Co-reporter:Xiaodong Qin, Gang Xu, Feihong Chen, Lei Fang, Shaohua Gou
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 8(Issue 8) pp:
Publication Date(Web):15 April 2017
DOI:10.1016/j.bmc.2017.03.007
•A series of multi-targeted platinum(IV) complexes conjugated with wogonin, a natural product, via a linker group were designed and synthesized to study their anticancer effects.•Compound 10, the conjugate containing wogonin and cisplatin units, displayed the best anticancer activity, comparable to cisplatin, by inhibiting COX and inducing the accumulation of ROS in addition to damaging DNA.Platinum-based complexes like cisplatin and oxaliplatin are well known the mainstay of chemotherapy regimens on clinic. Wogonin, a natural product that possesses wide biological activities, is now in phase I clinical test as an anticancer agent in China. Herein reported are a series of novel Pt(IV) complexes that conjugated a wogonin derivative (compound 3) to the axial position via a linker group. After being tethered to the platinum(IV) complexes, the wogonin derivative provided multiple anticancer effects, especially in compound 10, a fusion containing wogonin and cisplatin units. Compound 10 not only inherited the genotoxicity from cisplatin, but also obtained the COX inhibitory property from the wogonin derivative. Further mechanistic investigation revealed that compound 10 caused the accumulation of ROS, decreased the mitochondrial membrane potential (ΔΨm) and then activated the p53 pathway. Overall, the research demonstrates that the “integrative” prodrug can be an effective strategy to promote the anticancer potency of Pt-based drugs for cancer treatment.Download high-res image (130KB)Download full-size image
Co-reporter:Zhikun Liu, Lei Fang, Huan Zhang, Shaohua Gou, Li Chen
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 8(Issue 8) pp:
Publication Date(Web):15 April 2017
DOI:10.1016/j.bmc.2017.02.049
Total sixteen tacrine-curcumin hybrid compounds were designed and synthesized for the purpose of searching for multifunctional anti-Alzheimer agents. In vitro studies showed that these hybrid compounds showed good cholinesterase inhibitory activity. Particularly, the potency of K3-2 is even beyond tacrine. Some of the compounds exhibited different selectivity on acetylcholinesterase or butyrylcholinesterase due to the structural difference. Thus, the structure and activity relationship is summarized and further discussed based on molecular modeling studies. The ORAC and MTT assays indicated that the hybrid compounds possessed pronounced antioxidant activity and could effectively protect PC12 cells from the H2O2/Aβ42-induced toxicity. Moreover, the hybrid compounds also showed positive metal ions-chelating ability in vitro, suggesting a potential to halt ion-induced Aβ aggregation. All the obtained results demonstrated that the tacrine-curcumin hybrid compounds, in particular compound K3-2, can be considered as potential therapeutic agents for Alzheimer’s disease.Download high-res image (124KB)Download full-size image
Co-reporter:Zhimei Wang, Haiyan Yu, Shaohua Gou, Feihong Chen, and Lei Fang
Inorganic Chemistry 2016 Volume 55(Issue 9) pp:4519-4528
Publication Date(Web):April 13, 2016
DOI:10.1021/acs.inorgchem.6b00361
A series of platinum(II) complexes, with N-monosubstituted 1R,2R-diaminocyclohexane bearing methoxy-substituted benzyl groups as carrier ligands, were designed and synthesized. The newly prepared compounds, with chloride anions as leaving groups, were found to be very active against the tested cancer cell lines, including a cisplatin-resistant cell line. Despite their efficacy against tumor cells, they also showed low toxicity to a human normal liver cell line. Among them, complex 1 had superior cytotoxic activity against A549, HCT-116, MCF-7, SGC7901, and SGC7901/CDDP cancer cell lines. The DNA binding assay is of further special interest, as an unusual monofunctional binding mode was found, due to the introduction of a rigid substituted aromatic ring in the 1R,2R-diaminocyclohexane framework as steric hindrance. The linkage of complex 1 with DNA was stable and insensitive to nucleophilic attack. Moreover, studies including cellular uptake, gel electrophoresis, apoptosis and cell cycle, and Western blot analysis have provided insight into the high potency of this compound.
Co-reporter:Fengfan Liu; Shaohua Gou; Feihong Chen; Lei Fang;Jian Zhao
Journal of Medicinal Chemistry 2015 Volume 58(Issue 16) pp:6368-6377
Publication Date(Web):August 6, 2015
DOI:10.1021/jm501952r
