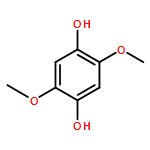
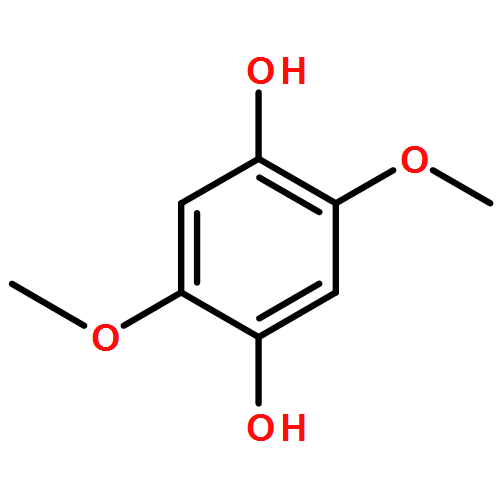
The 2,2′-azobis(isobutyronitrile)(AIBN)-induced autoxidation of γ-terpinene (TH) at 50 °C produces p-cymene and hydrogen peroxide in a radical-chain reaction having HOO. as one of the chain-carrying radicals. The kinetics of this reaction in cyclohexane and tert-butyl alcohol show that chain termination involves the formal HOO. + HOO. self-reaction over a wide range of γ-terpinene, AIBN, and O2 concentrations. However, in acetonitrile this termination process is accompanied by termination via the cross-reaction of the terpinenyl radical, T., with the HOO. radical under conditions of relatively high [TH] (140–1000 mM) and low [O2] (2.0–5.5 mM). This is because the formal HOO. + HOO. reaction is comparatively slow in acetonitrile (2k∼8×107 M−1 s−1), whereas, this reaction is almost diffusion-controlled in tert-butyl alcohol and cyclohexane, 2k∼6.5×108 and 1.3×109 M−1 s−1, respectively. Three mechanisms for the bimolecular self-reaction of HOO. radicals are considered: 1) a head-to-tail hydrogen-atom transfer from one radical to the other, 2) a head-to-head reaction to form an intermediate tetroxide, and 3) an electron-transfer between HOO. and its conjugate base, the superoxide radical anion, O2−.. The rate constant for reaction by mechanism (1) is shown to be dependent on the hydrogen bond (HB) accepting ability of the solvent; that by mechanism (2) is shown to be too slow for this process to be of any importance; and that by mechanism (3) is dependent on the pH of the solvent and its ability to support ionization. Mechanism (3) was found to be the main termination process in tert-butyl alcohol and acetonitrile. In the gas phase, the rate constant for the HOO. + HOO. reaction (mechanism (1)) is about 1.8×109 M−1 s−1 but in water at pH≤2 where the ionization of HOO. is completely suppressed, this rate constant is only 8.6×105 M−1 s−1. The very large retarding effect of water on this reaction has not previously been explained. We find that it can be quantitatively accounted for by using Abraham's HB acceptor parameter, , for water of 0.38 and an estimated HB donor parameter, , for HOO. of about 0.87. These Abraham parameters allow us to predict a rate constant for the HOO. + HOO. reaction in water at 25 °C of 1.2×106 M−1 s−1 in excellent agreement with experiment.