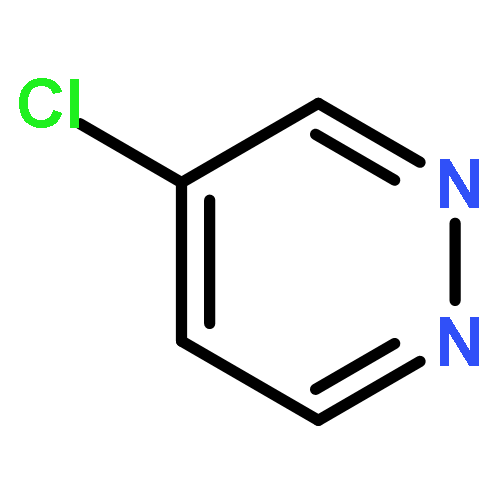
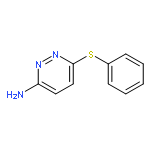
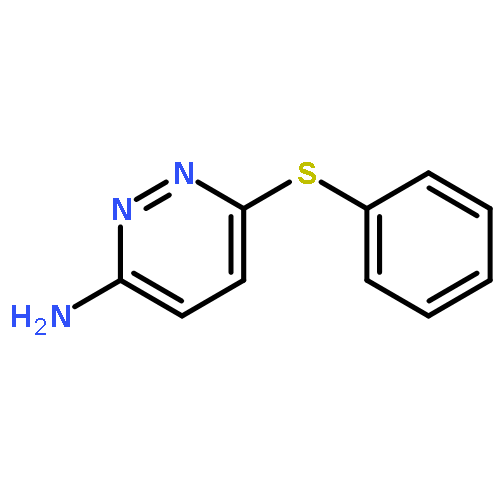
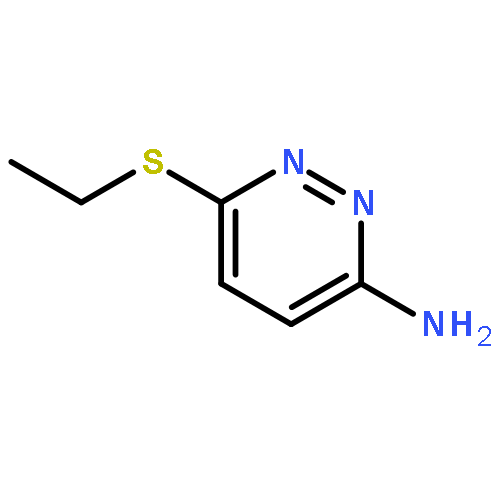
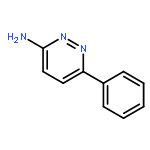
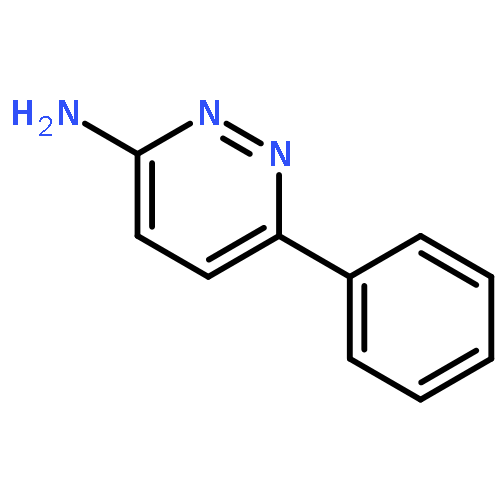
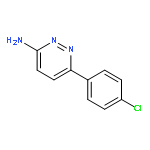
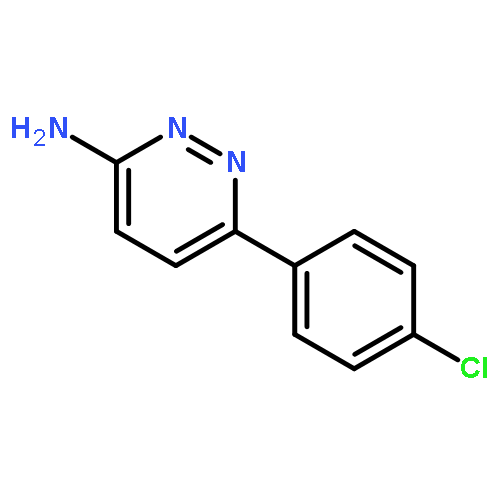
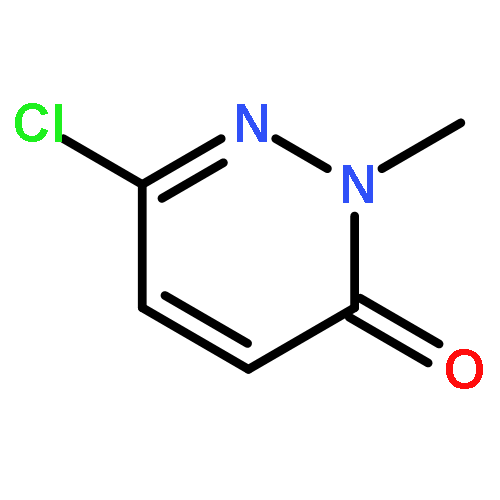
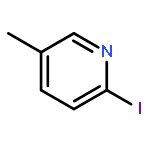
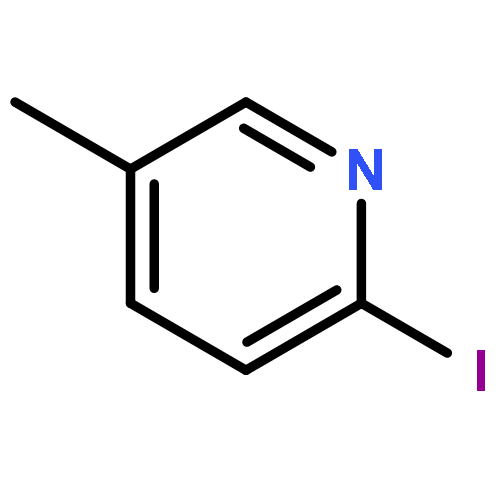
Co-reporter:Hans Sterckx;Carlo Sambiagio;Vincent Médran-Navarrete
Advanced Synthesis & Catalysis 2017 Volume 359(Issue 18) pp:3226-3236
Publication Date(Web):2017/09/18
DOI:10.1002/adsc.201700588
AbstractA copper-catalyzed aerobic oxidation of benzylpyridine N-oxides is reported. The N-oxide moiety acts as a built-in activator for the benzylic methylene oxidation, without requirement of additives. Reaction conditions were identified which suppress undesired benzoylpyridine formation via N-deoxygenation involving intermolecular oxygen transfer. The versatility of the N-oxide group of the benzoylpyridine N-oxide reaction products for post-functionalization of the pyridine ring is demonstrated through efficient C–C, C–N, C–O and C–Cl bond forming procedures, with both nucleophiles and electrophiles. Finally, the applicability of the new synthetic methodology is demonstrated in an alternative route towards the antihistaminic drug Acrivastine via three consecutive N-oxide activated C–H functionalization processes, starting from picoline N-oxide.
Co-reporter:Yan-Ping Zhu;Pieter Mampuys;Sergey Sergeyev;Steven Ballet
Advanced Synthesis & Catalysis 2017 Volume 359(Issue 14) pp:2481-2498
Publication Date(Web):2017/07/17
