Co-reporter:Michael Grasso, Michelle A. Estrada, Christian Ventocilla, Minu Samanta, Jasna Maksimoska, Jessie Villanueva, Jeffrey D. Winkler, and Ronen Marmorstein
ACS Chemical Biology 2016 Volume 11(Issue 10) pp:2876
Publication Date(Web):August 29, 2016
DOI:10.1021/acschembio.6b00529
The BRAF kinase, within the mitogen activated protein kinase (MAPK) signaling pathway, harbors activating mutations in about half of melanomas and to a significant extent in many other cancers. A single valine to glutamic acid substitution at residue 600 (BRAFV600E) accounts for about 90% of these activating mutations. While BRAFV600E-selective small molecule inhibitors, such as debrafenib and vemurafenib, have shown therapeutic benefit, almost all patients develop resistance. Resistance often arises through reactivation of the MAPK pathway, typically through mutation of upstream RAS, downstream MEK, or splicing variants. RAF kinases signal as homo- and heterodimers, and another complication associated with small molecule BRAFV600E inhibition is drug-induced allosteric activation of a wild-type RAF subunit (BRAF or CRAF) of the kinase dimer, a process called “transactivation” or “paradoxical activation.” Here, we used BRAFV600E and vemurafenib as a model system to develop chemically linked kinase inhibitors to lock RAF dimers in an inactive conformation that cannot undergo transactivation. This structure-based design effort resulted in the development of Vem-BisAmide-2, a compound containing two vemurafenib molecules connected by a bis amide linker. We show that Vem-BisAmide-2 has comparable inhibitory potency as vemurafenib to BRAFV600E both in vitro and in cells but promotes an inactive dimeric BRAFV600E conformation unable to undergo transactivation. The crystal structure of a BRAFV600E/Vem-BisAmide-2 complex and associated biochemical studies reveal the molecular basis for how Vem-BisAmide-2 mediates selectivity for an inactive over an active dimeric BRAFV600E conformation. These studies have implications for targeting BRAFV600E/RAF heterodimers and other kinase dimers for therapy.
Co-reporter:Adegoke O. Adeniji ; Barry M. Twenter ; Michael C. Byrns ; Yi Jin ; Mo Chen ; Jeffrey D. Winkler ;Trevor M. Penning
Journal of Medicinal Chemistry 2012 Volume 55(Issue 5) pp:2311-2323
Publication Date(Web):January 20, 2012
DOI:10.1021/jm201547v
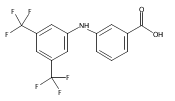
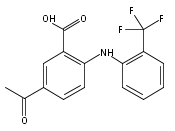
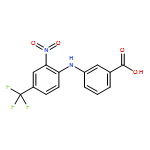
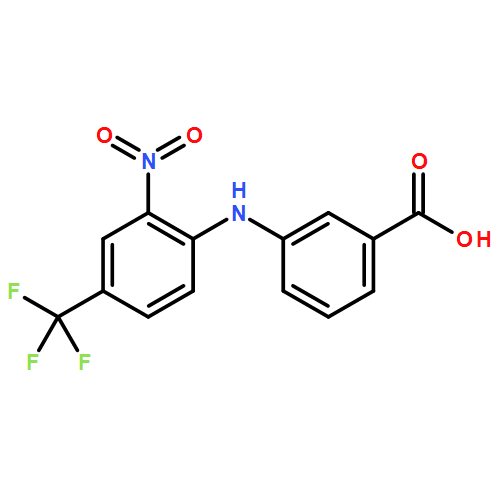
Aldo–keto reductase 1C3 (AKR1C3; type 5 17β-hydroxysteroid dehydrogenase) is overexpressed in castration resistant prostate cancer (CRPC) and is implicated in the intratumoral biosynthesis of testosterone and 5α-dihydrotestosterone. Selective AKR1C3 inhibitors are required because compounds should not inhibit the highly related AKR1C1 and AKR1C2 isoforms which are involved in the inactivation of 5α-dihydrotestosterone. NSAIDs, N-phenylanthranilates in particular, are potent but nonselective AKR1C3 inhibitors. Using flufenamic acid, 2-{[3-(trifluoromethyl)phenyl]amino}benzoic acid, as lead compound, five classes of structural analogues were synthesized and evaluated for AKR1C3 inhibitory potency and selectivity. Structure–activity relationship (SAR) studies revealed that a meta-carboxylic acid group relative to the amine conferred pronounced AKR1C3 selectivity without loss of potency, while electron withdrawing groups on the phenylamino B-ring were optimal for AKR1C3 inhibition. Lead compounds did not inhibit COX-1 or COX-2 but blocked the AKR1C3 mediated production of testosterone in LNCaP-AKR1C3 cells. These compounds offer promising leads toward new therapeutics for CRPC.
Co-reporter:Jie Qin ; Peng Xie ; Christian Ventocilla ; Guoqiang Zhou ; Adina Vultur ; Quan Chen ; Qin Liu ; Meenhard Herlyn ; Jeffrey Winkler ;Ronen Marmorstein
Journal of Medicinal Chemistry 2012 Volume 55(Issue 11) pp:5220-5230
Publication Date(Web):April 26, 2012
DOI:10.1021/jm3004416
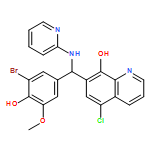
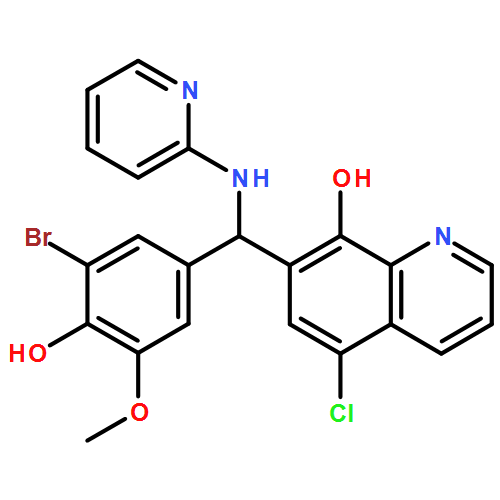
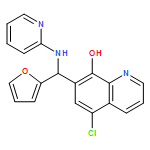
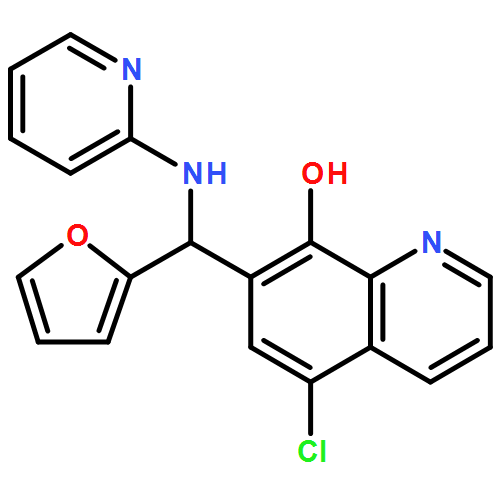
The BRAF oncoprotein is mutated in about half of malignant melanomas and other cancers, and a kinase activating single valine to glutamate substitution at residue 600 (BRAFV600E) accounts for over 90% of BRAF-mediated cancers. Several BRAFV600E inhibitors have been developed, although they harbor some liabilities, thus motivating the development of other BRAFV600E inhibitor options. We report here the use of an ELISA based high-throughput screen to identify a family of related quinolol/naphthol compounds that preferentially inhibit BRAFV600E over BRAFWT and other kinases. We also report the X-ray crystal structure of a BRAF/quinolol complex revealing the mode of inhibition, employ structure-based medicinal chemistry efforts to prepare naphthol analogues that inhibit BRAFV600E in vitro with IC50 values in the 80–200 nM range under saturating ATP concentrations, and demonstrate that these compounds inhibit MAPK signaling in melanoma cells. Prospects for improving the potency and selectivity of these inhibitors are discussed.
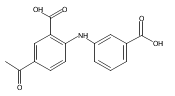
Co-reporter:Mo Chen, Adegoke O. Adeniji, Barry M. Twenter, Jeffrey D. Winkler, David W. Christianson, Trevor M. Penning
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 10) pp:3492-3497
Publication Date(Web):15 May 2012
DOI:10.1016/j.bmcl.2012.03.085
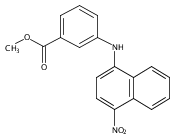
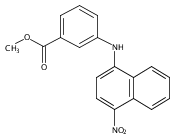
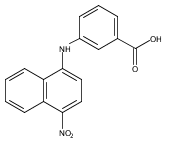
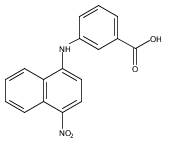
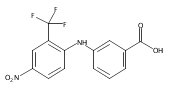
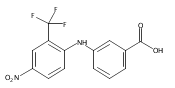
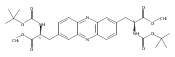
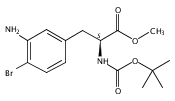
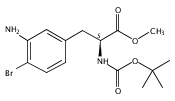
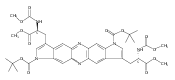
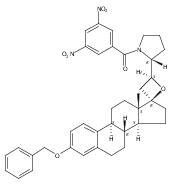
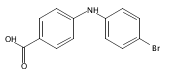
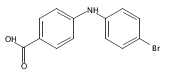
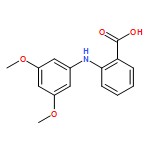
Castrate resistant prostate cancer (CRPC) is associated with increased androgen receptor (AR) signaling often brought about by elevated intratumoral androgen biosynthesis and AR amplification. Inhibition of androgen biosynthesis and/or AR antagonism should be efficacious in the treatment of CRPC. AKR1C3 catalyzes the formation of potent AR ligands from inactive precursors and is one of the most upregulated genes in CRPC. AKR1C3 inhibitors should not inhibit the related isoforms, AKR1C1 and AKR1C2 that are involved in 5α-dihydrotestosterone inactivation in the prostate. We have previously developed a series of flufenamic acid analogs as potent and selective AKR1C3 inhibitors [Adeniji, A. O. et al., J. Med. Chem.2012, 55, 2311]. Here we report the X-ray crystal structure of one lead compound 3-((4-(trifluoromethyl)phenyl) amino)benzoic acid (1) in complex with AKR1C3. Compound 1 adopts a similar binding orientation as flufenamic acid, however, its phenylamino ring projects deeper into a subpocket and confers selectivity over the other AKR1C isoforms. We exploited the observation that some flufenamic acid analogs also act as AR antagonists and synthesized a second generation inhibitor, 3-((4-nitronaphthalen-1-yl)amino)benzoic acid (2). Compound 2 retained nanomolar potency and selective inhibition of AKR1C3 but also acted as an AR antagonist. It inhibited 5α-dihydrotestosterone stimulated AR reporter gene activity with an IC50 = 4.7 μM and produced a concentration dependent reduction in androgen receptor levels in prostate cancer cells. The in vitro and cell-based effects of compound 2 make it a promising lead for development of dual acting agent for CRPC. To illuminate the structural basis of AKR1C3 inhibition, we also report the crystal structure of the AKR1C3·NADP+·2 complex, which shows that compound 2 forms a unique double-decker structure with AKR1C3.Superimposed structures of AKR1C3 in complex with two N-aryl(amino)benzoic acid analogs, compound 1 (magenta) and compound 2 (blue).
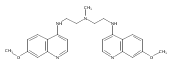
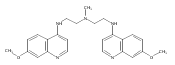
Co-reporter:Quentin McAfee;Zhihui Zhang;John P. Lynch;Antonia R. Sepulveda;Ravi K. Amaravadi;Takeshi Uehara;Lisa E. Davis;Arabinda Samanta;Shengfu Piao;Xiao-Hong Ma;Samuel M. Levi
PNAS 2012 Volume 109 (Issue 21 ) pp:8253-8258
Publication Date(Web):2012-05-22
DOI:10.1073/pnas.1118193109
Autophagy is a lysosome-dependent degradative process that protects cancer cells from multiple stresses. In preclinical models,
autophagy inhibition with chloroquine (CQ) derivatives augments the efficacy of many anticancer therapies, but CQ has limited
activity as a single agent. Clinical trials are underway combining anticancer agents with hydroxychloroquine (HCQ), but concentrations
of HCQ required to inhibit autophagy are not consistently achievable in the clinic. We report the synthesis and characterization
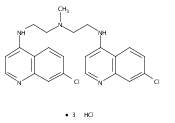
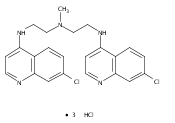
of bisaminoquinoline autophagy inhibitors that potently inhibit autophagy and impair tumor growth in vivo. The structural
motifs that are necessary for improved autophagy inhibition compared with CQ include the presence of two aminoquinoline rings
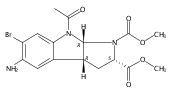
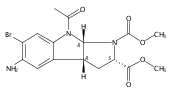
