamine exhibit very small reaction rates with thiols, and no consecutive addition is observed.
Co-reporter:CharlesE. Hoyle ;ChristopherN. Bowman
Angewandte Chemie 2010 Volume 122( Issue 9) pp:1584-1617
Publication Date(Web):
DOI:10.1002/ange.200903924
Abstract
Nach Sharpless’ visionärer Charakterisierung verschiedener idealisierter Reaktionen als Klick-Reaktionen hat man innerhalb der Materialwissenschaften und Synthesechemie große Anstrengungen unternommen, um solche Reaktionen zu finden und zu implementieren. In diesem Aufsatz diskutieren wir die radikalvermittelte Thiol-En-Reaktion, die über all die Merkmale einer Klick-Reaktion verfügt: hohe Effizienz, einfache Durchführung, keine Nebenprodukte, hohe Reaktionsgeschwindigkeiten, hohe Ausbeuten. Darüber hinaus besteht die Möglichkeit, die Thiol-En-Reaktion photoinitiiert auszuführen, was insbesondere für Photopolymerisationen zur Synthese extrem einheitlicher Polymernetzwerke genutzt wird. Der Reaktionsmechanismus wird nach dem aktuellen Kenntnisstand erläutert, und zentrale Anwendungen der Thiol-En-Reaktion in der Material- und Molekülsynthese, der Biofunktionalisierung, der Polymersynthese und der Oberflächen- und Polymermodifizierung werden zusammengefasst.
Co-reporter:Hironori Matsushima;Junghwan Shin;Charles E. Hoyle
Journal of Polymer Science Part A: Polymer Chemistry 2010 Volume 48( Issue 15) pp:3255-3264
Publication Date(Web):
DOI:10.1002/pola.24102
Abstract
Thiol-isocyanate-acrylate ternary networks were formed by the combination of thiol-isocyanate coupling, thiol-acrylate Michael addition, and acrylate homopolymerization. This hybrid polymerization reaction sequence was preferentially controlled by using phosphine catalyst systems in combination with photolysis. The reaction kinetics of the phosphine/acrylate thiol-isocyanate coupling reactions were systematically investigated by evaluating model, small molecule reactions. The thiol-isocyanate reaction was completed within 1 min while the thiol-acrylate Michael addition reaction required ∼10 min. Both thiol-isocyanate coupling and thiol-acrylate Michael addition reactions involving two-step anionic processes were found to be both quantitative and efficient. However, the thiol-isocyanate coupling reaction was much more rapid than the thiol-acrylate Michael addition, promoting initial selectivity of the thiol-isocyanate reaction in a medium containing thiol, isocyanate, and acrylate functional groups. Films were prepared from thiol-isocyanate-acrylate ternary mixtures using 2-acryloyloxyethylisocyanate and di-, tri-, and tetra-functional thiols. The sequential thiol-isocyanate, thiol-acrylate, and acrylate homopolymerization reactions were monitored by infrared spectroscopy during film formation, whereas thermal and mechanical properties of the films were evaluated as a function of the chemical composition following polymerization. The results indicate that the network structures and material properties are tunable over a wide range of properties (Tg ∼ 14–100 °C, FWHM ∼ 8–46 °C), while maintaining nearly quantitative reactions, simply by controlling the component compositions. © 2010 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 48: 3255–3264, 2010
Co-reporter:Christopher J. Kloxin, Timothy F. Scott, Brian J. Adzima, and Christopher N. Bowman
Macromolecules 2010 Volume 43(Issue 6) pp:2643-2653
Publication Date(Web):February 23, 2010
DOI:10.1021/ma902596s
Polymer networks possessing reversible covalent cross-links constitute a novel material class with the capacity for adapting to an externally applied stimulus. These covalent adaptable networks (CANs) represent a trend in polymer network fabrication toward the rational design of structural materials possessing dynamic characteristics for specialty applications. Herein, we discuss the unique attributes of CANs that must be considered when designing, fabricating, and characterizing these smart materials that respond to either thermal or photochemical stimuli. While there are many reversible reactions which to consider as possible cross-link candidates in CANs, there are very few that are readily and repeatedly reversible. Furthermore, characterization of the mechanical properties of CANs requires special consideration owing to their unique attributes. Ultimately, these attributes are what lead to the advantageous properties displayed by CANs, such as recyclability, healability, tunability, shape changes, and low polymerization stress. Throughout this Perspective, we identify several trends and future directions in the emerging field of CANs that demonstrate the progress to date as well as the essential elements that are needed for further advancement.
Co-reporter:CharlesE. Hoyle ;ChristopherN. Bowman
Angewandte Chemie International Edition 2010 Volume 49( Issue 9) pp:1540-1573
Publication Date(Web):
DOI:10.1002/anie.200903924
Abstract
Following Sharpless′ visionary characterization of several idealized reactions as click reactions, the materials science and synthetic chemistry communities have pursued numerous routes toward the identification and implementation of these click reactions. Herein, we review the radical-mediated thiol–ene reaction as one such click reaction. This reaction has all the desirable features of a click reaction, being highly efficient, simple to execute with no side products and proceeding rapidly to high yield. Further, the thiol–ene reaction is most frequently photoinitiated, particularly for photopolymerizations resulting in highly uniform polymer networks, promoting unique capabilities related to spatial and temporal control of the click reaction. The reaction mechanism and its implementation in various synthetic methodologies, biofunctionalization, surface and polymer modification, and polymerization are all reviewed.
Co-reporter:Hee Young Park, Christopher J. Kloxin, Timothy F. Scott, and Christopher N. Bowman
Macromolecules 2010 Volume 43(Issue 24) pp:10188-10190
Publication Date(Web):November 24, 2010
DOI:10.1021/ma1020209
Co-reporter:Ryan R. Hansen, Leah M. Johnson, Christopher N. Bowman
Analytical Biochemistry 2009 Volume 386(Issue 2) pp:285-287
Publication Date(Web):15 March 2009
DOI:10.1016/j.ab.2008.12.009
Polymerization-based signal amplification offers sensitive visualization of biotinylated biomolecules functionalized to glass microarrays in a manner suitable for point-of-care use. Here we report using this method for visual detection of multiplexed nucleic acid hybridizations from complex media and develop an application toward point mutation detection and single nucleotide polymorphism (SNP) typing. Primer extension reactions were employed to label selectively and universally all complementary surface DNA hybrids with photoinitiators, permitting simultaneous and dynamic photopolymerization from positive sites to 0.5-nM target concentrations. Dramatic improvements in signal ratios between complementary and mismatched hybrids enabled visual discrimination of single base differences in KRAS codon-12 biomarkers.
Co-reporter:Peter M. Johnson;Jeffrey W. Stansbury
Macromolecular Reaction Engineering 2009 Volume 3( Issue 9) pp:522-528
Publication Date(Web):
DOI:10.1002/mren.200900029
Co-reporter:Tai Yeon Lee;Neil B. Cramer;Charles E. Hoyle;Jeffrey W. Stansbury
Journal of Polymer Science Part A: Polymer Chemistry 2009 Volume 47( Issue 10) pp:2509-2517
Publication Date(Web):
DOI:10.1002/pola.23327
Abstract
In this study, vinyl ester monomers were synthesized by an amine catalyzed Michael addition reaction between a multifunctional thiol and the acrylate double bond of vinyl acrylate. The copolymerization behavior of both methacrylate/vinyl ester and acrylate/vinyl ester systems was studied with near-infrared spectroscopy. In acrylate/vinyl ester systems, the acrylate groups polymerize faster than the vinyl ester groups resulting in an overall conversion of 80% for acrylate double bonds in the acrylate/vinyl ester system relative to only 50% in the bulk acrylate system. In the methacrylate/vinyl ester systems, the difference in reactivity is even more pronounced resulting in two distinguishable polymerization regimes, one dominated by methacrylate polymerization and a second dominated by vinyl ester polymerization. A faster polymerization rate and higher overall conversion of the methacrylate double bonds is thus achieved relative to polymerization of the pure methacrylate system. The methacrylate conversion in the methacrylate/vinyl ester system is near 100% compared to only ∼60% in the pure methacrylate system. Utilizing hydrophilic vinyl ester and hydrophobic methacrylate monomers, polymerization-induced phase separation is observed. The phase separated domain size is in the order of ∼1 μm under the polymerization conditions. The phase separated domains become larger and more distinct with slower polymerization and correspondingly increased time for diffusion. © 2009 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 47: 2509–2517, 2009
Co-reporter:Kathryn A. Berchtold, Bilge Hacioğlu, Jun Nie, Neil B. Cramer, Jeffrey W. Stansbury and Christopher N. Bowman
Macromolecules 2009 Volume 42(Issue 7) pp:2433-2437
Publication Date(Web):March 13, 2009
DOI:10.1021/ma802406j
A cyclic acetal-functionalized urethane acrylate monomer is synthesized here and polymerized in a crystalline state without the polymerization kinetics being deleteriously affected by the solid state. Depending on the processing conditions, the cyclic acetal urethane acrylate monomer exists in either a metastable liquid state or a crystalline state at ambient conditions. Because of mobility restrictions, extremely poor polymerization kinetics and functional group conversions are typically achieved in solid-state polymerizations. However, the solid-state photopolymerization of a cyclic acetal urethane acrylate results in nearly identical polymerization rates and ultimately higher conversion in the crystalline state than in the liquid state under otherwise identical conditions. We conclude that the crystallization process occurs in such a manner as to template the acrylic double bonds in a structure that facilitates rapid, minimally activated propagation.
Co-reporter:Benjamin D. Fairbanks, Timothy F. Scott, Christopher J. Kloxin, Kristi S. Anseth and Christopher N. Bowman
Macromolecules 2009 Volume 42(Issue 1) pp:211-217
Publication Date(Web):December 10, 2008
DOI:10.1021/ma801903w
Radical-mediated thiol−yne step-growth photopolymerizations are utilized to form highly cross-linked polymer networks. This reaction mechanism is shown to be analogous to the thiol−ene photopolymerization; however, each alkyne functional group is capable of consecutive reaction with two thiol functional groups. The thiol−yne reaction involves the sequential propagation of a thiyl radical with either an alkyne or a vinyl functional group followed by chain transfer of the radical to another thiol. The rate of thiyl radical addition to the alkyne was determined to be approximately one-third of that to the vinyl. Chain-growth polymerization of alkyne and vinyl functionalities was only observed for reactions in which the alkyne was originally in excess. Analysis of initial polymerization rates demonstrated a near first-order dependence on thiol concentration, indicating that chain transfer is the rate-determining step. Further analysis revealed that the polymerization rate scaled with the initiation rate to an exponent of 0.65, deviating from classical square root dependence predicted for termination occurring exclusively by bimolecular reactions. A tetrafunctional thiol was photopolymerized with a difunctional alkyne, forming an inherently higher cross-link density than an analogous thiol−ene resin, displaying a higher glass transition temperature (48.9 vs −22.3 °C) and rubbery modulus (80 vs 13 MPa). Additionally, the versatile nature of this chemistry facilitates postpolymerization modification of residual reactive groups to produce materials with unique physical and chemical properties.
Co-reporter:Leah M. Johnson, Benjamin D. Fairbanks, Kristi S. Anseth and Christopher N. Bowman
Biomacromolecules 2009 Volume 10(Issue 11) pp:
Publication Date(Web):October 12, 2009
DOI:10.1021/bm900846m
A rapid, water-soluble enzyme-mediated radical chain initiation system involving glucose oxidase and Fe2+ generated hydrogels within minutes at 25 °C and in ambient oxygen. The initiation components were evaluated for their effect on polymerization rates of hydroxyethyl acrylate-poly(ethylene glycol)575 diacrylate comonomer solutions using near-infrared spectroscopy. Increasing glucose concentration increased polymerization rates until reaching a rate plateau above 1 × 10−3 M of glucose. A square root dependence of the initial polymerization rate on Fe2+ concentration was observed between 1.0 × 10−4 M and 5.0 × 10−4 M of Fe2+, whereupon excess Fe2+ reduced final acrylate conversions. The glucose oxidase-mediated initiation system was employed for encapsulation of fibroblasts (NIH3T3s) into a poly(ethylene glycol) tetra-acrylate (Mn ∼ 20000) hydrogel scaffold demonstrating 96% (±3%) viability at 24 h postencapsulation. This first use of enzyme-mediated redox radical chain initiation for cellular encapsulation demonstrates polymerization of hydrogels in situ with kinetic control, minimal oxygen inhibition issues, and utilization of low initiator concentrations.
Co-reporter:Heather J. Avens
Journal of Polymer Science Part A: Polymer Chemistry 2009 Volume 47( Issue 22) pp:6083-6094
Publication Date(Web):
DOI:10.1002/pola.23649
Abstract
A visible light photoinitiator, eosin, in combination with a tertiary amine coinitiator is found to initiate polymerization despite the presence of at least 1000-fold excess dissolved oxygen, which functions as an inhibitor of radical polymerizations. Additionally, 0.4 μM eosin is able to overcome 100-fold excess (40 μM) 2,2,6,6-Tetramethyl-1-piperidinyloxy (TEMPO) inhibitor, initiating polymerization after only a 2 min inhibition period. In contrast, 40 μM Irgacure-2959, a standard cleavage-type initiator, is unable to overcome even an equivalent amount of inhibitor (40 μM TEMPO). Through additional comparisons of these two initiation systems, a reaction mechanism is developed which is consistent with the kinetic data and provides an explanation for eosin's relative insensitivity to oxygen, TEMPO, and other inhibitors. A cyclic mechanism is proposed in which semireduced eosin radicals react by disproportionation with radical inhibitors and radical intermediates in the inhibition process to regenerate eosin and effectively consume inhibitor. In behavior similar to that of eosin, rose bengal, fluorescein, and riboflavin are also found to initiate polymerization despite the presence of excess TEMPO, indicating that cyclic regeneration likely enhances the photoinitiation kinetics of many dye photosensitizers. Selection of such dye initiation systems constitutes a valuable strategy for alleviating inhibitory effects in radical polymerizations. © 2009 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 47: 6083–6094, 2009