A series of platinum(II) complexes, characteristic of chiral trans-bicyclo[2.2.2]octane-7,8-diamine as ligand possessing dicyclic steric hindrance, were designed and synthesized. Biological evaluation showed that almost all complexes had cytotoxic activity against the tested cancer cell lines, among which most of chiral (R,R)-enantiomeres had stronger cytotoxicity than their (S,S)-counterparts, and 2a, [trans-bicyclo[2.2.2]octane-7R,8R-diamine](oxalato-O,O′)platinum(II), is the most effective agent. Significantly, its counterpart, 2b, was much more sensitive to cisplatin resistant SGC7901/CDDP cancer cell line at a higher degree than 2a. Docking study and agarose gel electrophoresis revealed that the interaction of 2a with DNA was similar to that of oxaliplatin. Western blot analysis demonstrated that 2a could induce a better effect than cisplatin on a mitochondrial-dependent apoptosis pathway. Kinetic study indicated that the dicyclic ligand can accelerate the reaction rate of the complex.
Co-reporter:Feng Cao, Yahan Gao, Meng Wang, Lei Fang, and Qineng Ping
Molecular Pharmaceutics 2013 Volume 10(Issue 4) pp:1378-1387
Publication Date(Web):January 22, 2013
DOI:10.1021/mp300647m
In our previous studies, ethylene glycol-linked amino acid diester prodrugs of oleanolic acid (OA), a Biopharmaceutics Classification System (BCS) class IV drug, designed to target peptide transporter 1 (PepT1) have been synthesized and evaluated. Unlike ethylene glycol, propylene glycol is of very low toxicity in vivo. In this study, propylene glycol was used as a linker to further compare the effect of the type of linker on the stability, permeability, affinity, and bioavailability of the prodrugs of OA. Seven diester prodrugs with amino acid/dipeptide promoieties containing l-Val ester (7a), l-Phe ester (7b), l-Ile ester (7c), d-Val-l-Val ester (9a), l-Val-l-Val ester (9b), l-Ala-l-Val ester (9c), and l-Ala-l-Ile ester (9d) were designed and successfully synthesized. In situ rat single-pass intestinal perfusion (SPIP) model was performed to screen the effective permeability (Peff) of the prodrugs. Peff of 7a, 7b, 7c, 9a, 9b, 9c, and 9d (6.7-fold, 2.4-fold, 1.24-fold, 1.22-fold, 4.15-fold, 2.2-fold, and 1.4-fold, respectively) in 2-(N-morpholino)ethanesulfonic acid buffer (MES) with pH 6.0 showed significant increase compared to that of OA (p < 0.01). In hydroxyethyl piperazine ethanesulfonic acid buffer (HEPES) of pH 7.4, except for 7c, 9a, and 9d, Peff of the other prodrugs containing 7a (5.2-fold), 7b (2.0-fold), 9b (3.1-fold), and 9c (1.7-fold) exhibited significantly higher values than that of OA (p < 0.01). In inhibition studies with glycyl-sarcosine (Gly-Sar, a typical substrate of PepT1), Peff of 7a (5.2-fold), 7b (2.0-fold), 9b (3.1-fold), and 9c (2.3-fold) had significantly reduced values (p < 0.01). Compared to the apparent permeability coefficient (Papp) of OA with Caco-2 cell monolayer, significant enhancement of the Papp of 7a (5.27-fold), 9b (3.31-fold), 9a (2.26-fold), 7b (2.10-fold), 7c (2.03-fold), 9c (1.87-fold), and 9d (1.39-fold) was also observed (p < 0.01). Inhibition studies with Gly-Sar (1 mM) showed that Papp of 7a, 9b, and 9c significantly reduced by 1.3-fold, 1.6-fold, and 1.4-fold (p < 0.01), respectively. These results may be attributed to PepT1-mediated transport and their differential affinity toward PepT1. According to the permeability and affinity, 7a and 9b were selected in the pharmacokinetic studies in rats. Compared with group OA, Cmax for group 7a and 9b was enhanced to 3.04-fold (p < 0.01) and 2.62-fold (p < 0.01), respectively. AUC0→24 was improved to 3.55-fold (p < 0.01) and 3.39-fold (p < 0.01), respectively. Compared to the ethylene glycol-linked amino acid diester prodrugs of OA in our previous work, results from this study revealed that part of the propylene glycol-linked amino acid/dipeptide diester prodrugs showed better stability, permeability, affinity, and bioavailability. In conclusion, propylene glycol-linked amino acid/dipeptide diester prodrugs of OA may be suitable for PepT1-targeted prodrugs of OA to improve the oral bioavailability of OA.Keywords: bioavailability; Caco-2; ester prodrug; in situ single-pass intestinal perfusion; oleanolic acid; peptide transporter 1; pharmacokinetics; propylene glycol linker;
Co-reporter:Xubin Fang, Lei Fang, Shaohua Gou, Lin Cheng
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 5) pp:1297-1301
Publication Date(Web):1 March 2013
DOI:10.1016/j.bmcl.2012.12.098
A series of dimethylaminomethyl-substituted curcumin derivatives/analogues were designed and synthesized. All compounds effectively inhibited HepG2, SGC-7901, A549 and HCT-116 tumor cell lines proliferation in MTT assay. Particularly, compounds 2a and 3d showed much better activity than curcumin against all of the four tumor cell lines. Antioxidant test revealed that these compounds had higher free radical scavenging activity than curcumin towards both DPPH and galvinoxyl radicals. Furthermore, the aqueous solubility and stability of the target compounds were also significantly improved compared with curcumin.