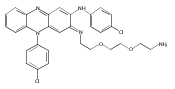
and a triamine linker and C-7 chlorine. The lead compound, Lys01, is a 10-fold more potent autophagy inhibitor than HCQ. Compared
with HCQ, Lys05, a water-soluble salt of Lys01, more potently accumulates within and deacidifies the lysosome, resulting in
impaired autophagy and tumor growth. At the highest dose administered, some mice develop Paneth cell dysfunction that resembles
the intestinal phenotype of mice and humans with genetic defects in the autophagy gene ATG16L1, providing in vivo evidence that Lys05 targets autophagy. Unlike HCQ, significant single-agent antitumor activity is observed
without toxicity in mice treated with lower doses of Lys05, establishing the therapeutic potential of this compound in cancer.
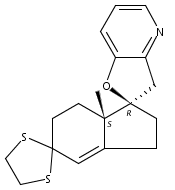
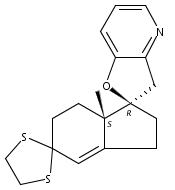
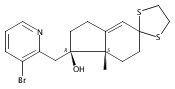
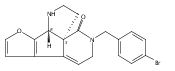
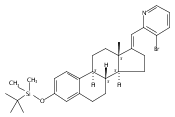
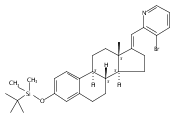
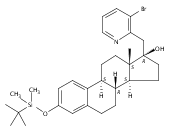
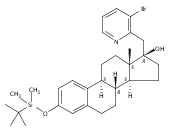
Co-reporter:André K. Isaacs, Chaomei Xiang, Valérie Baubet, Nadia Dahmane, and Jeffrey D. Winkler
Organic Letters 2011 Volume 13(Issue 19) pp:5140-5143
Publication Date(Web):September 9, 2011
DOI:10.1021/ol202020c
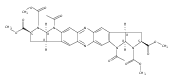
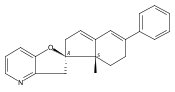
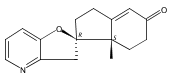
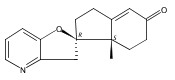
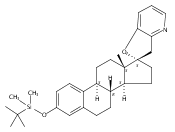
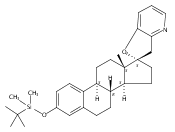
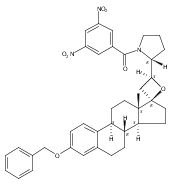
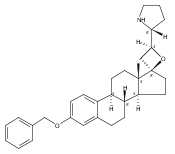
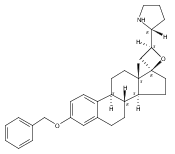
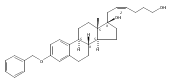
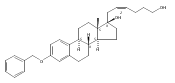
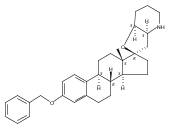
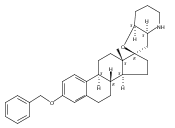
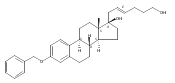
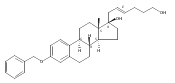
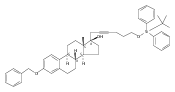
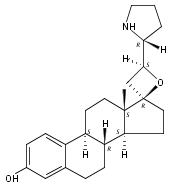
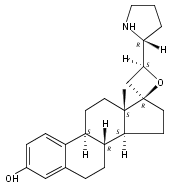
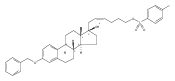
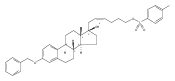
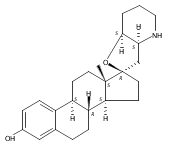
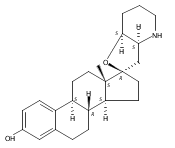
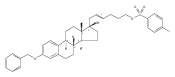
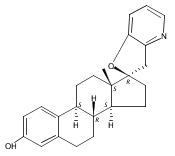
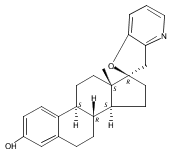
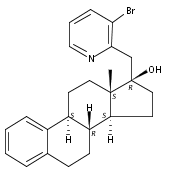
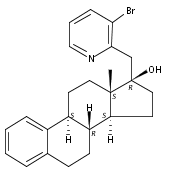
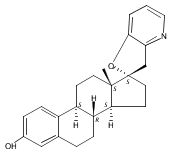
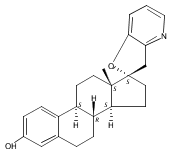
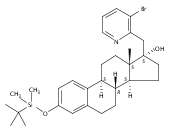
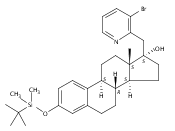
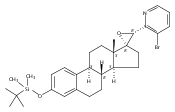
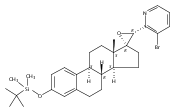
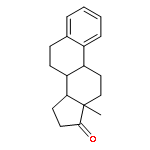
Previous work from our laboratory has established that the readily available steroid-based analog 2 of cyclopamine 1 is, like 1, a highly potent inhibitor of Hedgehog signaling. The first structure–activity relationship studies on 2, i.e., the synthesis and biological evaluation of both the C-17 epi analog 4 and the C-3 deoxy analog 11, both of which are more potent than cyclopamine 1, are described. The implications of these results for the emerging pharmacophore of these Sonic Hedgehog signaling inhibitors are discussed.
Co-reporter:Adegoke O. Adeniji, Barry M. Twenter, Michael C. Byrns, Yi Jin, Jeffrey D. Winkler, Trevor M. Penning
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 5) pp:1464-1468
Publication Date(Web):1 March 2011
DOI:10.1016/j.bmcl.2011.01.010
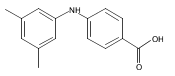
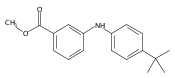
Aldo-keto reductase 1C3 (AKR1C3) also known as type 5 17β-hydroxysteroid dehydrogenase has been implicated as one of the key enzymes driving the elevated intratumoral androgen levels observed in castrate resistant prostate cancer (CRPC). AKR1C3 inhibition therefore presents a rational approach to managing CRPC. Inhibitors should be selective for AKR1C3 over other AKR1C enzymes involved in androgen metabolism. We have synthesized 2-, 3-, and 4-(phenylamino)benzoic acids and identified 3-(phenylamino)benzoic acids that have nanomolar affinity and exhibit over 200-fold selectivity for AKR1C3 versus other AKR1C isoforms. The AKR1C3 inhibitory potency of the 4′-substituted 3-(phenylamino)benzoic acids shows a linear correlation with both electronic effects of substituents and the pKa of the carboxylic acid and secondary amine groups, which are interdependent. These compounds may be useful in treatment and/or prevention of CRPC as well as understanding the role of AKR1C3 in endocrinology.
Co-reporter:Thomas Haimowitz, Mark E. Fitzgerald, Jeffrey D. Winkler
Tetrahedron Letters 2011 Volume 52(Issue 17) pp:2162-2164
Publication Date(Web):27 April 2011
DOI:10.1016/j.tetlet.2010.11.134
The application of a Pummerer-initiated tandem reaction cascade leads to the highly stereoselective formation of the tetracyclic core of nakadomarin A.
Co-reporter:Jeffrey D. Winkler, André K. Isaacs, Chaomei Xiang, Valérie Baubet, Nadia Dahmane
Tetrahedron 2011 67(52) pp: 10261-10266
Publication Date(Web):
DOI:10.1016/j.tet.2011.10.028
Co-reporter:Hyunil Jo, Mark E. Fitzgerald and Jeffrey D. Winkler
Organic Letters 2009 Volume 11(Issue 8) pp:1685-1687
Publication Date(Web):March 20, 2009
DOI:10.1021/ol900186y
Irradiation of the enone benzothiazoline 3 leads to the formation of cyclobutane 5. Preliminary mechanistic studies establish the intermediacy of an enecarbamate 14 in this photochemical transformation, which could be the result of sulfur extrusion from an episulfide intermediate. Photocycloaddition of the enecarbamate intermediate 14 leads to the formation of “crossed” photoadducts, i.e., 5, in excellent yield, with high levels of regio- and stereochemical control.
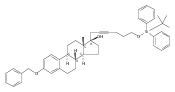
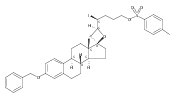
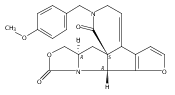
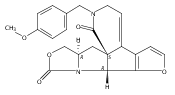
Co-reporter:Jeffrey D. Winkler, André Isaacs, Laura Holderbaum, Valérie Tatard and Nadia Dahmane
Organic Letters 2009 Volume 11(Issue 13) pp:2824-2827
Publication Date(Web):June 4, 2009
DOI:10.1021/ol900974u
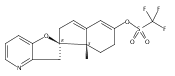
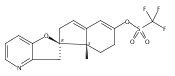
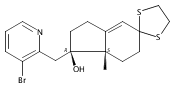
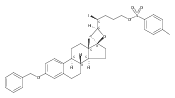
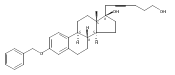
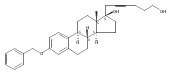
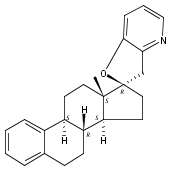
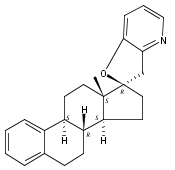
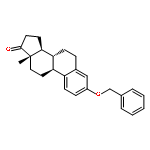
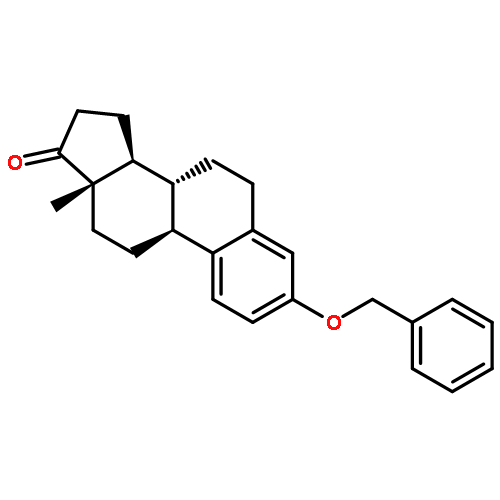
The synthesis and biological evaluation of structurally simplified, metabolically stable cyclopamine-like Sonic Hedgehog (SHH) signaling inhibitors, i.e., 5, is described in four chemical steps from commercially available steroidal precursors. Biological evaluation of this cyclopamine analogue in two different systems establishes the high potency of 5 as a SHH signaling inhibitor. This approach provides important new lead structures for the development of new cancer chemotherapeutic agents based on the inhibition on SHH signaling.
Co-reporter:Robert J. Doerksen, Bin Chen, Jing Yuan, Jeffrey D. Winkler and Michael L. Klein
Chemical Communications 2003 (Issue 20) pp:2534-2535
Publication Date(Web):23 Sep 2003
DOI:10.1039/B309584C
For a novel family of oxanorbornene β-peptides, density functional theory computations of the three-dimensional structure and 1H NMR chemical shifts predict that the dimer and trimer form consecutive 8-membered hydrogen-bonded ring helices, which is supported by excellent agreement with experimental solution NMR data.
Co-reporter:Jeffrey D. Winkler Dr.;Evgueni L. Piatnitski;John Mehlmann;Jiri Kasparec;Paul H. Axelsen Dr.
Angewandte Chemie 2001 Volume 113(Issue 4) pp:
Publication Date(Web):15 FEB 2001
DOI:10.1002/1521-3757(20010216)113:4<765::AID-ANGE7650>3.0.CO;2-X
.jpg)
![N-(3-(5-Bromo-1-(2,6-dichlorobenzoyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluorophenyl)propane-1-sulfonamide](http://img.cochemist.com/ccimg/1263000/1262985-24-9.png)
![N-(3-(5-Bromo-1-(2,6-dichlorobenzoyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluorophenyl)propane-1-sulfonamide](http://img.cochemist.com/ccimg/1263000/1262985-24-9_b.png)
![N-(3-(5-(4-Chlorophenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluorophenyl)propane-1-sulfonamide](http://img.cochemist.com/ccimg/918600/918504-65-1.png)
![N-(3-(5-(4-Chlorophenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluorophenyl)propane-1-sulfonamide](http://img.cochemist.com/ccimg/918600/918504-65-1_b.png)
![1,3,5-Naphthalenetrisulfonicacid,8,8'-[carbonylbis[imino-3,1-phenylenecarbonylimino(4-methyl-3,1-phenylene)carbonylimino]]bis-](http://img.cochemist.com/ccimg/200/145-63-1.png)
![1,3,5-Naphthalenetrisulfonicacid,8,8'-[carbonylbis[imino-3,1-phenylenecarbonylimino(4-methyl-3,1-phenylene)carbonylimino]]bis-](http://img.cochemist.com/ccimg/200/145-63-1_b.png)
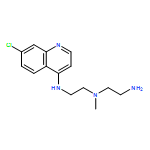
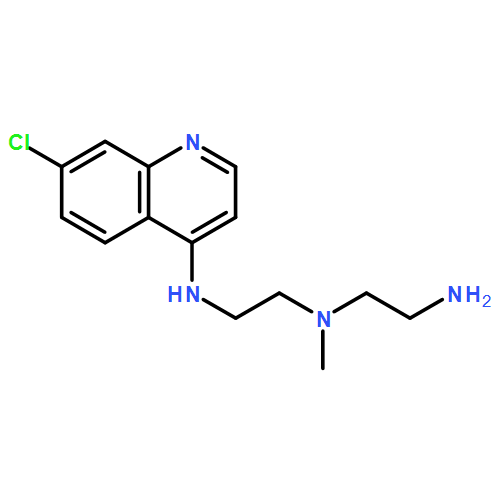
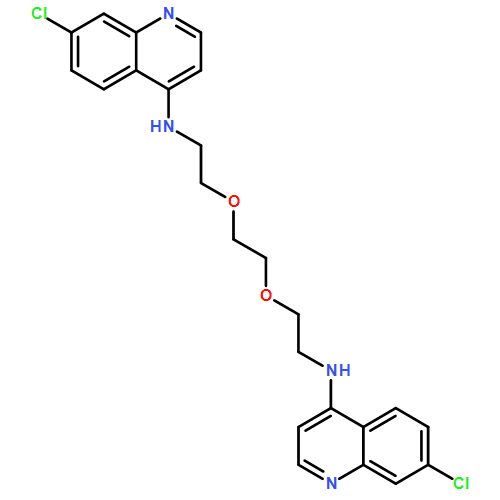
![N2-(7-Chloro-4-quinolinyl)-N1-[2-[(7-chloro-4-quinolinyl)amino]ethyl]-N1-methyl-1,2-ethanediamine](http://img.cochemist.com/ccimg/1391500/1391426-22-4.png)
![N2-(7-Chloro-4-quinolinyl)-N1-[2-[(7-chloro-4-quinolinyl)amino]ethyl]-N1-methyl-1,2-ethanediamine](http://img.cochemist.com/ccimg/1391500/1391426-22-4_b.png)
























