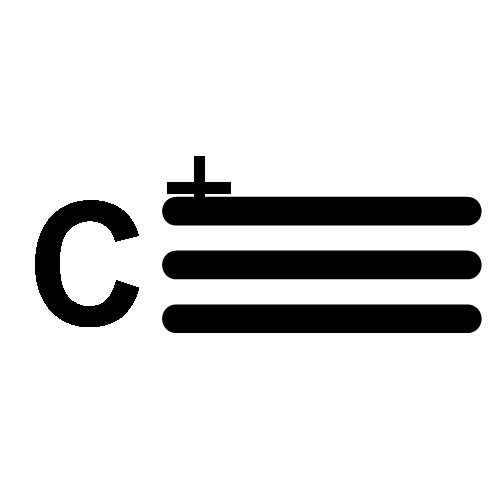
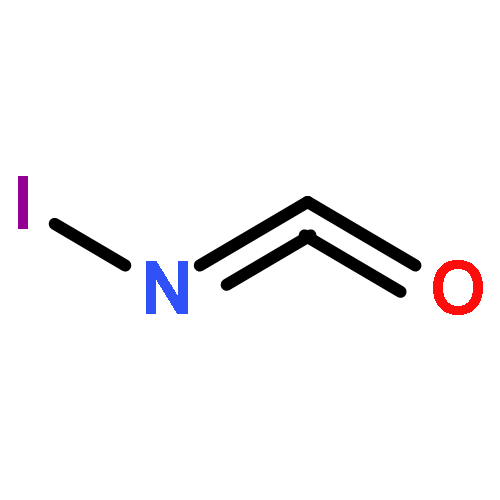
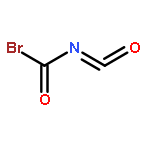
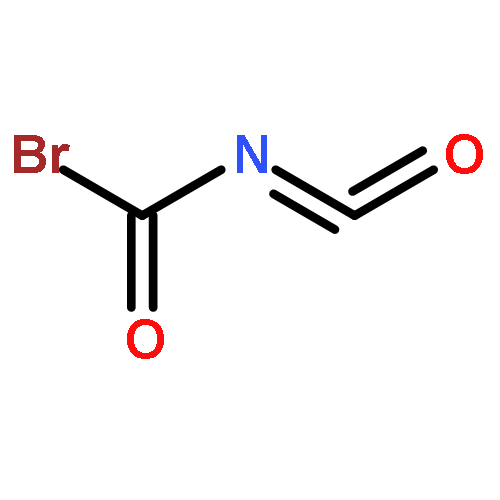
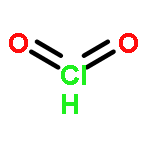
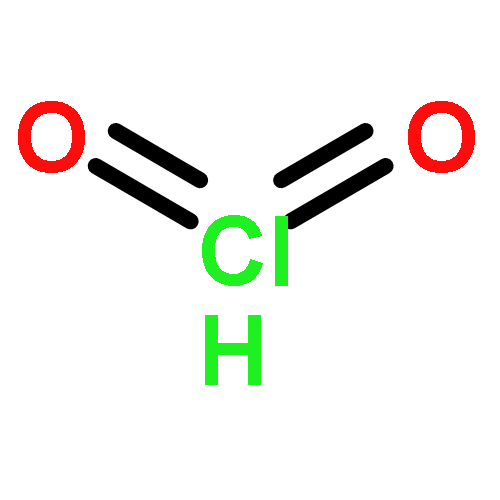
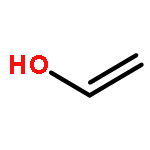
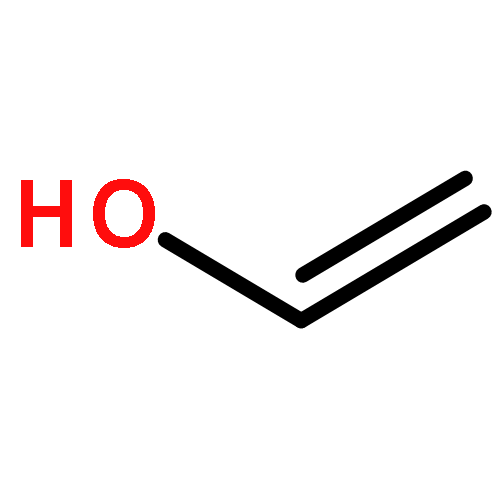
Co-reporter:James D. Fletcher;Dr. Michael A. Parkes ; Stephen D. Price
Chemistry - A European Journal 2013 Volume 19( Issue 33) pp:10965-10970
Publication Date(Web):
DOI:10.1002/chem.201301861
Abstract
Time-of-flight mass spectrometry reveals that atomic and small molecular triply charged cations exhibit extensive bond-forming chemistry, following gas-phase collisions with neutral molecules. These experiments show that at collision energies of a few eV, I3+ reacts with a variety of small molecules to generate molecular monocations and molecular dications containing iodine. Xe3+ and CS23+ react in a similar manner to I3+, undergoing bond-forming reactions with neutrals. A simple model, involving relative product energetics and electrostatic interaction potentials, is used to account for the observed reactivity.