![2-Propenoic acid, 3-[4-[(ethoxycarbonyl)oxy]-3-methoxyphenyl]-, (E)-](http://img.cochemist.com/ccimg/62700/62621-20-9.png)
![2-Propenoic acid, 3-[4-[(ethoxycarbonyl)oxy]-3-methoxyphenyl]-, (E)-](http://img.cochemist.com/ccimg/62700/62621-20-9_b.png)











![2H-Indol-2-one, octahydro-1-[(4-methylphenyl)sulfonyl]-, (3aR,7aS)-rel-](http://img.cochemist.com/ccimg/853300/853269-32-6.png)
![2H-Indol-2-one, octahydro-1-[(4-methylphenyl)sulfonyl]-, (3aR,7aS)-rel-](http://img.cochemist.com/ccimg/853300/853269-32-6_b.png)
![Cyclohexanone, 2,6-bis[(4-hydroxyphenyl)methylene]-, (2E,6E)-](http://img.cochemist.com/ccimg/247100/247051-33-8.png)
![Cyclohexanone, 2,6-bis[(4-hydroxyphenyl)methylene]-, (2E,6E)-](http://img.cochemist.com/ccimg/247100/247051-33-8_b.png)
![Cyclohexanone, 2,6-bis[(4-hydroxy-3-methoxyphenyl)methylene]-,(2E,6E)-](http://img.cochemist.com/ccimg/214600/214558-93-7.png)
![Cyclohexanone, 2,6-bis[(4-hydroxy-3-methoxyphenyl)methylene]-,(2E,6E)-](http://img.cochemist.com/ccimg/214600/214558-93-7_b.png)












![PLATINUM, DICHLORO(1,2-CYCLOHEXANEDIAMINE-N,N')-, [SP-4-2-(1S-TRANS)]-](http://img.cochemist.com/ccimg/61900/61848-62-2.png)
![PLATINUM, DICHLORO(1,2-CYCLOHEXANEDIAMINE-N,N')-, [SP-4-2-(1S-TRANS)]-](http://img.cochemist.com/ccimg/61900/61848-62-2_b.png)
![Benzene, [[2-bromo-1-(chloromethyl)ethoxy]methyl]-](http://img.cochemist.com/ccimg/54400/54307-67-4.png)
![Benzene, [[2-bromo-1-(chloromethyl)ethoxy]methyl]-](http://img.cochemist.com/ccimg/54400/54307-67-4_b.png)





![[1,1'-Biphenyl]-2,2',6,6'-tetracarboxylicacid](http://img.cochemist.com/ccimg/4400/4371-27-1.png)
![[1,1'-Biphenyl]-2,2',6,6'-tetracarboxylicacid](http://img.cochemist.com/ccimg/4400/4371-27-1_b.png)






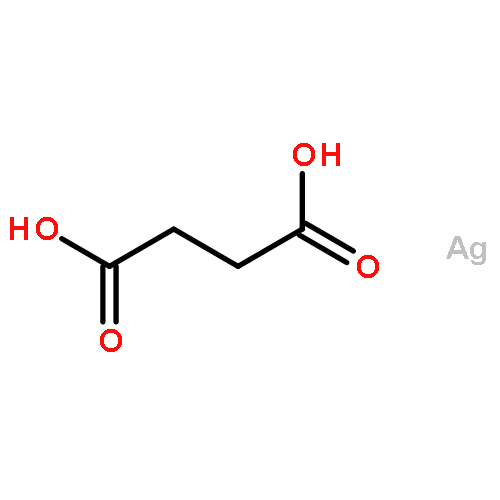
![Silver, [m-[ethanedioato(2-)-kO1,kO2':kO1',kO2]]di-](http://img.cochemist.com/ccimg/600/533-51-7.png)
![Silver, [m-[ethanedioato(2-)-kO1,kO2':kO1',kO2]]di-](http://img.cochemist.com/ccimg/600/533-51-7_b.png)