Co-reporter:Christopher J. Kloxin, Timothy F. Scott and Christopher N. Bowman
Macromolecules 2009 Volume 42(Issue 7) pp:2551-2556
Publication Date(Web):March 10, 2009
DOI:10.1021/ma802771b
Allyl sulfide addition−fragmentation chain transfer was employed concurrently with the radical-mediated formation of a thiol-ene network to enable network adaptation and mitigation of polymerization-induced shrinkage stress. This result represents the first demonstration of simultaneous polymerization and network adaptation in covalently cross-linked networks with significant implications for the fabrication of low-stress polymer networks. For comparison, analogous networks incorporating propyl sulfide moieties, incapable of addition−fragmentation, were synthesized and evaluated in parallel. At the highest irradiation intensity, the allyl sulfide-containing material demonstrated a >75% reduction in the final stress when compared with the propyl sulfide-containing material. Analysis of the conversion evolution revealed that allyl sulfide addition−fragmentation decreased the polymerization rate owing to thiyl radical sequestration. Slow consumption of the allyl sulfide functional group suggests that intramolecular hydrolytic substitution occurs by a stepwise, rather than concerted, mechanism. Simultaneous stress and conversion measurements demonstrated that the initial stress evolution was identical for both the allyl and propyl sulfide-containing materials but diverged after gelation. Whereas addition−fragmentation chain transfer was found to occur throughout the polymerization, its effect on the stress evolution was concentrated toward the end of polymerization when network rearrangement becomes the dominant mechanism for stress relaxation. Even after the polymerization reaction was completed, the polymerization-induced shrinkage stress in the allyl sulfide-containing material continued to decrease, exhibiting a maximum in the stress evolution and demonstrating the potential for continuing, longer-term stress relaxation.
Co-reporter:Vaibhav S. Khire;Youngwoo Yi;Noel A. Clark
Advanced Materials 2008 Volume 20( Issue 17) pp:3308-3313
Publication Date(Web):
DOI:10.1002/adma.200800672
Co-reporter:Ryan R. Hansen, Hadley D. Sikes and Christopher N. Bowman
Biomacromolecules 2008 Volume 9(Issue 1) pp:
Publication Date(Web):December 1, 2007
DOI:10.1021/bm700672z
DNA biochip technology holds potential for highly parallel, rapid, and sensitive genetic diagnostic screening of target pathogens and disease biomarkers. A primary limitation involves a simultaneous, sequence-specific identification of low copy number target polynucleotides using a clinically appropriate detection methodology that implements only inexpensive detection instrumentation. Here, a rapid (20 min), nonenzymatic method of signal amplification utilizing surface-initiated photopolymerization is presented in glass microarray format. Visible light photoinitiators covalently coupled to streptavidin were used to bind biotin-labeled capture sequences. Amplification was achieved through subsequent contact with a monomer solution and the appropriate light exposure to generate 20−240-nm-thick hydrogel layers exclusively from spots containing the biotin-labeled DNA. An amplification factor of 106 to 107 was observed as well as a detectable response generated from as low as ∼104 labeled oligonucleotides using minimal instrumentation, such as an optical microscope or CCD camera.
Co-reporter:Ryan R. Hansen;Heather J. Avens;Raveesh Shenoy
Analytical and Bioanalytical Chemistry 2008 Volume 392( Issue 1-2) pp:
Publication Date(Web):2008 September
DOI:10.1007/s00216-008-2259-6
Quantitative evaluation of minimal polynucleotide concentrations has become a critical analysis among a myriad of applications found in molecular diagnostic technology. Development of high-throughput, nonenzymatic assays that are sensitive, quantitative and yet feasible for point-of-care testing are thus beneficial for routine implementation. Here, we develop a nonenzymatic method for quantifying surface concentrations of labeled DNA targets by coupling regulated amounts of polymer growth to complementary biomolecular binding on array-based biochips. Polymer film thickness measurements in the 20–220 nm range vary logarithmically with labeled DNA surface concentrations over two orders of magnitude with a lower limit of quantitation at 60 molecules/μm2 (∼106 target molecules). In an effort to develop this amplification method towards compatibility with fluorescence-based methods of characterization, incorporation of fluorescent nanoparticles into the polymer films is also evaluated. The resulting gains in fluorescent signal enable quantification using detection instrumentation amenable to point-of-care settings.
Co-reporter:Vaibhav S. Khire;April M. Kloxin;Charles L. Couch;Kristi S. Anseth
Journal of Polymer Science Part A: Polymer Chemistry 2008 Volume 46( Issue 20) pp:6896-6906
Publication Date(Web):
DOI:10.1002/pola.22999
Abstract
This study investigates the formation of linear polymer grafts using thiol-acrylate conjugate addition reactions on nanoparticle surfaces. Silica nanoparticles were first modified with an amine functionality, followed by the attachment of a photocleavable acrylate. Dithiol-diacrylate films were attached to the particles through the surface acrylate groups at various stoichiometric ratios of thiol to acrylate by conducting amine-catalyzed conjugate addition polymerizations. The particles were then exposed to UV light to release the grafted polymer by photocleavage. The cleaved, grafted polymers were analyzed using infrared spectroscopy and gel permeation chromatography and compared to polymers formed in the bulk, which remained unattached to the particles. The measured number and weight average molecular weights were similar for both polymer types within experimental error and increased from 2000 to 5000 g/mol and 4000 to 10,000 g/mol, respectively, as the ratio of limiting to excess functionality increased from 0.8 to 1. Both number and weight average molecular weights followed the trend of step growth polymers with the highest molecular weight achieved for stoichiometric monomeric mixtures. Surface coverage of the nanoparticles was estimated using the molecular weight and thermogravimetric data and was found to be uniform (∼0.15 chains/nm2) irrespective of the stoichiometry of the reacting monomers. © 2008 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 46: 6896–6906, 2008
Co-reporter:Brian J. Adzima, H. Alan Aguirre, Christopher J. Kloxin, Timothy F. Scott and Christopher N. Bowman
Macromolecules 2008 Volume 41(Issue 23) pp:9112-9117
Publication Date(Web):November 4, 2008
DOI:10.1021/ma801863d
A network polymer, incorporating dynamic and reversible cross-links, was synthesized using the Diels−Alder reaction. Fourier transform infrared (FTIR) spectroscopy was used to characterize the reaction rate and thermodynamic equilibrium over a broad temperature range. Equilibrium conversion of the furan and maleimide varied from 74% at 85 °C to 24% at 155 °C, demonstrating significant depolymerization via the retro-Diels−Alder reaction. The gel point temperature, as determined by rheometry using the Winter−Chambon criterion, was 92 °C, corresponding to a gel-point conversion of 71%, consistent with the Flory−Stockmayer equation. The scaling exponents for the complex moduli, viscosity, and plateau modulus, in the vicinity of the gel-point, were determined and compared with experimental and theoretical literature values. Further, the material exhibited a low frequency relaxation owing to dynamic rearrangement of cross-links by the Diels−Alder and retro-Diels−Alder reactions.
Co-reporter:Kathryn A. Berchtold, Jun Nie, Jeffrey W. Stansbury and Christopher N. Bowman
Macromolecules 2008 Volume 41(Issue 23) pp:9035-9043
Publication Date(Web):November 11, 2008
DOI:10.1021/ma801644j
The tremendous diversity of materials properties available with polymers is due in large part to the ability to design structures from the monomeric state. The ease of use of comonomer mixtures only expands this versatility. While final polymer properties are obviously important in the selection or development of a material for a given purpose, for a number of applications, such as optical fiber coatings, photolithography, and microelectronics, the additional requirement of a very rapid polymerization process may be equally critical. A class of unusually reactive mono(meth)acrylate monomers bearing secondary functionality that includes carbonates, carbamates, and oxazolidones, has been demonstrated but not fully explained. Here, the influence of an integral cyclic carbonate functional group on (meth)acrylate photopolymerization kinetics is examined in detail with respect to monomers with a wide variety of alternative secondary functionality structure as well as in comparison to conventional mono- and di(meth)acrylates. The kinetic results from full cure studies of several cyclic carbonate-containing monomers clearly highlight specific structural variations that effectively promote monomer reactivity. Copolymerizations with tetrahydrofurfuryl methacrylate reflect similar dramatic kinetic effects associated with the novel monomers, while partial cure homopolymerization studies reveal exceptional dark cure behavior linked to observations of uncommonly low ratios of termination to propagation rates throughout the conversion profile. Temperature effects on reaction kinetics, including both reaction rate and the individual kinetic parameters, as well as the temperature dependence of hydrogen bonding interactions specifically involving the secondary functional groups, are probed as a means to understand better the fundamentally interesting and practically important behavior of these monomers.
Co-reporter:Vaibhav S. Khire, Tai Yeon Lee and Christopher N. Bowman
Macromolecules 2008 Volume 41(Issue 20) pp:7440-7447
Publication Date(Web):September 25, 2008
DOI:10.1021/ma8008965
The formation of linear polymer films using thiol−ene photopolymerizations was investigated by grafting thiol−ene films on the surface with subsequent cleavage and polymer analysis. Silica nanoparticles were first modified with an acrylated silane molecule, and then a dithiol was reacted to this surface using a base catalyzed thiol−acrylate conjugate addition reaction to prepare a uniform surface presenting thiol groups attached to the surface with a cleavable ester linkage. Photopolymerization of difunctional thiol and ene monomers present in various stoichiometric ratios was carried out in the presence of the nanoparticles, and a thiol−ene film was attached to the surface. The polymer film was cleaved from the surface using acid-catalyzed hydrolysis and separated from the particles. It was found that the particle presence does not affect the polymerization of thiol−ene polymers in bulk; however, the presence of surface thiols changes the relative stoichiometry of the thiol and ene monomers at the surface and hence strongly affects the molecular weight of the attached polymer. The number average and the weight average molecular weight of the unattached polymers ranged from 900 to 12,600 g/mol and 1500 to 25,700 g/mol, respectively, as the thiol:ene ratio in the bulk increased from 0.6 to 1. The highest molecular weight of the grafted polymers (Mn = 1600 g/mol, Mw = 2700 g/mol) was achieved when the thiol:ene ratio was close to 0.77:1 in the bulk and decreased monotonically for both higher and lower stoichiometric ratios. When the polymerization rate was decreased and the time scales for reaction increased, the grafted polymer molecular weight increased for stoichiometric thiol−ene monomer mixtures, because the relative effect of surface thiols was decreased.
Co-reporter:Timothy F. Scott, Christopher J. Kloxin, Rocky B. Draughon and Christopher N. Bowman
Macromolecules 2008 Volume 41(Issue 9) pp:2987-2989
Publication Date(Web):April 9, 2008
DOI:10.1021/ma8002505
Co-reporter:Peter M. Johnson, Jeffrey W. Stansbury and Christopher N. Bowman
ACS Combinatorial Science 2007 Volume 9(Issue 6) pp:1149
Publication Date(Web):October 26, 2007
DOI:10.1021/cc700110p
Copolymerizations of hexanediol diacrylate with three monoacrylates were analyzed using high-throughput conversion analysis to elucidate the effects of varying alkyl pendant groups at different compositions. Each analyzed copolymerization system contained hexanediol diacrylate (HDDA), and copolymerizations with 30–60 wt % monoacrylate reached nearly complete conversion after 30 s of exposure time. For higher amounts of monoacrylate, the photopolymerization kinetics of the hexyl acrylate (HA) copolymerization were significantly slower than the copolymerization with either ethylhexyl acrylate (EHA) or dodecyl acrylate (DDA). With 20 wt % HDDA, conversion at 30 s with a comonomer of HA was 62 ± 3%, as compared to 76 ± 3% and 84 ± 3% when copolymerized with EHA and DDA, respectively. Model kinetic parameters were estimated for all four monomer systems, with HDDA monomer parameters found to be within the same error when estimated from any of the copolymerizations. With kinetic parameters for each monomer, comparison maps showing the difference in conversion between two copolymerizations were generated. These comparison maps allow for an assessment of two comonomer systems to determine the optimal photopolymerization conditions. Slower photopolymerization kinetics for HA occur at nearly all compositions containing monoacrylate, with the largest reduction occurring between 20 and 40 wt % monoacrylate.
Co-reporter:Jacquelyn A. Carioscia;Lauren Schneidewind;Casey O'Brien;Robert Ely;Caitlin Feeser;Neil Cramer
Journal of Polymer Science Part A: Polymer Chemistry 2007 Volume 45(Issue 23) pp:5686-5696
Publication Date(Web):23 OCT 2007
DOI:10.1002/pola.22318