![9H-Pyrido[3,4-b]indole-1-carboxaldehyde](http://img.cochemist.com/ccimg/20200/20127-63-3.png)
![9H-Pyrido[3,4-b]indole-1-carboxaldehyde](http://img.cochemist.com/ccimg/20200/20127-63-3_b.png)




































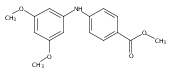
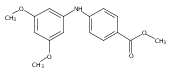
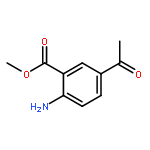
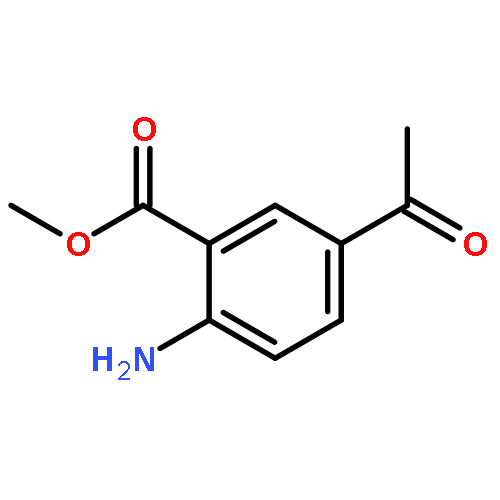
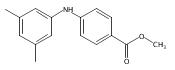
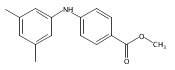
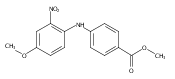

