Co-reporter:Yaping Wen, Huiqing Yang, Danning Zheng, Kenan Sun, Li Wang, and Jinglai Zhang
The Journal of Physical Chemistry C July 6, 2017 Volume 121(Issue 26) pp:14019-14019
Publication Date(Web):June 19, 2017
DOI:10.1021/acs.jpcc.7b03409
The aggregation effect of five A–D(π)–A organic dyes, 1–5, with different anchoring groups is investigated by a combination of first-principles and molecular dynamics (MD). The dye with CSSH anchoring group (2) exhibits improved optoelectronic properties with the wider absorption range and larger absorption strength as compared with the other four dyes. More importantly, the stacking dimer of 2 has smaller or equal electronic coupling as compared with 1, indicating more efficient electron injection from dye to TiO2. Besides the isolated dyes, the monomer and two H-aggregation configurations of 1 and 2 are adsorbed onto the TiO2 anatase (101) surface to evaluate the injection time and variation of the band gap. The aggregation would be suppressed by choice of the suitable anchoring groups. Even if the aggregation is formed, it is not totally detrimental for the photovoltaic efficiency. The influence of aggregation is complicated and should be evaluated from many aspects.
Co-reporter:Xiaolin Wang, Yuan Ma, Songyan Feng, Li Wang, Junfeng Li, Jinglai Zhang
Organic Electronics 2017 Volume 41() pp:251-258
Publication Date(Web):February 2017
DOI:10.1016/j.orgel.2016.11.012
•Develop novel Ir(III) complexes bearing different ancillary ligands.•Follow or violate Kasha emissive behavior.•Emissive spectra are estimated by both vertical and 0-0 transition energy.•The kr is calculated by QR-TDDFT method.•The quantum yield is evaluated by semi-quantitative method.The complexity of emissive process for five heteroleptic Ir(III) complexes (dfpypy)2Ir(LˆX), where dfpypy = 4-methyl-2',6'-difluoro-2,3'-bipyridine and LˆX = picolinate (1), dipivaloylmethanate (2), picolinic acid N-oxide (3), N,N'-di-tert-butylbenzamidinate (4), or 5-(4′-methylpyridine-2'-yl)-3-trifluoromethyl-1,2,4-triazole (5) (See Fig. 1), is unveiled by density functional theory (DFT) and quadratic response (QR) time-dependent (TD)DFT calculations including spin-orbit coupling (SOC). Besides the emission wavelength, we would like to pay intense attention on the emissive rule. It is found that the emission likely originates from different triplet states rather than only from the lowest Kasha state for complexes 1, 2, 3, and 5, which indicates they obey dual emission scenarios. In contrast, complex 4 follows the Kasha rule. Different from the total qualitative study, the quantum yield is semi-quantitatively determined in this work. The radiative decay rate constants (kr) from possible emissive states are quantitatively determined by the quadratic response method. The triplet potential energy surfaces are constructed to elucidate the factors that affect the temperature-dependent nonradiative rate constants (knr). Complex 4 have the higher quantum yields in all the investigated complexes because of the larger kr and smaller knr. The metal-centered (3MC) triplet state in the deactivation pathways is confirmed to play a vital role in determining the quantum yield.
Co-reporter:Xin Wang, Danning Zheng, Songyan Feng, Li Wang, Junfeng Li, Jinglai Zhang
Journal of Luminescence 2017 Volume 183() pp:178-184
Publication Date(Web):March 2017
DOI:10.1016/j.jlumin.2016.11.033
The phosphorescent process of two heteroleptic ((DMDPI)2Ir(tftap) and (tftap)2Ir(DMDPI)) and one homoleptic (Ir(DMDPI)3) Ir(III) complexes (See Fig. 1) is theoretically investigated by density functional theory (DFT) and quadratic response (QR) time-dependent density functional theory (TDDFT) calculations including spin-orbit coupling (SOC). Two or three triplet excited states are confirmed for three complexes, respectively. On the basis of the respective optimized triplet geometry, the emissive wavelength is determined by the ΔSCF-DFT method. Furthermore, the radiative rate constant (kr) is also calculated corresponding to each triplet state. Combination of kr and emissive energy, the emission rule is determined. It is found that complex (DMDPI)2Ir(tftap) follows the dual emission scenarios, while complexes (tftap)2Ir(DMDPI) and Ir(DMDPI)3 obey the Kasha rule. The nonradiative rate constant (knr) is qualitatively evaluated by the construction of triplet potential surface via metal centered (3MC d-d) state. Finally, the sequence of quantum yield is compared by both kr and knr. The quantum yield of homoleptic Ir(III) complex Ir(DMDPI)3 is higher than that of heteroleptic Ir(III) complexes (DMDPI)2Ir(tftap) and (tftap)2Ir(DMDPI). However, the emissive wavelength of Ir(DMDPI)3 is in the red color region rather than blue color.
Co-reporter:Yue Zhang, Yuanyuan Li, Ci Chen, Li Wang, Jinglai Zhang
Organic Electronics 2017 Volume 49(Volume 49) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.orgel.2017.06.064
•Two organic HTMs are computer-assisted designed.•The performance of HTMs is obviously improved by introduction of phenyl in core.•The solubility and stability of HTMs are estimated.•The possible synthesized route is proposed.The electronic, optical, and hole transporting mobility of three organic hole transporting materials (HTMs), X59, X59-P, and X59-T, are investigated by combination of first principle and molecular dynamics associated with Marcus theory and Einstein equation. As compared with the experimental reported X59, the new designed X59-P has more stable HOMO energy level. Moreover, the latter has smaller reorganization energy and larger hole transfer integral resulting in the larger hole transporting mobility. Besides the hole transporting mobility, the solubility and stability of two designed molecules are also evaluated by comparison with X59, which are two important items to determine the cost and performance in real application of solar cell. More importantly, they would be synthesized in a benign condition without expensive materials. Our studies introduce a possible pathway to explore the efficient HTMs by suitable combination mode rather than development of new groups.Download high-res image (159KB)Download full-size image
Co-reporter:Jieqiong Li, Li Wang, Xin Wang, Jinglai Zhang, Xiaofeng Cui, Youwei Li, Bingkun Han
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2017 Volume 171() pp:425-431
Publication Date(Web):15 January 2017
DOI:10.1016/j.saa.2016.08.021
•The absorption and phosphorescence spectra of Ir(III) complexes are investigated.•Their Φem, IP, EA, and reorganization energy are compared.•The effect of different substituents on phosphorescent properties is explored.•Three possible blue-emitting Ir(III) complexes are designed by DFT.The phosphorescent properties of a series of potential blue-emitting Ir(III) complexes (C^N)2Ir(N^N′) are studied by means of the density functional theory/time-dependent density functional theory (DFT/TDDFT). Their possibilities to be blue-emitting phosphors are theoretically evaluated by the electroluminescence (EL) performance and phosphorescence quantum yield. The effect of two different substituents attached on the difluorophenyl ring is explored by comparison of the complexes in groups I (1a–4a) and II (1b–4b). Furthermore, to explore the influence of the stronger electron-donating/withdrawing group substituted on the primary ligand, the properties of complexes 1c and 1d are estimated. All the substituents are added on the para-position of the corresponding ring. The comparable radiative rate constant (kr) and nonradiative rate constant (knr) result in the similar quantum yield for complexes in two groups. Besides, the balance of the reorganization energies for complexes 2b–4b is better than others.The geometric and electronic structures, absorption and emission spectra, quantum yields, and the organic light-emitting diode (OLED) performance of ten iridium(III) complexes containing substituted 2-phenylpyridine primary ligands and pyridyltriazole ancillary ligands are investigated using the density functional theory/time-dependent density functional theory.
Co-reporter:Kenan Sun, Yuan Ma, Weiyi Zhang, Yaping Wen, Li Wang, Jinglai Zhang
Dyes and Pigments 2017 Volume 139(Volume 139) pp:
Publication Date(Web):1 April 2017
DOI:10.1016/j.dyepig.2016.11.051
•Six new dyes with “asymmetric” butterfly structure are computationally designed.•Interfacial properties between the dye and TiO2 clusters are explored.•The influence of nature and extent of π-linker is similar for all studied dyes.•Two phenyl groups are more favorable to be used as π group in the right “wing”.A series of carbazole-based organic dyes with asymmetric butterfly structure are designed and their efficiency is theoretically estimated. In the left “wing”, benzene and thiophene are employed as the π group. For respective left “wing”, three different right “wings” are constructed. The influence of nature and number of different π group on the efficiency is investigated. On the basis of the isolated dyes, the short-circuit current density (Jsc), open-circuit voltage (Voc), and reorganization energy (λtotal) are determined to evaluate the efficiency of the solar cell. Moreover, the structure of the dye adsorbed on the (TiO2)38 cluster is optimized by the first-principle. The adsorption energy and the density of states (DOS) are calculated to further estimate the efficiency. Combining all factors, the employment of two phenyl groups as π group of the right “wing” is favorable to improve the efficiency.Download high-res image (172KB)Download full-size image
Co-reporter:Beibei An, Songyan Feng, Keke Wen, Wenpeng Wu, Huijuan Yuan, Qiuling Zhu, Xugeng Guo, Jinglai Zhang
Organic Electronics 2017 Volume 45(Volume 45) pp:
Publication Date(Web):1 June 2017
DOI:10.1016/j.orgel.2017.02.034
•The single fluorescence emissions were observed in a series of amino-type hydrogen-bonding molecules.•All the targeted molecules could undergo an ultrafast ESIPT process.•A new ESIPT dye molecule was designed theoretically.Excited-state intramolecular proton transfer (ESIPT) has many important potential applications. In this paper, the geometries of a series of amide-based NH⋯N hydrogen-bonding compounds in their ground S0 states and first excited singlet S1 states were optimized with density functional theory (DFT) and time-dependent density functional theory (TD-DFT) approaches. Both topological analysis and noncovalent interactions analysis show strong intramolecular hydrogen-bonds in the studied five systems and the electron density function ρ(r) exhibits good linear relationship with the distance of H⋯N2. The potential energy curves of the S0 and S1 states were scanned by using of DFT and TD-DFT to elucidate the ESIPT process. It reveals that all the systems considered here can undergo an ultrafast ESIPT reaction, with energy barrier of less than 0.05 eV, giving rise to the single fluorescence emission from the proton-transfer tautomer. It is also found that the shorter the hydrogen bond in the normal-form S1 state, the easier the ESIPT takes place.Download high-res image (140KB)Download full-size image
Co-reporter:Yuan Ma, Ci Chen, Tengfei Wang, Jingshun Zhang, JiaJia Wu, Xiangdong Liu, Tiegang Ren, Li Wang, Jinglai Zhang
Applied Catalysis A: General 2017 Volume 547(Volume 547) pp:
Publication Date(Web):25 October 2017
DOI:10.1016/j.apcata.2017.09.009
•Dialkylpyrazolium ILs are firstly used as catalysts for the fixation of CO2.•Good yields are achieved under a mild condition without solvent or co-catalyst.•The mechanism with a synergistic effect is found to promote the reaction.•They would be reused by five times without decrease of catalytic activity.The efficient fixation of CO2 without co-catalyst and solvent under metal-free condition is still an urgent topic in sustainable chemistry. In this work, a series of dialkylpyrazolium ionic liquids are employed to promote the cycloaddition of CO2 and PO to produce PC. They would be easily synthesized by a simple one-pot reaction. The effect of alkyl chain length in cation and different anion is explored. Diethylpyrazolium iodide presents the excellent catalytic activity with the product yield of 96% and selectivity of 99% in a benign condition. Moreover, the catalyst could be reused for at least five times without significant loss of catalytic activity. An intensive structure-activity research testifies that the cycloaddition of CO2 with PO is activated by a synergistic effect from both cation and anion of ILs. To confirm it, the detailed mechanism is investigated by density functional theory associated with the non-covalent interactions and atoms in molecule analysis. Besides the electrostatic interaction between cation of ionic liquid and PO, the noncovalent interaction, especially for hydrogen bond, plays a vital role in promoting the reaction.Download high-res image (136KB)Download full-size image
Co-reporter:Tengfei Wang, Danning Zheng, Yuan Ma, Jiayi Guo, Zhipeng He, Bin Ma, Lianhuan Liu, Tiegang Ren, Li Wang, Jinglai Zhang
Journal of CO2 Utilization 2017 Volume 22(Volume 22) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.jcou.2017.09.009
•Eight benzyl substituted imidazolium ionic liquids are firstly synthesized.•They are efficient homogeneous catalysts for cycloaddition of CO2 with PO.•The reaction condition is benign with high product yield.•No solvent and co-catalyst are required.•The reaction mechanism is elucidated by DFT calculation.A series of low-cost and easily prepared benzyl substituted imidazolium ionic liquids are firstly synthesized and employed as catalyst for the cycloaddition of carbon dioxide with epoxides without any solvent and co-catalyst. The synthesized imidazolium ionic liquids are characterized by 1H NMR, HRMS, and MS. The influence of different substituted groups in cation and reaction parameters on catalytic activity is investigated. The highest conversion yield of cyclic carbonate (94.89%) could be achieved with slight amount of catalyst (0.25 mol%) under 130 °C and 2.0 MPa during 4 h. Meanwhile, the mechanisms of cycloaddition of carbon dioxide with epoxides catalyzed by four benzyl substituted ionic liquids with different substituted groups are investigated by theoretical calculations. The role of hydrogen bond and other noncovalent interactions played in catalytic process is further uncovered to deeply understand the difference of various catalysts from atomistic level.
