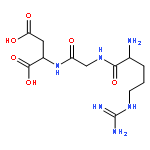
Co-reporter:Kerstin M. Galler, Lorenzo Aulisa, Katherine R. Regan, Rena N. D’Souza and Jeffrey D. Hartgerink
Journal of the American Chemical Society March 10, 2010 Volume 132(Issue 9) pp:3217-3223
Publication Date(Web):February 16, 2010
DOI:10.1021/ja910481t
Multidomain peptides are a class of amphiphilic self-assembling peptides with a modular ABA block motif in which the amphiphilic B block drives self-assembly while the flanking A blocks, which are electrostatically charged, control the conditions under which assembly takes place. Previously we have shown that careful selection of the amino acids in the A and B blocks allow one to control the self-assembled fiber length and viscoelastic properties of formed hydrogels. Here we demonstrate how the modular nature of this peptide assembler can be designed for biological applications. With control over fiber length and diameter, gelation conditions, and viscoelastic properties, we can develop suitable materials for biological applications. Going beyond a simple carrier for cell delivery, a biofunctional scaffold will interact with the cells it carries, promoting advantageous cell−matrix interactions. We demonstrate the design of a multidomain peptide into a bioactive variant by incorporation of a matrix metalloprotease 2 (MMP-2) specific cleavage site and cell adhesion motif. Gel formation and rheological properties were assessed and compared to related peptide hydrogels. Proteolytic degradation by collagenase IV was observed in a gel weight loss study and confirmed by specific MMP-2 degradation monitored by mass spectrometry and cryo-transmission electron microscopy (cryo-TEM). Combination of this cleavage site with the cell adhesion motif RGD resulted in increased cell viability and cell spreading and encouraged cell migration into the hydrogel matrix. Collectively the structural, mechanical, and bioactive properties of this multidomain peptide hydrogel make it suitable as an injectable material for a variety of tissue engineering applications.
Co-reporter:I-Che Li and Jeffrey D. Hartgerink
Journal of the American Chemical Society June 14, 2017 Volume 139(Issue 23) pp:8044-8044
Publication Date(Web):June 5, 2017
DOI:10.1021/jacs.7b04655
A great deal of effort has been invested in the design and characterization of systems which spontaneously assemble into nanofibers. These systems are interesting for their fundamental supramolecular chemistry and have also been shown to be promising materials, particularly for biomedical applications. Multidomain peptides are one such assembler, and in previous work we have demonstrated the reversibility of their assembly under mild and easily controlled conditions, along with their utility for time-controlled drug delivery, protein delivery, cell encapsulation, and cell delivery applications. Additionally, their highly compliant criteria for sequence selection allows them to be modified to incorporate protease susceptibility and biological-recognition motifs for cell adhesion and angiogenesis. However, control of their assembly has been limited to the formation of disorganized nanofibers. In this work, we expand our ability to manipulate multidomain-peptide assembly into parallel-aligned fiber bundles. Albeit this alignment is achieved by the shearing forces of syringe delivery, it is also dependent on the amino acid sequence of the multidomain peptide. The incorporation of the amino acid DOPA (3,4-dihydroxyphenylalanine) allows the self-assembled nanofibers to form an anisotropic hydrogel string under modest shear stress. The hydrogel string shows remarkable birefringence, and highly aligned nanofibers are visible in scanning electronic microscopy. Furthermore, the covalent linkage induced by DOPA oxidation allows covalent capture of the aligned nanofiber bundles, enhancing their birefringence and structural integrity.
Co-reporter:Katherine A. Clements, Amanda M. Acevedo-Jake, Douglas R. Walker, and Jeffrey D. Hartgerink
Biomacromolecules 2017 Volume 18(Issue 2) pp:
Publication Date(Web):January 18, 2017
DOI:10.1021/acs.biomac.6b01808
Osteogenesis imperfecta typically results from missense mutations in the collagen genome where the required glycine residues are replaced with another amino acid. Many models have attempted to replicate the structure of mutated collagen on the triple helix level. However, composition and register control of the triple helix is complicated and requires extreme precision, especially when these destabilizing mutations are present. Here we present mutations to a composition- and register-controlled AAB helix where one of the requisite glycines in the A chain of the triple helix is changed to serine or alanine. We see a loss of compositional control when the A chain is mutated, resulting in an A′BB composition that minimizes the number of mutations included in the triple helix. However, when both A and B chains are mutated and no nonmutated peptide chains are available, the designed A′A′B′ composition is reestablished. Our work shows the ability of the mutations to influence and alter the composition and register of the collagen triple helix.
Co-reporter:Amanda M. Acevedo-Jake, Katherine A. Clements, and Jeffrey D. Hartgerink
Biomacromolecules 2016 Volume 17(Issue 3) pp:
Publication Date(Web):February 9, 2016
DOI:10.1021/acs.biomac.5b01562
Osteogenesis imperfecta (OI) is a disease caused primarily by mutations of glycine in the standard (Xaa-Yaa-Gly)n repeat of a type I collagen triple helix. Type I collagen is an AAB heterotrimer which means that, depending on whether the A or B chain is mutated, the glycine substitution will appear once or twice. In this work we use designed axial charged pairs to self-assemble an AAB triple helix with controlled composition and register. We then substitute a single glycine of the B chain with alanine, serine, valine, aspartate, or arginine and assess the impact on the structure and folding of this OI mimic by CD, NMR, and restraint-guided modeling. We find that alanine and serine substitutions are tolerated, resulting in localized disruptions to the triple helix structure, while bulkier amino acids result in alternatively folded structures. This work demonstrates the potential of axial charged pairs to control the structure of low stability triple helices and also helps to elucidate the structure and folding challenges associated with OI-type mutations in collagen.
Co-reporter:I-Che Li, Amanda N. Moore, and Jeffrey D. Hartgerink
Biomacromolecules 2016 Volume 17(Issue 6) pp:
Publication Date(Web):June 2, 2016
DOI:10.1021/acs.biomac.6b00309
The clinical administration of many small molecule hydrophobic drugs is challenged by the insolubility of these drugs under physiological conditions. Because of this, the development of biocompatible scaffolds capable of effectively delivering hydrophobic drug molecules is of particular interest. Multidomain peptides (MDPs) provide biocompatible hydrogel scaffolds that are injectable and space-conforming, allowing for in situ delivery of a variety of drugs. Here we demonstrate that through manipulation of peptide primary sequence, a molecular cavity can be incorporated into the hydrophobic core of these peptide nanofibers allowing for encapsulation and delivery of small molecule drugs with poor water solubility. Using SN-38, daunorubicin, diflunisal, etodolac, levofloxacin, and norfloxacin, we demonstrate drug encapsulation and release from multidomain peptide fibers. Steady-state fluorescence and drug release studies show that hydrogels loaded with SN-38, diflunisal, and etodolac exhibit prolonged drug release profiles due to intrafibrillar drug encapsulation. This study establishes multidomain peptides as promising carriers for localized in situ delivery of small molecule drugs with poor water solubility.