Co-reporter:Jessica F. Lockyear, Claire L. Ricketts, Michael A. Parkes and Stephen D. Price
Chemical Science 2011 vol. 2(Issue 1) pp:150-156
Publication Date(Web):24 Sep 2010
DOI:10.1039/C0SC00344A
The nitrogen molecular dication (N22+) has been proposed as a minor but significant component of the ionosphere of Saturn's moon Titan with an abundance comparable to that of several key monocations. It has also been suggested that the reactions of N22+ with H2 can provide a source of N2H2+ in Titan's atmosphere. This paper reports the results from experiments, using a position-sensitive coincidence technique, which reveal the chemical reactions forming pairs of monocations following collisions of the N22+ dication with H2(D2) at a centre-of-mass collision energy of 0.9(1.8) eV. These experiments show, in addition to single electron-transfer processes, a bond-forming pathway forming NH+ + H+ + N and allow an estimate to be made of the reaction cross section and the rate coefficient for this reaction. The correlations between the product velocities revealed by the coincidence experiments show that NH+ is formed via N atom loss from a primary encounter complex [N2H2]2+ to form NH22+, with this triatomic daughter dication then fragmenting to yield NH+ + H+. A computational investigation of stationary points on the lowest energy singlet and triplet [N2H2]2+ potential energy surfaces confirms the mechanistic deductions from the experiments and indicates that the formation of NH+ occurs solely, and efficiently, from the reaction of the c3Σ+u excited electronic state of N22+.
Co-reporter:Michael A. Parkes, Jessica F. Lockyear, Detlef Schröder, Jana Roithová and Stephen D. Price
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 41) pp:18386-18392
Publication Date(Web):12 Aug 2011
DOI:10.1039/C1CP21612K
The single-electron transfer reaction between NO2+ and NO, which initially forms a pair of NO+ ions, has been studied using a position-sensitive coincidence technique. The reactivity in this class of collision system, which involves the interaction of a dication with its neutral precursor, provides a sensitive test of recent ideas concerning electronic state selectivity in dicationic single-electron transfer reactions. In stark contrast to the recently observed single-electron transfer reactivity in the analogous CO22+/CO2 and O22+/O2 collision systems, electron transfer between NO2+ and NO generates two product NO+ ions which behave in an identical manner, whether the ions are formed from NO2+ or NO. This observed behaviour is in excellent accord with the recently proposed rationalization of the state selectivity in dication-molecule SET reactions using simple propensity rules involving one-electron transitions.
Co-reporter:Kevin M. Douglas, Stephen D. Price
International Journal of Mass Spectrometry 2011 Volume 303(2–3) pp:147-153
Publication Date(Web):1 June 2011
DOI:10.1016/j.ijms.2011.01.023
Precursor-specific relative partial ionization cross sections, for all the different fragment ions formed by electron ionization of hydrogen sulfide, have been measured using time-of-flight mass spectrometry coupled with a two-dimensional ion coincidence technique. Relative cross sections are reported for ionizing energies from 30 to 200 eV. These cross sections allow the contribution from single, double and triple ionization to the individual fragment ion yields, following ionization of hydrogen sulfide, to be quantified. To compare our data with the literature we reduce our precursor-specific cross sections, by summing them for each fragment ion, to generate relative partial ionization cross sections. Following this data reduction, good agreement is found between our data and one set of recently published absolute partial ionization cross sections, but discrepancies are observed with another set of recently published data. Our analysis shows that the contribution of double ionization to the total ion yield reaches a maximum of 20% at 100 eV. Given the lack of available information on the fate of the excited electronic states of H2S2+, we have extracted kinetic energy releases for the various dicationic fragmentation channels from our coincidence data. From these kinetic energy releases, estimates of the energies of the electronic states of H2S2+ which are responsible for the different fragmentation channels can be made. These estimates, in comparison with other data on the electronic states of H2S2+, reveal the population of excited states of H2S2+ at electron energies above 50 eV. At ionizing electron energies above 50 eV, a significant proportion of the major dissociation channels of H2S2+ appear to involve the population of excited electronic states.Graphical abstractResearch highlights► The various fragment ions formed by electron ionization of H2S have been quantified. ► The contribution of multiple ionization to the fragment ion yield is measured. ► Energetics and fate of the excited electronic states of H2S2+ are revealed. ► Above 50 eV, most H2S2+ fragmentations involve excited dicationic states.