Co-reporter:Huiqing Yang, Jiayi Guo, Yaping Wen, Tiegang Ren, Li Wang, Jinglai Zhang
Molecular Catalysis 2017 Volume 441(Volume 441) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.mcat.2017.08.009
•Solvent effect of ionic liquids on the fixation of CO2 is studied by ONIOM and MD.•Critical hydrogen bond and C-Br interaction are analyzed by NCI and AIM.•ONIOM model displays the more reasonable result than PCM model.•Which one is more important, nucleophilic or electrophilic activation?The solvent effect of ionic liquids on CO2 fixation catalyzed by hydroxyl-functionalized quaternary ammonium-based ionic liquids is thoroughly investigated by ONIOM method associated with the molecular dynamics simulation. As compared with the catalytic activity determined in experiment, the catalytic activity estimated by ONIOM model is more credible than previous result calculated by the polarized continuum model. The critical hydrogen bond and C-Br interaction are analyzed by non-covalent interaction and the atoms in molecules methods. The analysis indicates that the electrophilic activation aroused by hydrogen bond provides more contribution than nucleophilic activation aroused by C-Br interaction in reaction catalyzed by trihydroxyethylethyl ammonium bromide. In contrast, the nucleophilic activation is more important in reactions catalyzed by other three ionic liquids. Choice of the suitable model is possible to obtain the reliable catalytic activity, which would be comparable with the experimental result.Download full-size image
Co-reporter:Beibei An, Huijuan Yuan, Qiuling Zhu, Yuanyuan Li, Xugeng Guo, Jinglai Zhang
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2017 Volume 175() pp:36-42
Publication Date(Web):15 March 2017
DOI:10.1016/j.saa.2016.12.020
•A stronger intramolecular hydrogen-bond leads to a lower energy barrier in the ESIPT reaction.•The fluorescence behaviors of three amino-type hydrogen-bonding molecules are significantly different.•There may exist a long-lived trans-tautomer species in their ground states.Excited-state intramolecular proton transfer (ESIPT) dynamics of the amino-type hydrogen-bonding compound 2-(2′-aminophenyl)benzothiazole (PBT-NH2) as well as its two derivatives 2-(5′-cyano-2′-aminophenyl)benzothiazole (CN-PBT-NH2) and 2-(5′-cyano-2′-tosylaminophenyl)benzothiazole (CN-PBT-NHTs) were studied by the time-dependent density functional theory (TD-DFT) approach with the B3LYP density functional, and their absorption and emission spectra were also explored at the same level of theory. A good agreement is observed between the theoretical simulations and experimental spectra, indicating that the present calculations are reasonably reliable. In addition, it is also found that the energy barriers of the first excited singlet state of the three targeted molecules along the ESIPT reaction are computed to be 0.38, 0.34 and 0.12 eV, respectively, showing the trend of gradual decrease, which implies that the introduction of the electron-withdrawing cyano or tosyl group can facilitate the occurrence of the ESIPT reaction of these amino-type H-bonding systems. Following the ESIPT, both CN-PBT-NH2 and CN-PBT-NHTs dye molecules can undergo the cis-trans isomerization reactions in the ground-state and excited-state potential energy curves along the C2-C3 bond between benzothiazole and phenyl moieties, where the energy barriers of the trans-tautomer → cis-tautomer isomerizations in the ground states are calculated to be 0.83 and 0.34 eV, respectively. According to our calculations, it is plausible that there may exist the long-lived trans-tautomer species in the ground states of CN-PBT-NH2 and CN-PBT-NHTs.
Co-reporter:Yaping Wen, Wenpeng Wu, Yuanyuan Li, Weiyi Zhang, Zhaoyang Zeng, Li Wang, Jinglai Zhang
Journal of Power Sources 2016 Volume 326() pp:193-202
Publication Date(Web):15 September 2016
DOI:10.1016/j.jpowsour.2016.07.001
•Explore the influence of π-linker on overall conversion efficiency.•The interfacial properties of dyes adsorbed on TiO2 surface are investigated.•The change of the performance before and after adsorbed is compared.Four organic compounds with different π-linkers are theoretically explored as potential photosensitizers for application in dye-sensitized solar cells (DSSCs). Besides the isolated dyes, the interfacial properties of dyes adsorbed on TiO2 anatase (101) surface are theoretically investigated. The overall conversion efficiency (η) of DSSCs is evaluated by the following items on the basis of the isolated dyes, including structures, absorption spectrum, energy gap, open-circuit voltage (Voc), short-circuit current density (Jsc), and reorganization energies (λtotal). After adsorbed on the TiO2 surface, the electron would be efficiently injected from dye into the TiO2 surface because of the increased of the lowest unoccupied molecular orbital (LUMO) energy level of the dyes, the decreased of the conduction band of TiO2 surface, and the narrowed band gaps for both dye and TiO2. Moreover, the injection times are in a reasonable range indicating that they are ideal dyes. Combination of all items, the performance of THI-2T-C (See Scheme 1, the sketch structures of all the investigated isolated dyes) stands out from the rest investigated dyes from the theoretical viewpoint. Only enlargement of the π-linker extent is not a smart choice, since the nature of π-linker plays a more important role in affecting the performance of DSSCs.
Co-reporter:Jieqiong Li, Li Wang, Kenan Sun and Jinglai Zhang
Dalton Transactions 2016 vol. 45(Issue 7) pp:3034-3047
Publication Date(Web):23 Dec 2015
DOI:10.1039/C5DT04209G
The electronic structures and photophysical properties of three homoleptic iridium(III) complexes IrL3 with C^N ligands, including 2a (L = 1-(2,6-diisopropylphenyl)-2-phenyl-1H-imidazole), 5a (L = 1-(2,6-dimethylphenyl)-2-phenyl-1H-imidazole), and 6a (L = 1-(3,5-diisopropylbiphenyl-4-yl)-2-phenyl-1H-imidazole), are investigated by means of the density functional method. Furthermore, seven new complexes are theoretically designed, including 1a (L = 1,2-diphenyl-1H-imidazole), 3a (L = 1-(2,6-dimethoxyphenyl)-2-phenyl-1H-imidazol), 4a (L = 2-(2-phenyl-1H-imidazol-1-yl)isophthalaldehyde), 1b (L = 2-(biphenyl-3-yl)-1H-imidazole), 2b (L = 2-(2′,6′-diisopropylbiphenyl-3-yl)-1H-imidazole), 3b (L = 2-(2′,6′-dimethoxybiphenyl-3-yl)-1H-imidazole), and 4b (L = 3′-(1H-imidazol-2-yl)biphenyl-2,6-dicarbaldehyde), to explore the influence of different substituents and different substituted positions on the electronic structures, phosphorescence properties, and organic light-emitting diode (OLED) performance. The HOMO–LUMO energy gap is greatly decreased by introduction of the –CHO group into the phenyl ring (4a and 4b see Scheme 1—sketched structures for all the investigated Ir(III) complexes). As a result, their absorption and emission spectra present red-shifting leading them to be potential red-emitting phosphors. Other complexes are all blue-emitting materials, indicating that the effect of the substituted position on the emitting color is negligible. However, the addition of the substituent on the para-position of the phenyl ring in the phenylimidazole ligand would increase the quantum yield and electroluminescence (EL) performance compared with that on the imidazole ring.
Co-reporter:Qing Zhang, Xiaolin Wang, Xin Wang, Li Wang, Jinglai Zhang
Organic Electronics 2016 Volume 33() pp:281-289
Publication Date(Web):June 2016
DOI:10.1016/j.orgel.2016.03.026
•The structure-property relationships of Ir(III) complexes are investigated.•The influence of substituents with different space volume is studied.•The potential energy profiles for the thermal deactivation pathway are explored.•Three novel Ir(III) complexes are theoretically designed.To provide a deeply understanding of the nature of the emissive origin as well as the radiative and nonradiative processes, theoretical studies have been performed on four amidinate/bis(pyridylphenyl) iridium(III) complexes. It has been testified that they have exhibited bright yellowishgreen phosphorescence emission with moderate photoluminescence quantum yields. Besides geometries, electronic structure, absorption and phosphorescence spectra, and the factors governing the radiative decay rate constants of the emissive state have been examined. Additionally, this work also explores the potential energy profiles of the deactivation pathway via the triplet mental-centered states. Among these complexes, complex 2, which contains the bulky t-butyl group on the amidinate nitrogen atoms, presents the highest internal quantum yield. To explore more efficient phosphors, three novel phosphors, 2a, 2b, and 2c have been designed on the basis of complex 2 by incorporation of substituents on the bis(pyridylphenyl) ligand with a slightly higher quantum yield.The geometric and electronic structures, absorption and emission spectra, phosphorescent properties as well as the potential energy profiles for the thermal deactivation pathway and the organic light-emitting diode (OLED) performance of four experimental reported and three newly designed iridium(III) complexes are theoretically investigated by means of the density functional theory/time-dependent density functional theory.
Co-reporter:Yaping Wen, Wenpeng Wu, Yuanyuan Li, Yang Li, Tengfei Qin, Yuan Tang, Li Wang, Jinglai Zhang
Organic Electronics 2016 Volume 38() pp:61-68
Publication Date(Web):November 2016
DOI:10.1016/j.orgel.2016.07.039
•The influence of extent of π-linker on isolated and adsorbed dyes is different.•The thieno[2,3-b]indole is employed as the donor.•The interfacial properties between the dye and TiO2 photonodes are explored.•The dye with the moderate π-linker is considered to be more suitable choice.A series of novel D-π-A type organic dyes with different π-linker are theoretically designed and investigated for their potential applications in dye-sensitized solar cells (DSSCs). We mainly focused on the influence of the extension of π-linker on the overall efficiency. The discussions are classified into two aspects: one is the isolated dyes and the other is the dye-TiO2 system. The calculated results indicate that the isolated dyes THI-2T-C, THI-4T-3C, and THI-6T-5C have the better overall efficiency. Further, the THI-4T-3C and THI-5T-4C have the acceptable performance if the dye-TiO2 system is considered. Combination of the isolated and the adsorbed systems, the THI-4T-3C with the moderate π-linker is considered to be more suitable choice for thieno[2,3-b]indole-based dyes rather than the longest π-linker.
Co-reporter:Xiaolin Wang, Yuanyuan Li, Li Wang, Jinglai Zhang
Organic Electronics 2016 Volume 35() pp:208-215
Publication Date(Web):August 2016
DOI:10.1016/j.orgel.2016.05.024
•Influence of different cyclometalated ligands on the binding interaction.•Comparison of the ππ-stacking between different aromatic ligands.•The effect of different coordinated ligands on the quantum yield is explored.The photophysical properties of four Pt(II) complexes [Pt(Lx)2], x = 1–4, (1–4), where Lx are 6-t-butyl-1-(3-trifluoro-methyl-1H-pyrazol-5-yl) isoquinoline (1), 3,5-di-t-butyl-1-(3-trifluoromethyl-1H-pyrazol-5-yl) isoquinoline (2), 6-(2,6-diisopropylphenyl)-1-(3-trifluoro-methyl-1H-pyrazol-5-yl) isoquinoline (3), and 4-(2,6-diisopropylphenyl)-1-(3-trifluoro-methyl-1H-pyrazol-5-yl) isoquinoline (4), are investigated by the density functional theory (DFT) and time-dependent density functional theory (TD-DFT). Furthermore, the binding interaction in Ptn stack is studied to discover the influence of different cyclometalated ligand. The calculated results rationalize that the complex 1 exhibits a stack of three molecules rather than the infinite aligned stack. Complexes 1 and 3 present the stronger tendency to form the aligned ππ-stacking interaction as compared with complexes 2 and 4. The dimers of other four complexes, 3a (Pt(L3)(Ma), Ma = 5-(2-pyridyl)-3-trifluoromethylpyrazole), 3b (Pt(L3)(Mb), Mb = 5-(4-phenyl-2-pyridyl)-3-trifluoromethylpyrazole), 3c (Pt(L3)(Mc), Mc = 5-(4-tert-butyl-2-pyridyl)-3-trifluoromethylpyrazole), and 5 (Pt(fppz)2 fppz = 5-(2-pyridyl)-3-trifluoromethylpyrazole), are also studied to investigate the effect of different aromatic ligand or substituents on the ππ-stacking interaction. The emissions of complexes 1–4 originate from various charge transfer states including the intraligand charge transfer (ILCT) and ligand-to-ligand charge transfer (LLCT) together with the metal-to-ligand charge transfer (MLCT). Finally, the items related with the radiative and nonradiative rate constants are examined. Besides the potential energy profile between the lowest triplet state (3MLCT) and metal centered state (3MC), the deactivation process of the 3MC state via the minimum energy crossing point (MECP) between the 3MC and the ground state (1S0) potential surfaces is also explored.