Co-reporter:Vivek A. Kumar; Siyu Shi; Benjamin K. Wang; I-Che Li; Abhishek A. Jalan; Biplab Sarkar; Navindee C. Wickremasinghe
Journal of the American Chemical Society 2015 Volume 137(Issue 14) pp:4823-4830
Publication Date(Web):April 1, 2015
DOI:10.1021/jacs.5b01549
Self-assembly of multidomain peptides (MDP) can be tailored to carry payloads that modulate the extracellular environment. Controlled release of growth factors, cytokines, and small-molecule drugs allows for unique control of in vitro and in vivo responses. In this study, we demonstrate this process of ionic cross-linking of peptides using multivalent drugs to create hydrogels for sustained long-term delivery of drugs. Using phosphate, heparin, clodronate, trypan, and suramin, we demonstrate the utility of this strategy. Although all multivalent anions result in good hydrogel formation, demonstrating the generality of this approach, suramin led to the formation of the best hydrogels per unit concentration and was studied in greater detail. Suramin ionically cross-linked MDP into a fibrous meshwork as determined by scanning and transmission electron microscopy. We measured material storage and loss modulus using rheometry and showed a distinct increase in G′ and G″ as a function of suramin concentration. Release of suramin from scaffolds was determined using UV spectroscopy and showed prolonged release over a 30 day period. Suramin bioavailability and function were demonstrated by attenuated M1 polarization of THP-1 cells compared to positive control. Overall, this design strategy has allowed for the development of a novel class of polymeric delivery vehicles with generally long-term release and, in the case of suramin, cross-linked hydrogels that can modulate cellular phenotype.
Co-reporter:Amanda M. Acevedo-Jake, Abhishek A. Jalan, and Jeffrey D. Hartgerink
Biomacromolecules 2015 Volume 16(Issue 1) pp:
Publication Date(Web):December 10, 2014
DOI:10.1021/bm501281a
The collagen triple helix consists of three supercoiled solvent-exposed polypeptide chains, and by dry weight it is the most abundant fold in mammalian tissues. Many factors affecting the structure and stability of collagen have been determined through the use of short synthetically prepared peptides, generally called collagen-mimetic peptides (CMPs). NMR (nuclear magnetic resonance spectroscopy) investigations into the molecular structure of CMPs have suffered from large amounts of signal overlap and degeneracy because of collagen’s repetitive primary sequence, the close and symmetric packing of the three chains and the identical peptide sequences found in homotrimers. In this paper a peptide library is prepared in which a single isotopic 15N-Gly label is moved sequentially along the peptide backbone. Our approach allows for a more explicit examination of local topology than available in past reports. This reveals larger regions of disorder at the C-terminus than previously detected by crystallographic or NMR studies, and here C-terminal fraying is seen to extend for six amino acids in a (POG)10 sequence. Furthermore, small sequence changes at the N-terminus greatly influence the degree of this localized disorder and may be useful in the future design of CMPs to maximize collagen’s interstrand hydrogen bonding pattern. Our approach and data serves as a reference for future CMP characterizations to determine the quality and extent of folding.
Co-reporter:Vivek A. Kumar, Nichole L. Taylor, Siyu Shi, Navindee C. Wickremasinghe, Rena N. D'Souza, Jeffrey D. Hartgerink
Biomaterials 2015 52() pp: 71-78
Publication Date(Web):
DOI:10.1016/j.biomaterials.2015.01.079
Co-reporter:Navindee C. Wickremasinghe, Vivek A. Kumar, Siyu Shi, and Jeffrey D. Hartgerink
ACS Biomaterials Science & Engineering 2015 Volume 1(Issue 9) pp:845
Publication Date(Web):August 20, 2015
DOI:10.1021/acsbiomaterials.5b00210
Multidomain peptide (MDP) nanofibers create scaffolds that can present bioactive cues to promote biological responses. Orthogonal self-assembly of MDPs and growth-factor-loaded liposomes generate supramolecular composite hydrogels. These composites can act as delivery vehicles with time-controlled release. Here we examine the controlled release of placental growth factor-1 (PlGF-1) for its ability to induce angiogenic responses. PlGF-1 was loaded either in MDP matrices or within liposomes bound inside MDP matrices. Scaffolds showed expected rapid infiltration of macrophages. When released through liposomes incorporated in MDP gels (MDP(Lipo)), PlGF-1 modulates HUVEC VEGF receptor activation in vitro and robust vessel formation in vivo. These loaded MDP(Lipo) hydrogels induce a high level of growth-factor-mediated neovascular maturity. MDP(Lipo) hydrogels offer a biocompatible and injectable platform to tailor drug delivery and treat ischemic tissue diseases.Keywords: angiogenesis; cellular infiltration; liposomal encapsulation; multidomain peptide; PlGF-1; self-assembly
Co-reporter:Vivek A. Kumar, Navindee C. Wickremasinghe, Siyu Shi, and Jeffrey D. Hartgerink
ACS Biomaterials Science & Engineering 2015 Volume 1(Issue 12) pp:1300
Publication Date(Web):October 22, 2015
DOI:10.1021/acsbiomaterials.5b00356
Controlling perioperative bleeding is of critical importance to minimize hemorrhaging and fatality. Patients on anticoagulant therapy such as heparin have diminished clotting potential and are at risk for hemorrhaging. Here we describe a self-assembling nanofibrous peptide hydrogel (termed SLac) that on its own can act as a physical barrier to blood loss. SLac was loaded with snake-venom derived Batroxobin (50 μg/mL) yielding a drug-loaded hydrogel (SB50). SB50 was potentiated to enhance clotting even in the presence of heparin. In vitro evaluation of fibrin and whole blood clotting helped identify appropriate concentrations for hemostasis in vivo. Batroxobin-loaded hydrogels rapidly (within 20s) stop bleeding in both normal and heparin-treated rats in a lateral liver incision model. Compared to standard of care, Gelfoam, and investigational hemostats such as Puramatrix, only SB50 showed rapid liver incision hemostasis post surgical application. This snake venom-loaded peptide hydrogel can be applied via syringe and conforms to the wound site resulting in hemostasis. This demonstrates a facile method for surgical hemostasis even in the presence of anticoagulant therapies.Keywords: hemostasis; multidomain peptide; self-assembly; supramolecular chemistry