The ability to prepare high Tg low shrinkage thiol–ene materials is attractive for applications such as coatings and dental restoratives. However, thiol and nonacrylated vinyl materials typically consist of a flexible backbone, limiting the utility of these polymers. Hence, it is of importance to synthesize and investigate thiol and vinyl materials of varying backbone chemistry and stiffness. Here, we investigate the effect of backbone chemistry and functionality of norbornene resins on polymerization kinetics and glass transition temperature (Tg) for several thiol–norbornene materials. Results indicate that Tgs as high as 94 °C are achievable in thiol–norbornene resins of appropriately controlled chemistry. Furthermore, both the backbone chemistry and the norbornene moiety are important factors in the development of high Tg materials. In particular, as much as a 70 °C increase in Tg was observed in a norbornene–thiol specimen when compared with a sample prepared using allyl ether monomer of analogous backbone chemistry. © 2007 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 45: 5686–5696, 2007
Co-reporter:T. F. Scott;R. B. Draughon;C. N. Bowman
Advanced Materials 2006 Volume 18(Issue 16) pp:2128-2132
Publication Date(Web):18 JUL 2006
DOI:10.1002/adma.200600379
Photoinduced polymer actuation by relieving stress unevenly through the thickness of a chemically crosslinked, rubbery polymer upon light exposure is demonstrated (see figure). The sensitivity of this method to light is greater than previously developed photoinduced actuation techniques, as the recurring chain-transfer reactions amplify the effects of each absorbed photon and subsequently generated radical on the stress-relief and actuation processes.
Co-reporter:Vaibhav S. Khire;Danielle S. W. Benoit;Kristi S. Anseth
Journal of Polymer Science Part A: Polymer Chemistry 2006 Volume 44(Issue 24) pp:7027-7039
Publication Date(Web):7 NOV 2006
DOI:10.1002/pola.21786
The application of surface-attached, thiol-ene polymer films for controlling material properties in a gradient fashion across a surface was investigated. Thiol-ene films were attached to the surface by first depositing a thiol-terminated self-assembled monolayer and performing a thiol-ene photopolymerization reaction on the surface. Property gradients were created either by creating and modifying a gradient in the surface thiol density in the SAM or by changing the polymerization conditions or both. Film thickness was modified across the substrate by changing either the density of the anchoring thiol functional groups or by changing the reaction conditions such as exposure time. Thicker films (1–11 nm) were obtained by polymerizing acrylate polymer brushes from the surface with varying exposure time (0–60 s). The two factors, that is, the surface thiol density and the exposure time, were combined in orthogonal directions to obtain thiol-ene films with a two-dimensional thickness gradient with the maximum thickness being 4 nm. Finally, a thiol-acrylate Michael type addition reaction was used to modify the surface thiol density gradient with the cell-adhesive ligand, Arg-Gly-Asp-Ser (RGDS), which subsequently yielded a gradient in osteoblast density on the surface. © 2006 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 44: 7027–7039, 2006
Co-reporter:K. Tommy Haraldsson, J. Brian Hutchison, Robert P. Sebra, Brian T. Good, Kristi S. Anseth, Christopher N. Bowman
Sensors and Actuators B: Chemical 2006 Volume 113(Issue 1) pp:454-460
Publication Date(Web):17 January 2006
DOI:10.1016/j.snb.2005.03.096
In this contribution, a new method for the fabrication of complex polymeric microfluidic devices is presented. The technology, contact liquid photolithographic polymerization (CLiPP), overcomes many of the drawbacks associated with other rapid prototyping schemes, such as limited materials choices and time-consuming microassembly protocols. CLiPP shares many traits with other photolithographic methods, but three distinct features: (i) liquid photoresists in contact with the photomask, (ii) readily removed sacrificial materials, and (iii) living radical processes, enable multiple polymeric chemistries and mechanical properties while simultaneously enabling facile fabrication of 3D geometries and surface chemistry control. This contribution details fabrication techniques and methods for the fabrication of high aspect ratio posts covalently bonded to a polymeric substrate, an array of independently stacked bars on top of perpendicular bars, multiple undercut structures fabricated simultaneously, and a complex 3D geometry with intertwined channels.
Co-reporter:Timothy F. Scott;Andrew D. Schneider;Wayne D. Cook
Science 2005 Vol 308(5728) pp:1615-1617
Publication Date(Web):10 Jun 2005
DOI:10.1126/science.1110505
Abstract
Chemically cross-linked polymers are inherently limited by stresses that are introduced by post-gelation volume changes during polymerization. It is also difficult to change a cross-linked polymer's shape without a corresponding loss of material properties or substantial stress development. We demonstrate a cross-linked polymer that, upon exposure to light, exhibits stress and/or strain relaxation without any concomitant change in material properties. This result is achieved by introducing radicals via photocleavage of residual photoinitiator in the polymer matrix, which then diffuse via addition-fragmentation chain transfer of midchain functional groups. These processes lead to photoinduced plasticity, actuation, and equilibrium shape changes without residual stress. Such polymeric materials are critical to the development of microdevices, biomaterials, and polymeric coatings.
Co-reporter:Helen M. Simms, Christopher M. Brotherton, Brian T. Good, Robert H. Davis, Kristi S. Anseth and Christopher N. Bowman
Lab on a Chip 2005 vol. 5(Issue 2) pp:151-157
Publication Date(Web):08 Dec 2004
DOI:10.1039/B412589D
Novel fabrication techniques and polymer systems are being explored to enable mass production of low cost microfluidic devices. In this contribution we discuss a new fabrication scheme for making microfluidic devices containing porous polymer components in situ. Contact lithography, a living radical photopolymer (LRPP) system and salt leaching were used to fabricate multilayer microfluidic devices rapidly with various channel geometries and covalently attached porous polymer plugs made of various photopolymerizable substrates. LRPP systems offer the advantages of covalent attachment of microfluidic device layers and facile surface modification via grafting. Several applications of the porous plugs are also explored, including a static mixer, a high surface area-to-volume reactor and a rapidly responding hydrogel valve. Quantitative and qualitative data show an increase in mixing of a fluorescein and a water stream for channels containing porous plugs relative to channels with no porous plugs. Confocal laser scanning microscopy images demonstrate the ability to graft a functional material onto porous plug surfaces. A reaction was carried out on the grafted pore surfaces, which resulted in fluorescent labelling of the grafted material throughout the pores of the plug. Homogenous fluorescence throughout the depth of the porous plug and along pore surfaces indicated that the porous plugs were surface modified by grafting and that reactions can be carried out on the pore surfaces. Finally, porous hydrogel valves were fabricated which swelled in response to contact with various pH solutions. Results indicate that a porous hydrogel valve will swell and close more rapidly than other valve geometries made with the same polymer formulation. The LRPP-salt leaching method provides a means for rapidly incorporating porous polymer components into microfluidic devices, which can be utilized for a variety of pertinent applications upon appropriate selection of porous plug materials and surface treatments.
Co-reporter:Hui Lu, Jacquelyn A. Carioscia, Jeffery W. Stansbury, Christopher N. Bowman
Dental Materials 2005 Volume 21(Issue 12) pp:1129-1136
Publication Date(Web):December 2005
DOI:10.1016/j.dental.2005.04.001
ObjectivesThe goal of this work was to investigate the feasibility of formulating novel dental restorative materials that utilize a step-growth thiol-ene photopolymerization. Particularly, we are aiming to significantly reduce the polymerization shrinkage and shrinkage stress while retaining adequate physical properties as compared to current dimethacrylatre-based systems.MethodsThe thiol-ene system is composed of a 4:3 molar mixture of triallyl-1,3,5-triazine-2,4,6-trione (TATATO) and pentaerythritol tetramercaptopropionate (PETMP). The simultaneous measurement of shrinkage stress and functional group conversion was performed. Solvent extraction of unreacted monomers and dynamic mechanical analysis on the polymer networks that were formed were also studied. Flexural strength was measured for both filled and unfilled PETMP/TATATO and Bis-GMA/TEGDMA systems.ResultsPhotopolymerization of PETMP/TATATO occurs at a much higher rate, with the maximum polymerization rate six times faster, than Bis-GMA/TEGDMA cured under the identical conditions. The results from the simultaneous measurement of shrinkage stress and conversion showed that the onset of shrinkage stress coincides with the delayed gel point conversion, which is predicted to be 41% for the 3:4 stoichiometric PETMP/TATATO resin composition. The maximum shrinkage stress developed for PETMP/TATATO was about 0.4 MPa, which was only approximately 14% of the maximum shrinkage stress of the Bis-GMA/TEGDMA system. Adequate flexural strength and flexural modulus values were obtained for both filled and unfilled PETMP/TATATO systems.SignificanceThe dramatically reduced shrinkage stress, increased polymerization rate, significance increased functional group conversion, and decreased leachable species are all benefits for the use-of thiol-ene systems as potential dental restorative materials.
Co-reporter:Jacquelyn A. Carioscia, Hui Lu, Jeffrey W. Stanbury, Christopher N. Bowman
Dental Materials 2005 Volume 21(Issue 12) pp:1137-1143
Publication Date(Web):December 2005
DOI:10.1016/j.dental.2005.04.002
ObjectiveThe aim of this work was to prereact thiol-ene monomers to create reactive thiol or vinyl (ene)-functionalized oligomers, and to investigate the use of these materials as novel dental restorative material. Investigation has focused on the application of oligomeric thiol-ene materials as dental restorative resins with lower polymerization shrinkage and polymerization stress as compared to monomeric thiol-ene systems and particularly with respect to current dimethacrylate-based systems.MethodsReactive thiol-functionalized oligomers were created via photopolymerization using triallyl-1,3,5-triazine-2,4,6-trione (TATATO), trimethylolpropane tris(3-mercaptopropionate) (trithiol) and pentaerythritol tetramercaptopropionate (tetrathiol). Kinetic and mechanical investigation of Bis-GMA/TEGDMA, and oligomeric and monomeric thiol-ene systems were conducted. More specifically, polymerization shrinkage and stress, polymerization kinetics, glass transition temperature, flexural strength and flexural modulus were evaluated.ResultsUpon evaluation, the polymerization stress of oligomeric thiol-ene systems was dramatically reduced by as much as 33% when compared with the stress exhibited by monomeric thiol-ene systems and as much as a 92% reduction in stress relative to the current dimethacrylate-based dental restorative materials. Furthermore, the flexural strength and modulus of the monomeric and oligomeric thiol-ene resins were not significantly different.SignificanceOligomeric thiol-ene systems offer potential as alternative dental restorative resins due to the significant reduction in polymerization shrinkage and stress while retaining the mechanical properties of monomer-based thiol-ene resins.
Co-reporter:Hui Lu, Jeffrey W. Stansbury, Jun Nie, Kathryn A. Berchtold, Christopher N. Bowman
Biomaterials 2005 Volume 26(Issue 12) pp:1329-1336
Publication Date(Web):April 2005
DOI:10.1016/j.biomaterials.2004.04.041
Reactive diluents such as triethyleneglycol-dimethacrylate (TEGDMA) have been widely used with bisphenol-A-glycidyl-dimethacrylate (Bis-GMA) to achieve restorative resins with appropriate viscosity and higher conversion. However, additional water sorption and polymerization shrinkage were also introduced. The aim of this work is to investigate whether the cure and material properties can be improved in dental resins containing novel mono-(meth)acrylates as reactive diluents so that these Bis-GMA-based copolymers have reduced polymerization shrinkage but higher overall double bond conversion. Several ultra-high-reactivity mono-(meth)acrylates that contain secondary functionalities have been synthesized and investigated. The polymerization rate and double bond conversion were monitored using photo-FTIR. Polymerization shrinkage, dynamic mechanical analysis, and flexural strength were characterized. Compared with the Bis-GMA/TEGDMA control, the Bis-GMA/mono-methacrylate systems studied showed higher final conversions, faster curing rates, and decreased polymerization shrinkage. Our optimum system Bis-GMA/morpholine carbamate methacrylate achieved 86% final conversion (vs. 65%), a polymerization rate 3.5 times faster, and a 30% reduction in polymerization volumetric shrinkage. These results indicate that certain highly reactive, novel mono-(meth)acrylates possess very promising potential to replace TEGDMA as reactive diluents and can readily be applied to develop superior dental resins.
Co-reporter:Sirish K. Reddy;Robert P. Sebra;Kristi S. Anseth
Journal of Polymer Science Part A: Polymer Chemistry 2005 Volume 43(Issue 10) pp:2134-2144
Publication Date(Web):7 APR 2005
DOI:10.1002/pola.20693
The formation of reactive substrates with iniferter-mediated living radical photopolymerization is a powerful technique for surface modification, which can readily be used to facilitate the incorporation of a variety of surface functionalities. In this research, the photopolymerization kinetics of novel bulk thiol–ene systems have been compared with those of typical acrylate and methacrylate systems when polymerized in the presence of the photoiniferter p-xylene bis(N,N-diethyl dithiocarbamate) (XDT). In the presence of XDT, the thiol–ene systems photopolymerize more quickly than the traditional acrylate and methacrylate systems by one to two orders of magnitude. Fourier transform infrared spectroscopy has been used to monitor the photografting kinetics of various monomers on dithiocarbamate-functionalized surfaces. Furthermore, this technique has been used to evaluate surface-initiation kinetics and to emphasize the influence of bulk substrate properties on grafting kinetics. Finally, photopatterning has been demonstrated on a dithiocarbamate-incorporated thiol–ene substrate with conventional photolithographic techniques. © 2005 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 43: 2134–2144, 2005