DOI:10.1002/adsc.201700134
AbstractN-arylamino acid amides have been synthesized via a novel method based on N-arylamine activation into isothioureas followed by reaction with amino acids under iron catalysis. The activated N-arylamines are easily prepared using a three-component reaction with commercial reagents, tert-butylisocyanide and S-phenyl benzenethiosulfonate. The protocol shows a broad functional group compatibility, with respect to side chain functionality of the amino acid (e. g. aliphatic and aromatic OH, (hetero)aromatic NH, amide NH, thioether), and the chiral amino acids do not undergo epimerization. The mechanism of the new amide synthesis has been studied.
Co-reporter:S. Abou-Shehada;P. Mampuys;B. U. W. Maes;J. H. Clark;L. Summerton
Green Chemistry (1999-Present) 2017 vol. 19(Issue 1) pp:249-258
Publication Date(Web):2017/01/03
DOI:10.1039/C6GC01928E
Multicomponent reactions (MCRs) are considered green and material efficient methods for the synthesis of organic compounds, however very few studies have investigated the metrics of the upstream processes involved to achieve the starting materials used in these reactions. A novel MCR-approach for the synthesis of N-cyclohexyl-S-methyl-N′-phenylisothiourea from S-methyl methanethiosulfonate, cyclohexyl isocyanide and aniline was appraised in terms of its green metrics using the CHEM21 green metrics toolkit and compared with those of the state of the art approach. The upstream process leading to the component starting materials for both approaches were also appraised to gain an insight into how the greenness of both methods translate from the raw materials to the final product.