Co-reporter:Vivek A. Kumar, Nichole L. Taylor, Siyu Shi, Benjamin K. Wang, Abhishek A. Jalan, Marci K. Kang, Navindee C. Wickremasinghe, and Jeffrey D. Hartgerink
ACS Nano 2015 Volume 9(Issue 1) pp:860
Publication Date(Web):January 13, 2015
DOI:10.1021/nn506544b
Major limitations of current tissue regeneration approaches using artificial scaffolds are fibrous encapsulation, lack of host cellular infiltration, unwanted immune responses, surface degradation preceding biointegration, and artificial degradation byproducts. Specifically, for scaffolds larger than 200–500 μm, implants must be accompanied by host angiogenesis in order to provide adequate nutrient/waste exchange in the newly forming tissue. In the current work, we design a peptide-based self-assembling nanofibrous hydrogel containing cell-mediated degradation and proangiogenic moieties that specifically address these challenges. This hydrogel can be easily delivered by syringe, is rapidly infiltrated by cells of hematopoietic and mesenchymal origin, and rapidly forms an extremely robust mature vascular network. Scaffolds show no signs of fibrous encapsulation and after 3 weeks are resorbed into the native tissue. These supramolecular assemblies may prove a vital paradigm for tissue regeneration and specifically for ischemic tissue disease.Keywords: angiogenesis; multidomain peptide; self-assembly; supramolecular chemistry;
Co-reporter:Abhishek A. Jalan ; Katherine A. Jochim
Journal of the American Chemical Society 2014 Volume 136(Issue 21) pp:7535-7538
Publication Date(Web):May 14, 2014
DOI:10.1021/ja5001246
In a canonical collagen triple helix, three peptides self-assemble into a supercoiled motif with a one-amino-acid offset between the peptide chains. Design of triple helices that contain more than one residue offset is lucrative, as it leaves the non-covalent interactions unsatisfied at the termini and renders the termini “sticky” to further self-assemble into collagen-like nanofibers. Here we use lysine–glutamate axial salt-bridges to design a heterotrimeric collagen triple helix, ABC-1, containing a non-canonical offset of four residues between the peptide chains. The four-residue offset is necessary to prevent aggregation, which would prevent characterization of the non-canonical chain arrangement at the molecular level by NMR spectroscopy. A second heterotrimer, ABC-2, also stabilized by axial salt-bridges, is designed containing a canonical one-amino-acid offset to facilitate comparison of structure and stability by CD and NMR. ABC-1 and ABC-2 demonstrate our ability to modulate chain offset in a collagen triple helix. This lays the groundwork to design longer, and therefore stickier, offsets allowing access to a new class of collagen-related nanostructures.
Co-reporter:Marci K. Kang, John S. Colombo, Rena N. D’Souza, and Jeffrey D. Hartgerink
Biomacromolecules 2014 Volume 15(Issue 6) pp:
Publication Date(Web):May 12, 2014
DOI:10.1021/bm500075r
Here we report three new nanofibrous, self-assembling multidomain peptide (MDP) sequences and examine the effect of sequence on the morphology and expansion of encapsulated Stem cells from Human Exfoliated Deciduous teeth (SHED). We modified our previously reported set of serine-based MDPs, changing the serine residues in the amphiphilic region to threonine. The three new threonine-based sequences self-assemble into antiparallel β-sheet nanofibers, confirmed by CD and IR. AFM and negative-stained TEM show that the nanofibers formed by the new sequences are more curved than their serine-containing predecessors. Despite this change in nanofiber morphology, SEM illustrates that all three new sequences still form porous hydrogels. K(TL)2SLRG(TL)3KGRGDS, with a designed cleavage site, is able to be degraded by Matrix Metalloprotease 2. We then examine SHED cell response to these new sequences as well as their serine-based predecessors. We observe faster cell attachment and spreading in hydrogels formed by K2(SL)6K2GRGDS and K(SL)3RG(SL)3KGRGDS. By day 3, the SHEDs in all of the serine-based sequences exhibit a fibroblast-like morphology. Additionally, the SHED cells expand more rapidly in the serine-based gels while the cell number remains relatively constant in the threonine-based peptides. In hydrogels formed by K2(TL)6K2GRGDS and K(TL)2SLRG(TL)3KGRGDS, this low expansion rate is accompanied by changes in morphology where SHEDs exhibit a stellate morphology after 3 days in culture; however, by day 7 they appear more fibroblast-shaped. Throughout the duration of the experiment, the SHED cells encapsulated in the K2(TL)6K2 hydrogels remain rounded. These results suggest that the basic MDP structure easily accommodates modifications in sequence and, for SHED cells, the threonine-containing gels require the integrin-binding RGDS sequence for cell attachment to occur, while the serine-based gels are less selective and support an increase in cell number, regardless of the presence or absence of RGDS.
Co-reporter:Vivek A. Kumar, Nichole L. Taylor, Abhishek A. Jalan, Lyahn K. Hwang, Benjamin K. Wang, and Jeffrey D. Hartgerink
Biomacromolecules 2014 Volume 15(Issue 4) pp:
Publication Date(Web):April 2, 2014
DOI:10.1021/bm500091e
Collagen is a major component of the extracellular matrix and plays a wide variety of important roles in blood clotting, healing, and tissue remodeling. Natural, animal derived, collagen is used in many clinical applications but concerns exist with respect to its role in inflammation, batch-to-batch variability, and possible disease transfection. Therefore, development of synthetic nanomaterials that can mimic the nanostructure and properties of natural collagen has been a heavily pursued goal in biomaterials. Previously, we reported on the design and multihierarchial self-assembly of a 36 amino acid collagen mimetic peptide (KOD) that forms nanofibrous triple helices that entangle to form a hydrogel. In this report, we utilize this nanofiber forming collagen mimetic peptide as a synthetic biomimetic matrix useful in thrombosis. We demonstrate that nanofibrous KOD synthetic collagen matrices adhere platelets, activate them (indicated by soluble P-selectin secretion), and clot plasma and blood similar to animal derived collagen and control surfaces. In addition to the thrombotic potential, THP-1 monocytes incubated with our KOD collagen mimetic showed minimal proinflammatory cytokine (TNF-α or IL-1β) production. Together, the data presented demonstrates the potential of a novel synthetic collagen mimetic as a hemostat.
