Co-reporter:Junhang Jiang; Canhui Zheng; Kongkai Zhu; Jia Liu; Nannan Sun; Chongqing Wang; Hualiang Jiang; Ju Zhu; Cheng Luo;Youjun Zhou
Journal of Medicinal Chemistry 2015 Volume 58(Issue 5) pp:2538-2546
Publication Date(Web):February 17, 2015
DOI:10.1021/acs.jmedchem.5b00118
A potent combretastatin A-4 (CA-4) like tubulin polymerization inhibitor 22b was found with strong antitumor activity previously. However, it easily undergoes cis–trans isomerization under natural light, and the resulting decrease in activity limits its further applications. In this study, we used quantum chemistry calculations to explore the molecular basis of its instability. Aided by the calculations, two rounds of structural optimization of 22b were conducted. Accelerated quantitative light stability testing confirmed that the stability of these designed compounds was significantly improved as predicted. Among them, compounds 1 and 3b displayed more potent inhibitory activity on tumor cell growth than 22b. In addition, the potent in vivo antitumor activity of compound 1 was confirmed. Quantum chemistry calculations were used in the optimization of stilbene-like molecules, providing new insight into stilbenoid optimization and important implications for the future development of novel CA-4-like tubulin polymerization inhibitors.
Co-reporter:Ran Zhou, Yiqian Xie, Hao Hu, Guang Hu, Viral Sanjay Patel, Jin Zhang, Kunqian Yu, Yiran Huang, Hualiang Jiang, Zhongjie Liang, Yujun George Zheng, and Cheng Luo
Journal of Chemical Information and Modeling 2015 Volume 55(Issue 12) pp:2623-2632
Publication Date(Web):November 12, 2015
DOI:10.1021/acs.jcim.5b00454
Protein arginine methyltransferases (PRMTs) catalyze the posttranslational methylation of arginine, which is important in a range of biological processes, including epigenetic regulation, signal transduction, and cancer progression. Although previous studies of PRMT1 mutants suggest that the dimerization arm and the N-terminal region of PRMT1 are important for activity, the contributions of these regions to the structural architecture of the protein and its catalytic methylation activity remain elusive. Molecular dynamics (MD) simulations performed in this study showed that both the dimerization arm and the N-terminal region undergo conformational changes upon dimerization. Because a correlation was found between the two regions despite their physical distance, an allosteric pathway mechanism was proposed based on a network topological analysis. The mutation of residues along the allosteric pathways markedly reduced the methylation activity of PRMT1, which may be attributable to the destruction of dimer formation and accordingly reduced S-adenosyl-L-methionine (SAM) binding. This study provides the first demonstration of the use of a combination of MD simulations, network topological analysis, and biochemical assays for the exploration of allosteric regulation upon PRMT1 dimerization. These findings illuminate the results of mechanistic studies of PRMT1, which have revealed that dimer formation facilitates SAM binding and catalytic methylation, and provided direction for further allosteric studies of the PRMT family.
Co-reporter:Yiqian Xie, Ran Zhou, Fulin Lian, Yan Liu, Limin Chen, Zhe Shi, Naixia Zhang, Mingyue Zheng, Bairong Shen, Hualiang Jiang, Zhongjie Liang and Cheng Luo
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 47) pp:9665-9673
Publication Date(Web):28 Oct 2014
DOI:10.1039/C4OB01591F
Protein arginine methylation is a common post-translational modification which is crucial for a variety of biological processes. Dysregulation of protein arginine methyltransferases (PRMTs) activity has been implicated in cancer and other serious diseases. Thus, small molecule inhibitors against PRMT have great potential for therapeutic development. Herein, through the combination of virtual screening and bioassays, six small molecular compounds were identified as PRMT1 inhibitors. Amongst them, the binding affinity of compounds DCLX069 and DCLX078 with PRMT1 was further validated by T1ρ and saturation transfer difference (STD) NMR experiments. Most important of all, both compounds effectively blocked cell proliferation in breast cancer, liver cancer and acute myeloid leukemia cell lines. The binding mode analysis from molecular docking simulations theoretically indicated that both inhibitors occupied the SAM binding pocket to exert the inhibitory effect. Taken together, our compounds enriched the structural scaffolds as PRMT1 inhibitors and afforded clues for further optimization.