Co-reporter:J. Brian Hutchison, K. Tommy Haraldsson, Brian T. Good, Robert P. Sebra, Ning Luo, Kristi S. Anseth and Christopher N. Bowman
Lab on a Chip 2004 vol. 4(Issue 6) pp:658-662
Publication Date(Web):24 Sep 2004
DOI:10.1039/B405985A
Microfluidic devices are commonly fabricated in silicon or glass using micromachining technology or elastomers using soft lithography methods; however, invariable bulk material properties, limited surface modification methods and difficulty in fabricating high aspect ratio devices prevent these materials from being utilized in numerous applications and/or lead to high fabrication costs. Contact Liquid Photolithographic Polymerization (CLiPP) was developed as an alternative microfabrication approach that uniquely exploits living radical photopolymerization chemistry to facilitate surface modification of device components, fabrication of high aspect ratio structures from many different materials with numerous covalently-adhered layers and facile construction of three-dimensional devices. This contribution describes CLiPP and demonstrates unique advantages of this new technology for microfabrication of polymeric microdevices. Specifically, the procedure for fabricating devices with CLiPP is presented, the living radical photopolymerization chemistry which enables this technology is described, and examples of devices made using CLiPP are shown.
Co-reporter:Neil B. Cramer;Sirish K. Reddy;Tsali Cross;Rishi Raj;Allison K. O' Brien
Macromolecular Symposia 2004 Volume 206(Issue 1) pp:361-374
Publication Date(Web):17 FEB 2004
DOI:10.1002/masy.200450228
A novel thiol-ene photopolymerization reaction involving copolymerization of tetrathiol monomer with vinyl silazane is experimentally characterized and is modeled successfully. The overall polymerization rate is found to be controlled by the ratio of the propagation to chain transfer kinetic parameters. The polymerization rate of this mixture, in the presence of added photoinitiator, is approximately first order in ene functionality and is independent of thiol functional group concentration. Initiation rates in this system, when cured utilizing a light centered around 365 nm light, and in the presence of no added photoinitiator, are shown to be proportional to the ene monomer concentration. When the mixture is polymerized utilizing light centered at 254 nm light, and without photoinitiator, the initiation rates are proportional to the thiol monomer concentrations. This novel reaction scheme is further utilized to form ultra rapidly polymerizable polymer derived ceramic structures with high aspect ratios.
Co-reporter:Hui Lu;Jeffrey W. Stansbury;Sabine H. Dickens;Frederick C. Eichmiller;Sabine H. Dickens;Hui Lu;Frederick C. Eichmiller;Jeffrey W. Stansbury
Journal of Biomedical Materials Research Part B: Applied Biomaterials 2004 Volume 71B(Issue 1) pp:206-213
Publication Date(Web):11 JUN 2004
DOI:10.1002/jbm.b.30088
This study probes the interrelationships between polymerization shrinkage stress development and the polymerization progress with a novel experimental technique. This technique is capable of real time, simultaneous measurement of double-bond conversion and shrinkage stress with the use of a noninvasive near-infrared fiber-optic system, along with a cantilever beam-based tensometer. The results from both filled and unfilled bis-GMA/TEGDMA (70:30 mass ratio) systems showed that the shrinkage stress buildup was concentrated in the latter stages of polymerization, with its dramatic increase linked to the asymptotic approach of conversion to its limiting value. The monotonic increase of shrinkage stress with conversion in the vitrified state is attributed to the dramatic increase of the sample's elastic modulus during the vitrification stage and a certain amount of cooling stress as the sample cools down from the temperature rise caused by the exothermic polymerization and light absorption. Excellent reproducibility of both the polymerization kinetics assessment and the shrinkage stress measurement has been achieved. © 2004 Wiley Periodicals, Inc. J Biomed Mater Res Part B: Appl Biomater 71B: 206–213, 2004
Co-reporter:Neil B. Cramer;Sirish K. Reddy;Michael Cole;Charles Hoyle
Journal of Polymer Science Part A: Polymer Chemistry 2004 Volume 42(Issue 22) pp:5817-5826
Publication Date(Web):7 OCT 2004
DOI:10.1002/pola.20419
Thiol–ene photopolymerizations without added photoinitiators were studied, and the kinetics accurately predicted by modeling for a wide range of different vinyl chemistries. Initiation rates in polymerizations initiated with light centered around 365 nm were found to be proportional to the concentration of ene functional groups. When 254 nm light was used as the initiating light source, initiation rates were proportional to the concentration of thiol functional groups. The mechanism of initiation at 254 nm was attributed to direct cleavage of thiol functional groups. An appropriate species or mechanism has not yet been found that is consistent with the experimental data for initiation when 365 nm light is used. © 2004 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 42: 5817–5826, 2004
Co-reporter:Lale G Lovell, Sheldon M Newman, Matthew M Donaldson, Christopher N Bowman
Dental Materials 2003 Volume 19(Issue 6) pp:458-465
Publication Date(Web):September 2003
DOI:10.1016/S0109-5641(02)00090-8
Objective. Two visible light sources (tungsten-quartz-halogen and xenon-arc plasma) with vastly different intensities (200 and 1800 mW/cm2) but similar spectral outputs, were used to examine the effects of light intensity on conversion and flexural strength of a model dental resin formulation (75/25 wt% bis-GMA/TEGDMA).Methods. The exact same polymer samples were used to correlate double bond conversion (measured with near-IR spectroscopy) to flexural strength, both immediately after light exposure and after storage.Results. In general, polymers which were irradiated with the high light intensity source exhibited greater double bond conversion. However, increasing the light intensity also increased the maximum temperature reached during polymerization. Therefore, the greater double bond conversion was caused by a combination of both photo and thermal effects. Regardless of the light intensity, a single linear relationship existed between conversion and final flexural strength (measured 4 days after cure) over the conversion range analyzed (50–80%). However, deviations from linearity were noted in several samples that were tested immediately after exposure.Significance. These findings illustrate that light intensity does not affect the final flexural strength of a dental resin as long as the final conversions are similar.
Co-reporter:Tara M. Lovestead, Allison K. O’Brien, Christopher N. Bowman
Journal of Photochemistry and Photobiology A: Chemistry 2003 Volume 159(Issue 2) pp:135-143
Publication Date(Web):14 July 2003
DOI:10.1016/S1010-6030(03)00178-3
Models for predicting multivinyl free radical photopolymerization that incorporate diffusion controlled propagation and termination are discussed. One model focuses on spatial effects by incorporating heat and mass transfer in photopolymerizing films. Temperature, species concentrations, e.g., oxygen and initiator, and light intensity are varied as a function of both time and depth. Specifically, the effect of using polychromatic initiation on oxygen inhibition was investigated. The model predicts that by utilizing initiating radiation of two distinct wavelengths, it is possible to overcome oxygen inhibition and achieve complete cure. The second model incorporates chain length dependent termination (CLDT) and chain transfer to polymer (CTP) in a homogenous, polymerizing system. Specifically, how CTP affects the polymerization rate (Rp), the reaction diffusion coefficient, the scaling relationship between polymerization rate and initiation rate (Ri), i.e., Rp∝Riα, and the transition from CLDT to reaction diffusion controlled termination was investigated. The model predicts that, in general, at low double bond conversion, CTP inclusion decreases the reaction diffusion coefficient and increases the polymerization rate. Additionally, increasing the CTP rate decreases the double bond conversion both at which the termination mechanism begins to transition from CLDT to reaction diffusion controlled termination and at which reaction diffusion controlled termination becomes the dominant termination mechanism. The model provides more insight into the termination mechanism and complex polymerization behavior.
Co-reporter:Ning Luo;J. Brian Hutchison;Kristi S. Anseth
Journal of Polymer Science Part A: Polymer Chemistry 2002 Volume 40(Issue 11) pp:1885-1891
Publication Date(Web):19 APR 2002
DOI:10.1002/pola.10272
(Methacryloyl ethylenedioxycarbonyl) benzyl N,N-diethyldithiocarbamate (HEMA-E-In) was synthesized and used as a monomer iniferter to develop a novel, photopatternable grafting technology. This molecule functions as both a methacrylic monomer and a photoiniferter (photoinitiator–transfer agent–terminator). The structure of HEMA-E-In was characterized by 1H NMR, Fourier transform infrared, and ultraviolet–visible spectroscopies. In the presence of the monomer iniferter, methyl methacrylate was polymerized by exposure to 365-nm ultraviolet radiation, confirming the initiation capability of HEMA-E-In. After the copolymerization of HEMA-E-In into a methacrylate-based polymer, attenuated total reflectance Fourier transform infrared spectra revealed that the photoiniferter functionality was present at the surface of this polymeric substrate. Photografting of poly(ethylene glycol) monomethacrylate monomer from the surface caused a significant change in the hydrophobicity of the surface as demonstrated by contact angle measurements. The novel monomer photoiniferter HEMA-E-In initiates the polymerization of bulk monomer and provides a reactive functionality that facilitates further initiation and polymer modification by the polymerization of different monomers. © 2002 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 40: 1885–1891, 2002
Co-reporter:J.E. Elliott, L.G. Lovell, C.N. Bowman
Dental Materials 2001 Volume 17(Issue 3) pp:221-229
Publication Date(Web):1 May 2001
DOI:10.1016/S0109-5641(00)00075-0
An optimal dental restorative polymeric material would have a homogeneous cross-linking density giving it consistent mechanical strength throughout the material. When multifunctional monomers are polymerized, a pendant double bond can react intramolecularly with the radical on its propagating chain to form a loop, which results in a primary cyclization reaction. Primary cyclization does not contribute to overall network structure, causes microgel formation, and leads to heterogeneity in the polymer. Knowledge of how cure conditions control the degree of primary cyclization and cross-linking in the polymer is important in developing better dental materials. To gain more understanding about the evolving polymer network, the photopolymerization of a typical dental resin (75/25 wt% bis-GMA/TEGDMA) is modeled using a first principals approach. The overall polymerization rate behavior of 75/25 wt% bis-GMA/TEGDMA is predicted using experimentally obtained propagation and termination kinetic rate constants. The effect of chain stiffness and light intensity on the polymerization kinetics is also explored. Furthermore, the model predicts the extent of cross-linking and primary cyclization in the growing polymer network. At 45% conversion, the fraction of bis-GMA and TEGDMA pendant double bonds created that have cycled is 11 and 33%, respectively. The model shows that using a stiff monomer, like bis-GMA, in dental resins diminishes the extent of cyclization and increases the cross-linking density of the polymer. Therefore, better mechanical properties are obtained than if more flexible monomers were used.
Co-reporter:Lale G Lovell, Hui Lu, Jeannine E Elliott, Jeffrey W Stansbury, Christopher N Bowman
Dental Materials 2001 Volume 17(Issue 6) pp:504-511
Publication Date(Web):November 2001
DOI:10.1016/S0109-5641(01)00010-0
Co-reporter:Lale G. Lovell;Kathryn A. Berchtold;Hui Lu;Jeannine E. Elliott
Polymers for Advanced Technologies 2001 Volume 12(Issue 6) pp:335-345
Publication Date(Web):8 JUN 2001
DOI:10.1002/pat.115
Dimethacrylate monomers are commonly used as the organic phase of dental restorative materials but many questions remain about the underlying kinetics and network formation in these highly crosslinked photopolymer systems. Several novel experimental and modeling techniques that have been developed for other multifunctional (meth)acrylates were utilized to gain further insight into these resin systems. Specifically, this work investigates the copolymerization behavior ofbis-GMA (2,2-bis[p-(2-hydroxy-3-methacryloxyprop-1-oxy)-phenyl]propane) and TEGDMA (triethylene glycol dimethacrylate), two monomers typically used for dental resin formulations. Near-infrared spectroscopy, electron paramagnetic resonance spectroscopy, as well as dynamic mechanical and dielectric analysis were used to characterize the kinetics, radical populations, and structural properties of this copolymer system. In addition, a kinetic model is described that provides valuable information about the network evolution during the formation of this crosslinked polymer. The results of these numerous studies illustrate that all of the aforementioned techniques can be readily applied to dental resin systems and consequently can be used to obtain a wealth of information about these systems. The application of these techniques provides insight into the complex polymerization kinetics and corresponding network formation, and as a result, a more complete understanding of the anomolous behaviors exhibited by these systems, such as diffusion controlled kinetics and conversion dependent network formation, is attained. Copyright © 2001 John Wiley & Sons, Ltd.
Co-reporter:Neil B. Cramer