Co-reporter:Navindee C. Wickremasinghe, Vivek A. Kumar, and Jeffrey D. Hartgerink
Biomacromolecules 2014 Volume 15(Issue 10) pp:
Publication Date(Web):September 9, 2014
DOI:10.1021/bm500856c
Progress in self-assembly and supramolecular chemistry has been directed toward obtaining macromolecular assemblies with higher degrees of complexity, simulating the highly structured environment in natural systems. One approach to this type of complexity are multistep, multicomponent, self-assembling systems that allow approaches comparable to traditional multistep synthetic organic chemistry; however, only a few examples of this approach have appeared in the literature. Our previous work demonstrated nanofibrous mimics of the extracellular matrix. Here we demonstrate the ability to create a unique hydrogel, developed by stepwise self-assembly of multidomain peptide fibers and liposomes. The two-component system allows for controlled release of bioactive factors at multiple time points. The individual components of the self-assembled gel and the composite hydrogel were characterized by TEM, SEM, and rheometry, demonstrating that peptide nanofibers and lipid vesicles both retain their structural integrity in the composite gel. The rheological robustness of the hydrogel is shown to be largely unaffected by the presence of liposomes. Release studies from the composite gels loaded with different growth factors EGF, MCP-1, and PlGF-1 showed delay and prolongation of release by liposomes entrapped in the hydrogel compared to more rapid release from the hydrogel alone. This bimodal release system may have utility in systems where timed cascades of biological signals may be valuable, such as in tissue regeneration.
Co-reporter:Abhishek A. Jalan ; Borries Demeler
Journal of the American Chemical Society 2013 Volume 135(Issue 16) pp:6014-6017
Publication Date(Web):April 10, 2013
DOI:10.1021/ja402187t
Hydroxyproline plays a major role in stabilizing collagenous domains in eukaryotic organisms. Lack of this modification is associated with significant lowering in the thermal stability of the collagen triple helix and may also affect fibrillogenesis and folding of the peptide chains. In contrast, even though bacterial collagens lack hydroxyproline, their thermal stability is comparable to that of fibrillar collagen. This has been attributed to the high frequency of charged amino acids found in bacterial collagen. Here we report a thermally stable hydroxyproline-free ABC heterotrimeric collagen mimetic system composed of decapositive and decanegative peptides and a zwitterionic peptide. None of the peptides contain hydroxyproline, and furthermore the zwitterionic peptide does not even contain proline. The heterotrimer is electrostatically stabilized via multiple interpeptide lysine-aspartate and lysine-glutamate salt bridges and maintains good thermal stability with a melting temperature of 37 °C. The ternary peptide mixture also populates a single composition ABC heterotrimer as confirmed by circular dichroism (CD) and nuclear magnetic resonance (NMR) spectroscopy. This system illustrates the power of axial salt bridges to direct and stabilize the self-assembly of a triple helix and may be useful in analogous designs in expression systems where the incorporation of hydroxyproline is challenging.
Co-reporter:Erica L. Bakota, Ozge Sensoy, Beytullah Ozgur, Mehmet Sayar, and Jeffrey D. Hartgerink
Biomacromolecules 2013 Volume 14(Issue 5) pp:
Publication Date(Web):March 12, 2013
DOI:10.1021/bm4000019
Self-assembling multidomain peptides have been shown to have desirable properties, such as the ability to form hydrogels that rapidly recover following shear-thinning and the potential to be tailored by amino acid selection to vary their elasticity and encapsulate and deliver proteins and cells. Here we describe the effects of substitution of aliphatic hydrophobic amino acids in the central domain of the peptide for the aromatic amino acids phenylalanine, tyrosine, and tryptophan. While the basic nanofibrous morphology is retained in all cases, selection of the particular core residues results in switching from antiparallel hydrogen bonding to parallel hydrogen bonding in addition to changes in nanofiber morphology and in hydrogel rheological properties. Peptide nanofiber assemblies are investigated by circular dichroism polarimetry, infrared spectroscopy, atomic force microscopy, transmission and scanning electron microscopy, oscillatory rheology, and molecular dynamics simulations. Results from this study will aid in designing next generation cell scaffolding materials.
Co-reporter:Fang Wei;Jorge A. Fallas
Macromolecular Rapid Communications 2012 Volume 33( Issue 17) pp:1445-1452
Publication Date(Web):
DOI:10.1002/marc.201200221
Abstract
In this study, we examine eight ABC heterotrimers whose self-assembly is directed through electrostatic interactions. Oppositely charged pairs of amino acids, with varying side chain length, were assessed for their ability to stabilize a triple helix. Aspartate-lysine was found to result in the most thermally stable helix followed by lysine-glutamate, ornithine-aspartate, and finally ornithine-glutamate. When the sequence position of these charged amino acids was reversed from what is normally observed in nature, triple helix stability and compositional purity were significantly reduced. We examine the effect of salt on triple helix stability and observe that increased salt concentration reduces the thermal stability of heterotrimers by an average of 5 °C, but does not disrupt helix assembly. It was also found that some highly positively charged homotrimers can be stabilized in the presence of phosphate anions.
Co-reporter:Jorge A. Fallas ; Michael A. Lee ; Abhishek A. Jalan
Journal of the American Chemical Society 2011 Volume 134(Issue 3) pp:1430-1433
Publication Date(Web):December 22, 2011
DOI:10.1021/ja209669u
Design of heterotrimeric ABC collagen triple helices is challenging due to the large number of competing species that may be formed. Given the required one amino acid stagger between adjacent peptide strands in this fold, a ternary mixture of peptides can form as many as 27 triple helices with unique composition or register. Previously we have demonstrated that electrostatic interactions can be used to bias the helix population toward a desired target. However, homotrimeric assemblies have always remained the most thermally stable species in solution and therefore comprised a significant component of the peptide mixture. In this work we incorporate complementary modifications to this triple-helical design strategy to destabilize an undesirable competing state while compensating for this destabilization in the desired ABC composition. The result of these modifications is a new ABC triple-helical system with high thermal stability and control over composition, as observed by NMR. An additional set of modifications, which exchanges aspartate for glutamate, results in an overall lowering of stability of the ABC triple helix yet shows further improvement in the system’s specificity. This rationally designed system helps to elucidate the rules governing the self-assembly of synthetic collagen triple helices and sheds light on the biological mechanisms of collagen assembly.