Co-reporter:Ben F. Van Steijvoort, Nadya Kaval, Artem A. Kulago, and Bert U. W. Maes
ACS Catalysis 2016 Volume 6(Issue 7) pp:4486
Publication Date(Web):June 3, 2016
DOI:10.1021/acscatal.6b00841
A protocol for the Pd-catalyzed C5(sp3)-H arylation of readily available 1-Boc-3-(picolinoylamino)piperidine with iodo(hetero)arenes is reported. The substrate can be obtained from a biorenewable feedstock, namely l-arginine. The use of the right N1 protective group is decisive to get arylation. The addition of a catalytic amount of 2,6-dimethylbenzoic acid and performing the reaction at high concentration are important to achieve a high conversion and yield. The procedure gives arylated 1-Boc-3-(picolinoylamino)piperidines in a regiospecific (C5) and stereospecific (cis) manner. Orthogonal cleavage of the amide over the carbamate group allows one to further selectively derivatize the amino moieties of the piperidine scaffold.Keywords: biorenewable; C(sp3)-H activation; cyclic amine; palladium catalysis; regiospecific; stereospecific
Co-reporter:Hans Sterckx, Johan De Houwer, Carl Mensch, Ignacio Caretti, Kourosch Abbaspour Tehrani, Wouter A. Herrebout, Sabine Van Doorslaer and Bert U. W. Maes
Chemical Science 2016 vol. 7(Issue 1) pp:346-357
Publication Date(Web):29 Sep 2015
DOI:10.1039/C5SC03530A
A mechanistic study of the copper-catalyzed oxidation of the methylene group of aryl(di)azinylmethanes was performed. Initial reaction rates were measured making use of in situ IR reaction monitoring and a kinetic analysis of the reaction was executed. The reaction proved to be first order in oxygen concentration. For substrate and acid concentration, saturation kinetics due to O2 mass transfer limitation were observed. The occurrence of mass transfer limitation was further confirmed by examining the effect of the stirring rate on the initial reaction rate. Interestingly, the effect of the concentration of the catalyst on the rate shows that higher loadings result in a maximal initial rate, followed initially by a steady decrease and subsequently a rate plateau when the concentration is increased further. Mass transfer limitation and increased concentration of dinuclear catalytically active species rationalizes this hitherto unprecedented rate behavior. Continuous-wave and pulsed electron paramagnetic resonance methods were used to characterize the catalytic species present in the solution during the reaction and confirmed the presence of both mono- and dinuclear copper species. Analysis of a diverse substrate scope points towards imine–enamine tautomerization as a crucial process in the oxidation reaction. DFT calculations of these equilibrium constants (pKeq) provided us with a qualitative tool to predict whether or not a substrate is viable for oxidation under the reaction conditions developed.
Co-reporter:Pieter Mampuys, Yanping Zhu, Sergey Sergeyev, Eelco Ruijter, Romano V. A. Orru, Sabine Van Doorslaer, and Bert U. W. Maes
Organic Letters 2016 Volume 18(Issue 12) pp:2808-2811
Publication Date(Web):June 8, 2016
DOI:10.1021/acs.orglett.6b01023
A new method for the synthesis of secondary thiocarbamates from readily available isocyanides and thiosulfonates with broad functional group tolerance is reported. The reaction proceeds under mild reaction conditions in isopropanol and is catalyzed by inexpensive sodium iodide.
Co-reporter:Yan-Ping Zhu, Sergey Sergeyev, Philippe Franck, Romano V. A. Orru, and Bert U. W. Maes
Organic Letters 2016 Volume 18(Issue 18) pp:4602-4605
Publication Date(Web):August 29, 2016
DOI:10.1021/acs.orglett.6b02247
A novel method for N-(hetero)arylamide synthesis based on rarely explored amine activation, rather than classical acid activation, is reported. The activated amines are easily prepared using a three-component reaction with commercial reagents. The new method shows a broad scope including challenging amides not (efficiently) accessible via classical protocols.
Co-reporter:Mattijs Baeten
Advanced Synthesis & Catalysis 2016 Volume 358( Issue 5) pp:826-833
Publication Date(Web):
DOI:10.1002/adsc.201501146
Co-reporter:Sergey Sergeyev; Ashok Kumar Yadav; Philippe Franck; Johan Michiels; Paul Lewi; Jan Heeres; Guido Vanham; Kevin K. Ariën; Christophe M. L. Vande Velde; Hans De Winter
Journal of Medicinal Chemistry 2016 Volume 59(Issue 5) pp:1854-1868
Publication Date(Web):January 19, 2016
DOI:10.1021/acs.jmedchem.5b01336
New non-nucleoside reverse transcriptase inhibitors (NNRTI), which are similar in structure to earlier described di(arylamino)pyrimidines but featuring a 2,6-di(arylamino)-3-fluoropyridine, 2,4-di(arylamino)-5-fluoropyrimidine, or 1,3-di(arylamino)-4-fluorobenzene moiety instead of a 2,4-disubstituted pyrimidine moiety, are reported. The short and practical synthesis of novel NNRTI relies on two sequential Pd-catalyzed aminations as the key steps. It is demonstrated through direct comparison with reference compounds that the presence of a fluorine atom increases the in vitro anti-HIV activity, both against the wild type virus and drug-resistant mutant strains.