Journal of Polymer Science Part A: Polymer Chemistry 2001 Volume 39(Issue 19) pp:3311-3319
Publication Date(Web):13 AUG 2001
DOI:10.1002/pola.1314
We used real-time Fourier transform infrared to monitor the conversion of both thiol and ene (vinyl) functional groups independently during photoinduced thiol–ene photopolymerizations. From these results, the stoichiometry of various thiol–ene and thiol–acrylate polymerizations was determined. For thiol–ene polymerizations, the conversion of ene functional groups was up to 15% greater than the conversion of thiol functional groups. For stoichiometric thiol–acrylate polymerizations, the conversion of the acrylate functional groups was roughly twice that of the thiol functional groups. With kinetic expressions for thiol–acrylate polymerizations, the acrylate propagation kinetic constant was found to be 1.5 times greater than the rate constant for hydrogen abstraction from the thiol. Conversions of thiol–acrylate systems of various initial stoichiometries were successfully predicted with this ratio of propagation and chain-transfer kinetic constants. Thiol–acrylate systems with different initial stoichiometries exhibited diverse network properties. Thiol–ene systems were initiated with benzophenone and 2,2-dimethoxy-2-phenylacetophenone as initiators and were also polymerized without a photoinitiator. © 2001 John Wiley & Sons, Inc. J Polym Sci Part A: Polym Chem 39: 3311–3319, 2001
Co-reporter:Han Byul Song, Nancy Sowan, Parag K. Shah, Austin Baranek, Alexander Flores, Jeffrey W. Stansbury, Christopher N. Bowman
Dental Materials (November 2016) Volume 32(Issue 11) pp:1332-1342
Publication Date(Web):November 2016
DOI:10.1016/j.dental.2016.07.014
ObjectivesPolymerization shrinkage stress and factors involved in the stress development such as volumetric shrinkage and modulus were investigated in photo-CuAAC (photo-initiated copper(I)-catalyzed azide-alkyne cycloaddition) polymerization and compared with conventional BisGMA-based methacrylate polymerization for their use as alternative dental resins.MethodsTri-functional alkyne and di-functional azide monomers were synthesized for photo-CuAAC polymerization. Conversion kinetics, stress development and polymerization shrinkage were determined with FTIR spectroscopy, tensometery, and with a linometer, respectively, for CuAAC and BisGMA-based monomer mixtures using a camphorquinone/amine visible light photoinitiator system. Thermo-mechanical properties for the cured polymer matrices were characterized by dynamic mechanical analysis and in three-point bending on a universal testing machine. Polymerization kinetics, polymerization shrinkage stress, dynamic volumetric shrinkage, glass transition temperature (Tg), flexural modulus, flexural strength, and flexural toughness were compared between the two different resin systems.ResultsA glassy CuAAC polymer (Tg = 62 °C) exhibited 15–25% lower flexural modulus of 2.5 ± 0.2 GPa and flexural strength of 117 ± 8 MPa compared to BisGMA-based polymer (Tg = 160 °C) but showed considerably higher energy absorption around 7.1 MJ × m−3 without fracture when strained to 11% via three-point bend compared to the flexural toughness of 2.7 MJ × m−3 obtained from BisGMA-based polymer. In contrast to BisGMA-based polymers at 75% functional group conversion, the CuAAC polymerization developed approximately three times lower shrinkage stress with the potential to achieve quantitative conversion under ambient temperature photocuring conditions. Moreover, relatively equivalent dynamic volumetric shrinkage of around 6–7% was observed via both CuAAC and dimethacrylate polymerization, suggesting that the low shrinkage stress of CuAAC polymerization was due to delayed gelation along with slower rate of polymerization and the formation of a more compliant network structure.SignificanceCuAAC crosslinked networks possessed high toughness and low polymerization shrinkage stress with quantitative conversion, which eliminated obstacles associated with BisGMA-based dental resins including limited conversion, unreacted extractable moieties, brittle failure, and high shrinkage stress.
Co-reporter:Han Byul Song, Nancy Sowan, Parag K. Shah, Austin Baranek, Alexander Flores, Jeffrey W. Stansbury, Christopher N. Bowman
Dental Materials (November 2016) Volume 32(Issue 11) pp:
Publication Date(Web):November 2016
DOI:10.1016/j.dental.2016.07.014
ObjectivesPolymerization shrinkage stress and factors involved in the stress development such as volumetric shrinkage and modulus were investigated in photo-CuAAC (photo-initiated copper(I)-catalyzed azide-alkyne cycloaddition) polymerization and compared with conventional BisGMA-based methacrylate polymerization for their use as alternative dental resins.MethodsTri-functional alkyne and di-functional azide monomers were synthesized for photo-CuAAC polymerization. Conversion kinetics, stress development and polymerization shrinkage were determined with FTIR spectroscopy, tensometery, and with a linometer, respectively, for CuAAC and BisGMA-based monomer mixtures using a camphorquinone/amine visible light photoinitiator system. Thermo-mechanical properties for the cured polymer matrices were characterized by dynamic mechanical analysis and in three-point bending on a universal testing machine. Polymerization kinetics, polymerization shrinkage stress, dynamic volumetric shrinkage, glass transition temperature (Tg), flexural modulus, flexural strength, and flexural toughness were compared between the two different resin systems.ResultsA glassy CuAAC polymer (Tg = 62 °C) exhibited 15–25% lower flexural modulus of 2.5 ± 0.2 GPa and flexural strength of 117 ± 8 MPa compared to BisGMA-based polymer (Tg = 160 °C) but showed considerably higher energy absorption around 7.1 MJ × m−3 without fracture when strained to 11% via three-point bend compared to the flexural toughness of 2.7 MJ × m−3 obtained from BisGMA-based polymer. In contrast to BisGMA-based polymers at 75% functional group conversion, the CuAAC polymerization developed approximately three times lower shrinkage stress with the potential to achieve quantitative conversion under ambient temperature photocuring conditions. Moreover, relatively equivalent dynamic volumetric shrinkage of around 6–7% was observed via both CuAAC and dimethacrylate polymerization, suggesting that the low shrinkage stress of CuAAC polymerization was due to delayed gelation along with slower rate of polymerization and the formation of a more compliant network structure.SignificanceCuAAC crosslinked networks possessed high toughness and low polymerization shrinkage stress with quantitative conversion, which eliminated obstacles associated with BisGMA-based dental resins including limited conversion, unreacted extractable moieties, brittle failure, and high shrinkage stress.
Co-reporter:Christopher J. Kloxin and Christopher N. Bowman
Chemical Society Reviews 2013 - vol. 42(Issue 17) pp:NaN7173-7173
Publication Date(Web):2013/04/12
DOI:10.1039/C3CS60046G
Covalently crosslinked materials, classically referred to as thermosets, represent a broad class of elastic materials that readily retain their shape and molecular architecture through covalent bonds that are ubiquitous throughout the network structure. These materials, in particular in their swollen gel state, have been widely used as stimuli responsive materials with their ability to change volume in response to changes in temperature, pH, or other solvent conditions and have also been used in shape memory applications. However, the existence of a permanent, unalterable shape and structure dictated by the covalently crosslinked structure has dramatically limited their abilities in this and many other areas. These materials are not generally reconfigurable, recyclable, reprocessable, and have limited ability to alter permanently their stress state, topography, topology, or structure. Recently, a new paradigm has been explored in crosslinked polymers – that of covalent adaptable networks (CANs) in which covalently crosslinked networks are formed such that triggerable, reversible chemical structures persist throughout the network. These reversible covalent bonds can be triggered through molecular triggers, light or other incident radiation, or temperature changes. Upon application of this stimulus, rather than causing a temporary shape change, the CAN structure responds by permanently adjusting its structure through either reversible addition/condensation or through reversible bond exchange mechanisms, either of which allow the material to essentially reequilibrate to its new state and condition. Here, we provide a tutorial review on these materials and their responsiveness to applied stimuli. In particular, we review the broad classification of these materials, the nature of the chemical bonds that enable the adaptable structure, how the properties of these materials depend on the reversible structure, and how the application of a stimulus causes these materials to alter their shape, topography, and properties.
Co-reporter:Weixian Xi, Matthias Krieger, Christopher J. Kloxin and Christopher N. Bowman
Chemical Communications 2013 - vol. 49(Issue 40) pp:NaN4506-4506
Publication Date(Web):2013/03/08
DOI:10.1039/C3CC41123K
The utilization of 2-(2-nitrophenyl)propyloxycarbonyl (NPPOC) as a photolabile primary amine cage enables the thiol-Michael ‘click’ reaction to be photo-triggered. The photolabile amine exhibits efficient catalytic activity upon UV irradiation and is shown to initiate the photopolymerization of tetrathiol and diacrylate comonomers viaMichael addition.
Co-reporter:Tao Gong, Brian J. Adzima and Christopher N. Bowman
Chemical Communications 2013 - vol. 49(Issue 72) pp:NaN7952-7952
Publication Date(Web):2013/07/25
DOI:10.1039/C3CC43637C
A novel copper(II) complex has been developed in which the counter anion, acylphosphinate, serves as a visible light photoinitiator. This molecule is inactive in the dark but, upon visible light exposure, both CuAAC and ATRP reactions are readily and rapidly initiated.
Co-reporter:Andrew B. Lowe, Charles E. Hoyle and Christopher N. Bowman
Journal of Materials Chemistry A 2010 - vol. 20(Issue 23) pp:NaN4750-4750
Publication Date(Web):2010/02/19
DOI:10.1039/B917102A
Radical mediated thiol-yne polymerization reactions complement the more well-known thiol-ene radical polymerization processes, with the added advantage of increased functionality. In one system studied, the rate constant for the addition of the thiol to the vinyl sulfide created by the initial reaction of the thiol with the alkyne is three times faster than the initial reaction. When hydrocarbon based dialkynes and dithiols were copolymerized, the resulting thiol-alkyne networks containing only hydrocarbon and sulfide linking groups exhibited refractive index values tunable above 1.65, with the refractive index directly related to the sulfur content. The thiol-yne reaction was also found to be useful in functionalizing thiol-terminated polymer chain ends via sequential Michael thiol-ene addition followed by the thiol-yne reaction: the result is the dual functionalization of the polymer chain end. A thermally responsive polymer hydrogel network was formed when an yne terminated water-soluble homopolymer was polymerized with a tetrafunctional thiol.
Co-reporter:Timothy F. Scott, Christopher J. Kloxin, Darren L. Forman, Robert R. McLeod and Christopher N. Bowman
Journal of Materials Chemistry A 2011 - vol. 21(Issue 37) pp:NaN14155-14155
Publication Date(Web):2011/07/08
DOI:10.1039/C1JM11915J
Optical direct write lithography (ODWL) has the capacity for generating three dimensional arbitrary patterns. Here we examine principles for voxel refinement and relate several techniques for achieving nanoscale resolution. The influence of optics, gelation, and polymerization scaling behavior are expounded, demonstrating the necessity for adopting a multidisciplinary mindset to control both voxel dimensions and minimize out-of-focus reactions. Aspects of two-photon ODWL are reviewed and recent multi-beam ODWL approaches that draw inspiration from STED microscopy are examined.
Co-reporter:Charles E. Hoyle, Andrew B. Lowe and Christopher N. Bowman
Chemical Society Reviews 2010 - vol. 39(Issue 4) pp:NaN1387-1387
Publication Date(Web):2010/02/09
DOI:10.1039/B901979K
The merits of thiol-click chemistry and its potential for making new forays into chemical synthesis and materials applications are described. Since thiols react to high yields under benign conditions with a vast range of chemical species, their utility extends to a large number of applications in the chemical, biological, physical, materials and engineering fields. This critical review provides insight into emerging venues for application as well as new mechanistic understanding of this exceptional chemistry in its many forms (81 references).
Co-reporter:Sheng Ye ; Neil B. Cramer ; Ian R. Smith ; Katerina R. Voigt
Macromolecules () pp:
Publication Date(Web):November 8, 2011
DOI:10.1021/ma2018809
Thiol–yne–methacrylate and thiol–yne–acrylate ternary systems were investigated for polymerization kinetics and material properties and compared to the analogous pure thiol–yne and (meth)acrylate systems. Both thiol–yne–methacrylate and thiol–yne–acrylate systems were demonstrated to reduce polymerization-induced shrinkage stress while simultaneously achieving high glass transition temperatures (Tg) and moduli. Formulations with 70 wt % methacrylate increased the Tg from 51 ± 2 to 75 ± 1 °C and the modulus from 1800 ± 100 to 3200 ± 400 MPa (44% increase) over the pure thiol–yne system. Additionally, the shrinkage stress was 1.2 ± 0.2 MPa, which is lower than that of the pure methacrylate, binary thiol–yne, and thiol–ene–methacrylate control systems which are all >2 MPa. Interestingly, with increasing methacrylate or acrylate concentration, a decrease and subsequent increase in the shrinkage stress values were observed. A minimum shrinkage stress value (1.0 ± 0.2 MPa) was observed in the 50 wt % methacrylate and 70 wt % acrylate systems. This tunable behavior results from the competitive reaction kinetics of the methacrylate or acrylate homopolymerization versus chain transfer to thiol and the accompanying thiol–yne step-growth polymerization. The cross-linking density of the networks and the amount of volumetric shrinkage that occurs prior to gelation relative to the total volumetric shrinkage were determined as two key factors that control the final shrinkage stress of the ternary systems.