Co-reporter:Lesley E. R. O’Leary ; Jorge A. Fallas
Journal of the American Chemical Society 2011 Volume 133(Issue 14) pp:5432-5443
Publication Date(Web):March 23, 2011
DOI:10.1021/ja111239r
Although collagen is the most abundant protein in the human body and has at least 28 types, research involving collagen mimetic systems only recently began to consider the innate ability of collagen to control helix composition and register. Collagen triple helices can be homotrimeric or heterotrimeric, and while some types of natural collagen form only one specific composition of helix, others can form multiple compositions. It is critical to fully understand and, if possible, reproduce the control that native collagen has on helix composition and register. In this Article, we utilize both positive and negative design for the assembly of specific AAB heterotrimers using charged amino acids to form intrahelix electrostatic interactions, which promote heterotrimer formation and simultaneously discourage homotrimers. Homotrimers are further discouraged by reducing hydroxyproline content, which would otherwise lead to nonspecific promotion of triple helix formation. We combine peptides in a 2:1 ratio in which the more abundant peptide has a charge 1/2 and opposite of the less abundant peptide, which can result in the formation of a zwitterionically neutral AAB heterotrimer. Using this approach, we are able to design collagen mimetic systems with full control over the composition of the resulting triple helix. All previous reports on synthetic collagen heterotrimers have shown mixed populations with respect to composition due to varying amounts of residual homotrimers. Our results yield a greater understanding of the self-assembly of collagenous sequences as well as provide a novel design scheme, both positive and negative, for the synthesis of extracellular matrix mimetics.
Co-reporter:Erica L. Bakota, Yin Wang, Farhad R. Danesh, and Jeffrey D. Hartgerink
Biomacromolecules 2011 Volume 12(Issue 5) pp:
Publication Date(Web):March 21, 2011
DOI:10.1021/bm200035r
Peptide hydrogels show immense promise as therapeutic materials. Here we present a rationally designed multidomain peptide that self-assembles into nanofibers approximately 8 nm wide, 2 nm high, and micrometers in length in the presence of Mg2+. At a concentration of 1% by weight, the peptide forms an extensive nanofibers network that results in a physically cross-linked viscoelastic hydrogel. This hydrogel undergoes shear thinning and then quickly recovers nearly 100% of its elastic modulus when the shearing force is released, making it ideal for use as an injectable material. When placed in the presence of human embryonic stem cells (ESCs), the nanofibrous hydrogel acts like a sponge, soaking up the vast array of growth factors and cytokines released by the ESCs. The peptide hydrogel sponge can then be removed from the presence of the ESCs and placed in a therapeutic environment, where it can subsequently release these components. In vitro experiments demonstrate that release of stem cell secretome from these hydrogels in the presence of glomerular epithelial cells treated with high glucose significantly decreased protein permeability in a model of diabetes-induced kidney injury. Tracking experiments were then performed to determine the fate of the hydrogel upon injection in vivo. Hydrogels labeled with a Gd3+ MRI contrast agent were injected into the abdominal cavity of mice and found to remain localized over 24 h. This implies that the hydrogel possesses sufficient rigidity to remain localized and release stem cell secretome over time rather than immediately dissolving in the abdominal cavity. Together, the shear thinning and recovery as observed by rheometry as well as secretome absorption and release in vivo demonstrate the potential of the nanofibrous multidomain peptide hydrogel as an injectable delivery agent.
Co-reporter:Erica L. Bakota, Lorenzo Aulisa, Kerstin M. Galler, and Jeffrey D. Hartgerink
Biomacromolecules 2011 Volume 12(Issue 1) pp:
Publication Date(Web):December 6, 2010
DOI:10.1021/bm1010195
The rheological properties of the environment in which a cell lives play a key role in how the cells will respond to that environment and may modify cell proliferation, morphology and differentiation. Effective means of modifying these properties are needed, particularly for peptide hydrogels which are generally relatively weak and soft. In this report we describe the enzymatic cross-linking of a nanofibrous multidomain peptide hydrogel. When this method was used, the storage modulus, G′, could be increased to over 4000 Pa without changes in hydrogel concentration and without dramatic changes in nanostructural architecture. Enzymatic cross-linking represents a mild and simple method for increasing the mechanical strength of peptide hydrogels in applications for which the robustness of the gel is essential. This method should be suitable for a broad array of peptide hydrogels containing lysine such as those currently under study by many different groups.
Co-reporter:Jorge A. Fallas, Lesley E. R. O'Leary and Jeffrey D. Hartgerink
Chemical Society Reviews 2010 vol. 39(Issue 9) pp:3510-3527
Publication Date(Web):30 Jul 2010
DOI:10.1039/B919455J
Collagen is a fascinating system of proteins that undergo a multi-step, hierarchical self-assembly which starts from individual peptide chains that assemble into a canonical triple helix. These triple helices then assemble into higher order structures which are often, but not always, fibrous in nature. While collagen is the most abundant protein in the human body, the details of its structure and mechanism of assembly are surprisingly poorly understood. This critical review will focus on small peptide systems, commonly referred to as collagen mimetic peptides (CMPs) which have been used successfully to help unravel some of the mystery of this complex structure. We will discuss homotrimeric CMPs, which are the most commonly researched subject in this field, and the structure of the collagen triple helix in detail and the factors that contribute to its stabilization. We will also cover how CMPs have been used to study breaks in triple helical domains as models for connective tissue diseases and, finally, how they have been used to understand the interactions of collagenous proteins with cell-surface receptors. Additionally, we will focus on heterotrimeric CMPs, a relatively new area of collagen research. Finally, we will deal with CMPs used as models for higher level self-assembly and also as materials that are designed to mimic the function of collagens in the extracellular matrix (178 references).
Co-reporter:Lesley E. Russell ; Jorge A. Fallas
Journal of the American Chemical Society 2010 Volume 132(Issue 10) pp:3242-3243
Publication Date(Web):January 8, 2010
DOI:10.1021/ja909720g
How collagen is able to obtain control of helix composition and register is poorly understood yet is critical for determining the structure and properties of the most abundant protein in the human body. In humans there are 28 known types of collagen that can form homotrimeric (AAA) or heterotrimeric (AAB and ABC) compositions. Additionally, because of a single amino acid offset between peptide chains in the triple helix, distinct heterotrimers of different registers can be formed. In this communication we describe an AAB collagen heterotrimer with controlled composition and register. This is the first report of a collagen heterotrimer whose thermal stability is greater than that of any of its component parts and therefore is the dominant species in solution. The design concept is simple: combination of peptides who follow the canonical (X-Y-Gly)n amino acid repeat in a 2:1 ratio in which the more abundant peptide has a charge 1/2 and opposite of the other should result in the formation of an AAB heterotrimeric collagen helix. This will be the dominant species because it is neutral (zwitterionic) while homotrimers should be destabilized because of charge repulsion. Here we show by circular dichroism, differential scanning calorimetry, and NMR that, in a 2:1 mixture of the peptides (EOGPOG)5 and (PRG)10, the AAB heterotrimer is the dominant structure in solution and melts 10 °C higher in temperature than the next most stable species.