Co-reporter:Pradip Debnath;Mattijs Baeten;Nicolas Lefèvre;Stijn Van Daele
Advanced Synthesis & Catalysis 2015 Volume 357( Issue 1) pp:197-209
Publication Date(Web):
DOI:10.1002/adsc.201400648
Co-reporter: Bert U. W. Maes;Dr. Stefan Verbeeck;Dr. Tom Verhelst;Audrey Ekomié;Niklas vonWolff;Dr. Guillaume Lefèvre;Dr. Emily A. Mitchell; Anny Jut
Chemistry - A European Journal 2015 Volume 21( Issue 21) pp:7858-7865
Publication Date(Web):
DOI:10.1002/chem.201406210
Abstract
The kinetics of the oxidative additions of haloheteroarenes HetX (X=I, Br, Cl) to [Pd0(PPh3)2] (generated from [Pd0(PPh3)4]) have been investigated in THF and DMF and the rate constants have been determined. In contrast to the generally accepted concerted mechanism, Hammett plots obtained for substituted 2-halopyridines and solvent effects reveal a reaction mechanism dependent on the halide X of HetX: an unprecedented SNAr-type mechanism for X=Br or Cl and a classical concerted mechanism for X=I. These results are supported by DFT studies.
Co-reporter:Marius Hervé;Dr. Guillaume Lefèvre;Dr. Emily A. Mitchell; Bert U. W. Maes;Dr. Anny Jut
Chemistry - A European Journal 2015 Volume 21( Issue 50) pp:18401-18406
Publication Date(Web):
DOI:10.1002/chem.201503309
Abstract
The mechanism of Stille reactions (cross-coupling of ArX with Ar′SnnBu3) performed in the presence of fluoride ions is established. A triple role for fluoride ions is identified from kinetic data on the rate of the reactions of trans-[ArPdBr(PPh3)2] (Ar=Ph, p-(CN)C6H4) with Ar′SnBu3 (Ar′=2-thiophenyl) in the presence of fluoride ions. Fluoride ions promote the rate-determining transmetallation by formation of trans-[ArPdF(PPh3)2], which reacts with Ar′SnBu3 (Ar′=Ph, 2-thiophenyl) at room temperature, in contrast to trans-[ArPdBr(PPh3)2], which is unreactive. However, the concentration ratio [F−]/[Ar′SnBu3] must not be too high, because of the formation of unreactive anionic stannate [Ar′Sn(F)Bu3]−. This rationalises the two kinetically antagonistic roles exerted by the fluoride ions that are observed experimentally, and is found to be in agreement with the kinetic law. In addition, fluoride ions promote reductive elimination from trans-[ArPdAr′(PPh3)2] generated in the transmetallation step.
Co-reporter:Tom Willemse;Karolien VanImp;Dr. Rebecca J. M. Goss;Dr. Herman W. T. VanVlijmen;Dr. Wim Schepens;Dr. Bert U. W. Maes;Dr. Steven Ballet
ChemCatChem 2015 Volume 7( Issue 14) pp:2055-2070
Publication Date(Web):
DOI:10.1002/cctc.201500190
Abstract
The Suzuki–Miyaura derivatisation of free amino acids, peptides and proteins is an attractive area with considerable potential utility for medicinal chemistry and chemical biology. Here we report the modification of unprotected and Boc-protected aromatic amino acids and dipeptides in aqueous media, enabling heteroarylation and vinylation. We systematically investigate the impact of the peptide backbone and adjacent amino acid residues upon the reaction. Our studies reveal that although asparagine and histidine hinder the reaction, by utilising dppf, a ferrocene-based bidentate phosphine ligand, cross coupling of halophenylalanine or halotryptophan adjacent to such a residue could be enabled. Our studies reveal dppf to have good compatibility with all unprotected, proteinogenic amino acid side chains.