![1-Propyne, 3,3'-[[2,2-bis[(2-propyn-1-yloxy)methyl]-1,3-propanediyl]bis(oxy)]bis-](/data/chemimg/2234900/127751-08-0.png)
![1-Propyne, 3,3'-[[2,2-bis[(2-propyn-1-yloxy)methyl]-1,3-propanediyl]bis(oxy)]bis-](/data/chemimg/2234900/127751-08-0_b.png)

![Benzoic acid, 4-[[6-[(1-oxo-2-propenyl)oxy]hexyl]oxy]-, 4-cyanophenylester](http://img.cochemist.com/ccimg/83900/83847-14-7.png)
![Benzoic acid, 4-[[6-[(1-oxo-2-propenyl)oxy]hexyl]oxy]-, 4-cyanophenylester](http://img.cochemist.com/ccimg/83900/83847-14-7_b.png)



![Ethanol, 2,2'-[sulfonylbis(2,1-ethanediylthio)]bis-](http://img.cochemist.com/ccimg/23200/23117-11-5.png)
![Ethanol, 2,2'-[sulfonylbis(2,1-ethanediylthio)]bis-](http://img.cochemist.com/ccimg/23200/23117-11-5_b.png)

![1-(allyloxy)-2,2-bis[(allyloxy)methyl]butane](http://img.cochemist.com/ccimg/700/682-08-6.png)
![1-(allyloxy)-2,2-bis[(allyloxy)methyl]butane](http://img.cochemist.com/ccimg/700/682-08-6_b.png)