Co-reporter:Kerstin M. Galler, Rena N. D'Souza and Jeffrey D. Hartgerink
Journal of Materials Chemistry A 2010 vol. 20(Issue 40) pp:8730-8746
Publication Date(Web):15 Sep 2010
DOI:10.1039/C0JM01207F
Engineering oral tissues as a multidisciplinary approach to build complex structures such as bone, teeth or soft dental tissues remains a challenging endeavor which will also require significant additional development of materials chemistry before it will be successful. We will highlight areas of recent success and describe major challenges which the materials chemistry community, in collaboration with clinicians, must still overcome. The isolation of stem cell populations from various sources in the oral cavity and advances in utilizing their differentiation potential has been driving the field forward. So far, bioinert materials have mainly been used as carriers and delivery vehicles, relying on the intrinsic cellular competence to form tissues. As this may not suffice to induce regeneration, there is a need for novel biomimetic scaffolds capable of providing chemical and mechanical cues to promote multiple specific interactions between cells and matrix. These signals can orchestrate processes such as cell adhesion, migration, differentiation, matrix synthesis, mineralization, and/or vasculogenesis. In this review, we give a brief description of oral anatomy and pathology, state-of-the-art treatment methods and their shortcomings. We provide an overview of current strategies to fabricate bioactive matrices, with an emphasis on nanostructured materials, and we suggest design principles for scaffolding systems specifically tailored towards dental tissue regeneration. In this review, we envision future approaches based on these emerging areas that rely on recent developments in tissue engineering and stem cell research. At the interface between material science and biology, cellular response can be controlled by materials chemistry, and potential applications for regenerative strategies are evolving.
Co-reporter:Lorenzo Aulisa, Nico Forraz, Colin McGuckin, Jeffrey D. Hartgerink
Acta Biomaterialia 2009 Volume 5(Issue 3) pp:842-853
Publication Date(Web):March 2009
DOI:10.1016/j.actbio.2008.11.002
Abstract
HOX genes encode conserved transcription factors that control the morphological diversification along the anteroposterior body axis. HOX proteins bind to DNA through a highly conserved 60 amino acid sequence called the homeodomain, and greater DNA binding specificity and stability are achieved when it forms complexes with cofactors such as PBX and MEIS in humans. In particular, HOX proteins from paralog groups 1–8, interact with PBX proteins via a specific and highly conserved hydrophobic six amino acid sequence localized in the N-terminal region of HOX. In several oncogenic transformations, deregulated HOX gene expression has been observed, indicating an involvement of these transcriptional regulators in carcinogenesis and metastasis. Inhibition of the HOX–PBX interaction could be a strategy to control the abnormal proliferation of these cancer cells. In this study we describe a small designed peptide amphiphile (PA) which self-assembles into micelles and shows inhibition of T3M4 pancreatic cancer cells, K562 leukemia cells and MJT1 melanoma cells while non-cancerous fibroblast NIH 3T3 cells are less affected. This molecule contains three critical regions: a 9-amino-acid sequence designed to disrupt HOX/PBX/DNA complex formation, a 16-amino-acid sequence to deliver the peptide into the cell and a 16-carbon-acyl chain which we show leads to the molecule’s self-assembly and significantly enhances the effectiveness of the molecule to slow cell proliferation.
Co-reporter:Erica L. Bakota, Lorenzo Aulisa, Dmitri A. Tsyboulski, R. Bruce Weisman and Jeffrey D. Hartgerink
Biomacromolecules 2009 Volume 10(Issue 8) pp:
Publication Date(Web):July 15, 2009
DOI:10.1021/bm900382a
We present a series of short, multidomain peptides as biocompatible solubilizing agents of single-walled carbon nanotubes (SWCNTs). These peptides are organized into an ABA block motif, where the A block is composed of charged amino acids, such as glutamic acid, and the B block is composed of alternating hydrophilic and hydrophobic residues. The hydrophobic amino acid residues interact with SWCNT sidewalls, while the hydrophilic residues interact primarily with water in an aqueous solution. When many peptides assemble along the length of the nanotube, it becomes effectively encapsulated within a peptide nanofiber. This noncovalent interaction between the peptide and the nanotube solubilizes SWCNTs while keeping the electronic structure of the nanotube intact, thereby preserving the optical and electrical properties that make SWCNTs promising for use in biological applications. To assess the toxicity of these peptide coatings, they were added to cultures of NIH 3T3 mouse fibroblasts and the effect on cell viability was measured. Toxicity was found to be far lower than for ionic surfactants typically used for SWCNT suspension and similar to Pluronics. The near-IR fluorescence intensity of SWCNTs in peptide suspensions was comparable to that in Pluronics. Five surfactants were tested for their effect on the proliferation of NIH 3T3 cells with and without SWCNTs. Although some differences were observed among surfactants, in no case did the presence of SWCNTs make a statistically significant difference. Based on their ability to solubilize SWCNTs, the fluorescence of the suspended tubes, their minimal impact on cell viability, and their potential for easy chemical modification, multidomain peptides have been found to have excellent potential as a biocompatible surfactant for suspension of SWCNTs.
Co-reporter:Lorenzo Aulisa, He Dong and Jeffrey D. Hartgerink
Biomacromolecules 2009 Volume 10(Issue 9) pp:
Publication Date(Web):August 25, 2009
DOI:10.1021/bm900634x
An important goal in supramolecular chemistry is to achieve controlled self-assembly of molecules into well-defined nanostructures and the subsequent control over macroscopic properties resulting from the formation of a nanostructured material. Particularly important to our lab is control over viscoelasticity and bioactivity. Recently we described a multidomain peptide motif that can self-assemble into nanofibers 2 × 6 × 120 nm. In this work we describe how sequence variations in this general motif can be used to create nanofibrous gels that have storage moduli, which range over 2 orders of magnitude and can undergo shear thinning and shear recovery while at the modest concentration of 1% by weight. Gel formation is controlled by addition of oppositely charged multivalent ions such as phosphate and magnesium and can be carried out at physiological pH. We also demonstrate how maximum strength can be obtained via covalent capture of the nanofibers through disulfide bond formation. Together these hydrogel properties are ideally suited as injectable materials for drug and cell delivery.