Co-reporter:Qing Zhang, Li Wang, Xin Wang, Yonghong Li, Jinglai Zhang
Organic Electronics 2016 Volume 28() pp:100-110
Publication Date(Web):January 2016
DOI:10.1016/j.orgel.2015.10.021
•The structure-property relationship of Ir(III) complexes are investigated.•The influence of different ancillary ligands is studied.•The effect of different substituent groups is explored.•Five new Ir(III) complexes are theoretically designed.A series of Ir(III) complexes [(CˆN)2Ir(PˆSiO)], where (CˆN)H is 2-phenylisoquinoline (1), 2-phenylpyridine (2) or 2-(2,4-difluorophenyl)pyridine (3), and (PˆSiO)H is an organosilanolate ancillary chelate with either diphenylsilyl (a) or dimethylsilyl (b) substituent, are investigated by means of the density functional theory/time-dependent density functional theory (DFT/TD-DFT). Their relationship between structure and property is evaluated by the geometries, electronic structure, and absorption and phosphorescence spectra associated with the internal quantum yield. The effect of different substitutions on the ancillary ligand is explored by compare of the complexes 1a (2a/3a) and 1b (2b/3b). Furthermore, five complexes, 2b-1, 2b-2, 2b-3, 2b-4, and 2b-5, are newly designed by introduction of the substitution groups on the phenyl rings of the 2b (See Fig. 1). The theoretical result estimates that the complexes 2b-1, 2b-2, 2b-4, and 2b-5 would be the blue-emitting phosphors. Especially, the complex 2b-1 has a higher quantum yield relative to 2b by comparison of the factors governing the radiative decay rate constants of the emissive state and the feasibility of the deactivation process from the T1 state via triplet metal-centered (3MC) state.The geometric and electronic structures, absorption and emission spectra, phosphorescent properties of six experimental reported iridium(III) complexes based on [(CˆN)2Ir(PˆSiO)] template are investigated by means of the density functional theory/time-dependent density functional theory. Furthermore, five iridium(III) complexes are newly designed by incorporation of the substituents and their phosphorescent properties are theoretically predicted.
Co-reporter:Qing Zhang, Yuanyuan Li, Xin Wang, Li Wang, Jinglai Zhang
Materials Chemistry and Physics 2016 Volume 177() pp:179-189
Publication Date(Web):1 July 2016
DOI:10.1016/j.matchemphys.2016.04.016
•The structure-property relationship of Ir(III) complexes are investigated.•The effect of different substituents/positions on properties is explored.•Do the emissions follow the Kasha or non-Kasha scenario?•Newly possible blue-emitting Ir(III) complexes are theoretically designed.Four iridium(III) complexes, (dfpmpy)2Ir(pic), (1), (dfpmpy)2Ir(EO2-pic) (2), (dfpmpy)2Ir(pic-N-O) (3), and (dfpmpy)2Ir(EO2-pic-N-O) (4) (dfpmpy = 2-(2,4-difluorophenyl)-4-methylpyridine, pic = picolinic acid, EO2-pic = 4-(2-ethoxyethoxy) picolinic acid, pic-N-O = picolinic acid N-oxide, and EO2-pic-N-O = 4-(2-ethoxyethoxy) picolinic acid N-oxide) are investigated by means of the density functional theory/time-dependent density functional theory (DFT/TD-DFT) to explore the influence of the ancillary ligand on the electronic structures, phosphorescent properties, and organic light-emitting diode (OLED) performance. Employing pic-N-O and EO2-pic-N-O as the ancillary ligands would decrease the vertical energy and result in the red-shifted wavelength. Then, other four iridium(III) complexes (2a-2d) (See Scheme 1) are designed by introduction of the phenyl and CHO substituents on the pyridine ring and phenyl ring of complex 2, respectively. As compared with complex 2, theoretical results show that newly designed complexes 2a-2c might be potential candidates for blue-emitting phosphors with better/comparable quantum yield and Δλ. Moreover, the performance of complexes 2a and 2c, i.e., introducing phenyl on the para-position of pyridine ring and phenyl ring in dfpmpy ligand, are better than that of 2b.
Co-reporter:Qing Zhang, Xin Wang, Yue Zhang, Li Wang, Junfeng Li, and Jinglai Zhang
The Journal of Physical Chemistry C 2016 Volume 120(Issue 48) pp:27523-27532
Publication Date(Web):November 7, 2016
DOI:10.1021/acs.jpcc.6b07562
Rare high-efficiency deep blue organometallic phosphors are one of the major roadblocks to develop the white organic light-emitting diodes (OLEDs). In this article, the phosphorescent properties of four potential blue-emitting cyclometalated (C^N) Ir(III) complexes (two experimental reported and two theoretical novel designed) are investigated by the density functional theory/time-dependent density functional theory (DFT/TDDFT) method to explore the cooperative effect of the electron-withdrawing substituent on the primary ligand associated with different ancillary ligands. The origins of emission are identified by means of DFT and TDDFT calculations including spin–orbit coupling (SOC). The theoretical results indicate that emissions from the higher-lying triplet state also have a contribution. The radiative rate constant (kr) is quantitatively determined. To further elucidate the phosphorescent decay process, the SOC matrix elements, singlet–triplet splitting energies, and transition dipole moments are calculated to evaluate the radiative process. Both the temperature-independent and temperature-dependent nonradiative decay processes are considered. The calculated results testify that the incorporation of the electron-withdrawing group heptafluoropropyl (HFP) is not insurance to improve the quantum yield. The substituents on the primary ligand should be combined with the suitable ancillary ligand to enhance the quantum yield.
Co-reporter:Jieqiong Li, Qing Zhang, Hongqing He, Li Wang and Jinglai Zhang
Dalton Transactions 2015 vol. 44(Issue 18) pp:8577-8589
Publication Date(Web):18 Feb 2015
DOI:10.1039/C5DT00048C
The geometric and electronic structures, phosphorescence properties and the organic light-emitting diode (OLED) performance of a series of Ir(III) complexes based on bis[(4,6-di-fluorophenyl)-pyridinato-N,C2′]picolinate (FIrpic) were investigated by using density functional theory/time-dependent density functional theory (DFT/TD-DFT), including Ir(III)bis[2-(2,4-difluorophenyl)-4-(tert-butyl)pyridinato-N,C2′]picolinate (1a), Ir(III)bis[2-(2,4-difluorophenyl)-4-(n-heptyl)pyridinato-N,C2′]picolinate (2a), Ir(III)bis[2-(2,4-difluorophenyl)-4-(3-ethylheptyl)pyridinato-N,C2′]picolinate (3a), Ir(III)bis[2-(2,4-difluorophenyl)-4-(2,4,6-trimethylphenyl)pyridinato-N,C2′]picolinate (5a), and Ir(III)bis[2-(2,4-difluoro-3-(2,4,6-trimethylphenyl)phenyl)-pyridinato-N,C2′] picolinate (5b). To explore the influence of the substituted positions on the optical and electronic properties of the Ir(III) complexes, seven other new complexes were designed by introducing the substituted groups on the difluorophenyl rings or pyridine rings. After introducing the phenyl substituted groups on the pyridine or difluorophenyl rings of cyclometalated ligands, the HOMO–LUMO energy gap is decreased. Thus, the absorption spectra of 4a and 4b undergo a red-shifting, especially for 4a, and have a stronger absorption strength that will be beneficial to improving their quantum yields, which is proved by the further evaluation of the radiative (kr) and non-radiative (knr) rate constants. The combinations of a larger 3MLCT-3MC energy gap, smaller ΔES1–T1, and higher contribution of 3MLCT in the emission process result in the higher quantum yields for complexes 4a and 4b. Besides, the designed complexes 4a and 4b are considered to be potential candidates as blue-emitting materials with better equilibrium between the hole transport (λhole) and electron transport (λelectron).
Co-reporter:Ya Li, Li Wang, Tengfei Huang, Jinglai Zhang, and Hongqing He
Industrial & Engineering Chemistry Research 2015 Volume 54(Issue 33) pp:8093-8099
Publication Date(Web):August 4, 2015
DOI:10.1021/acs.iecr.5b01409
Exploring a high-efficiency catalyst for the coupling reaction of carbon dioxide (CO2) with epoxide (PO) is still a challenging project. Ionic liquid (IL) is one of the most ideal catalysts since it could catalyze the coupling reaction in a benign environment in the absence of metal and organic solvent. The catalytic activity of a series of pyridinium-based ILs is theoretically investigated. The influences of the nature of cation, methylene chain length, and anion on the catalytic performance are explored. It has been proven that the catalytic activity of pyridinium-based IL is better than that of imidazolium-based and quaternary ammonium-based ILs. Since the properties of IL could be regulated by variation of cation and anion, four new ILs are designed by introduction of the −COOH, −OH, −SO3H, and −NH2 functional groups into the traditional pyridinium-based IL, respectively. Subsequently, the catalytic performance of four newly designed functionalized pyridinium-based ILs is compared with that of the traditional pyridinium-based IL. Only the carboxyl-functionalized pyridinium-based IL has better catalytic activity than the traditional pyridinium-based IL. It is expected that the theoretical investigation might provide helpful clues for further experiments.
Co-reporter:Jieqiong Li, Li Wang, Qing Zhang, Wenpeng Wu, Chaozheng He, Jinglai Zhang
Organic Electronics 2015 Volume 22() pp:108-116
Publication Date(Web):July 2015
DOI:10.1016/j.orgel.2015.03.038
•The properties of carbazole-based DSSCs are theoretically investigated.•The effect of the nature and length of π-linker on overall PCE is explored.•Five new dyes are designed and their performance is predicted.Three synthesized and five new designed carbazole-based organic dye sensitizers before and after bonding to the TiO2 are theoretically investigated by density functional theory and time-dependent density functional theory methods. The influences of inserting different groups between donor and acceptor and augmenting π-spacers on the performance are explored by optimized geometries, electronic structures, simulated absorption spectra, and other parameters. The efficiency of all dyes as sensitizers in dye-sensitized solar cells is evaluated by the key parameters, including HOMO–LUMO energy gap, distance of charge transfer upon excitation from the ground to excited state, light-harvesting efficiency, injection driving force, and total reorganization energy related with the short-circuit current density and open-circuit photovoltage. It has been testified that both the nature and the number of π-linker units are important factors for the performance of dye-sensitized solar cells.
Co-reporter:Tengfei Huang, Lei Fang, Ya Li, Hongqing He, Li Wang and Jinglai Zhang
RSC Advances 2015 vol. 5(Issue 67) pp:54266-54274
Publication Date(Web):08 Jun 2015
DOI:10.1039/C5RA05544J
To explore the reason for the high activity of the cycloaddition reaction of PO (propylene oxide) with CO2 catalyzed by ZnBr2/CH (choline chloride) co-catalyst, the mechanism has been constructed using a DFT (density functional theory) method. The combination of CH and ZnBr2 will generate three new stable complexes, i.e., [Ch]2[ZnBr2Cl2], Ch+ZnBr2Cl−, and Ch+ZnBrCl2−. The latter two are derived from the dissociation of [Ch]2[ZnBr2Cl2]. The detailed mechanism of a coupling reaction catalyzed by the more stable complex Ch+ZnBrCl2− is explored. It has been elucidated that the attack from the Zn complex and the Br− anion is the major factor in promoting the cleavage of the C–O bond of PO. Finally, the performance of [Ch]2[ZnBr2Cl2] is also investigated, providing less activity, indicating that it should dissociate to gain better catalytic effect.