Co-reporter:Jessica F. Lockyear;Dr. Michael A. Parkes ; Stephen D. Price
Angewandte Chemie International Edition 2011 Volume 50( Issue 6) pp:1322-1324
Publication Date(Web):
DOI:10.1002/anie.201006486
Co-reporter:Jessica F. Lockyear;Dr. Michael A. Parkes ; Stephen D. Price
Angewandte Chemie 2011 Volume 123( Issue 6) pp:1358-1360
Publication Date(Web):
DOI:10.1002/ange.201006486
Co-reporter:Michael A. Parkes, Jessica F. Lockyear, Stephen D. Price, Detlef Schröder, Jana Roithová and Zdenek Herman
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 23) pp:6233-6243
Publication Date(Web):15 Apr 2010
DOI:10.1039/B926049H
The single electron transfer reactions between 13CO22+ and 12CO2 and between 18O22+ and 16O2 have been studied, using a position-sensitive coincidence technique, to test recently proposed explanations for the preferential dissociation of the 13CO2+ ion (the capture monocation) formed following electron transfer to 13CO22+. In our studies of the carbon dioxide collision system, in agreement with previous work, the capture monocation shows a greater propensity to dissociate than the monocation formed from the neutral, 12CO2+ (the ejection monocation). The coincidence data clearly show that the dissociation pathways of the 13CO2+ and 12CO2+ ions are different and are consistent with the ejection monocation dissociating via population of the C2Σ+g state, whilst the capture ion is predominantly directly formed in dissociative quartet states. This state assignment is in accord with an expected preference for one-electron transitions in the electron transfer process. A propensity for one-electron transitions also rationalizes our observation that following dissociative single electron transfer between 18O22+ and 16O2 the ejection monocation (16O2+) preferentially dissociates; the opposite situation to that observed for carbon dioxide. The coincidence results for this reaction indicate the 16O2+ dissociation results from population of the B(2Σ−g) state. The less favoured dissociation of the capture monocation clearly involves population of a different electronic state(s) to those populated in the ejection ion. Indeed, the experimental data are consistent with the dissociation of the capture monocation via predissociated levels of the b(4Σ−g) state. Since the population of the B(2Σ−g) state from the neutral O2 molecule involves a one-electron transition, and the population of the valence dissociative states of O2+ from the dication are multi-electron processes, the preferential dissociation of the ejection monocation in this collision system can be rationalized by the same principles used to explain the electron transfer reactivity of CO22+ with CO2.
Co-reporter:Michael A. Parkes, Jessica F. Lockyear, Stephen D. Price
International Journal of Mass Spectrometry 2009 280(1–3) pp: 85-92
Publication Date(Web):
DOI:10.1016/j.ijms.2008.07.027
Co-reporter:Daniela Ascenzi, Paolo Tosi, Jana Roithová, Claire L. Ricketts, Detlef Schröder, Jessica F. Lockyear, Michael A. Parkes and Stephen D. Price
Physical Chemistry Chemical Physics 2008 vol. 10(Issue 47) pp:7121-7128
Publication Date(Web):15 Oct 2008
DOI:10.1039/B810398D
Using doubly ionized acetylene as a superelectrophilic reagent, the new rare-gas compounds HCCAr2+ and HCCKr2+ have been prepared for the first time in hyperthermal collisions of mass-selected C2H22+ with neutral rare gases (Rg). However, electron transfer from the rare gas to the acetylene dication as well as proton transfer from C2H22+ to the rare gas efficiently compete with formation of HCCRg2+. The computational investigations show that the formation of HCCRg2+ from acetylene dication is endothermic with Rg = He, Ne, Ar and Kr and only weakly exothermic with Xe. These energetic factors, as well as the pronounced competition with the other reactive channels help to explain why HCCRg2+ is only observed with Rg = Ar and Kr.