Co-reporter:WilliamC. Pomerantz;ViranyM. Yuwono;ClaireL. Pizzey;JefferyD. Hartgerink ;NicholasL. Abbott ;SamuelH. Gellman
Angewandte Chemie 2008 Volume 120( Issue 7) pp:1261-1264
Publication Date(Web):
DOI:10.1002/ange.200704372
Co-reporter:WilliamC. Pomerantz;ViranyM. Yuwono;ClaireL. Pizzey;JefferyD. Hartgerink ;NicholasL. Abbott ;SamuelH. Gellman
Angewandte Chemie International Edition 2008 Volume 47( Issue 7) pp:1241-1244
Publication Date(Web):
DOI:10.1002/anie.200704372
Co-reporter:Leah S. Witus, John-David R. Rocha, Virany M. Yuwono, Sergey E. Paramonov, R. Bruce Weisman and Jeffrey D. Hartgerink
Journal of Materials Chemistry A 2007 vol. 17(Issue 19) pp:1909-1915
Publication Date(Web):13 Mar 2007
DOI:10.1039/B700174F
Methods which solubilize single-walled carbon nanotubes (SWNTs) in water as individuals, not bundles, while retaining their unique electronic, photonic and mechanical properties are highly desirable. Furthermore, functionalization with a diverse array of selectable chemical moieties would allow the range of useful applications to be significantly extended and may permit the designed assembly of SWNT networks. This paper presents a series of peptides that non-covalently solubilize carbon nanotubes in water using a design motif that combines a combinatorial library sequence to bind to nanotubes with a rationally designed section to create environmentally tuned solubility characteristics. The ability of the peptides to individually disperse carbon nanotubes without altering their electronic structure is shown by vis-NIR absorbance, fluorescence, and regular and vitreous ice cryo-TEM. Identification of the species composition of each sample by NIR fluorescence reveals that the peptides exhibit some diameter selectivity. Additionally, one of the rationally designed modifications addresses the poor stability of non-covalently solubilized SWNT suspensions by including cysteine residues for covalent crosslinking between adjacent peptides.
Co-reporter:Ho-Wook Jun, Sergey E. Paramonov and Jeffrey D. Hartgerink
Soft Matter 2006 vol. 2(Issue 3) pp:177-181
Publication Date(Web):24 Jan 2006
DOI:10.1039/B516805H
Peptide-amphiphiles, peptides to which a non-peptidic hydrophobic moiety has been added to the N or C terminal end, have been demonstrated to be a versatile method for simultaneously controlling nanostructure and chemical functionality. These amphiphiles are able to self-assemble, in a controlled fashion, into nanofibers with diameter between 6–10 nm and with length in excess of 1000 nm. At proper concentration these nanofibers form a viscoelastic gel capable of entrapping living cells and eliciting specific responses from them. Because of the flexibility of the display of chemical functionality on a controlled nanofibrous scaffold, applications for peptide-amphiphiles have been proposed including heterogeneous catalysis, nanoelectronics, drug delivery, and tissue engineering.
Co-reporter:Vivek A. Kumar, Qi Liu, Navindee C. Wickremasinghe, Siyu Shi, Toya T. Cornwright, Yuxiao Deng, Alon Azares, Amanda N. Moore, Amanda M. Acevedo-Jake, Noel R. Agudo, Su Pan, Darren G. Woodside, Peter Vanderslice, James T. Willerson, Richard A. Dixon, Jeffrey D. Hartgerink
Biomaterials (August 2016) Volume 98() pp:113-119
Publication Date(Web):August 2016
DOI:10.1016/j.biomaterials.2016.04.032
For a proangiogenic therapy to be successful, it must promote the development of mature vasculature for rapid reperfusion of ischemic tissue. Whole growth factor, stem cell, and gene therapies have yet to achieve the clinical success needed to become FDA-approved revascularization therapies. Herein, we characterize a biodegradable peptide-based scaffold engineered to mimic VEGF and self-assemble into a nanofibrous, thixotropic hydrogel, SLanc. We found that this injectable hydrogel was rapidly infiltrated by host cells and could be degraded while promoting the generation of neovessels. In mice with induced hind limb ischemia, this synthetic peptide scaffold promoted angiogenesis and ischemic tissue recovery, as shown by Doppler-quantified limb perfusion and a treadmill endurance test. Thirteen-month-old mice showed significant recovery within 7 days of treatment. Biodistribution studies in healthy mice showed that the hydrogel is safe when administered intramuscularly, subcutaneously, or intravenously. These preclinical studies help establish the efficacy of this treatment for peripheral artery disease due to diminished microvascular perfusion, a necessary step before clinical translation. This peptide-based approach eliminates the need for cell transplantation or viral gene transfection (therapies currently being assessed in clinical trials) and could be a more effective regenerative medicine approach to microvascular tissue engineering.
Co-reporter:Vivek A. Kumar, Qi Liu, Navindee C. Wickremasinghe, Siyu Shi, Toya T. Cornwright, Yuxiao Deng, Alon Azares, Amanda N. Moore, Amanda M. Acevedo-Jake, Noel R. Agudo, Su Pan, Darren G. Woodside, Peter Vanderslice, James T. Willerson, Richard A. Dixon, Jeffrey D. Hartgerink
Biomaterials (August 2016) Volume 98() pp:113-119
Publication Date(Web):August 2016
DOI:10.1016/j.biomaterials.2016.04.032
Co-reporter:Biplab Sarkar ; Lesley E. R. O’Leary
Journal of the American Chemical Society () pp:
Publication Date(Web):October 6, 2014
DOI:10.1021/ja504377s
Mimicking the multistep self-assembly of the fibrillar protein collagen is an important design challenge in biomimetic supramolecular chemistry. Utilizing the complementarity of oppositely charged domains in short collagen-like peptides, we have devised a strategy for the self-assembly of these peptides into fibers. The strategy depends on the formation of a staggered triple helical species facilitated by interchain charged pairs, and is inspired by similar sticky-ended fibrillation designs applied in DNA and coiled coil fibers. We compare two classes of collagen mimetic peptides with the same composition but different domain arrangements, and show that differences in their proposed nucleation events differentiates their fibrillation capabilities. Larger nucleation domains result in rapid fiber formation and eventual precipitation or gelation while short nucleation domains leave the peptide soluble for long periods of time. For one of the fiber-forming peptides, we elucidate the packing parameters by X-ray diffraction.