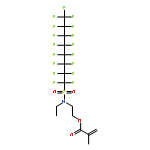
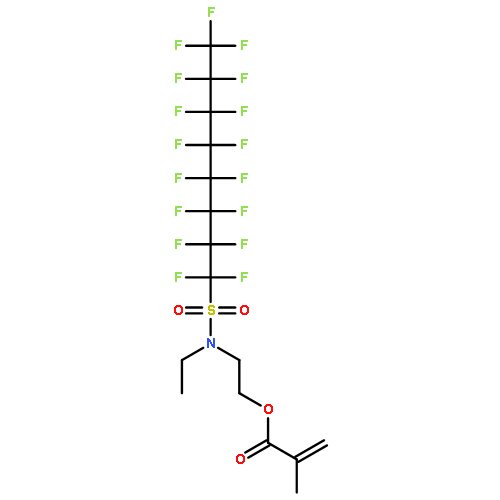


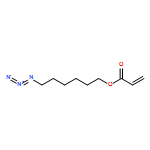
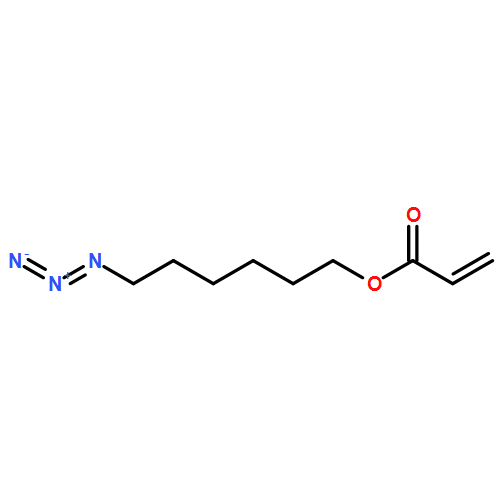
![2-Propenoic acid, [1-(phenylmethyl)-1H-1,2,3-triazol-4-yl]methyl ester](/data/chemimg/3722500/1262236-96-3.png)
![2-Propenoic acid, [1-(phenylmethyl)-1H-1,2,3-triazol-4-yl]methyl ester](/data/chemimg/3722500/1262236-96-3_b.png)
dimethyl-](http://img.cochemist.com/ccimg/886500/886450-14-2.png)
dimethyl-](http://img.cochemist.com/ccimg/886500/886450-14-2_b.png)
![Silane, [[4-(chloromethyl)phenyl]methoxy](1,1-dimethylethyl)dimethyl-](http://img.cochemist.com/ccimg/874900/874883-18-8.png)
![Silane, [[4-(chloromethyl)phenyl]methoxy](1,1-dimethylethyl)dimethyl-](http://img.cochemist.com/ccimg/874900/874883-18-8_b.png)
![Propanoic acid, 3-[(2-butoxy-2-oxoethyl)thio]-, ethyl ester](http://img.cochemist.com/ccimg/500000/499987-74-5.png)
![Propanoic acid, 3-[(2-butoxy-2-oxoethyl)thio]-, ethyl ester](http://img.cochemist.com/ccimg/500000/499987-74-5_b.png)