Co-reporter:Philip Champkin, Nikolas Kaltsoyannis, Stephen D Price
Journal of Electron Spectroscopy and Related Phenomena 1999 Volume 105(Issue 1) pp:21-28
Publication Date(Web):November 1999
DOI:10.1016/S0368-2048(99)00018-3
A mechanism for the dissociation of the ozone dication formed by double ionisation of neutral O3 is determined by ab initio calculations. The dication ground singlet state is found to have a linear equilibrium geometry that is stable with respect to dissociation to O+ and O2+. However, at the Franck-Condon zone for formation of O32+ from the neutral molecule the singlet potential energy surface intersects with a dissociative triplet state. We propose that crossing to this dissociative triplet state can account for the absence of any long-lived O32+ ions in the electron-impact mass spectrum of ozone. Further calculations of the kinetic energy release for the fragmentation of O32+ to O++O2+ indicate that considerable vibrational excitation may be present in the O2+ ion.
Co-reporter:Nurun Tafadar, Nikolas Kaltsoyannis, Stephen D Price
International Journal of Mass Spectrometry 1999 Volume 192(1–3) pp:205-214
Publication Date(Web):27 September 1999
DOI:10.1016/S1387-3806(99)00093-7
We have recorded the relative intensities of the product ions formed following collisions of CF32+ with Ar at collision energies between 1.8 and 4.4 eV in the centre of mass frame. These experiments show that electron-transfer and neutral-loss reactions dominate the ion yield. The neutral-loss reaction produces CF22+ whilst the electron-transfer reactions produce CF+ and CF2+ together with Ar+. The variation of the neutral-loss ion yield with the collision energy provides a first estimate for the bond energy of the weak CF22+–F bond as 58 kJ mol−1. Unrestricted Hartree Fock/second order Moller-Plesset ab initio calculations indicate that the ground state of CF32+ adopts a C2v equilibrium geometry. Complete active space self-consistent field/multireference configuration interaction calculations of the electronic states of CF3+ at the C2v geometry of the dication have also been performed. Using these calculated state energies, together with Landau-Zener theory, to try to rationalise the electron-transfer reactivity, it appears likely that at least two electronic states of CF32+ are present in the dication beam. The ground state of CF32+ is predicted to react via electron transfer to form predominantly CF2+. An excited state of CF32+ lying approximately 5 eV above the ground state is hence required to explain the presence of CF+ ions that we observe in the experiments.
Co-reporter:Jessica F. Lockyear, Claire L. Ricketts, Michael A. Parkes and Stephen D. Price
Chemical Science (2010-Present) 2011 - vol. 2(Issue 1) pp:NaN156-156
Publication Date(Web):2010/09/24
DOI:10.1039/C0SC00344A
The nitrogen molecular dication (N22+) has been proposed as a minor but significant component of the ionosphere of Saturn's moon Titan with an abundance comparable to that of several key monocations. It has also been suggested that the reactions of N22+ with H2 can provide a source of N2H2+ in Titan's atmosphere. This paper reports the results from experiments, using a position-sensitive coincidence technique, which reveal the chemical reactions forming pairs of monocations following collisions of the N22+ dication with H2(D2) at a centre-of-mass collision energy of 0.9(1.8) eV. These experiments show, in addition to single electron-transfer processes, a bond-forming pathway forming NH+ + H+ + N and allow an estimate to be made of the reaction cross section and the rate coefficient for this reaction. The correlations between the product velocities revealed by the coincidence experiments show that NH+ is formed via N atom loss from a primary encounter complex [N2H2]2+ to form NH22+, with this triatomic daughter dication then fragmenting to yield NH+ + H+. A computational investigation of stationary points on the lowest energy singlet and triplet [N2H2]2+ potential energy surfaces confirms the mechanistic deductions from the experiments and indicates that the formation of NH+ occurs solely, and efficiently, from the reaction of the c3Σ+u excited electronic state of N22+.