Co-reporter:Kerstin M. Galler ; Lorenzo Aulisa ; Katherine R. Regan ; Rena N. D’Souza
Journal of the American Chemical Society () pp:
Publication Date(Web):February 16, 2010
DOI:10.1021/ja910481t
Multidomain peptides are a class of amphiphilic self-assembling peptides with a modular ABA block motif in which the amphiphilic B block drives self-assembly while the flanking A blocks, which are electrostatically charged, control the conditions under which assembly takes place. Previously we have shown that careful selection of the amino acids in the A and B blocks allow one to control the self-assembled fiber length and viscoelastic properties of formed hydrogels. Here we demonstrate how the modular nature of this peptide assembler can be designed for biological applications. With control over fiber length and diameter, gelation conditions, and viscoelastic properties, we can develop suitable materials for biological applications. Going beyond a simple carrier for cell delivery, a biofunctional scaffold will interact with the cells it carries, promoting advantageous cell−matrix interactions. We demonstrate the design of a multidomain peptide into a bioactive variant by incorporation of a matrix metalloprotease 2 (MMP-2) specific cleavage site and cell adhesion motif. Gel formation and rheological properties were assessed and compared to related peptide hydrogels. Proteolytic degradation by collagenase IV was observed in a gel weight loss study and confirmed by specific MMP-2 degradation monitored by mass spectrometry and cryo-transmission electron microscopy (cryo-TEM). Combination of this cleavage site with the cell adhesion motif RGD resulted in increased cell viability and cell spreading and encouraged cell migration into the hydrogel matrix. Collectively the structural, mechanical, and bioactive properties of this multidomain peptide hydrogel make it suitable as an injectable material for a variety of tissue engineering applications.
Co-reporter:Jorge A. Fallas, Lesley E. R. O'Leary and Jeffrey D. Hartgerink
Chemical Society Reviews 2010 - vol. 39(Issue 9) pp:NaN3527-3527
Publication Date(Web):2010/07/30
DOI:10.1039/B919455J
Collagen is a fascinating system of proteins that undergo a multi-step, hierarchical self-assembly which starts from individual peptide chains that assemble into a canonical triple helix. These triple helices then assemble into higher order structures which are often, but not always, fibrous in nature. While collagen is the most abundant protein in the human body, the details of its structure and mechanism of assembly are surprisingly poorly understood. This critical review will focus on small peptide systems, commonly referred to as collagen mimetic peptides (CMPs) which have been used successfully to help unravel some of the mystery of this complex structure. We will discuss homotrimeric CMPs, which are the most commonly researched subject in this field, and the structure of the collagen triple helix in detail and the factors that contribute to its stabilization. We will also cover how CMPs have been used to study breaks in triple helical domains as models for connective tissue diseases and, finally, how they have been used to understand the interactions of collagenous proteins with cell-surface receptors. Additionally, we will focus on heterotrimeric CMPs, a relatively new area of collagen research. Finally, we will deal with CMPs used as models for higher level self-assembly and also as materials that are designed to mimic the function of collagens in the extracellular matrix (178 references).
Co-reporter:Kerstin M. Galler, Rena N. D'Souza and Jeffrey D. Hartgerink
Journal of Materials Chemistry A 2010 - vol. 20(Issue 40) pp:NaN8746-8746
Publication Date(Web):2010/09/15
DOI:10.1039/C0JM01207F
Engineering oral tissues as a multidisciplinary approach to build complex structures such as bone, teeth or soft dental tissues remains a challenging endeavor which will also require significant additional development of materials chemistry before it will be successful. We will highlight areas of recent success and describe major challenges which the materials chemistry community, in collaboration with clinicians, must still overcome. The isolation of stem cell populations from various sources in the oral cavity and advances in utilizing their differentiation potential has been driving the field forward. So far, bioinert materials have mainly been used as carriers and delivery vehicles, relying on the intrinsic cellular competence to form tissues. As this may not suffice to induce regeneration, there is a need for novel biomimetic scaffolds capable of providing chemical and mechanical cues to promote multiple specific interactions between cells and matrix. These signals can orchestrate processes such as cell adhesion, migration, differentiation, matrix synthesis, mineralization, and/or vasculogenesis. In this review, we give a brief description of oral anatomy and pathology, state-of-the-art treatment methods and their shortcomings. We provide an overview of current strategies to fabricate bioactive matrices, with an emphasis on nanostructured materials, and we suggest design principles for scaffolding systems specifically tailored towards dental tissue regeneration. In this review, we envision future approaches based on these emerging areas that rely on recent developments in tissue engineering and stem cell research. At the interface between material science and biology, cellular response can be controlled by materials chemistry, and potential applications for regenerative strategies are evolving.
Co-reporter:Leah S. Witus, John-David R. Rocha, Virany M. Yuwono, Sergey E. Paramonov, R. Bruce Weisman and Jeffrey D. Hartgerink
Journal of Materials Chemistry A 2007 - vol. 17(Issue 19) pp:NaN1915-1915
Publication Date(Web):2007/03/13
DOI:10.1039/B700174F
Methods which solubilize single-walled carbon nanotubes (SWNTs) in water as individuals, not bundles, while retaining their unique electronic, photonic and mechanical properties are highly desirable. Furthermore, functionalization with a diverse array of selectable chemical moieties would allow the range of useful applications to be significantly extended and may permit the designed assembly of SWNT networks. This paper presents a series of peptides that non-covalently solubilize carbon nanotubes in water using a design motif that combines a combinatorial library sequence to bind to nanotubes with a rationally designed section to create environmentally tuned solubility characteristics. The ability of the peptides to individually disperse carbon nanotubes without altering their electronic structure is shown by vis-NIR absorbance, fluorescence, and regular and vitreous ice cryo-TEM. Identification of the species composition of each sample by NIR fluorescence reveals that the peptides exhibit some diameter selectivity. Additionally, one of the rationally designed modifications addresses the poor stability of non-covalently solubilized SWNT suspensions by including cysteine residues for covalent crosslinking between adjacent peptides.
.jpg)
![1,3,5-Naphthalenetrisulfonicacid,8,8'-[carbonylbis[imino-3,1-phenylenecarbonylimino(4-methyl-3,1-phenylene)carbonylimino]]bis-](http://img.cochemist.com/ccimg/200/145-63-1.png)
![1,3,5-Naphthalenetrisulfonicacid,8,8'-[carbonylbis[imino-3,1-phenylenecarbonylimino(4-methyl-3,1-phenylene)carbonylimino]]bis-](http://img.cochemist.com/ccimg/200/145-63-1_b.png)