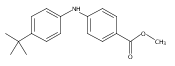
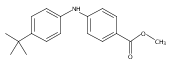
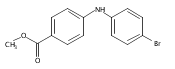
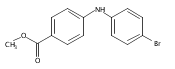
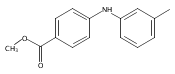
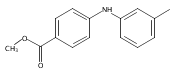
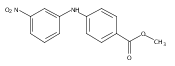
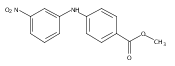
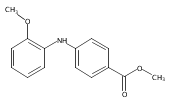
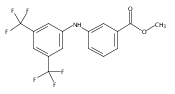
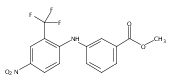
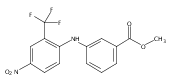
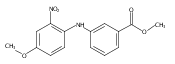
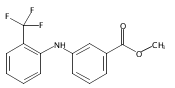
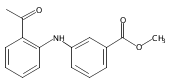
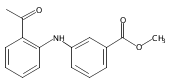
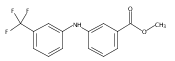
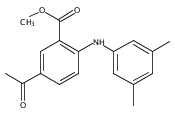
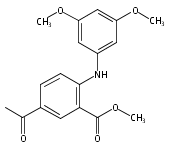
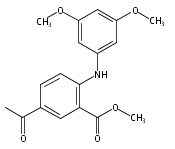
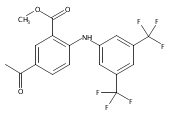
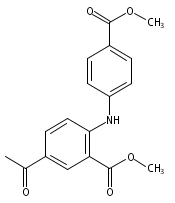
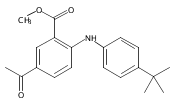
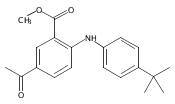
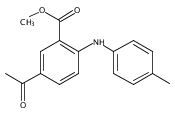
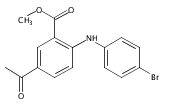
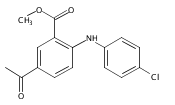
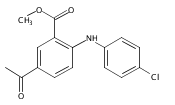
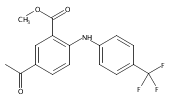
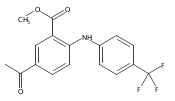
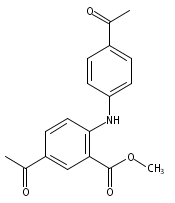
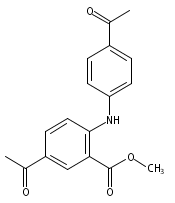
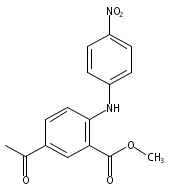
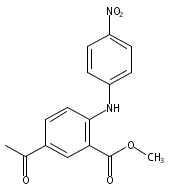
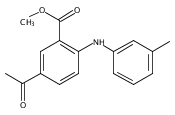
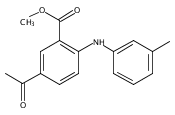
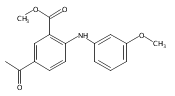
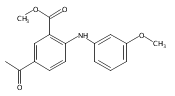
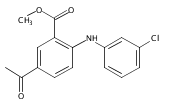
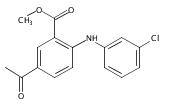
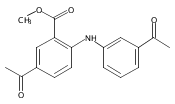
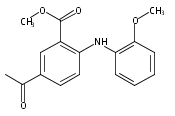
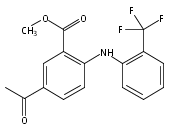
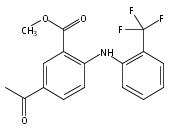
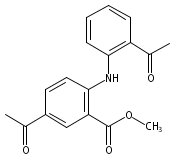
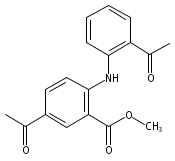
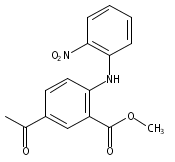
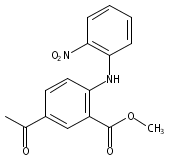
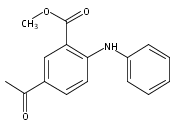
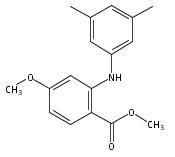
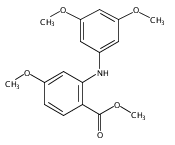
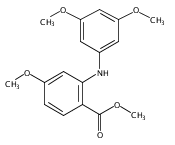
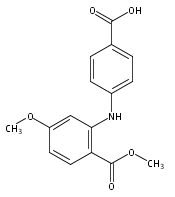
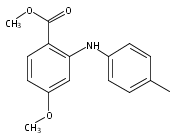
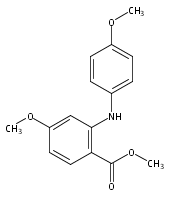
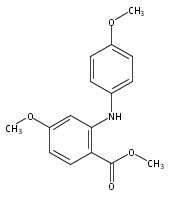
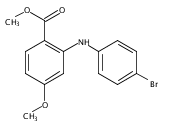
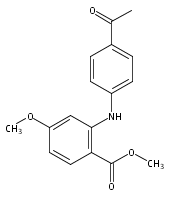
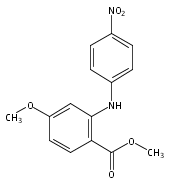
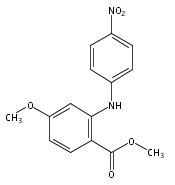
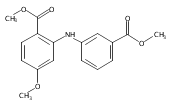
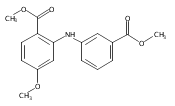
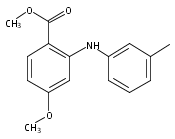
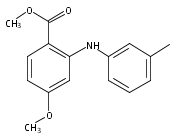
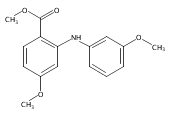
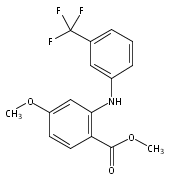
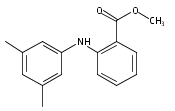
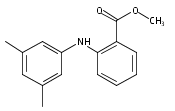
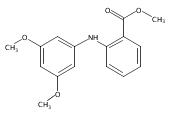
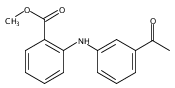
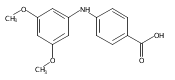
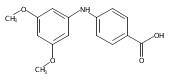
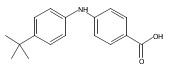
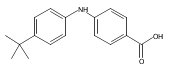
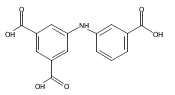
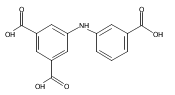
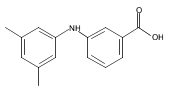
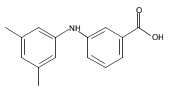
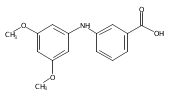
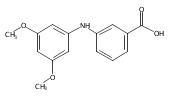
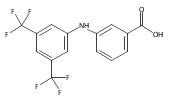
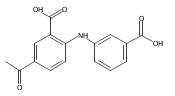
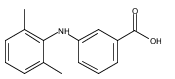
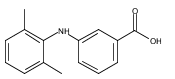
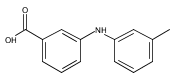
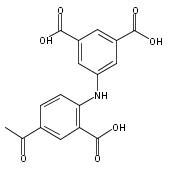
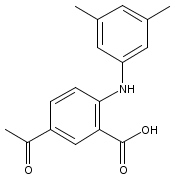
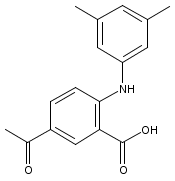
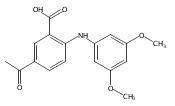
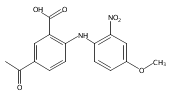
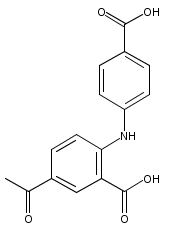
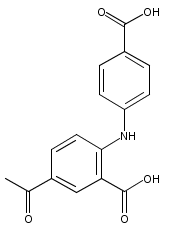
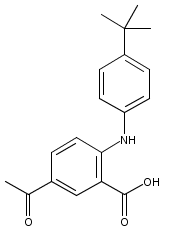
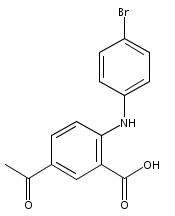
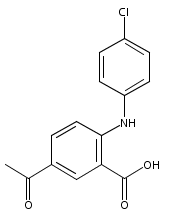
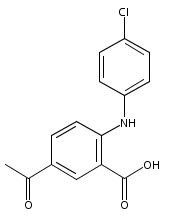
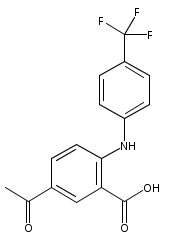
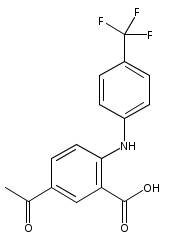
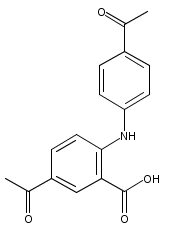
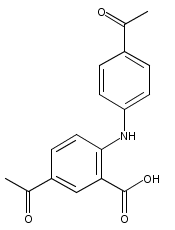
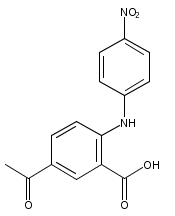
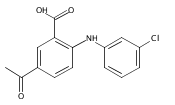
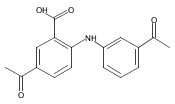
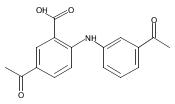
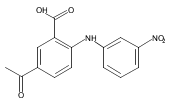

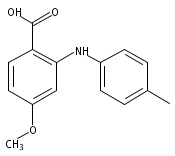
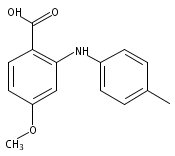
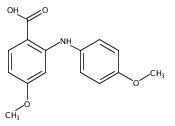

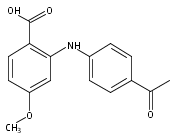
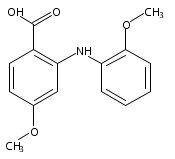
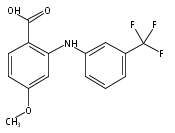
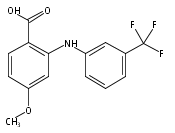


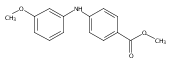
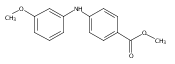
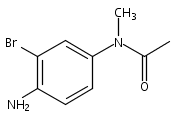
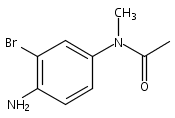
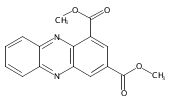
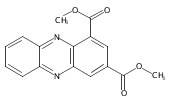
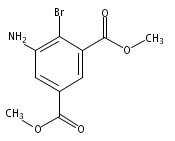
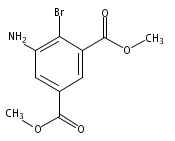
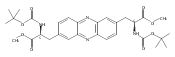
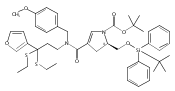
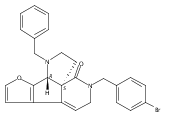
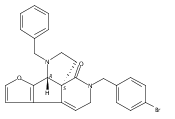
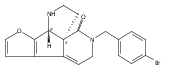
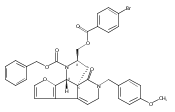
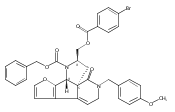
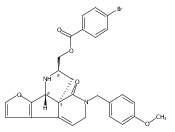
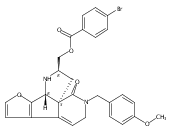
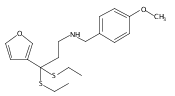
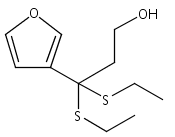
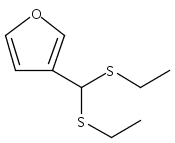
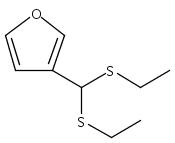
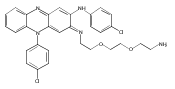
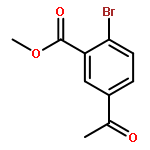
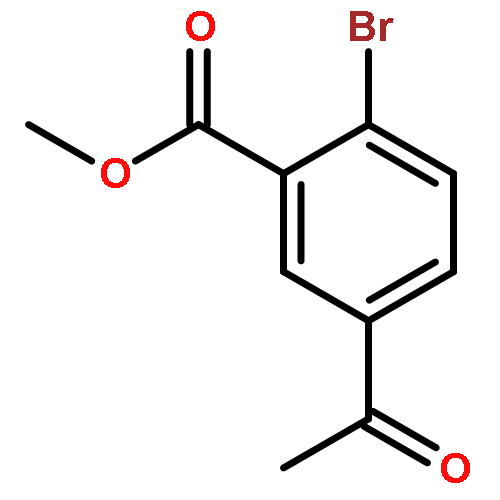

























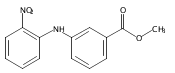
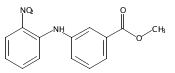
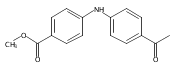
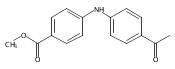
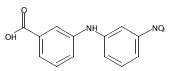
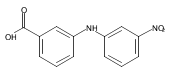
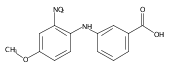
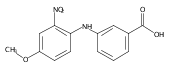
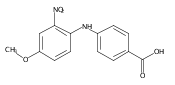
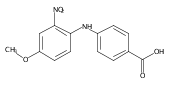
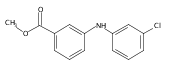
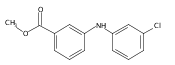
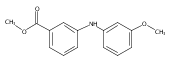
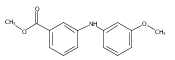