Co-reporter:Daniela Ascenzi, Paolo Tosi, Jana Roithová, Claire L. Ricketts, Detlef Schröder, Jessica F. Lockyear, Michael A. Parkes and Stephen D. Price
Physical Chemistry Chemical Physics 2008 - vol. 10(Issue 47) pp:NaN7128-7128
Publication Date(Web):2008/10/15
DOI:10.1039/B810398D
Using doubly ionized acetylene as a superelectrophilic reagent, the new rare-gas compounds HCCAr2+ and HCCKr2+ have been prepared for the first time in hyperthermal collisions of mass-selected C2H22+ with neutral rare gases (Rg). However, electron transfer from the rare gas to the acetylene dication as well as proton transfer from C2H22+ to the rare gas efficiently compete with formation of HCCRg2+. The computational investigations show that the formation of HCCRg2+ from acetylene dication is endothermic with Rg = He, Ne, Ar and Kr and only weakly exothermic with Xe. These energetic factors, as well as the pronounced competition with the other reactive channels help to explain why HCCRg2+ is only observed with Rg = Ar and Kr.
Co-reporter:Michael A. Parkes, Jessica F. Lockyear, Stephen D. Price, Detlef Schröder, Jana Roithová and Zdenek Herman
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 23) pp:NaN6243-6243
Publication Date(Web):2010/04/15
DOI:10.1039/B926049H
The single electron transfer reactions between 13CO22+ and 12CO2 and between 18O22+ and 16O2 have been studied, using a position-sensitive coincidence technique, to test recently proposed explanations for the preferential dissociation of the 13CO2+ ion (the capture monocation) formed following electron transfer to 13CO22+. In our studies of the carbon dioxide collision system, in agreement with previous work, the capture monocation shows a greater propensity to dissociate than the monocation formed from the neutral, 12CO2+ (the ejection monocation). The coincidence data clearly show that the dissociation pathways of the 13CO2+ and 12CO2+ ions are different and are consistent with the ejection monocation dissociating via population of the C2Σ+g state, whilst the capture ion is predominantly directly formed in dissociative quartet states. This state assignment is in accord with an expected preference for one-electron transitions in the electron transfer process. A propensity for one-electron transitions also rationalizes our observation that following dissociative single electron transfer between 18O22+ and 16O2 the ejection monocation (16O2+) preferentially dissociates; the opposite situation to that observed for carbon dioxide. The coincidence results for this reaction indicate the 16O2+ dissociation results from population of the B(2Σ−g) state. The less favoured dissociation of the capture monocation clearly involves population of a different electronic state(s) to those populated in the ejection ion. Indeed, the experimental data are consistent with the dissociation of the capture monocation via predissociated levels of the b(4Σ−g) state. Since the population of the B(2Σ−g) state from the neutral O2 molecule involves a one-electron transition, and the population of the valence dissociative states of O2+ from the dication are multi-electron processes, the preferential dissociation of the ejection monocation in this collision system can be rationalized by the same principles used to explain the electron transfer reactivity of CO22+ with CO2.
Co-reporter:Michael A. Parkes, Jessica F. Lockyear, Detlef Schröder, Jana Roithová and Stephen D. Price
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 41) pp:NaN18392-18392
Publication Date(Web):2011/08/12
DOI:10.1039/C1CP21612K
The single-electron transfer reaction between NO2+ and NO, which initially forms a pair of NO+ ions, has been studied using a position-sensitive coincidence technique. The reactivity in this class of collision system, which involves the interaction of a dication with its neutral precursor, provides a sensitive test of recent ideas concerning electronic state selectivity in dicationic single-electron transfer reactions. In stark contrast to the recently observed single-electron transfer reactivity in the analogous CO22+/CO2 and O22+/O2 collision systems, electron transfer between NO2+ and NO generates two product NO+ ions which behave in an identical manner, whether the ions are formed from NO2+ or NO. This observed behaviour is in excellent accord with the recently proposed rationalization of the state selectivity in dication-molecule SET reactions using simple propensity rules involving one-electron transitions.