Co-reporter:Jieqiong Li, Li Wang, Xin Wang, Chaozheng He, Jinglai Zhang
Materials Chemistry and Physics 2015 Volume 163() pp:545-553
Publication Date(Web):1 August 2015
DOI:10.1016/j.matchemphys.2015.08.011
•The absorption and phosphorescence spectra of Pt(II) complexes are investigated.•Their Φem, IP, EA, and reorganization energy are compared.•Three new Pt(II) complexes are designed.The phosphorescent properties of three synthesized and three new designed platinum(II) complexes are focused on in this work. To reveal their structure–property relationships, a density functional theory/time-dependent density functional theory (DFT/TDDFT) investigation is performed on the geometric and electronic structures, absorption and emission spectra. The electroluminescent (EL) properties are evaluated by the ionization potential (IP), electron affinity (EA), and reorganization energy (λ). Furthermore, the radiative rate constant (kr) is qualitatively elucidated by various factors including the strength of the SOC interaction between the higher-lying singlet excited states (Sn) and the T1 state, the oscillator strength (f) of the Sn states that can couple with the T1 state, and the energy separation between the coupled states. A combined analysis of various elements that could affect the phosphorescent efficiency is beneficial to exploring efficient triplet phosphors in OLEDs. Consequently, complexes Pt-1 and 1 would be more suitable blue-emitting phosphorescent materials with balance of EL properties and acceptable quantum yields.
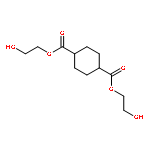
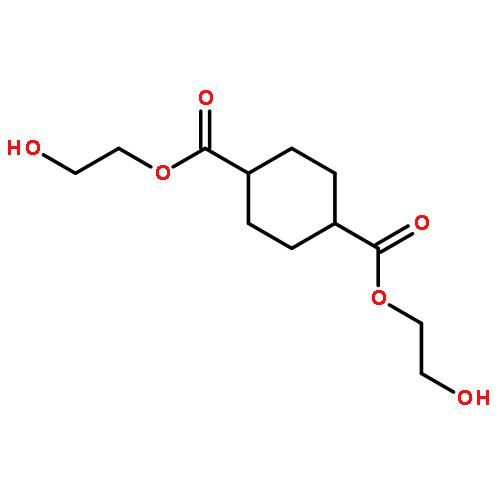
Co-reporter:Li Wang, Yuanyuan Li, Yanxin Zhang, Hongqing He, Jinglai Zhang
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2015 Volume 137() pp:259-266
Publication Date(Web):25 February 2015
DOI:10.1016/j.saa.2014.08.063
Co-reporter:Li Wang, Jinmiao Wen, Hongqing He and Jinglai Zhang
Dalton Transactions 2014 vol. 43(Issue 7) pp:2849-2858
Publication Date(Web):11 Nov 2013
DOI:10.1039/C3DT52616J
DFT and TD-DFT methods have been employed to theoretically investigate the properties of three recently synthesized green-emitting platinum(II) complexes (1–3) bearing tetradentate O^N^C^N ligands (O^C^N^C = 2-(4-(3,5-di-tert-butylphenyl)-6-(3-(pyridin-2-yl)phenyl)-pyridin-2-yl)phenolate and its derivatives) that have been testified to be good emitters in organic light-emitting diodes (OLEDs), especially for complex 3. The effect of the variation of the substituents on the electronic and optical properties is emphatically explored. Our calculation results reveal that the introduction of an electron-releasing group on one phenyl ring of the O^C^N^C ligand (complex 2) does not result in a distinct alteration of the spectra. However, the incorporation of the norbornane group to the O^C^N^C ligand (complex 3) leads to a blue-shift in the absorption and emission spectra as compared with 1. In addition, how the absorption and emission spectra are affected by the solvent polarity is studied. Both the absorption and the emission spectra display red-shifts of various degrees with the decrease of the solvent polarity. The different phosphorescent quantum yields of the three complexes are compared. It is reasonable to believe that the high 3MLCT (metal-to-ligand charge transfer) contribution and high percentage of the metallic character (Mc, %) in the emission process, as well as the largest vertical transition energy for 3, result in the highest quantum efficiency.
Co-reporter:Li Wang, Xiangfeng Jin, Ping Li, and Jinglai Zhang , Hongyan He and Suojiang Zhang
Industrial & Engineering Chemistry Research 2014 Volume 53(Issue 20) pp:8426-8435
Publication Date(Web):2017-2-22
DOI:10.1021/ie501063f
The mechanism of cycloaddition reaction between carbon dioxide and epoxide, catalyzed by HEMIMC (1-(2-hydroxyl-ethyl)-3-methylimidazolium chloride), was investigated using the DFT (density functional theory) method. In the presence of HEMIMC, the reaction mechanism changed from single-step to multipath. Seven reaction pathways are reported here, including two steps, epoxide ring-opening and ring-closure of cyclic carbonate, or three main steps, epoxide ring-opening, carbon dioxide insertion, and ring-closure of cyclic carbonate. The catalytic activity of HEMIMC was studied, and the catalytic mechanism was elucidated. The nucleophilic attack of anion and hydrogen bond are two of the most important factors to promote the cycloaddition reaction, especially the OH functional group in HEMIMC. Finally, the influence of different anions on the catalytic activity was investigated.
Co-reporter:Li Wang, Jinmiao Wen, Hongqing He, Jinglai Zhang
Journal of Fluorine Chemistry 2014 Volume 164() pp:1-9
Publication Date(Web):August 2014
DOI:10.1016/j.jfluchem.2014.04.009
•The mechanisms of CH3−nFnOCF2CHFCl (n = 0, 1, and 2) with OH radical are studied theoretically.•The effect of fluorine substitution on the reactivity is explored.•The total rate constants will decrease with the increasing of the fluorine atoms.The mechanism and dynamic properties for the multi-channel reactions of CH3OCF2CHFCl + OH (R1), CH2FOCF2CHFCl + OH (R2), and CHF2OCF2CHFCl + OH (R3) were carried out theoretically. The geometric parameters were optimized at the BMK/6-311+G(d,p) level. Subsequently, the energies were refined at the BMC-CCSD level. Based on the information of partial potential energy surface, the rate constants were evaluated by using the canonical variational transition state theory (CVT) with a small-curvature tunneling correction (SCT) method. For every reaction, there are two possible H-abstraction positions, i.e., CH3−nFn (n = 0, 1, and 2) group and CHFCl group. The major reaction channel and the effect of fluorine substitution on the reactivity are explored. Both questions are elucidated by analyzing the reaction energy, barrier height, bond dissociation energy, and rate constants.By using dual-level direct dynamics method (X//Y), schematic pathways and rate constants for the reaction CH3−nFnOCF2CHFCl (n = 0, 1, and 2) + OH are obtained. The predominant reaction pathway is hydrogen abstraction rather than displacement process because of higher barrier.
Co-reporter:Li Wang, Na Wang, Yanxin Zhang, Hongqing He, Jinglai Zhang
Synthetic Metals 2014 Volume 194() pp:160-169
Publication Date(Web):August 2014
DOI:10.1016/j.synthmet.2014.04.027
•The spectral nature is correlated with structural variation.•Conjugation length of ligands affects the color tuning and quantum yield.•Luminescent performance is evaluated by combining quantum yield and EL efficiency.•The potential of Ir(III) complexes to be promising OLED materials are explored.A DFT/TD-DFT theoretical study has been performed to elucidate the properties of a series of Ir(III) complexes {(xbi)2Ir(acac)}, where bi = cyclometalated benzoimidazole; acac = acetylacetonate; x = phenyl (pbi) (1); naphthalene (nbi) (2); phenanthrene (pnbi) (3); ph-cyclopentadiene-ph (fbi) (4); ph-pyrrol-ph (cbi) (5); ph-pyridine-ph (pybi) (6); ph-thiophene-ph (tabi) (7); thiophene (tbi) (8). By changing the conjugation length of benzoimidazole ligand and substitution on the benzoimidazole, the emitting color can be switched from red to green. The theoretical investigations shed some light on the reasons that phosphorescence performance of 1, 4, and 5 is better than others. These new structure–property relationships can guide an improved design and optimization of Ir-based emitting complexes.
Co-reporter:Li Wang, Ting Zhang, Jinmiao Wen, Hongqing He, Jinglai Zhang
Journal of Fluorine Chemistry 2014 Volume 168() pp:25-33
Publication Date(Web):December 2014
DOI:10.1016/j.jfluchem.2014.08.024
•Kinetics of the gas phase reaction of CF3CF2CH2OCHF2 + OH was investigated.•The reactivity of displacement and H-abstraction reaction is compared.•The rate constants of the H-abstraction pathways are calculated.A theoretical study on the mechanism and dynamics properties of the gas-phase reaction of CF3CF2CH2OCHF2 + OH is performed by using BMK method with the 6-311+G(d,p) basis set. The energy is further refined by the BMC-CCSD theory on the basis of the optimized structures. There are two kinds of H-abstraction channels and two displacement channels. The reactivity of displacement and H-abstraction reaction is compared from both the thermodynamic and kinetic viewpoints. The contributions from displacement channels are negligible. For both H-abstraction channels, the H-abstraction from CH2 site is more favorable than that from CHF2 site. The rate constants of title reaction are firstly determined by theoretical method in a wide temperature range.By using dual-level direct dynamics method (X//Y), schematic pathways and rate constants for the reaction CF3CF2CH2OCHF2 + OH are obtained. The predominant reaction pathway is hydrogen abstraction rather than displacement process.
Co-reporter:Li Wang, Yanxin Zhang, Yuanyuan Li, Hongqing He, Jinglai Zhang
Journal of Photochemistry and Photobiology A: Chemistry 2014 Volume 294() pp:14-19
Publication Date(Web):15 November 2014
DOI:10.1016/j.jphotochem.2014.07.025
•The geometrical and spectral properties of HPO are investigated.•Both excited- and ground-state PESs are explored for elucidation of ESIPT.•The overall scenario for ESIPT and possible mechanism for DF are discussed.To gain an in-depth knowledge on the enol–keto photoisomerization of an oxazole derivative, namely, 2-(2′-hydroxyphenyl)phenanthro[9,10-d]oxazole (HPO), a thorough theoretical investigation was performed on the geometrical and electronic spectroscopic properties. The present results suggest that the bond length exclusively dominates in the photoinduced process with all involved critical points keeping planar geometries. The predicted absorptions centered at 3.82 and 3.07 eV account for experimental bands at 3.61 and 3.42 eV corresponding to enol and keto forms, respectively. Orbital analysis shows that both absorption and emission bands in the title molecule essentially stem from HOMO to LUMO with typical ππ* transition character. Furthermore, both the ground- and excited-state energy profiles after photo-excitation were explored for exhaustive elucidation of the excited state intramolecular proton transfer (ESIPT). In addition, the delayed fluorescence (DF) behavior at low temperature of 77 K in experiment was tentatively attributed to triplet–triplet annihilation (TTA).Scheme of the photoinduced decay mechanism suggested by the present quantum-chemical calculations.
Co-reporter:Li Wang, Jianxiang Zhao, Hongqing He, Jinglai Zhang
Journal of Fluorine Chemistry 2013 Volume 149() pp:72-81
Publication Date(Web):May 2013
DOI:10.1016/j.jfluchem.2013.02.015
The hydrogen abstraction and displacement process of OH radicals with CHF2CF2OCH3 (R1) and CH2FCF2OCH3 (R2) are studied theoretically. Moreover, the secondary reactions of their products (CHF2CF2OCH2, CF2CF2OCH3, CH2FCF2OCH2, and CHFCF2OCH3) with OH radical are also carried out. Electronic structures of all stationary points are calculated at the BMK/6-311+G(d,p) level. And the single point energy calculation is performed using BMC-CCSD method to refine the reaction enthalpy and barrier height for each reaction channel. The correlation between the barrier height and the corresponding bond dissociation energy is studied. Using the canonical variational transition-state theory (CVT) including the small-curvature tunneling (SCT) correction, the rate constants for CHF2CF2OCH3/CH2FCF2OCH3 + OH reactions are calculated within 200–2000 K. The total rate constants calculated from the sum of the individual rate constants taking into account the Boltzmann distribution of each conformer. A good agreement is obtained between theoretical and experimental rate constants. Finally, the theoretical rate constants are fitted to four-parameter expression to assist further experimental study.Graphical abstractSchematic pathways for the reaction CHF2CF2OCH3 (SC11) + OH. Relative energies with ZPE at the BMC-CCSD//BMK/6-311+G(d,p) level are in kcal mol−1.Highlights► The mechanisms of CHF2CF2OCH3/CH2FCF2OCH3 with OH radical are studied theoretically. ► The displacement processes are also considered. ► The dynamics properties are calculated by dual-level (X//Y) direct dynamics methods.