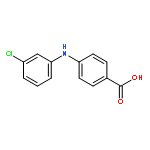
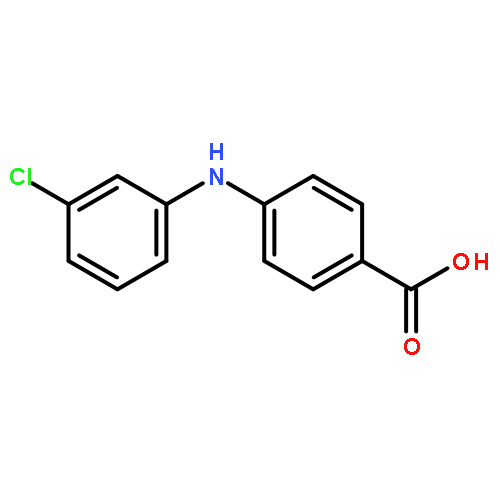
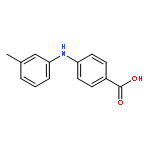
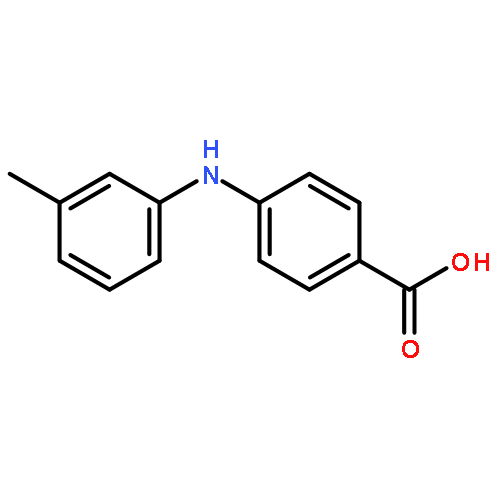
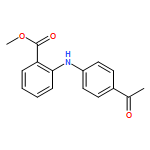
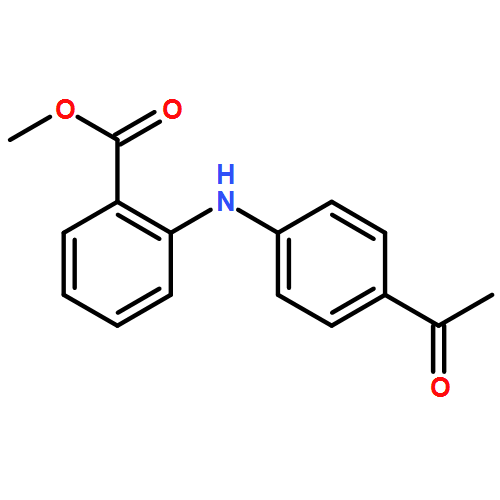
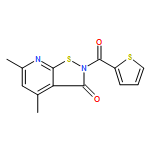
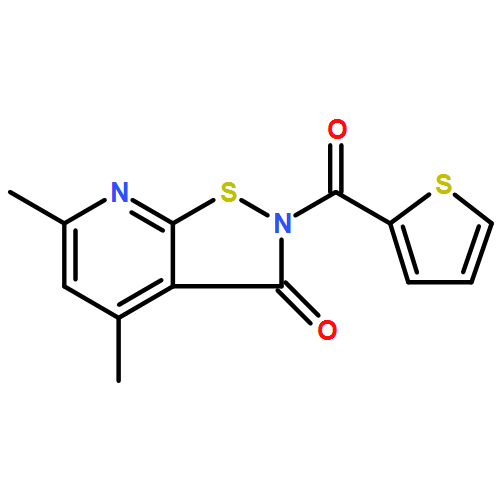
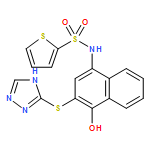

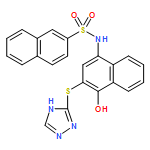
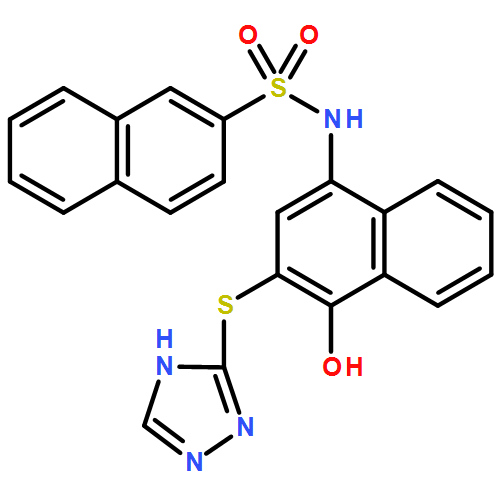
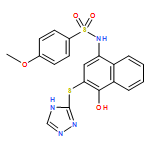
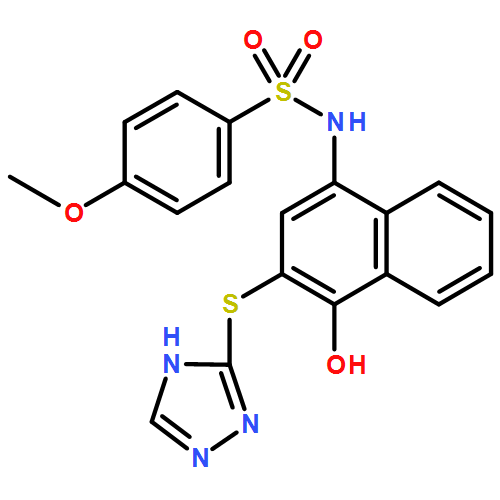
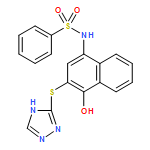
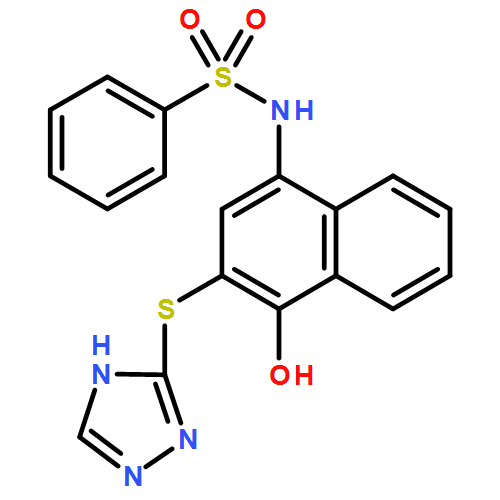
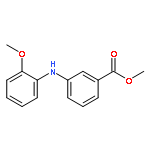
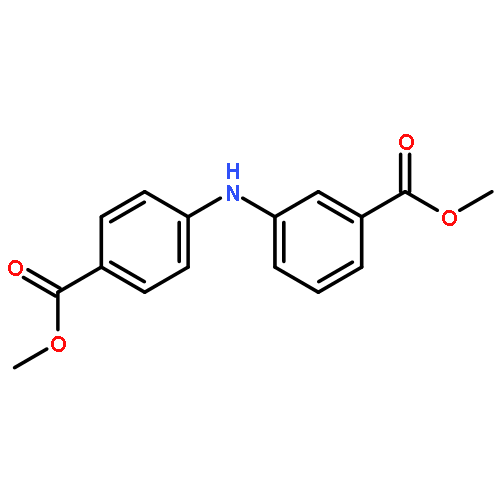
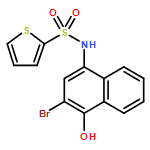
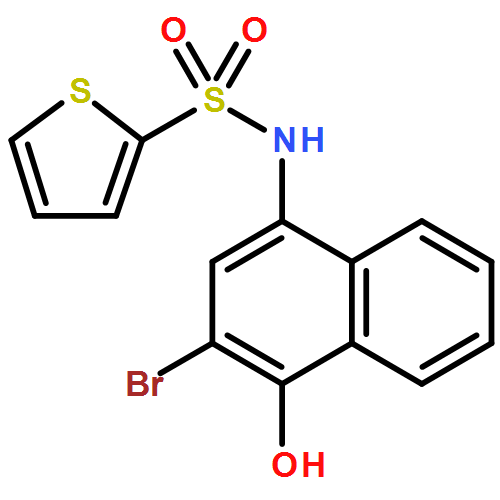
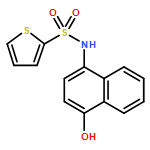
![Benzoic acid, 5-acetyl-2-[[(trifluoromethyl)sulfonyl]oxy]-, methyl ester](http://img.cochemist.com/ccimg/343400/343338-97-6.png)
![Benzoic acid, 5-acetyl-2-[[(trifluoromethyl)sulfonyl]oxy]-, methyl ester](http://img.cochemist.com/ccimg/343400/343338-97-6_b.png)