Co-reporter:Tom Willemse;Karolien VanImp;Dr. Rebecca J. M. Goss;Dr. Herman W. T. VanVlijmen;Dr. Wim Schepens;Dr. Bert U. W. Maes;Dr. Steven Ballet
ChemCatChem 2015 Volume 7( Issue 14) pp:
Publication Date(Web):
DOI:10.1002/cctc.201500742
Co-reporter:Artem A. Kulago;Ben F. VanSteijvoort;Emily A. Mitchell;Lieven Meerpoel
Advanced Synthesis & Catalysis 2014 Volume 356( Issue 7) pp:1610-1618
Publication Date(Web):
DOI:10.1002/adsc.201400117
Co-reporter:Pieter Mampuys;Dr. Yanping Zhu;Dr. Tjøstil Vlaar;Dr. Eelco Ruijter;Dr. Romano V. A. Orru;Dr. Bert U. W. Maes
Angewandte Chemie International Edition 2014 Volume 53( Issue 47) pp:12849-12854
Publication Date(Web):
DOI:10.1002/anie.201406717
Abstract
Multiple applications of isothioureas as fine chemicals (or their precursors) are known, but a general sustainable method for their synthesis was hitherto unavailable. We report a novel general approach towards S-alkyl and S-aryl isothioureas through a copper(I)-catalyzed three-component reaction between amines, isocyanides, and thiosulfonates. The formal synthesis of a superpotent sweetener further illustrates the applicability of our method.
Co-reporter:Veerle Smout, Aldo Peschiulli, Stefan Verbeeck, Emily A. Mitchell, Wouter Herrebout, Patrick Bultinck, Christophe M. L. Vande Velde, Didier Berthelot, Lieven Meerpoel, and Bert U. W. Maes
The Journal of Organic Chemistry 2013 Volume 78(Issue 19) pp:9803-9814
Publication Date(Web):September 6, 2013
DOI:10.1021/jo401521y
Two strategies, “hydrogenation–hydride reduction” and “quaternization–hydride reduction”, are reported that make use of mild reaction conditions (room temperature) to efficiently remove the N-pyridin-2-yl directing group from a diverse set of C-2-substituted piperidines that were synthesized through directed Ru-catalyzed sp3 C–H functionalization. The deprotected products are obtained in moderate to good overall yields irrespective of the strategy followed, indicating that both methods are generally equally effective. Only in the case of 2,6-disubstituted piperidines, could the “quaternization–hydride reduction” strategy not be used. The “hydrogenation–hydride reduction” protocol was successfully applied to trans- and cis-2-methyl-N-(pyridin-2-yl)-6-undecylpiperidine in a short synthetic route toward (±)-solenopsin A (trans diastereoisomer) and (±)-isosolenopsin A (cis diastereoisomer). The absolute configuration of the enantiomers of these fire ant alkaloids could be determined via VCD spectroscopy.
Co-reporter:Hans Sterckx, Johan De Houwer, Carl Mensch, Ignacio Caretti, Kourosch Abbaspour Tehrani, Wouter A. Herrebout, Sabine Van Doorslaer and Bert U. W. Maes
Chemical Science (2010-Present) 2016 - vol. 7(Issue 1) pp:NaN357-357
Publication Date(Web):2015/09/29
DOI:10.1039/C5SC03530A
A mechanistic study of the copper-catalyzed oxidation of the methylene group of aryl(di)azinylmethanes was performed. Initial reaction rates were measured making use of in situ IR reaction monitoring and a kinetic analysis of the reaction was executed. The reaction proved to be first order in oxygen concentration. For substrate and acid concentration, saturation kinetics due to O2 mass transfer limitation were observed. The occurrence of mass transfer limitation was further confirmed by examining the effect of the stirring rate on the initial reaction rate. Interestingly, the effect of the concentration of the catalyst on the rate shows that higher loadings result in a maximal initial rate, followed initially by a steady decrease and subsequently a rate plateau when the concentration is increased further. Mass transfer limitation and increased concentration of dinuclear catalytically active species rationalizes this hitherto unprecedented rate behavior. Continuous-wave and pulsed electron paramagnetic resonance methods were used to characterize the catalytic species present in the solution during the reaction and confirmed the presence of both mono- and dinuclear copper species. Analysis of a diverse substrate scope points towards imine–enamine tautomerization as a crucial process in the oxidation reaction. DFT calculations of these equilibrium constants (pKeq) provided us with a qualitative tool to predict whether or not a substrate is viable for oxidation under the reaction conditions developed.