Co-reporter:Li Wang, Xiaohan Yu, Jianxiang Zhao, Yanxin Zhang, Hongqing He, Jinglai Zhang
Synthetic Metals 2013 Volume 175() pp:174-182
Publication Date(Web):1 July 2013
DOI:10.1016/j.synthmet.2013.05.016
•Ground-state geometric structures of all species are optimized at M06 level of theory.•The optical properties of all species are calculated by means of TD-M06 method.•Detailed analysis for electronic transitions is performed to elucidate the UV spectra.To elucidate the structure information and spectroscopic properties, the ground structures of N1,N5-bis[pyridine-2-methylene]-thiocarbohydrazone ligand (L) and their corresponding Zn (II) complex ((H2L)ZnCl2) (A) and Ni (II) complex ((H2L)NiCl2) (B) are optimized at the M06 method. Two conformers, i.e., cis- or trans-, are considered for every species. The ligand presents almost planar structure; while two metal complexes show distorted tetrahedral structure. Subsequently, the spectroscopic properties are calculated using time-dependent density functional theory (TD-DFT) with the polarized continuum model (PCM). For the ligand L, all UV–vis absorption peaks are originated from (p, π) → π* with a character of intraligand charge-transfer (ILCT). With respect to complex A ((H2L)ZnCl2), the lowest-energy absorption peak is correlated with HOMO → LUMO transition, which is ascribed to (p, π) → π* with a mixed ligand–ligand charge-transfer and intraligand charge-transfer (LL/ILCT) character. Compared with complex A, the maximum absorption wavelength of complex B ((H2L)NiCl2) exhibits red shift.The theoretical spectra of both cis- and trans-ligand are in good accordance with the experimental measurements. The maximum absorption wavelengths of both ligands are almost the same.
Co-reporter:Li Wang, Yanxin Zhang, Hongqing He, Jinglai Zhang
Synthetic Metals 2013 Volume 167() pp:51-63
Publication Date(Web):1 March 2013
DOI:10.1016/j.synthmet.2013.02.004
To explore the spectroscopic properties, a series of palladium complexes are investigated theoretically through density functional theory (DFT) and time-dependent density functional theory (TD-DFT) calculations. Full geometry optimizations of 1-allyl-3-(2-pyridyl)thiourea (APTU) and its corresponding Pd (II) complexes [Pd(APTU)Cl2] (A), trans-[Pd(APTU)2]2+ (B), cis-[Pd(APTU)2]2+ (C), together with two thiolic tautomeric APTU-based Pd (II) complex cis-[Pd(APTU)2] (D) and trans-[Pd(APTU)2] (E) are carried out at the B3LYP, B3PW91, and M06 levels. All of them present slightly distorted square-planar geometries. On the basis of the optimized geometries, the absorption spectra in ethanol are evaluated by using three TD-DFT methods with the PCM model. Experimental absorption spectra are well reproduced by our time-dependent density functional theory calculations. Both UV–vis absorption peaks at 219.64 and 254.80 nm for complex A are mainly attributed to (p,π,d) → π* with a mixed character of intraligand charge-transfer (ILCT)/ligand–ligand charge-transfer (LLCT)/metal-ligand charge-transfer (MLCT) transitions. Another weak absorption band of complex A is located at 313.17 nm largely originated from HOMO−6 to LUMO transition. There are two isomers with small energy difference for [Pd(APTU)2]2+, i.e., trans-[Pd(APTU)2]2+ (B) and cis-[Pd(APTU)2]2+ (C). Only when both isomers are considered, four absorption peaks can be obtained, which is in line with the experimental results. The absorption peaks of complexes D and E present obvious red shift that is associated with their lower HOMO–LUMO gap (ΔE|HOMO–LUMO|) among five complexes. Differences and similarities in molecular orbitals and electronic transition characters for five complexes are discussed in detail.Highlights► Ground-state geometric structures of five complexes are optimized by three DFT methods. ► The spectroscopic properties of five complexes are explored by means of TD-DFT computations. ► Comparison between theoretical values and experimental observations are made in detail. ► Different conformers are considered.
Co-reporter:Xin Chen, Xiaohan Yu, Yafang Liu, Jinglai Zhang
Journal of Molecular Graphics and Modelling 2013 Volume 46() pp:83-92
Publication Date(Web):November 2013
DOI:10.1016/j.jmgm.2013.09.012
•A stepwise molecular design of peptide to functionalize the large-diameter SWCNT.•Two kinds of functional peptides were designed for the armchair (24, 24) SWCNT.•The residues with aromatic rings and long alkylene chains were found to be most effective in the adsorption of peptide on the SWCNT surface.•The predominant driving force of protein/peptide onto the SWCNT surface was VDW interaction.•The adsorption manner of the protein/peptide was affected by the flexibility of size chains.Single-walled carbon nanotube (SWCNT) is one of the most popular low-dimensional carbon materials in material science, nanomedicine, and nanoscale electronics. Yet the application of the SWCNTs was hindered by the self-aggregation. To purify and disperse the SWCNTs, non-covalent wrapping is one of the effective options to overcome such defects. In this work, two kinds of short peptides were designed to facilitate the modification of large-diameter SWCNT. The design of the peptide was carried out in a stepwise manner. The effective residues of helix-rich and sheet-rich proteins on SWCNT were studied at the first step, and then a coarse model peptide composed of the key adsorption residues above was built to investigate the adsorption dynamics on SWCNT. In the end, the residues include long alkyl side chain and that include aromatic rings were found to play key roles on the adsorption of protein/peptide on hydrophobic SWCNT. And two peptides rich in the long alkyl chain and aromatic rings were constructed respectively. The predominant adsorption capabilities of the two kinds of peptides were discerned by the adsorption details.
Co-reporter:Pan Ning, Tiegang Ren, Yanxin Zhang, Jinglai Zhang
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2013 Volume 115() pp:464-468
Publication Date(Web):November 2013
DOI:10.1016/j.saa.2013.06.063
•The ground state of the two groups compounds were optimizated based on 6-31G (d) basis set of B3LYP method.•UV absorption spectra are simulated through TD-DFT method with PCM model.•Explained the red shift behavior in the electronic spectra of products.•The reaction products of two sets of experiments are confirmed reasonably with theoretical calculations.The spectroscopic properties of 8-hydroxyquinoline derivatives are theoretically investigated by means of density functional theory (DFT) and time-dependent density functional theory (TD-DFT) methods. The target molecules are divided into two groups: group (I): (E)-2-(2-(3,5-dimethyl-1-phenyl-1H-pyrazol-4-yl)vinyl)quinolin-8-ol (A), together with corresponding potential reaction products of A with acetic acid, i.e., (E)-2-(2-(3,5-dimethyl-1-phenyl-1H-pyrazol-4-yl)vinyl)quinolin-8-yl acetate (AR1), and (E)-2-(2-(3,5-dimethyl-1-phenyl-1H-pyrazol-4-yl)vinyl)-8-hydroxyquinolinium (AR2); group (II): (E)-2-(2-(1-(4-chlorophenyl)-3,5-dimethyl-1H-pyrazol-4-yl)vinyl)quinolin-8-ol (B), as well as potential reaction products of B with acetic acid, i.e., (E)-2-(2-(1-(4-chlorophenyl)-3,5-dimethyl-1H-pyrazol-4-yl)vinyl)quinolin-8-yl acetate (BR1), and (E)-2-(2-(1-(4-chlorophenyl)-3,5-dimethyl-1H-pyrazol-4-yl)vinyl)-8-hydroxyquinolinium (BR2). The geometries are optimized by B3LYP and M06 methods. The results indicate that product molecules tend to be effectively planar compared with reactants. Subsequently, UV absorption spectra are simulated through TD-DFT method with PCM model to further confirm the reasonable products of two reactions. AR2 and BR2 are identified as the target molecules through the experimental spectra for the real products. It is worth noting that the maximum absorption wavelengths of compounds AR2 and BR2 present prominent red shift compared the initial reactants A and B, respectively, which should be ascribed to the enhancive planarity of products that mentioned above and the decreased HOMO–LUMO energy gap. Geometric structures and optical properties for corresponding compounds are discussed in detail.Graphical abstract
Co-reporter:Yanxin Zhang, Pan Ning, Jinglai Zhang
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2013 Volume 101() pp:283-293
Publication Date(Web):15 January 2013
DOI:10.1016/j.saa.2012.09.100
The B3LYP, CAM-B3LYP, and coupled cluster CCSD(T) calculations have been utilized to determine the equilibrium structures of linear carbon radicals CnH (n = 5–12) in their ground states, as well as the CASSCF method used to optimize the ground and selected low-lying excited states. DFT-calculations show that even-n radicals C2nH have polyacetylene-like structures with significant single-triple bond length alternation, whereas the odd-numbered analogues C2n+1H exhibit a trend from polyacetylene-like characters into cumulenic-like arrangement towards C ends along the carbon chains. The stabilities of the system under study have been evaluated by analyses of the vibrational frequencies and incremental binding energies. For the whole CnH (n = 5–12) series, the vertical excitation energies and oscillator strengths have been calculated at the CASPT2/cc-pVTZ level of theory. At the B3LYP optimized geometries, the lowest 12Δ ← X2Π transitions for C5H and C7H occur at 2.36 and 2.14 eV, respectively, comparing well with the observed values of 2.33 and 2.09 eV. Moreover, the strongest 22Π ← X2Π transitions for C2nH (n = 3–6) are predicted to be at 2.39, 2.00, 1.80, and 1.64 eV, respectively, which are in agreement with the experimental observations. Additionally, the possible dissociation channels and the fragmentation energies of CnH (n = 5–12) series are discussed in the paper.Graphical abstractHighlights► Properties of ground- and excited-states for CnH (n = 5–12) radicals are discussed. ► Even-n radicals are more stable than odd-n analogues with the parity alternation. ► A comparison between the CASPT2-predicted vertical excitation energies and experimental observations has been performed. ► Similarly non-linear ΔE–n relationships are found in different transition systems.
Co-reporter:Yanxin Zhang, Jia Guo, Jinglai Zhang
International Journal of Mass Spectrometry 2012 Volume 309() pp:56-62
Publication Date(Web):1 January 2012
DOI:10.1016/j.ijms.2011.08.025
With unrestricted B3LYP and CAM-B3LYP calculations, we have investigated the linear HCnN+ (n = 2–14) clusters focusing on the ground-state geometries, vibrational frequencies, rotational constants, dipole moments, energy differences (ΔEn), and incremental binding energies (ΔEI). The results indicate that the odd-numbered HCnN+ clusters have polyacetylene-like structures with significant single–triple bond length alternation, while the even-numbered analogues exhibit some sort of cumulenic character, with the former being more stable than the latter. The CASPT2/cc-pVTZ approach has been employed to estimate the vertical excitation energies for the dipole-allowed (2, 3)2Π ← X2Π and dipole-forbidden 12Φ ← X2Π transitions in HCnN+ (n = 5–14) clusters. The predicted excitation energies of 22Π ← X2Π transitions for HCnN+ (n = 5–13) clusters are 2.27, 2.33, 2.04, 2.02, 1.84, 1.76, 1.62, 1.58, and 1.49 eV, respectively, in good agreement with the available observed values (2.12, 2.17, 1.84, 1.89, 1.61, 1.66, 1.42, 1.49, and 1.27 eV). In addition, the higher electronic transitions of HCnN+ (n = 5–14) are also calculated. We expect that calculations for 32Π ← X2Π transitions in odd-n clusters are able to contribute to further experimental and theoretical researches because of their relatively higher oscillator strengths. Finally, in the paper are discussed the possible dissociation channels and corresponding fragmentation energies of HCnN+ clusters.Graphical abstractHighlights► Properties of ground- and excited-states for HCnN+ (n = 2–14) are discussed. ► Odd-n clusters are more stable than even-n analogues with the parity alternation. ► The CASPT2 predicted vertical excitation energies agree well with observed values. ► Notably linear λ–n relationships are found in the (2, 3)2Π ← X2Π transitions.
Co-reporter:Li Wang, Yanxin Zhang, Xiaohan Yu, Hongqing He, Jinglai Zhang
Synthetic Metals 2012 Volume 162(Issue 23) pp:2138-2148
Publication Date(Web):December 2012
DOI:10.1016/j.synthmet.2012.09.012
The ground structures of pyridinylimine derivatives (L1-a = N-(pyridin-2-ylmethylene)aniline and L1-b = N1,N1-(dimethyl-N4-pyridin-2-ylmethylene)benzene-1,4-diamine) and pyridinylmethylamine derivatives (L2-a = N-(pyridin-2-ylmethyl)aniline and L2-b = N1,N1-(dimethyl-N4-pyridin-2-ylmethy)benzene-1,4-diamine) and their corresponding Zn (II) complexes ([Zn(L1-a)Cl2] (A), [Zn(L1-b)Cl2] (B), [Zn(L2-a)Cl2] (C), and [Zn(L2-b)Cl2] (D)) are optimized at three DFT levels, i.e., B3LYP, B3PW91, and M06. The spectroscopic properties are calculated using time-dependent density functional theory (TD-DFT) in gas and in solution. The occupied orbitals involved in the transitions have mixed character of the Cl atom p orbital and ligand-based π orbital, while the lowest unoccupied molecular orbital (LUMO) presents π* orbital character. Two UV–Vis absorption peaks located at 252 and 353 nm are assigned to (p, π) → π* transition with mixed intraligand charge-transfer (ILCT)/ligand–ligand charge-transfer (LLCT) character and π → π* transition with ILCT character for complex A, respectively. With respect to complex B, the absorption bands show red shift with two peaks at 281 and 470 nm with the appearance of an electron-releasing group (–N(CH3)2) because the energy gap (ΔE|HOMO–LUMO|) is decreased. Red shift phenomenon is also observed between other two complexes C and D. Comparison between theoretical and experimental results for the structures and spectra is discussed in detail.Highlights► Ground-state geometric structures of titled complexes are optimized by three DFT methods. ► The spectroscopic properties of titled complexes are explored by means of TD-DFT computations. ► Comparison between theoretical values and experimental observations are made in detail.