![1-Propanone,3-[(1-methylethyl)(phenylmethyl)amino]-1-(2-naphthalenyl)-](http://img.cochemist.com/ccimg/273800/273727-89-2.png)
![1-Propanone,3-[(1-methylethyl)(phenylmethyl)amino]-1-(2-naphthalenyl)-](http://img.cochemist.com/ccimg/273800/273727-89-2_b.png)
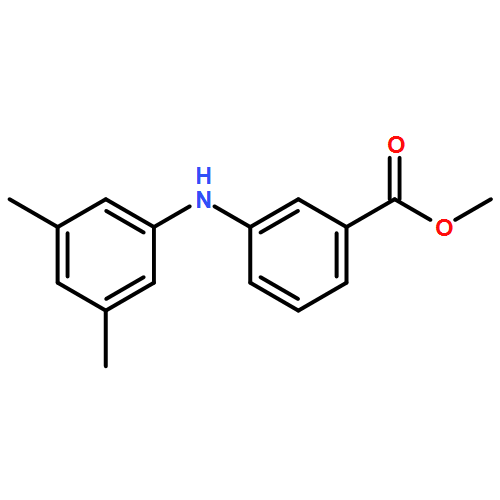
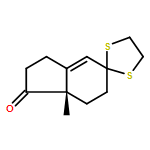
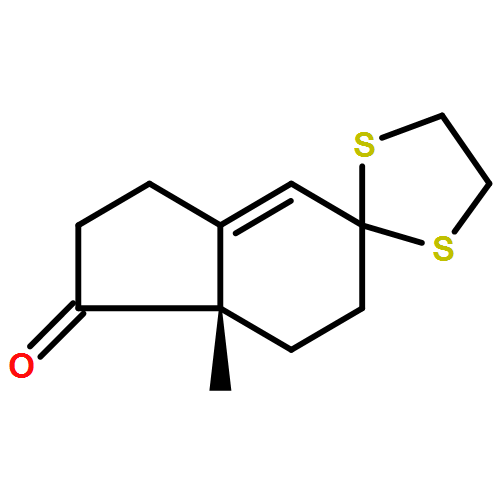
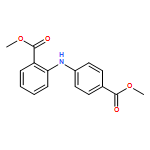
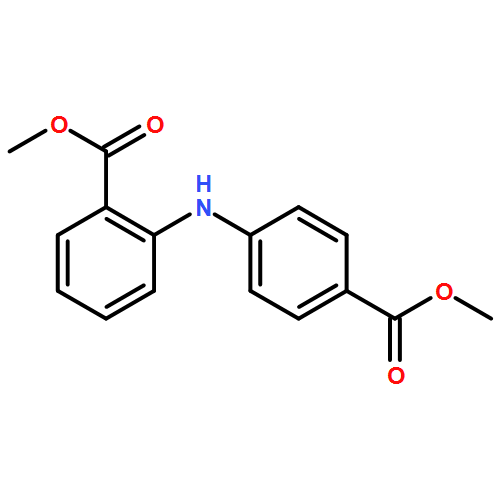
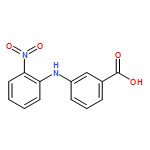
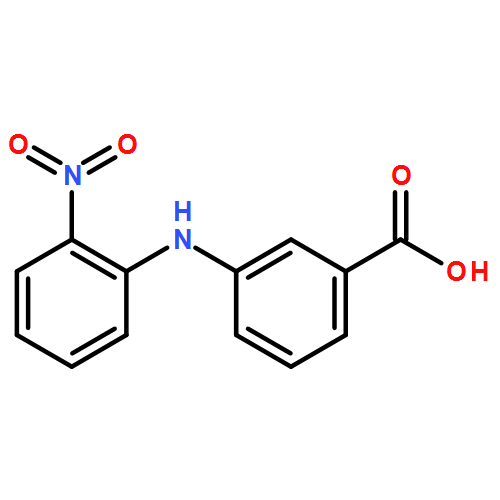
![Benzoic acid, 4-methoxy-2-[[(trifluoromethyl)sulfonyl]oxy]-, methyl ester](http://img.cochemist.com/ccimg/216800/216768-18-2.png)
![Benzoic acid, 4-methoxy-2-[[(trifluoromethyl)sulfonyl]oxy]-, methyl ester](http://img.cochemist.com/ccimg/216800/216768-18-2_b.png)
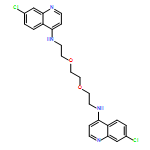
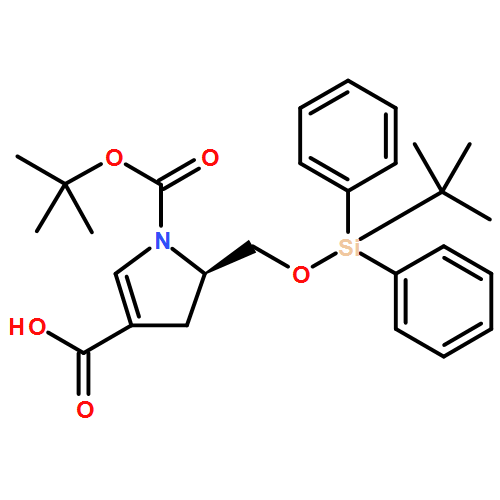




![Benzenepropanenitrile, a-[bis(methylthio)methylene]-b-oxo-3-(trifluoromethyl)-](http://img.cochemist.com/ccimg/116500/116492-97-8.png)
![Benzenepropanenitrile, a-[bis(methylthio)methylene]-b-oxo-3-(trifluoromethyl)-](http://img.cochemist.com/ccimg/116500/116492-97-8_b.png)
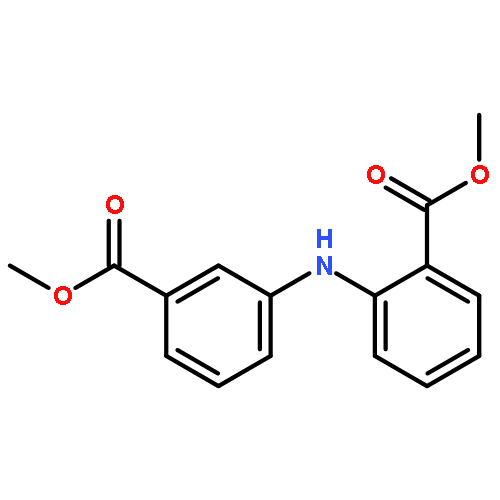
![Benzoic acid, 3-[[(trifluoromethyl)sulfonyl]oxy]-, methyl ester](/data/chemimg/118600/107658-28-6.png)
![Benzoic acid, 3-[[(trifluoromethyl)sulfonyl]oxy]-, methyl ester](/data/chemimg/118600/107658-28-6_b.png)


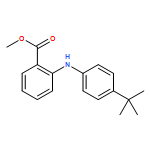
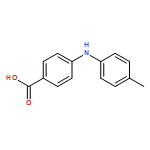
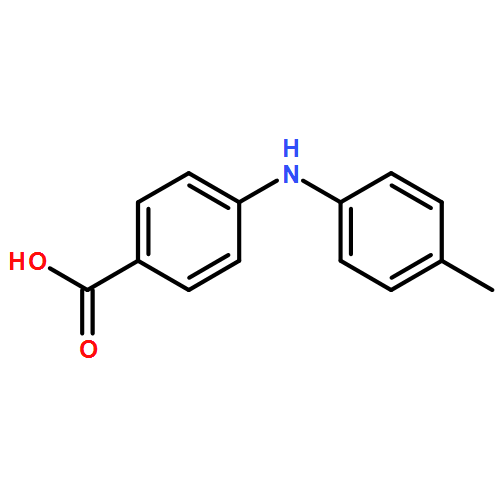
![Benzoic acid,4-[(4-methylphenyl)amino]-, methyl ester](http://img.cochemist.com/ccimg/101100/101089-83-2.png)
![Benzoic acid,4-[(4-methylphenyl)amino]-, methyl ester](http://img.cochemist.com/ccimg/101100/101089-83-2_b.png)
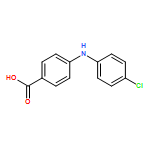
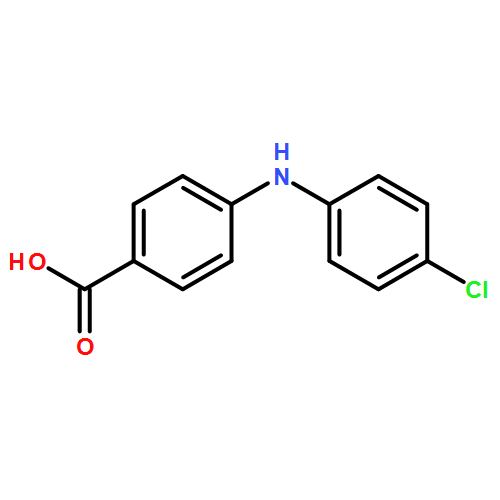