![6-Oxa-9-azaspiro[4.5]decane](http://img.cochemist.com/ccimg/130700/130643-07-1.png)
![6-Oxa-9-azaspiro[4.5]decane](http://img.cochemist.com/ccimg/130700/130643-07-1_b.png)
![Dibenzo[f,h]phthalazin-1(2H)-one, 2-(phenylmethyl)-](http://img.cochemist.com/ccimg/603200/603128-17-2.png)
![Dibenzo[f,h]phthalazin-1(2H)-one, 2-(phenylmethyl)-](http://img.cochemist.com/ccimg/603200/603128-17-2_b.png)
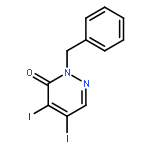
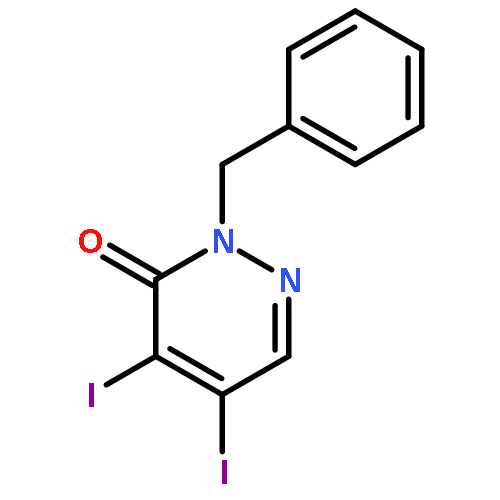
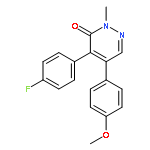
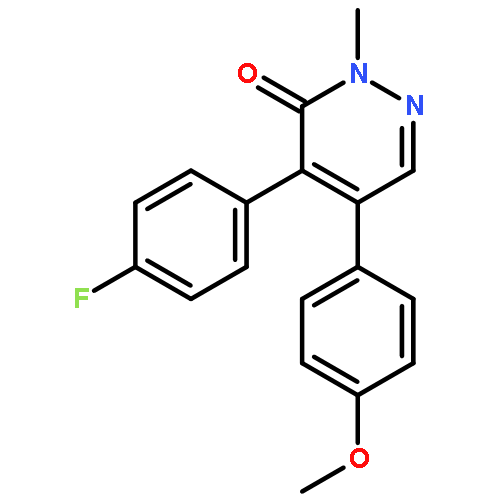
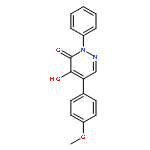
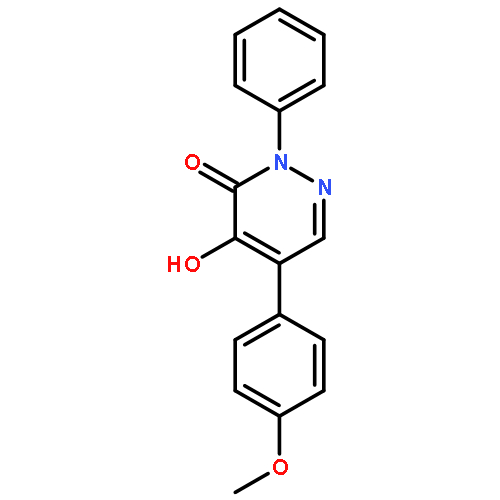
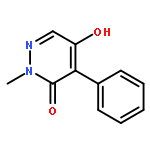
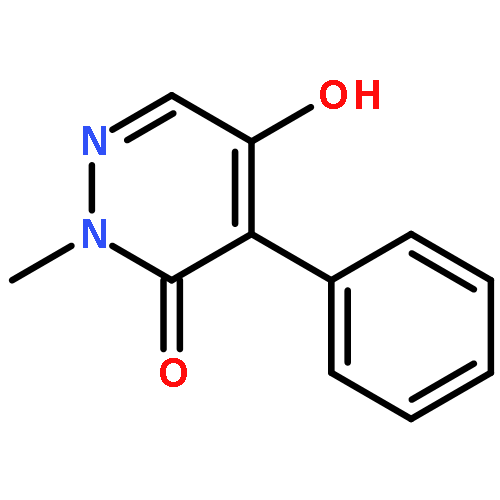
![3(2H)-Pyridazinone, 5-hydroxy-2-methyl-4-[4-(methylthio)phenyl]-](http://img.cochemist.com/ccimg/503500/503454-53-3.png)
![3(2H)-Pyridazinone, 5-hydroxy-2-methyl-4-[4-(methylthio)phenyl]-](http://img.cochemist.com/ccimg/503500/503454-53-3_b.png)
![3(2H)-Pyridazinone, 4-[4-(methylthio)phenyl]-5-phenyl-2-(phenylmethyl)-](http://img.cochemist.com/ccimg/503500/503454-64-6.png)
![3(2H)-Pyridazinone, 4-[4-(methylthio)phenyl]-5-phenyl-2-(phenylmethyl)-](http://img.cochemist.com/ccimg/503500/503454-64-6_b.png)
![DIBENZO[F,H]CINNOLIN-3(2H)-ONE, 2-METHYL-](http://img.cochemist.com/ccimg/603200/603128-22-9.png)