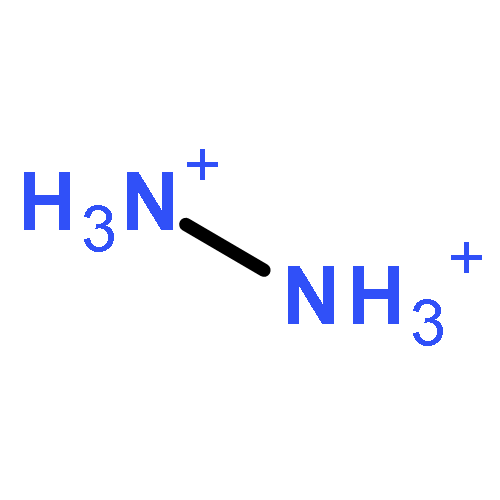
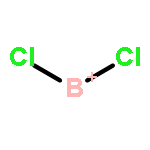
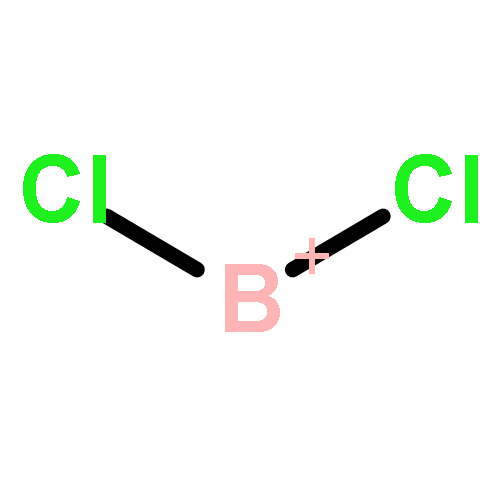
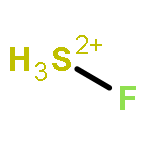
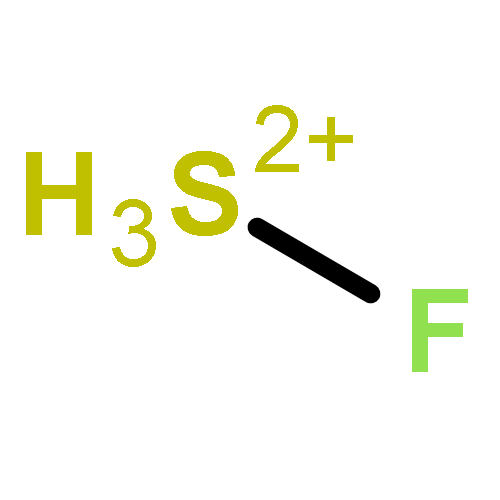
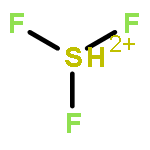
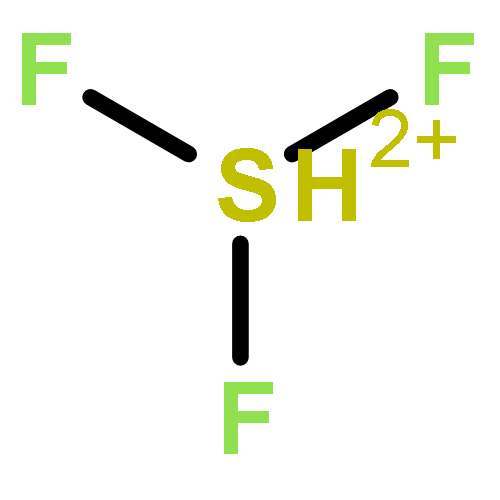
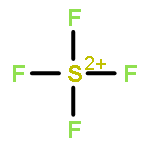
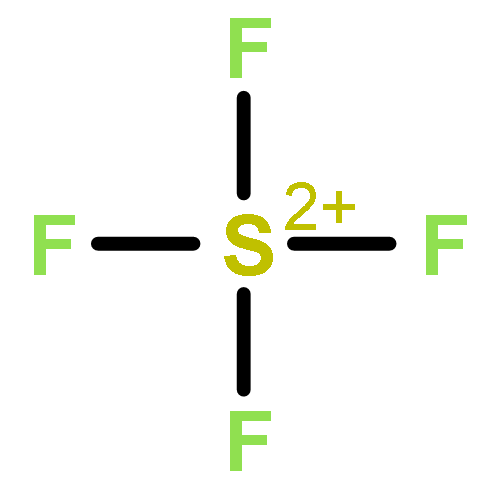
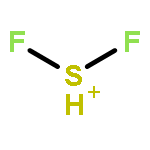
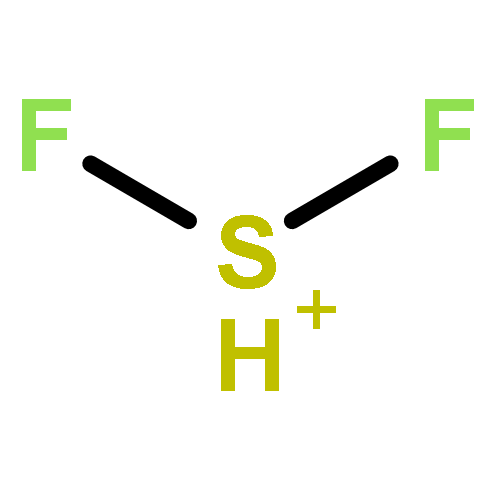
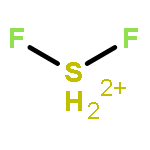
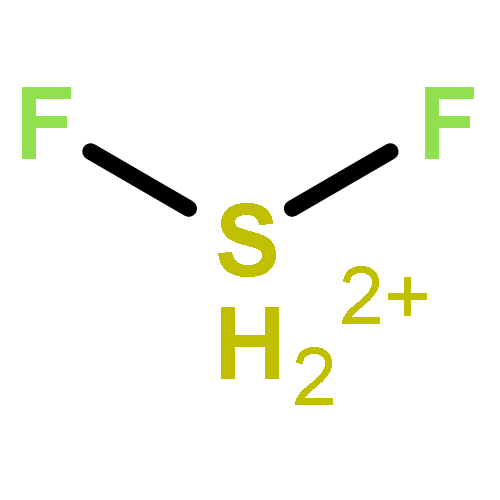