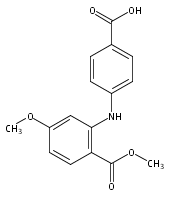
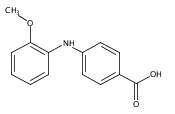
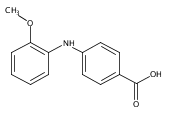
![Benzoic acid, 2-[(4-carboxyphenyl)amino]-4-methoxy-](http://img.cochemist.com/ccimg/86700/86611-38-3.png)
![Benzoic acid, 2-[(4-carboxyphenyl)amino]-4-methoxy-](http://img.cochemist.com/ccimg/86700/86611-38-3_b.png)
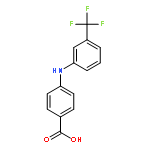
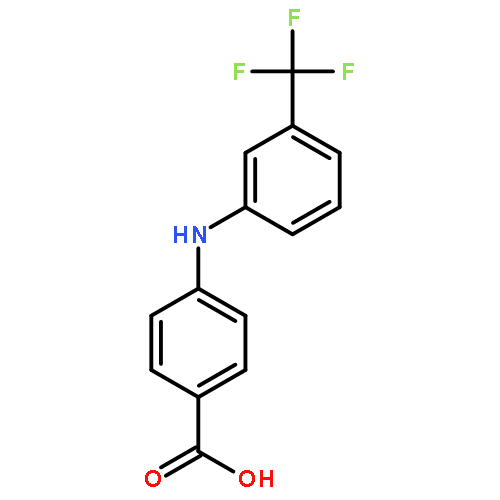
![Benzoic acid, 3-[[3-(trifluoromethyl)phenyl]amino]-](http://img.cochemist.com/ccimg/85100/85010-04-4.png)
![Benzoic acid, 3-[[3-(trifluoromethyl)phenyl]amino]-](http://img.cochemist.com/ccimg/85100/85010-04-4_b.png)
![2-Phenylbenzo[d][1,2]selenazol-3(2H)-one](http://img.cochemist.com/ccimg/61000/60940-34-3.png)
![2-Phenylbenzo[d][1,2]selenazol-3(2H)-one](http://img.cochemist.com/ccimg/61000/60940-34-3_b.png)
![Imidazo[2,1-b]naphtho[2,3-d]thiazole-5,10-dione](http://img.cochemist.com/ccimg/59800/59774-66-2.png)
![Imidazo[2,1-b]naphtho[2,3-d]thiazole-5,10-dione](http://img.cochemist.com/ccimg/59800/59774-66-2_b.png)
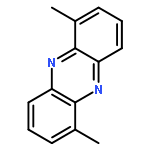
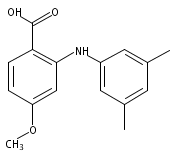
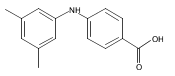
![BENZOIC ACID, 2-[(3,5-DIMETHYLPHENYL)AMINO]-](http://img.cochemist.com/ccimg/55700/55602-36-3.png)
![BENZOIC ACID, 2-[(3,5-DIMETHYLPHENYL)AMINO]-](http://img.cochemist.com/ccimg/55700/55602-36-3_b.png)
![2-[(3-acetylphenyl)amino]benzoic acid](http://img.cochemist.com/ccimg/27700/27696-28-2.png)
![2-[(3-acetylphenyl)amino]benzoic acid](http://img.cochemist.com/ccimg/27700/27696-28-2_b.png)
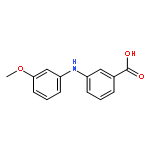
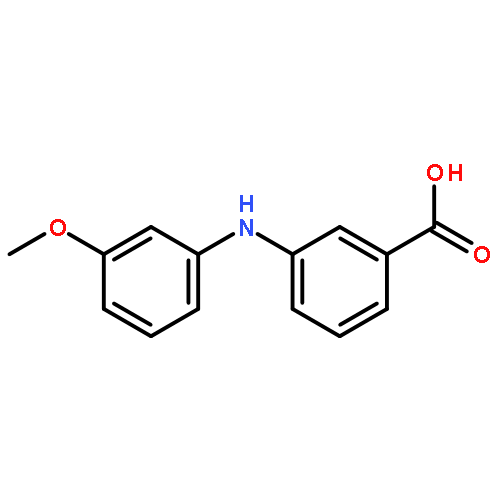
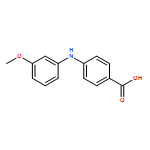
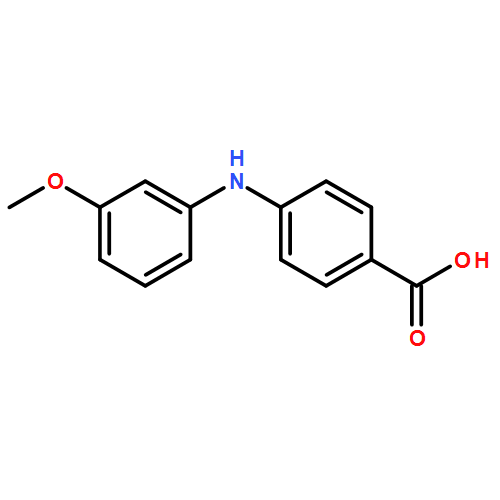
![Benzoic acid,2-[(3-methoxyphenyl)amino]-](http://img.cochemist.com/ccimg/27700/27693-73-8.png)
![Benzoic acid,2-[(3-methoxyphenyl)amino]-](http://img.cochemist.com/ccimg/27700/27693-73-8_b.png)
![Benzoic acid,2-[(3-nitrophenyl)amino]-](http://img.cochemist.com/ccimg/27700/27693-70-5.png)
![Benzoic acid,2-[(3-nitrophenyl)amino]-](http://img.cochemist.com/ccimg/27700/27693-70-5_b.png)
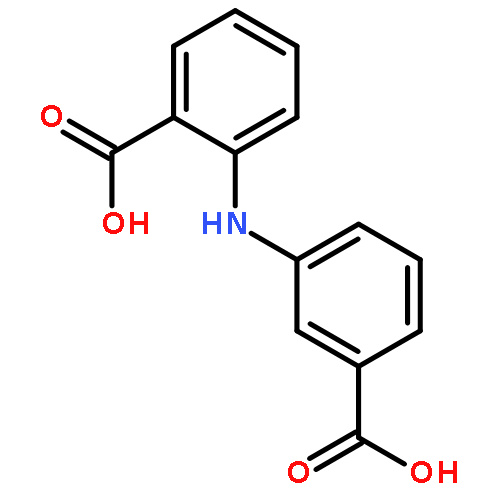
![Benzoic acid, 2-[(4-chlorophenyl)amino]-, methyl ester](http://img.cochemist.com/ccimg/23900/23868-23-7.png)
![Benzoic acid, 2-[(4-chlorophenyl)amino]-, methyl ester](http://img.cochemist.com/ccimg/23900/23868-23-7_b.png)
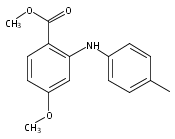
![Benzoic acid, 2-[(4-methylphenyl)amino]-, methyl ester](http://img.cochemist.com/ccimg/23900/23868-15-7.png)
![Benzoic acid, 2-[(4-methylphenyl)amino]-, methyl ester](http://img.cochemist.com/ccimg/23900/23868-15-7_b.png)
![Benzoic acid, 2-[(3-methylphenyl)amino]-, methyl ester](http://img.cochemist.com/ccimg/23900/23868-14-6.png)
![Benzoic acid, 2-[(3-methylphenyl)amino]-, methyl ester](http://img.cochemist.com/ccimg/23900/23868-14-6_b.png)
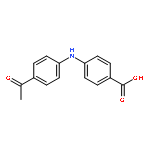
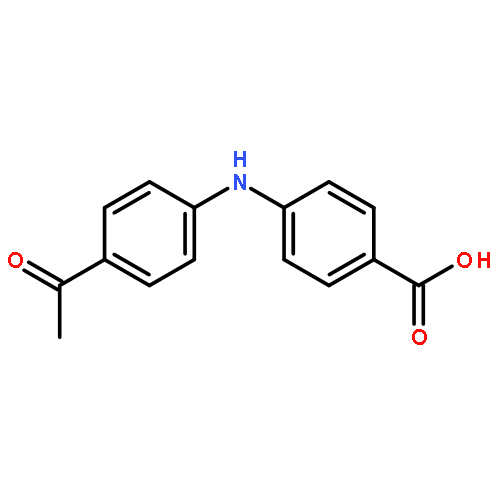
![Benzoic acid, 2-[(4-acetylphenyl)amino]-](http://img.cochemist.com/ccimg/23700/23600-82-0.png)
![Benzoic acid, 2-[(4-acetylphenyl)amino]-](http://img.cochemist.com/ccimg/23700/23600-82-0_b.png)
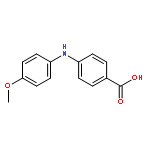
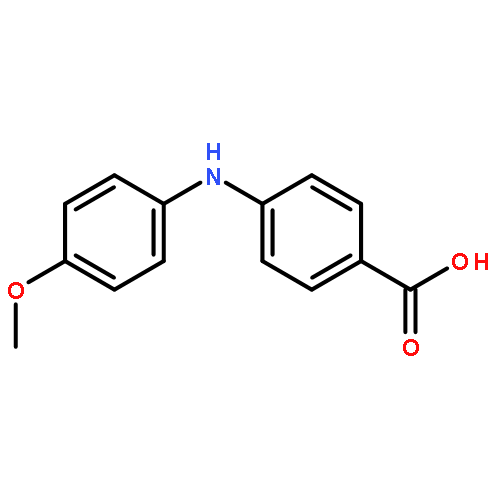

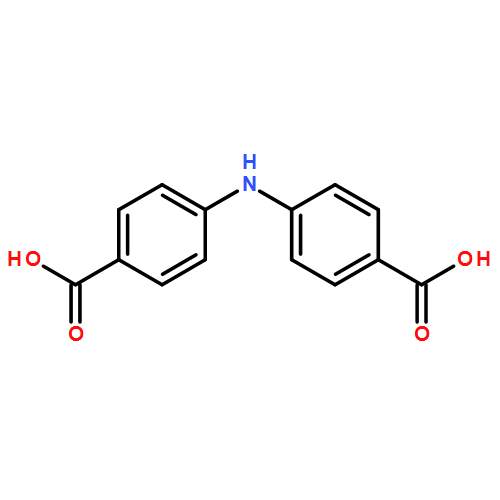
![Benzoic acid, 4-[[(trifluoromethyl)sulfonyl]oxy]-, methyl ester](http://img.cochemist.com/ccimg/17800/17763-71-2.png)
![Benzoic acid, 4-[[(trifluoromethyl)sulfonyl]oxy]-, methyl ester](http://img.cochemist.com/ccimg/17800/17763-71-2_b.png)
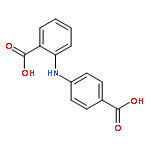
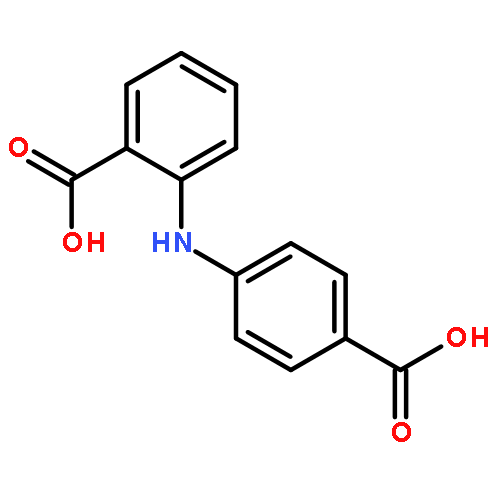
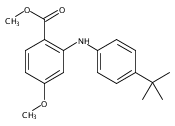
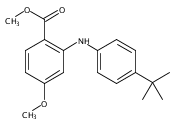
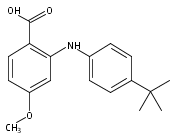
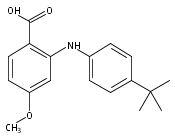
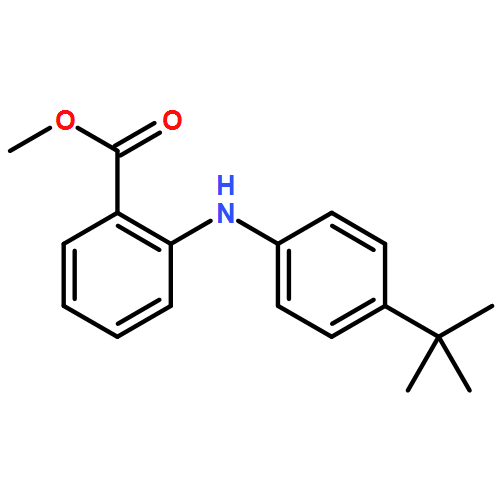
![Benzoic acid,2-[[4-(1,1-dimethylethyl)phenyl]amino]-](http://img.cochemist.com/ccimg/17400/17332-56-8.png)
![Benzoic acid,2-[[4-(1,1-dimethylethyl)phenyl]amino]-](http://img.cochemist.com/ccimg/17400/17332-56-8_b.png)
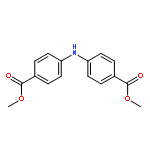
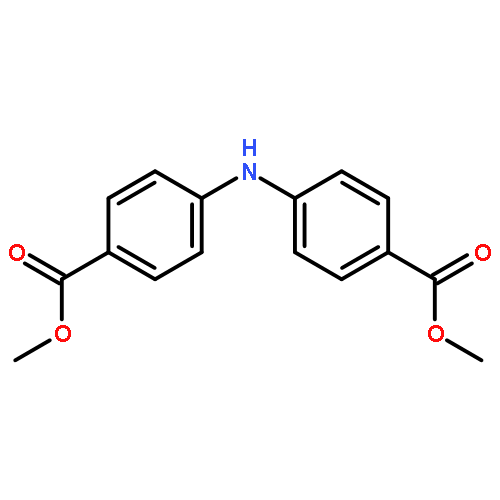
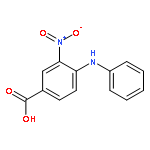
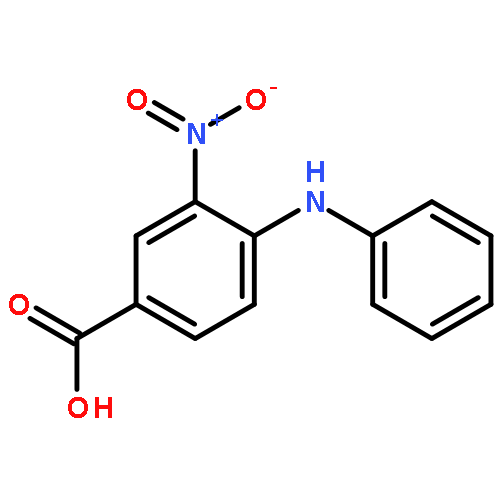