Co-reporter:Yuan Zhao, Jia Guo, Jinglai Zhang
Chemical Physics 2011 Volume 386(1–3) pp:23-28
Publication Date(Web):28 July 2011
DOI:10.1016/j.chemphys.2011.05.013
Abstract
The B3LYP, CAM-B3LYP, and RCCSD(T) theories have been used to calculate the ground state equilibrium geometries of the linear cationic chains NC2n+1N+ (n = 1–6). Compared with the system NC2nN+, the odd-n cationic chains are more susceptible to fragmentation than the even-n cationic chains. The complete active space self-consistent-field method has been utilized to determine the stationary structure of the ground state (X2Пg/u) and first excited state (12Пu/g). The complete active space second-order perturbation theory has been used to compute the vertical excitation energies for the dipole-allowed (1, 2, 3)2Пu/g ← X2Пg/u transitions as well as the dipole-forbidden 12Φu/g ← X2Пg/u transitions. The calculated transition energies of 12Пu/g ← X2Пg/u in the gas phase are 2.61, 2.37, 2.07, 1.88, 1.64, and 1.34 eV, respectively, which accord well with the available experimental values. Moreover, the absorption spectra of 22Пu/g ← X2Пg/u may be detected more easily among the selected four transitions.
Co-reporter:Li Wang, Yuan Zhao, Jinglai Zhang
Journal of Fluorine Chemistry 2011 Volume 132(Issue 3) pp:216-221
Publication Date(Web):March 2011
DOI:10.1016/j.jfluchem.2011.01.009
The rate constants of the hydrogen abstraction reactions of CF3CHFCF3 + H (R1) and CF3CF2CHF2 + H (R2) have been calculated by means of the dual-level direct dynamics method. Optimized geometries and frequencies of stationary points and extra points along the minimum-energy path (MEP) are obtained at the MPW1K/6-311+G(d,p) level, and the classical energetic information is further corrected with the interpolated single-point energy (ISPE) approach by the G3(MP2) level of theory. Using the canonical variational transition state theory (CVT) with small-curvature tunneling corrections (SCT), the rate constants are evaluated over a wide temperature range of 200–2000 K. The calculated CVT/SCT rate constants are in good agreement with available experimental values. It is found that the variational effect is very small and almost negligible over the whole temperature region. However, the small-curvature tunneling correction plays an important role in the lower temperature range. Furthermore, the heats of formation of species CF3CF2CHF2 (SC1 or SC2) and CF3CF2CF2 are studied using isodesmic reactions to further elucidate the thermodynamic properties.Graphical abstractPlot of the CVT/SCT rate constants calculated at the G3(MP2)//MPW1K/6-311+G(d,p) level along with the available experimental values versus 1000/T between 200 and 2000 K for the reaction CF3CF2CHF2 + H → CF3CF2CF2 + H2 (R2).Research highlights► By the dual-level direct dynamics method, the rate constants of the hydrogen abstraction reactions of CF3CHFCF3 + H (R1) and CF3CF2CHF2 + H (R2) are calculated. ► The small-curvature tunneling correction plays an important role in the lower temperature range. ► The heats of formation of species CF3CF2CHF2 (SC1 or SC2) and CF3CF2CF2 are firstly studied.
Co-reporter:Li Wang;Yuan Zhao;Jing Zhang;Yanna Dai
Theoretical Chemistry Accounts 2011 Volume 128( Issue 2) pp:183-189
Publication Date(Web):2011 January
DOI:10.1007/s00214-010-0813-8
The kinetic properties of the hydrogen abstraction reactions of CF3CH2F + F → CF3CHF + HF (R1) and CF3CH2Cl + F → CF3CHCl + HF (R2) have been studied by dual-level direct dynamics method. Optimized geometries and frequencies of all the stationary points and extra points along the minimum-energy path (MEP) were obtained at the B3LYP/6-311 + G(2d,2p) level. Two complexes with energies less than that of the reactants were located in the reactant side of each reaction. The energy profiles were further refined with the interpolated single-point energies (ISPE) method at the G3(MP2) level of theory. Using canonical variational transition state theory (CVT) with the small-curvature tunneling correction (SCT) method, the rate constants were evaluated over a wide temperature range of 200–2,000 K. Our calculations have shown that C–H bond activity decreases when one hydrogen atom of CF3CH3 is substituted by a fluorine atom, than when substituted with a chlorine atom. This is in good agreement with the experimental results.
Co-reporter:Li Wang;Yuan Zhao
Theoretical Chemistry Accounts 2011 Volume 129( Issue 1) pp:73-84
Publication Date(Web):2011 May
DOI:10.1007/s00214-011-0892-1
The reactions of OH (OD) radicals with CF2ClCClFH (R1), CF2ClCCl2H (R2), CFCl2CClFH (R3), and CFCl2CCl2H (R4) have been investigated theoretically by a dual-level direct dynamics method. The optimized geometries and frequencies of the stationary points are calculated at the MPW1K/6-311+G(d,p) level. To improve the reaction enthalpy and potential barrier of each reaction channel, the single-point energy calculation is made by the MC-QCISD method. The enthalpies of formation of the species CF2ClCClFH, CF2ClCCl2H, CFCl2CClFH, CFCl2CCl2H, CF2ClCClF, CF2ClCCl2, CFCl2CClF, and CFCl2CCl2 are evaluated by two sets of isodesmic reactions. Using canonical variational transition state theory (CVT) with the small-curvature tunneling correction (SCT) method, the rate constants of OH and OD radicals with CF2ClCClXH (X = F, Cl) and CFCl2CClXH (X = F, Cl) are evaluated over a wide temperature range of 100–2,000 K at the MC-QCISD//MPW1K/6-311+G(d,p) level. The calculated CVT/SCT rate constants are consistent with available experimental data. The results show that the tunneling correction has an important contribution in the calculation of rate constants at lower temperatures. For the above-mentioned four reactions, the kinetic isotope effects are also calculated. Finally, the effect of fluorine or chlorine substitution on reactivity of the C–H bond is discussed.
Co-reporter:Yuan Zhao;Jia Guo
Theoretical Chemistry Accounts 2011 Volume 129( Issue 6) pp:793-801
Publication Date(Web):2011 August
DOI:10.1007/s00214-011-0937-5
The ground-state equilibrium geometries of the linear carbon chain cations NC2nN+ (n = 1–7) have been investigated with B3LYP, CAM-B3LYP, and RCCSD(T) calculations. The ground state (X2Пg/u) and excited state (12Пu/g) have been optimized by using the complete active space self-consistent field method. The present study reveals that these linear cations generally have the characteristic of bond length alternation in both electronic states. The vertical excited energies for the dipole-allowed (1, 2, 3)2Пu/g ← X2Пg/u transitions as well as the dipole-forbidden 12Φu/g ← X2Пg/u transitions have been computed with the complete active space second-order perturbation theory. The calculated transition energies of 12Пu/g ← X2Пg/u for NC2nN+ (n = 1–6) in the gas phase are 2.26, 2.09, 1.91, 1.72, 1.56, and 1.39 eV, respectively, which mutually agree well with the available experimental values of 2.11, 2.07, 1.88, 1.67, 1.49, and 1.34 eV. Moreover, the corresponding absorption wavelengths are predicted to have the significant nonlinear size dependence, which is different from the bands origin in NC2nN (n = 1–7).
Co-reporter:Li Wang, Yuan Zhao, Zhi-qiao Wang, Cheng-gong Ju, Ya-li Feng, Jing-lai Zhang
Journal of Molecular Structure: THEOCHEM 2010 Volume 959(1–3) pp:101-105
Publication Date(Web):15 November 2010
DOI:10.1016/j.theochem.2010.08.013
The hydrogen abstraction reactions of CF3CHCl2 + F (R1) and CF3CHClF + F (R2) are investigated by dual-level direct dynamics method. The optimized geometries and frequencies of the stationary points are calculated at the B3LYP/6-311G(d,p) and MP2/6-311G(d,p) levels. Higher-level energies are obtained at the G3(MP2) method using the B3LYP and MP2-optimized geometries, respectively. Complexes with energies lower than those of the reactants are located at the entrance of these two reactions at the B3LYP level, respectively. Using the variational transition-state theory (VTST) with the inclusion of the small-curvature tunneling correction, the rate constants are calculated over a wide temperature range of 200–2000 K. The agreement between theoretical and experimental rate constants is good. In addition, the effect of fluorine substitution on reactivity of the C–H bond is discussed. Our calculations show that the fluorine substitution deactivates the C–H bond reactivity.
Co-reporter:Lianbin Wang, Wenpeng WU, Jinglai Zhang, Zexing Cao
Acta Physico-Chimica Sinica 2006 Volume 22(Issue 9) pp:1079-1084
Publication Date(Web):September 2006
DOI:10.1016/S1872-1508(06)60048-X
Equilibrium geometries of cis-HOOOH and trans-HOOOH have been investigated using the density functional theory (DFT), complete active space self-consistent-field (CASSCF), and coupled cluster with single and double replacement (CCSD) approaches. The harmonic vibrational frequencies on the optimized geometries were calculated using the DFT theory. The potential energy curve of the isomerization between the trans-HOOOH and cis-HOOOH was obtained by DFT calculations. The time-dependent density functional theory (TD-DFT) and the complete active space perturbation theory of second order (CASPT2) calculations have been performed to obtain the vertical excitation energies of selected low-lying singlet and triplet excited states. Computed results show that (1)trans-isomer is more stable than cis-isomer; (2) there are two pathways of the conversion between the trans-HOOOH and cis-HOOOH; (3) the vertical excitation energies of the lowest singlet and triplet excited states in trans-HOOOH are lower than those in cis-isomer; (4) in the singlet excited states, 21A state in trans-HOOOH and 21A″ state in cis-HOOOH have the longest lifetimes of 2.07×10−5s and 1.44×10−5s with the excitation energies of 165.52 and 167.43 nm, respectively.
Co-reporter:Lianbin Wang, Wenpeng Wu, Jinglai Zhang, Zexing Cao
Journal of Molecular Structure: THEOCHEM 2006 Volume 773(1–3) pp:81-86
Publication Date(Web):30 October 2006
DOI:10.1016/j.theochem.2006.07.006
Optimized geometries of small carbon clusters C3Cl and its ions in their ground and selected excited states have been obtained by CASSCF approach. The vertical excitation energies for the electronic excitations to low-lying excited states have been calculated by the CASPT2 method using cc-pVTZ basis set. The predicted vertical excitation energies are in good agreement with available experimental values. The spin–orbit effect on the spin-forbidden transitions is generally small, with oscillator strengths in the magnitude of 10−7–10−13. The vertical emission energies for the selected excited states were calculated at CASPT2/cc-pVTZ level of theory based on the CASSCF optimized geometries of the corresponding excited states.
Co-reporter:Wenpeng Wu, Jinglai Zhang, Zexing Cao
Journal of Molecular Structure: THEOCHEM 2006 Volume 765(1–3) pp:137-141
Publication Date(Web):15 June 2006
DOI:10.1016/j.theochem.2006.03.023
Structures of linear valence isoelectronic carbon chain anions C4O−, C4S− and C4Se− in their ground states have been investigated by density functional theory (DFT-B3LYP) and coupled cluster with single–double substitution (CCSD) approach. Complete-active-space-self-consistent-field (CASSCF) method has been used for geometry optimization of selected low-lying states. The vertical excitation energies from the ground state to selected low-lying excited states have been carried out with complete-active-space-second-order-perturbation-theory (CASPT2). All of them have a similar set of excited states. In comparison with available experimental observations, the predicted excitation energies for the allowed transitions have an accuracy of no more than 0.12 eV. Moreover, the calculations here confirm that the previous assignments of the observed electronic absorption bands of C4O− and C4S− are reliable. The excitation energies of 22Π←X2Π and 12Σ−←X2Π transitions in C4Se− are predicted to be 2.09 and 3.90 eV, with oscillator strengths 0.1941 and 0.0105, respectively, which may be observed in future experiment. CASPT2 also evaluated the vertical electron detachment energies for C4O−, C4S− and C4Se− to be 3.55, 3.40 and 3.40 eV, respectively.