![DIBENZO[F,H]CINNOLIN-3(2H)-ONE, 2-METHYL-](http://img.cochemist.com/ccimg/603200/603128-22-9_b.png)
![DIBENZO[F,H]PHTHALAZIN-1(2H)-ONE](http://img.cochemist.com/ccimg/603200/603128-18-3.png)
![DIBENZO[F,H]PHTHALAZIN-1(2H)-ONE](http://img.cochemist.com/ccimg/603200/603128-18-3_b.png)
![3(2H)-Pyridazinone, 5-methoxy-2-methyl-4-[4-(methylthio)phenyl]-](http://img.cochemist.com/ccimg/335400/335349-33-2.png)
![3(2H)-Pyridazinone, 5-methoxy-2-methyl-4-[4-(methylthio)phenyl]-](http://img.cochemist.com/ccimg/335400/335349-33-2_b.png)
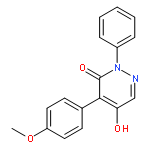
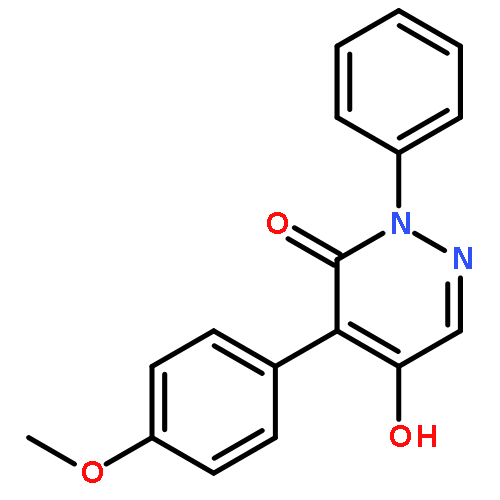

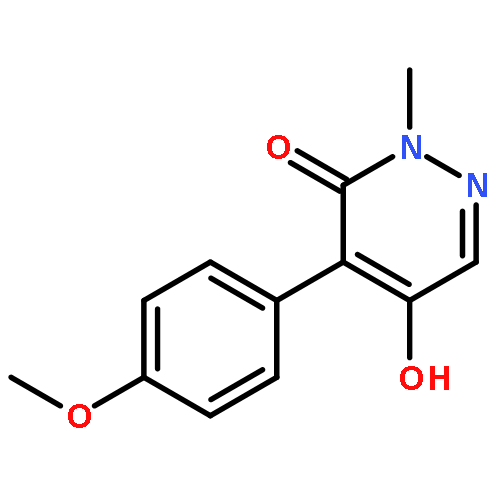
![3(2H)-PYRIDAZINONE, 5-HYDROXY-2-METHYL-4-[3-(TRIFLUOROMETHYL)PHENYL]-](http://img.cochemist.com/ccimg/503500/503454-54-4.png)
![3(2H)-PYRIDAZINONE, 5-HYDROXY-2-METHYL-4-[3-(TRIFLUOROMETHYL)PHENYL]-](http://img.cochemist.com/ccimg/503500/503454-54-4_b.png)
![4H-PYRIDAZINO[4,5-B]INDOL-4-ONE, 3,5-DIHYDRO-3-METHYL-1-PHENYL-](http://img.cochemist.com/ccimg/518100/518050-97-0.png)
![4H-PYRIDAZINO[4,5-B]INDOL-4-ONE, 3,5-DIHYDRO-3-METHYL-1-PHENYL-](http://img.cochemist.com/ccimg/518100/518050-97-0_b.png)
![3H-[2]BENZOPYRANO[3,4-D]PYRIDAZINE-4,6-DIONE, 3-(PHENYLMETHYL)-](http://img.cochemist.com/ccimg/503500/503454-71-5.png)