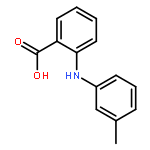
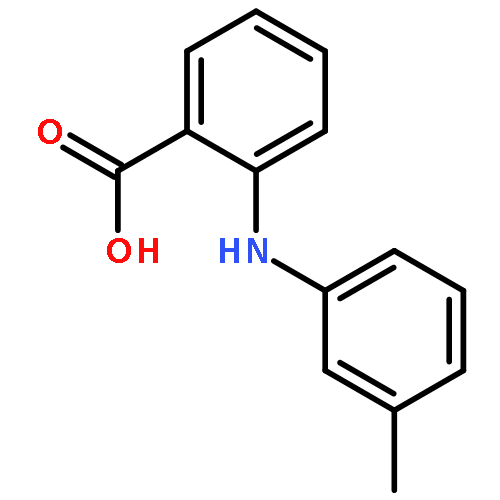
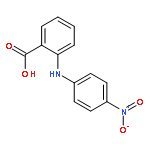
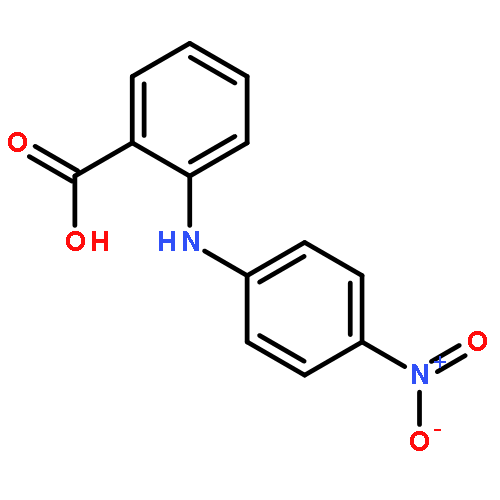
![Benzoic acid,4-[(4-nitrophenyl)amino]-](http://img.cochemist.com/ccimg/16200/16124-69-9.png)
![Benzoic acid,4-[(4-nitrophenyl)amino]-](http://img.cochemist.com/ccimg/16200/16124-69-9_b.png)












![[1,1'-Biphenyl]-4-sulfonamide](http://img.cochemist.com/ccimg/4400/4371-23-7.png)
![[1,1'-Biphenyl]-4-sulfonamide](http://img.cochemist.com/ccimg/4400/4371-23-7_b.png)


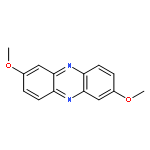
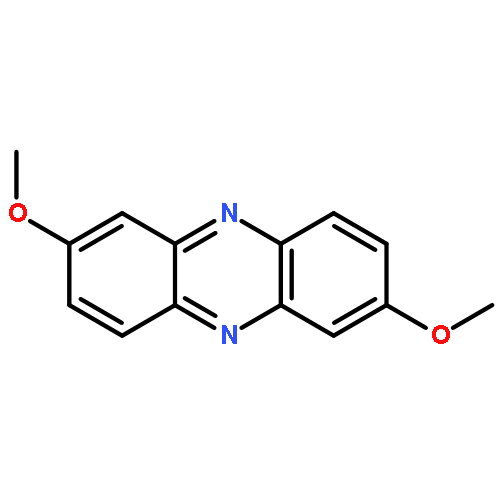
![Dibenzo[a,h]phenazine](http://img.cochemist.com/ccimg/300/226-47-1.png)
![Dibenzo[a,h]phenazine](http://img.cochemist.com/ccimg/300/226-47-1_b.png)



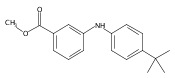
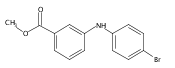
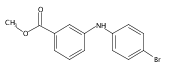
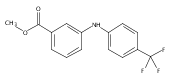
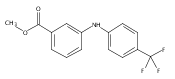
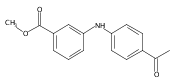
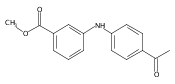
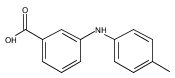
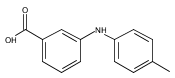
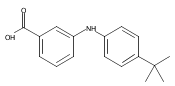
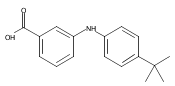
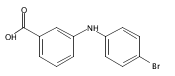
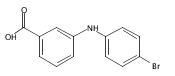
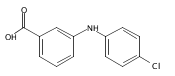
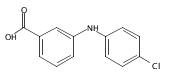
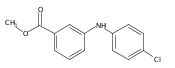

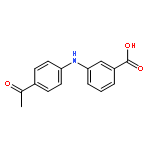
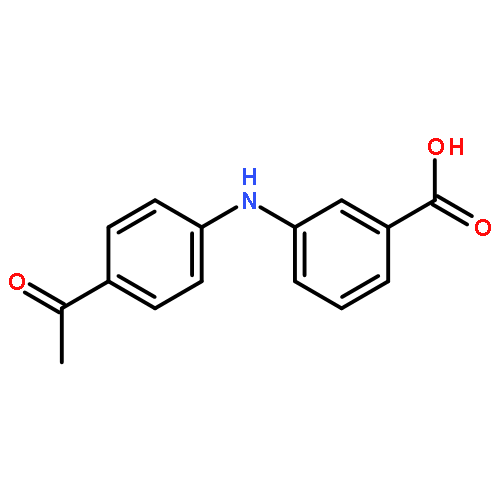
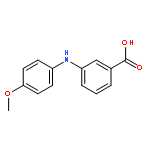
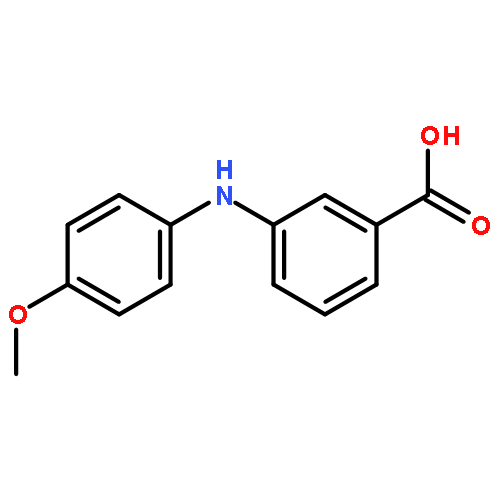
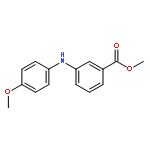
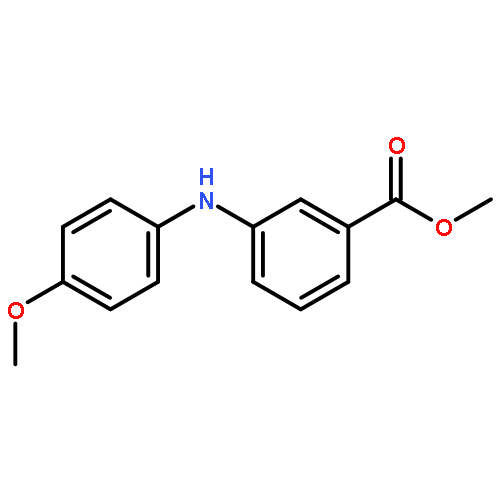
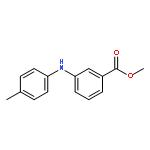
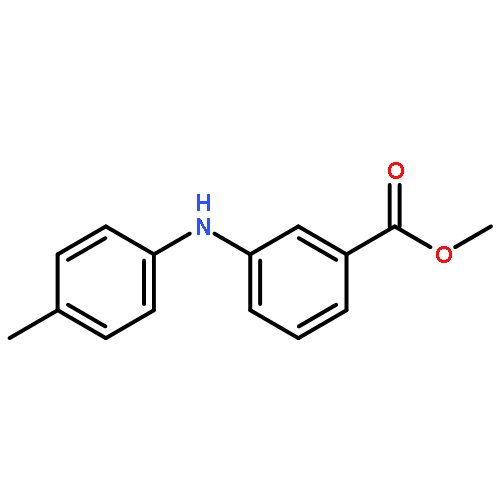


![Benzoic acid, 2-[[(trifluoromethyl)sulfonyl]oxy]-, methyl ester](http://img.cochemist.com/ccimg/17800/17763-70-1.png)
![Benzoic acid, 2-[[(trifluoromethyl)sulfonyl]oxy]-, methyl ester](http://img.cochemist.com/ccimg/17800/17763-70-1_b.png)
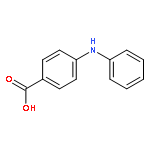
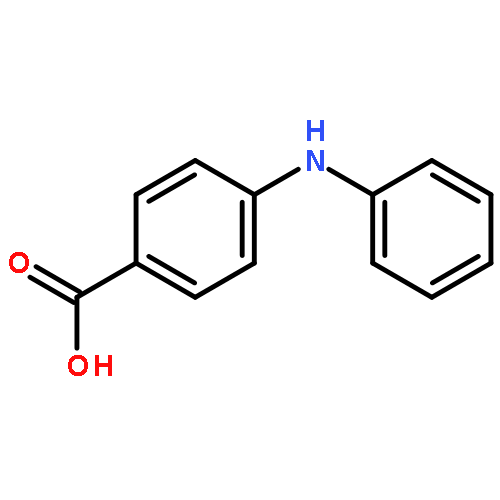





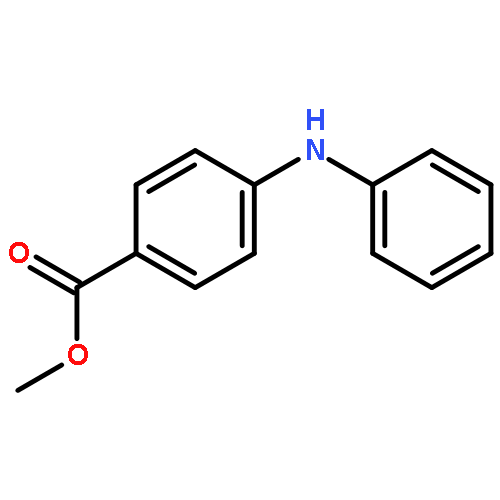
![Benzoic acid,2-[[3-(trifluoromethyl)phenyl]amino]-, methyl ester](http://img.cochemist.com/ccimg/2800/2765-91-5.png)
![Benzoic acid,2-[[3-(trifluoromethyl)phenyl]amino]-, methyl ester](http://img.cochemist.com/ccimg/2800/2765-91-5_b.png)