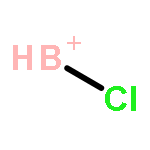
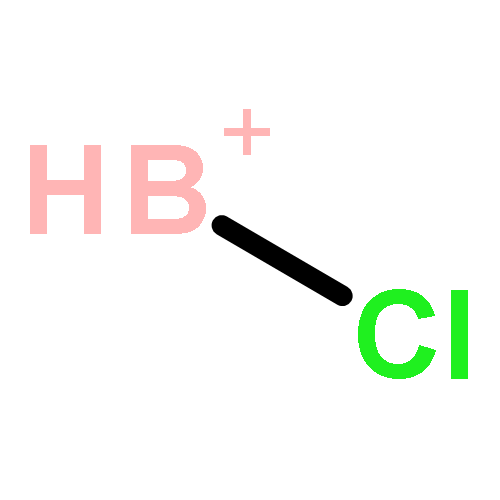
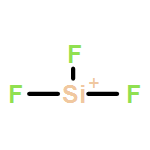
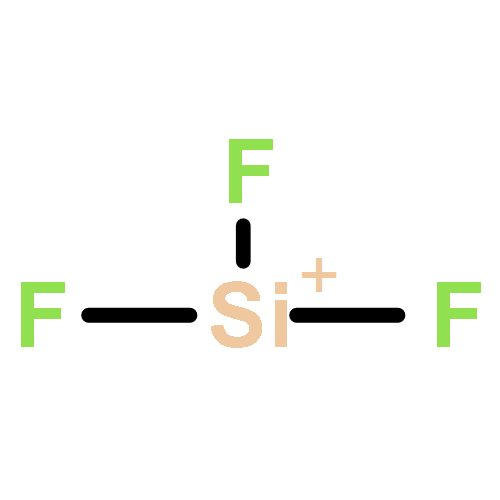















![2-{[(4-METHOXY-3,5-DIMETHYL-2-PYRIDINYL)METHYL]SULFINYL}-5-[(<SUP>2</SUP>H<SUB>3</SUB>)METHYLOXY]-1H-BENZIMIDAZOLE](http://img.cochemist.com/ccimg/7800/7791-21-1.png)
![2-{[(4-METHOXY-3,5-DIMETHYL-2-PYRIDINYL)METHYL]SULFINYL}-5-[(<SUP>2</SUP>H<SUB>3</SUB>)METHYLOXY]-1H-BENZIMIDAZOLE](http://img.cochemist.com/ccimg/7800/7791-21-1_b.png)