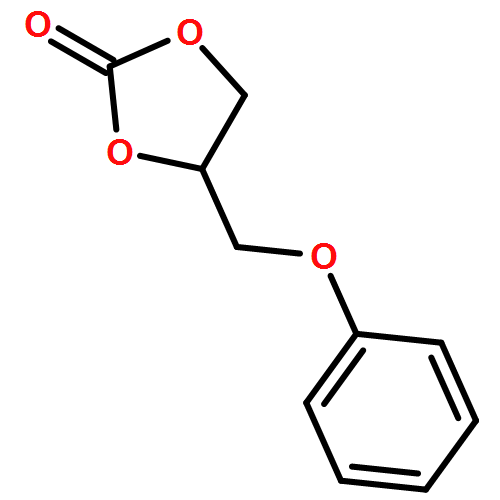
Co-reporter:Ci Chen, Yuan Ma, Danning Zheng, Li Wang, Junfeng Li, Jinglai Zhang, Hongyan He, Suojiang Zhang
Journal of CO2 Utilization (March 2017) Volume 18() pp:156-163
Publication Date(Web):1 March 2017
DOI:10.1016/j.jcou.2017.01.026
•The mechanism of amino-functionalized imidazolium-based IL for fixation of CO2.•The mechanism catalyzed by multi-species is considered.•Positive charge is not the sole item to determine the electrophilic attack.•Ring-opening is promoted by both weak interaction and electrostatic interaction.The mechanism of coupling reaction of CO2 with propylene oxide (PO) catalyzed by the amino-functionalized imidazolium-based ionic liquid is theoretically investigated by the density functional theory (DFT). Although the mechanisms of the fixation of CO2 catalyzed by various room-temperature ionic liquids or functionalized ionic liquids have been theoretically elucidated in previous literatures, it is not totally suitable for the amino-functionalized ionic liquid. First, the 1-(3-aminopropyl)-3-methylimidazolium chloride ([APmim]Cl) would react with CO2 to produce the 1-(3-carbamic acid propyl)-3-methylimidazolium chloride ([CAPmim]Cl). Then, [APmim]Cl, [CAPmim]Cl, and combination of them would be employed as the catalyst leading to nine possible routes. Different from the previous work, this work allows a better comprehension of the mechanism by means of a new model that the cooperative effect of two same or different components is considered. Both the interaction between catalyst and reactant and the influence between different catalytic components are considered. Besides the nucleophilic attack of the Cl− anion, the [CAPmim]+ cation is taken as the main component to activate the O atom of PO directly leading to the ring-opening. The [APmim]+ cation is utilized to stabilize the [CAPmim]+, which is the most favorable route. The noncovalent interactions (NCI) plot is employed as a tool to analyze the reason of the higher catalytic efficiency of top three favorable routes. The hydrogen bond and dispersive attractive interaction is confirmed to play a determining role in the catalytic activity.Download high-res image (106KB)Download full-size image
Co-reporter:Qiuling Zhu, Keke Wen, Songyan Feng, Wenpeng Wu, Beibei An, Huijuan Yuan, Xugeng Guo, Jinglai Zhang
Dyes and Pigments (June 2017) Volume 141() pp:
Publication Date(Web):June 2017
DOI:10.1016/j.dyepig.2017.02.019
•A stronger intramolecular hydrogen-bond leads to a lower energy barrier in the ESIPT reaction.•The absorption and fluorescence properties of five amino-type hydrogen-bonding dye molecules are calculated.•A new ESIPT dye molecule is theoretically designed.Structures of a series of 10-aminobenzo[h]quinoline (ABQ) derivatives in their ground states and first excited singlet states were optimized with density functional theory (DFT) and time-dependent density functional theory (TD-DFT) approaches. Their electronic absorption spectra and fluorescence emission spectra were simulated on the basis of the calculations on the vertical excitation and emission energies, which agree very well with the corresponding experimental spectra. The potential energy curves of their ground state and first excited singlet state were calculated by TD-DFT to elucidate the mechanisms of the excited-state intramolecular proton transfer (ESIPT) processes. It reveals that all the systems considered here can undergo a fast or even ultrafast ESIPT reaction, giving rise to the single fluorescence emission from their tautomer forms, as observed experimentally.
Co-reporter:Li Wang, Tengfei Huang, Ci Chen, Jinglai Zhang, Hongyan He, Suojiang Zhang
Journal of CO2 Utilization (June 2016) Volume 14() pp:61-66
Publication Date(Web):1 June 2016
DOI:10.1016/j.jcou.2016.02.006
•Elucidate the mechanism of guanidinium bromide/ZnBr2-catalyzed reaction.•Investigate the influence of cations on the performance of ionic liquids/ZnBr2.•Identify the effect of the bulk of guanidinium salt.•Compare the activity of different binary catalysts.Numerous binary catalysts of IL/Lewis acid have been developed for the coupling reaction of carbon dioxide and epoxides to form cyclic carbonates with high catalytic activity under benign environment. However, the mechanism is still obscure for most of catalysts. The catalytic mechanism of the binary catalysts hexaalkylguanidinium bromide/ZnBr2 is elucidated by theoretical method in this work to obtain the reason of their high catalytic activity. Owing to the complicated of the binary catalysts, there are lots of possible attack forms. Finally, it is confirmed that the electrophilic attack from the Zn complex and the nucleophilic attack from the Br− anion are the essential factors to promote the ring opening of PO. Following the most favorable route, the catalytic activity of different binary catalysts, including the ILs/ZnBr2 and NBu4Br/Zn(salphen), is compared. Moreover, the influence of the bulk of hexaalkylguanidinium salt on the catalytic activity is studied. The catalytic activity is enhanced with the increased bulk of the hexaalkylguanidinium salt. It is expected that our theoretical study would provide valuable clues to further refine the binary catalysts.Download high-res image (71KB)Download full-size image
Co-reporter:Li Wang, Ping Li, Xiangfeng Jin, Jinglai Zhang, Hongyan He, Suojiang Zhang
Journal of CO2 Utilization (June 2015) Volume 10() pp:113-119
Publication Date(Web):1 June 2015
DOI:10.1016/j.jcou.2015.02.006
•Elucidate the mechanism of catalyzed cycloaddition of CO2 with epoxide.•Understand the reasons for high reactivity of hydroxyl-functionalized ionic liquid.•Explore the influence of different anions on the reactivity.Utilization of CO2 (carbon dioxide) to synthesize useful organic compounds has attracted much attentions from the academia and industry. Recently, the fixation of CO2 catalyzed by using four hydroxyl-functionalized ionic liquids (ILs) has been successfully achieved. However, the detailed mechanism is still ambiguous. We performed a theoretical study on the mechanism of the fixation of CO2 catalyzed by 2-hydroxyl-ethyl-triethylammonium bromide (HETEAB) using the DFT method. The structural and energetic information for each possible route were obtained. Furthermore, the similarities and differences of two different catalysts, HETEAB and 2-hydroxyl-ethyl-tributylammonium bromide (HETBAB), were discussed in detail. In addition, the result shows that the OH functional group is one essential factor to improve the catalyst ability. Except that, including active H atom in the IL is the other major factor to refine the catalytic performance.Download full-size image
Co-reporter:Yuanyuan Li, Yue Zhang, Yuan Ma, Tiegang Ren, Li Wang, Jinglai Zhang
Organic Electronics (April 2017) Volume 43() pp:96-104
Publication Date(Web):April 2017
DOI:10.1016/j.orgel.2017.01.013
Co-reporter:Kenan Sun, Yuanyuan Li, Qing Zhang, Li Wang, Jinglai Zhang, Xin Zhou
Applied Surface Science (31 May 2017) Volume 405() pp:
Publication Date(Web):31 May 2017
DOI:10.1016/j.apsusc.2017.02.071
•Highly active photocatalytic Ta3N5 loaded with CoOx and NiOx.•Exploring the stable structure and the electronic properties by DFT calculations.•Investigating the behavior of water adsorption on the surface of photocatalytic.Experiments found that cocatalysts play an important role in influencing the efficiency of photocatalysis and photoelectrochemical water splitting. However, how to choose proper cocatalysts in certain photocatalytic system is still a challenging question. In this work, the first-principles density functional theory is employed to explore two photocatalysts with different photocatalytic activity, CoOx and NiOx loaded Ta3N5, including finding the stable structure of metal oxide adsorbed surface, analyzing the electronic properties, and investigating the behavior of water adsorption. Our results indicate that the structural match between cluster and surface and less distortion of interfacial structure are benefit to the stability of the whole system. Water dissociation tends to occur at the interface between metal oxide cluster and Ta3N5 surface. Combining observations in experiments with our calculated results, we propose that the obvious difference of photocatalytic activities in CoOx/Ta3N5 and NiOx/Ta3N5 is possibly related to whether there are impurity states located in the middle of band gap, which has adverse effect on the separation of photo-generated electrons and holes.Figure optionsDownload full-size imageDownload high-quality image (159 K)Download as PowerPoint slide
Co-reporter:Xin Wang, Songyan Feng, Xiaolin Wang, Chunzhang Wang, Li Wang, Jinglai Zhang
Computational and Theoretical Chemistry (1 April 2017) Volume 1105() pp:69-76
Publication Date(Web):1 April 2017
DOI:10.1016/j.comptc.2017.02.026
Co-reporter:Jieqiong Li, Li Wang, Kenan Sun and Jinglai Zhang
Dalton Transactions 2016 - vol. 45(Issue 7) pp:NaN3047-3047
Publication Date(Web):2015/12/23
DOI:10.1039/C5DT04209G
The electronic structures and photophysical properties of three homoleptic iridium(III) complexes IrL3 with C^N ligands, including 2a (L = 1-(2,6-diisopropylphenyl)-2-phenyl-1H-imidazole), 5a (L = 1-(2,6-dimethylphenyl)-2-phenyl-1H-imidazole), and 6a (L = 1-(3,5-diisopropylbiphenyl-4-yl)-2-phenyl-1H-imidazole), are investigated by means of the density functional method. Furthermore, seven new complexes are theoretically designed, including 1a (L = 1,2-diphenyl-1H-imidazole), 3a (L = 1-(2,6-dimethoxyphenyl)-2-phenyl-1H-imidazol), 4a (L = 2-(2-phenyl-1H-imidazol-1-yl)isophthalaldehyde), 1b (L = 2-(biphenyl-3-yl)-1H-imidazole), 2b (L = 2-(2′,6′-diisopropylbiphenyl-3-yl)-1H-imidazole), 3b (L = 2-(2′,6′-dimethoxybiphenyl-3-yl)-1H-imidazole), and 4b (L = 3′-(1H-imidazol-2-yl)biphenyl-2,6-dicarbaldehyde), to explore the influence of different substituents and different substituted positions on the electronic structures, phosphorescence properties, and organic light-emitting diode (OLED) performance. The HOMO–LUMO energy gap is greatly decreased by introduction of the –CHO group into the phenyl ring (4a and 4b see Scheme 1—sketched structures for all the investigated Ir(III) complexes). As a result, their absorption and emission spectra present red-shifting leading them to be potential red-emitting phosphors. Other complexes are all blue-emitting materials, indicating that the effect of the substituted position on the emitting color is negligible. However, the addition of the substituent on the para-position of the phenyl ring in the phenylimidazole ligand would increase the quantum yield and electroluminescence (EL) performance compared with that on the imidazole ring.
Co-reporter:Li Wang, Jinmiao Wen, Hongqing He and Jinglai Zhang
Dalton Transactions 2014 - vol. 43(Issue 7) pp:NaN2858-2858
Publication Date(Web):2013/11/11
DOI:10.1039/C3DT52616J
DFT and TD-DFT methods have been employed to theoretically investigate the properties of three recently synthesized green-emitting platinum(II) complexes (1–3) bearing tetradentate O^N^C^N ligands (O^C^N^C = 2-(4-(3,5-di-tert-butylphenyl)-6-(3-(pyridin-2-yl)phenyl)-pyridin-2-yl)phenolate and its derivatives) that have been testified to be good emitters in organic light-emitting diodes (OLEDs), especially for complex 3. The effect of the variation of the substituents on the electronic and optical properties is emphatically explored. Our calculation results reveal that the introduction of an electron-releasing group on one phenyl ring of the O^C^N^C ligand (complex 2) does not result in a distinct alteration of the spectra. However, the incorporation of the norbornane group to the O^C^N^C ligand (complex 3) leads to a blue-shift in the absorption and emission spectra as compared with 1. In addition, how the absorption and emission spectra are affected by the solvent polarity is studied. Both the absorption and the emission spectra display red-shifts of various degrees with the decrease of the solvent polarity. The different phosphorescent quantum yields of the three complexes are compared. It is reasonable to believe that the high 3MLCT (metal-to-ligand charge transfer) contribution and high percentage of the metallic character (Mc, %) in the emission process, as well as the largest vertical transition energy for 3, result in the highest quantum efficiency.