![[1,1'-BIPHENYL]-4-OL, 4'-MERCAPTO-](http://img.cochemist.com/ccimg/197000/196940-30-4.png)
![[1,1'-BIPHENYL]-4-OL, 4'-MERCAPTO-](http://img.cochemist.com/ccimg/197000/196940-30-4_b.png)
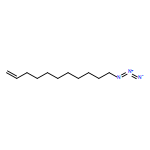
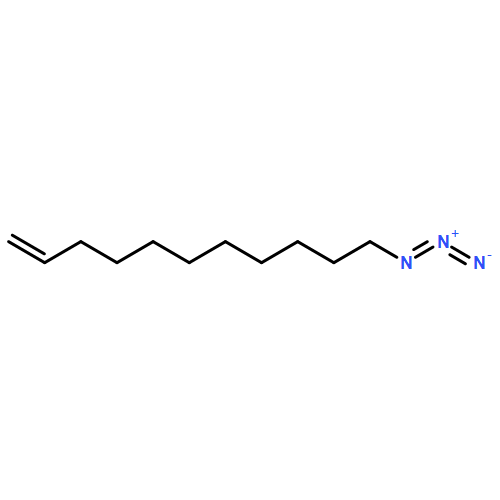
![Propanoic acid, 3-[(3-methoxy-3-oxopropyl)thio]-2-methyl-, methyl ester](http://img.cochemist.com/ccimg/184700/184647-78-7.png)
![Propanoic acid, 3-[(3-methoxy-3-oxopropyl)thio]-2-methyl-, methyl ester](http://img.cochemist.com/ccimg/184700/184647-78-7_b.png)


![Silane, [[4-(bromomethyl)phenyl]methoxy](1,1-dimethylethyl)dimethyl-](http://img.cochemist.com/ccimg/125400/125320-20-9.png)
![Silane, [[4-(bromomethyl)phenyl]methoxy](1,1-dimethylethyl)dimethyl-](http://img.cochemist.com/ccimg/125400/125320-20-9_b.png)

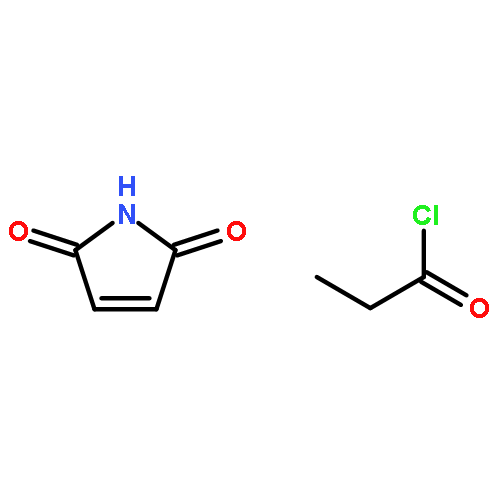
![Bicyclo[2.2.1]hept-5-ene-2-carboxylic acid, anhydride](http://img.cochemist.com/ccimg/110800/110773-97-2.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxylic acid, anhydride](http://img.cochemist.com/ccimg/110800/110773-97-2_b.png)



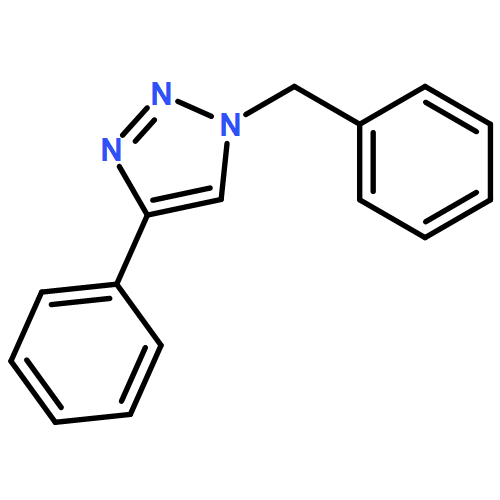
![Carbamodithioic acid,N,N-diethyl-, S,S''-[1,4-phenylenebis(methylene)] ester](http://img.cochemist.com/ccimg/90000/89964-93-2.png)
![Carbamodithioic acid,N,N-diethyl-, S,S''-[1,4-phenylenebis(methylene)] ester](http://img.cochemist.com/ccimg/90000/89964-93-2_b.png)




![Spiro[isobenzofuran-1(3H),9'-[9H]xanthen]-3-one,2',4',5',7'-tetrabromo-3',6'-dihydroxy-5-isothiocyanato-](http://img.cochemist.com/ccimg/60600/60520-47-0.png)
![Spiro[isobenzofuran-1(3H),9'-[9H]xanthen]-3-one,2',4',5',7'-tetrabromo-3',6'-dihydroxy-5-isothiocyanato-](http://img.cochemist.com/ccimg/60600/60520-47-0_b.png)
![1-Butanol, 2,2-bis[(3-mercaptopropoxy)methyl]-](http://img.cochemist.com/ccimg/57700/57654-66-7.png)
![1-Butanol, 2,2-bis[(3-mercaptopropoxy)methyl]-](http://img.cochemist.com/ccimg/57700/57654-66-7_b.png)

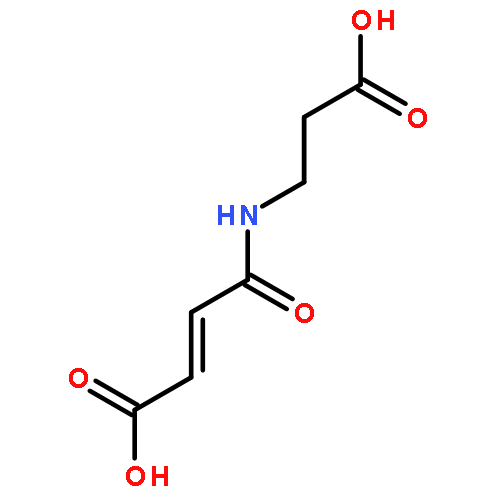
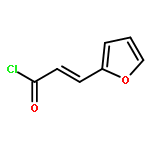
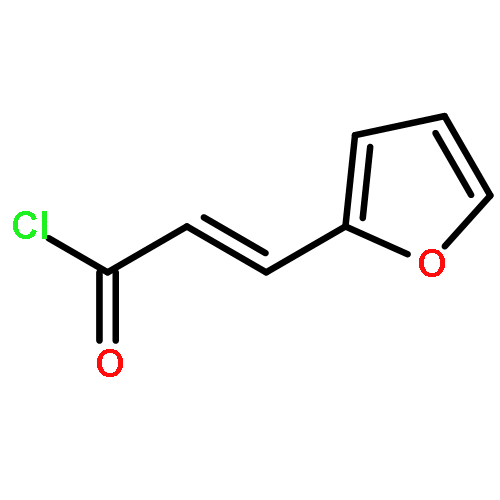




![2-Propenoic acid,2-methyl-, 1,1'-[1,2-ethanediylbis(oxy-2,1-ethanediyl)] ester, polymer with1,1'-[(1-methylethylidene)bis[4,1-phenyleneoxy(2-hydroxy-3,1-propanediyl)]]bis(2-methyl-2-propenoate)](http://img.cochemist.com/ccimg/26500/26426-05-1.png)
![2-Propenoic acid,2-methyl-, 1,1'-[1,2-ethanediylbis(oxy-2,1-ethanediyl)] ester, polymer with1,1'-[(1-methylethylidene)bis[4,1-phenyleneoxy(2-hydroxy-3,1-propanediyl)]]bis(2-methyl-2-propenoate)](http://img.cochemist.com/ccimg/26500/26426-05-1_b.png)
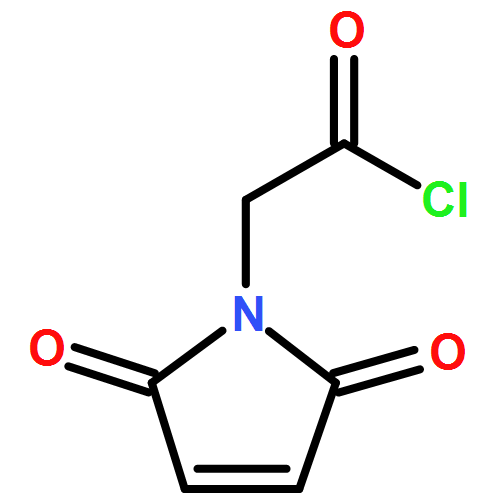
![Bicyclo[2.2.1]hept-2-ene, 5-(bromomethyl)-](http://img.cochemist.com/ccimg/17100/17016-12-5.png)
![Bicyclo[2.2.1]hept-2-ene, 5-(bromomethyl)-](http://img.cochemist.com/ccimg/17100/17016-12-5_b.png)


![Aluminum,hydroxy[29H,31H-phthalocyaninato(2-)-kN29,kN30,kN31,kN32]-, (SP-5-12)-](http://img.cochemist.com/ccimg/15600/15554-15-1.png)
![Aluminum,hydroxy[29H,31H-phthalocyaninato(2-)-kN29,kN30,kN31,kN32]-, (SP-5-12)-](http://img.cochemist.com/ccimg/15600/15554-15-1_b.png)


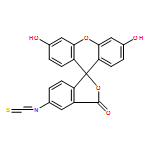




![Ethanamine,2-[(triphenylmethyl)thio]-](http://img.cochemist.com/ccimg/1100/1095-85-8.png)
![Ethanamine,2-[(triphenylmethyl)thio]-](http://img.cochemist.com/ccimg/1100/1095-85-8_b.png)



![2-Propanol, 1,1'-[(1-methylethylidene)bis(4,1-phenyleneoxy)]bis[3-azido-](/data/chemimg/3722800/1000416-64-7.png)
![2-Propanol, 1,1'-[(1-methylethylidene)bis(4,1-phenyleneoxy)]bis[3-azido-](/data/chemimg/3722800/1000416-64-7_b.png)


![Propanoic acid, 2,2'-[carbonothioylbis(thio)]bis[2-methyl-](/data/chemimg/104200/355120-40-0.png)
![Propanoic acid, 2,2'-[carbonothioylbis(thio)]bis[2-methyl-](/data/chemimg/104200/355120-40-0_b.png)