![3H-[2]BENZOPYRANO[3,4-D]PYRIDAZINE-4,6-DIONE, 3-(PHENYLMETHYL)-](http://img.cochemist.com/ccimg/503500/503454-71-5_b.png)
![3(2H)-PYRIDAZINONE, 5-[(1E)-3-OXO-3-PHENYL-1-PROPENYL]-2-PHENYL-](http://img.cochemist.com/ccimg/878000/877932-17-7.png)
![3(2H)-PYRIDAZINONE, 5-[(1E)-3-OXO-3-PHENYL-1-PROPENYL]-2-PHENYL-](http://img.cochemist.com/ccimg/878000/877932-17-7_b.png)
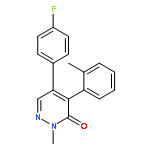

![6-[3-(trifluoromethyl)phenyl]-3-Pyridazinamine](http://img.cochemist.com/ccimg/102700/102627-28-1.png)
![6-[3-(trifluoromethyl)phenyl]-3-Pyridazinamine](http://img.cochemist.com/ccimg/102700/102627-28-1_b.png)
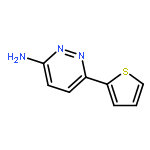
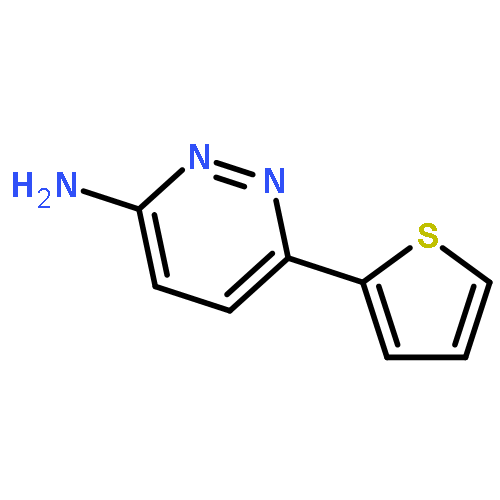
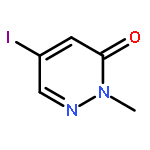
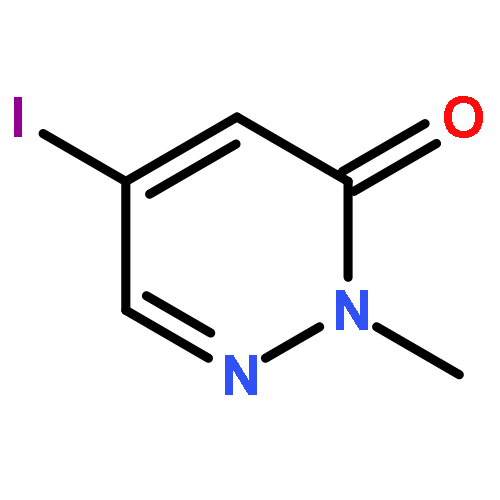

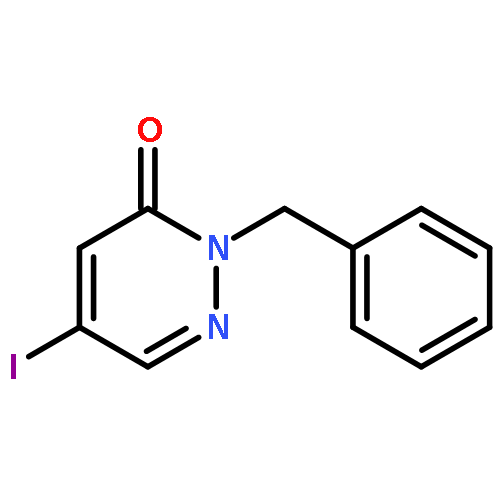
![3(2H)-Pyridazinone, 2-methyl-5-[(1E)-3-oxo-3-phenyl-1-propenyl]-](http://img.cochemist.com/ccimg/825700/825634-12-6.png)
![3(2H)-Pyridazinone, 2-methyl-5-[(1E)-3-oxo-3-phenyl-1-propenyl]-](http://img.cochemist.com/ccimg/825700/825634-12-6_b.png)
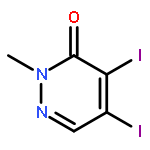
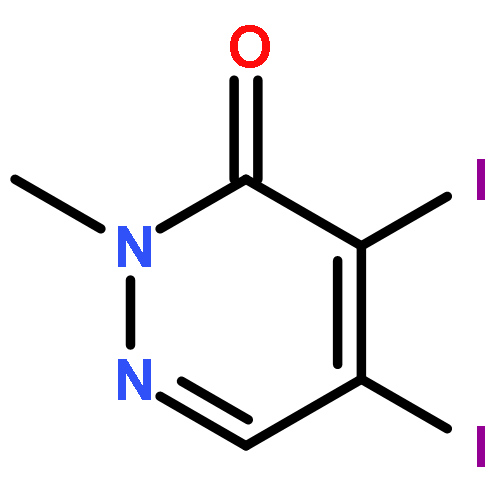
![3-Pyridazinamine, 6-[4-(methylthio)phenyl]-](http://img.cochemist.com/ccimg/271800/271784-03-3.png)
![3-Pyridazinamine, 6-[4-(methylthio)phenyl]-](http://img.cochemist.com/ccimg/271800/271784-03-3_b.png)