Co-reporter:Jieqiong Li, Qing Zhang, Hongqing He, Li Wang and Jinglai Zhang
Dalton Transactions 2015 - vol. 44(Issue 18) pp:NaN8589-8589
Publication Date(Web):2015/02/18
DOI:10.1039/C5DT00048C
The geometric and electronic structures, phosphorescence properties and the organic light-emitting diode (OLED) performance of a series of Ir(III) complexes based on bis[(4,6-di-fluorophenyl)-pyridinato-N,C2′]picolinate (FIrpic) were investigated by using density functional theory/time-dependent density functional theory (DFT/TD-DFT), including Ir(III)bis[2-(2,4-difluorophenyl)-4-(tert-butyl)pyridinato-N,C2′]picolinate (1a), Ir(III)bis[2-(2,4-difluorophenyl)-4-(n-heptyl)pyridinato-N,C2′]picolinate (2a), Ir(III)bis[2-(2,4-difluorophenyl)-4-(3-ethylheptyl)pyridinato-N,C2′]picolinate (3a), Ir(III)bis[2-(2,4-difluorophenyl)-4-(2,4,6-trimethylphenyl)pyridinato-N,C2′]picolinate (5a), and Ir(III)bis[2-(2,4-difluoro-3-(2,4,6-trimethylphenyl)phenyl)-pyridinato-N,C2′] picolinate (5b). To explore the influence of the substituted positions on the optical and electronic properties of the Ir(III) complexes, seven other new complexes were designed by introducing the substituted groups on the difluorophenyl rings or pyridine rings. After introducing the phenyl substituted groups on the pyridine or difluorophenyl rings of cyclometalated ligands, the HOMO–LUMO energy gap is decreased. Thus, the absorption spectra of 4a and 4b undergo a red-shifting, especially for 4a, and have a stronger absorption strength that will be beneficial to improving their quantum yields, which is proved by the further evaluation of the radiative (kr) and non-radiative (knr) rate constants. The combinations of a larger 3MLCT-3MC energy gap, smaller ΔES1–T1, and higher contribution of 3MLCT in the emission process result in the higher quantum yields for complexes 4a and 4b. Besides, the designed complexes 4a and 4b are considered to be potential candidates as blue-emitting materials with better equilibrium between the hole transport (λhole) and electron transport (λelectron).

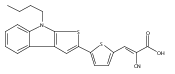
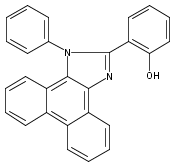
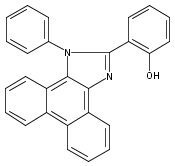

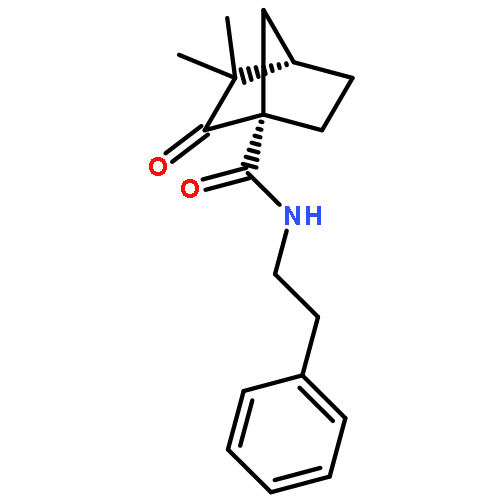
![GOLD, [[4-(PHENYLETHYNYL)PHENYL]ETHYNYL](TRIMETHYLPHOSPHINE)-](http://img.cochemist.com/ccimg/633400/633302-42-8.png)
![GOLD, [[4-(PHENYLETHYNYL)PHENYL]ETHYNYL](TRIMETHYLPHOSPHINE)-](http://img.cochemist.com/ccimg/633400/633302-42-8_b.png)



![Benzenesulfonamide, N-[2-(2-benzothiazolyl)phenyl]-4-methyl-](http://img.cochemist.com/ccimg/111200/111195-94-9.png)
![Benzenesulfonamide, N-[2-(2-benzothiazolyl)phenyl]-4-methyl-](http://img.cochemist.com/ccimg/111200/111195-94-9_b.png)



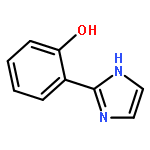
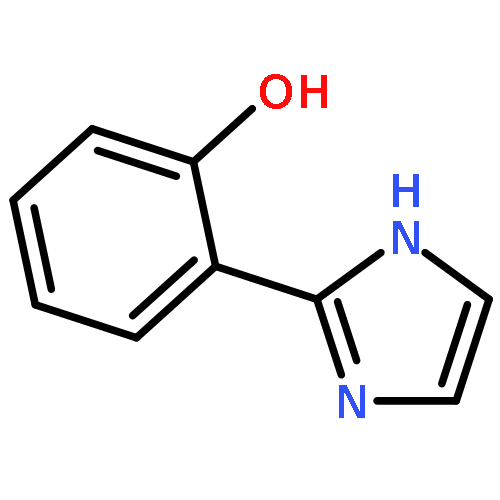
![dibenzo[a,h]phenazine-1,8-diol](http://img.cochemist.com/ccimg/26900/26846-41-3.png)
![dibenzo[a,h]phenazine-1,8-diol](http://img.cochemist.com/ccimg/26900/26846-41-3_b.png)
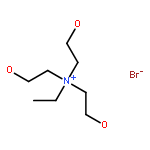
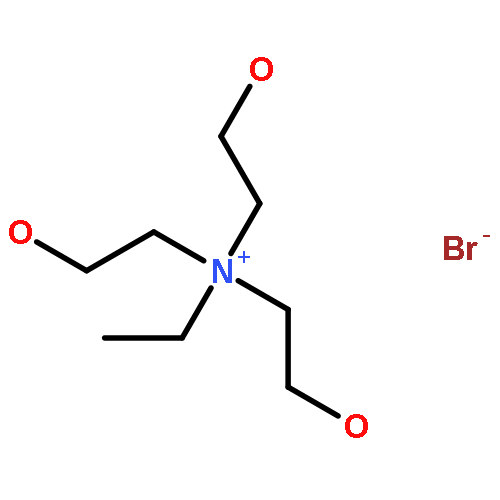
![6-(1,3-dihydro-2H-phenanthro[9,10-d]imidazol-2-ylidene)cyclohexa-2,4-dien-1-one](http://img.cochemist.com/ccimg/24300/24293-67-2.png)
![6-(1,3-dihydro-2H-phenanthro[9,10-d]imidazol-2-ylidene)cyclohexa-2,4-dien-1-one](http://img.cochemist.com/ccimg/24300/24293-67-2_b.png)


![1-(2,5-dichlorophenyl)-3-(piperidin-1-ylmethyl)-4,5-dihydro-2h-pyrrolo[2,3-c]pyrazole](http://img.cochemist.com/ccimg/5100/5073-63-2.png)
![1-(2,5-dichlorophenyl)-3-(piperidin-1-ylmethyl)-4,5-dihydro-2h-pyrrolo[2,3-c]pyrazole](http://img.cochemist.com/ccimg/5100/5073-63-2_b.png)
![2-(1H-Benzo[d]imidazol-2-yl)phenol](http://img.cochemist.com/ccimg/3000/2963-66-8.png)
![2-(1H-Benzo[d]imidazol-2-yl)phenol](http://img.cochemist.com/ccimg/3000/2963-66-8_b.png)




![Acetamide,N-[2-(5-hydroxy-1H-indol-3-yl)ethyl]-](http://img.cochemist.com/ccimg/1300/1210-83-9.png)
![Acetamide,N-[2-(5-hydroxy-1H-indol-3-yl)ethyl]-](http://img.cochemist.com/ccimg/1300/1210-83-9_b.png)









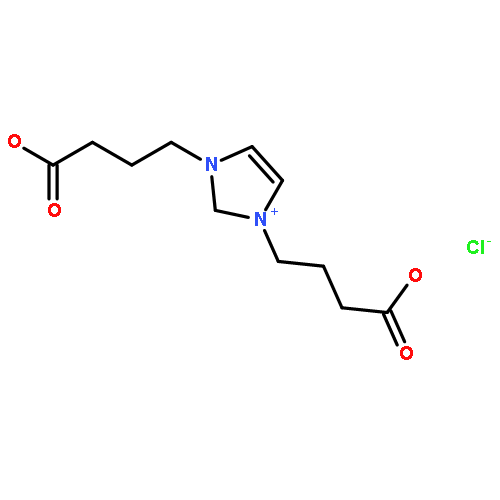
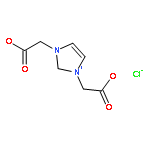
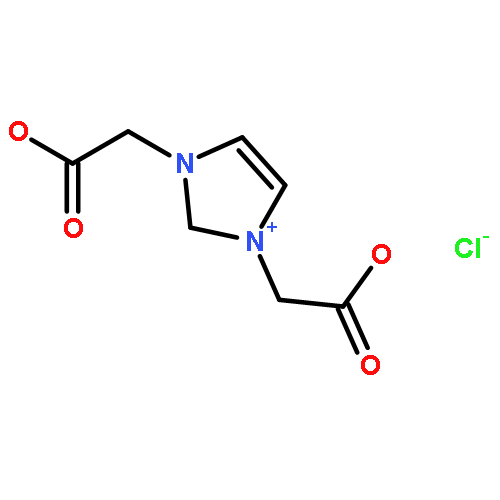
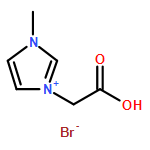
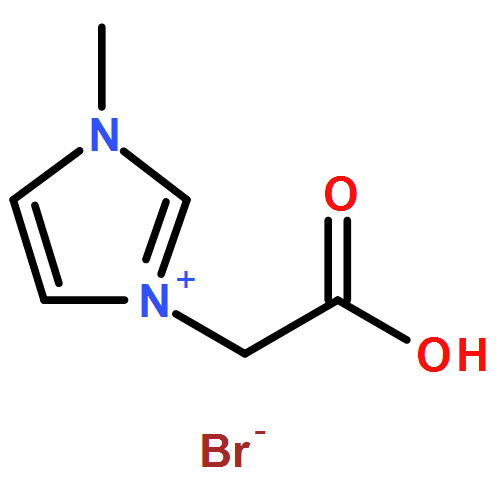
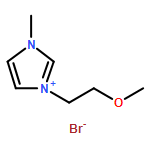
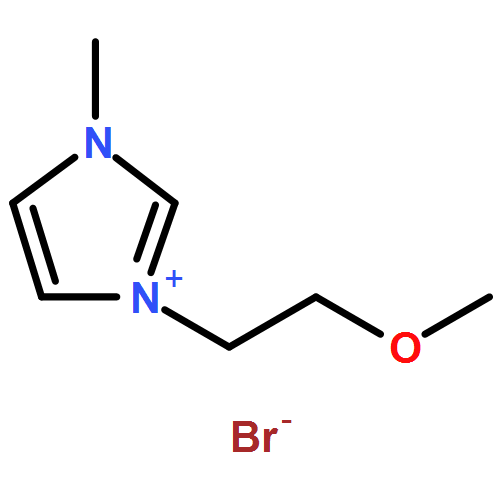


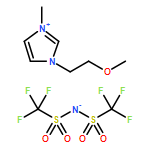


![Cyclohexanol, 1-[1-(phenylmethyl)-1H-1,2,3-triazol-4-yl]-](/data/chemimg/3722400/83027-66-1.png)
![Cyclohexanol, 1-[1-(phenylmethyl)-1H-1,2,3-triazol-4-yl]-](/data/chemimg/3722400/83027-66-1_b.png)





![1,1'-(benzene-1,4-diylbis{[(1E)-3-oxoprop-1-ene-1,3-diyl]benzene-3,1-diyl})dipyrrolidine-2,5-dione](http://img.cochemist.com/ccimg/5200/5187-14-4.png)
![1,1'-(benzene-1,4-diylbis{[(1E)-3-oxoprop-1-ene-1,3-diyl]benzene-3,1-diyl})dipyrrolidine-2,5-dione](http://img.cochemist.com/ccimg/5200/5187-14-4_b.png)