Co-reporter:Zhongjie Liang, Jing Ai, Xiao Ding, Xia Peng, Dengyou Zhang, Ruihan Zhang, Ying Wang, Fang Liu, Mingyue Zheng, Hualiang Jiang, Hong Liu, Meiyu Geng, and Cheng Luo
ACS Medicinal Chemistry Letters 2013 Volume 4(Issue 4) pp:408
Publication Date(Web):February 25, 2013
DOI:10.1021/ml4000047
The aberrant function of c-Met kinase signaling pathway is ubiquitously involved in a broad spectrum of human cancers; thus, a strong rationale exists for targeting the kinase pathway in cancer therapy. Via integration of computational and experimental studies, anthraquinone derivatives were identified for the first time as potent c-Met kinase inhibitors in this research. The aberrant activation of the c-Met kinase pathway results from (TPR)-Met, MET gene mutation, or amplification and a hepatocyte growth factor (HGF)/scatter factor-dependent autocrine or paracrine mechanism. However, anthraquinone derivatives exclusively suppressed c-Met phosphorylation stimulated by HGF in A549 cells, indicating that the compounds possess the ability to block the extracellular HGF-dependent pathway. A surface plasmon resonance assay revealed that the most potent compound, 2a, shows a high binding affinity for HGF with an equilibrium dissociation constant of 1.95 μM. The dual roles of compound 2a demonstrate the potency of anthraquinone derivatives and provide a new design solution for the c-Met kinase signaling pathway.Keywords: Anthraquinone derivatives; binding affinity with HGF; c-Met kinase inhibitors
Co-reporter:Dengyou Zhang, Xiaowei Zhang, Jing Ai, Yun Zhai, Zhongjie Liang, Ying Wang, Yi Chen, Chunpu Li, Fei Zhao, Hualiang Jiang, Meiyu Geng, Cheng Luo, Hong Liu
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 21) pp:6804-6820
Publication Date(Web):1 November 2013
DOI:10.1016/j.bmc.2013.07.032
A series of 2-amino-N-benzylpyridine-3-carboxnamides, 2-amino-N-benzylpyridine-3-sulfonamides and 2-amino-3-benzylthiopyridines against c-Met were designed by means of bioisosteric replacement and docking analysis. Optimization of the 2-amino-3-benzylthiopyridine scaffold led to the identification of compound (R)-10b displaying c-Met inhibition with an IC50 up to 7.7 nM. In the cytotoxic evaluation, compound (R)-10b effectively inhibited the proliferation of c-Met addictive human cancer cell lines (IC50 from 0.19 to 0.71 μM) and c-Met activation-mediated cell metastasis. At a dose of 100 mg/Kg, (R)-10b evidently inhibited tumor growth (45%) in NIH-3T3/TPR-Met xenograft model. Of note, (R)-10b could overcome c-Met-activation mediated gefitinib-resistance, which indicated its potential use for drug combination. Taken together, 2-amino-3-benzylthiopyridine scaffold was first disclosed and exhibited promising pharmacological profiles against c-Met, which left room for further exploration.A series of 2-amino-N-benzylpyridine-3-carboxnamides, 2-amino-N-benzylpyridine-3-sulfonamides and 2-amino-3-benzylthiopyridines against c-Met were designed by means of bioisosteric replacement and docking analysis. Optimization of the 2-amino-3-benzylthiopyridine scaffold identified compound (R)-10b displaying c-Met inhibition with an IC50 up to 7.7 nM. In the cytotoxic evaluation, compound (R)-10b effectively inhibited the proliferation of c-Met addictive human cancer cell lines (IC50 from 0.19 to 0.71 μM) and c-Met activation-mediated cell metastasis. Of note, (R)-10b could overcome c-Met-activation mediated gefitinib-resistance. (R)-10b could exhibit moderate inhibition of tumor growth (45%) in NIH-3T3/TPR-Met xenograft model probably due to its high clearance and low bioavailability.
Co-reporter:Yunfeng Xie, Xianjie Chen, Jie Qin, Xiangqian Kong, Fei Ye, Yuren Jiang, Hong Liu, Hualiang Jiang, Ronen Marmorstein, Cheng Luo
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 8) pp:2306-2312
Publication Date(Web):15 April 2013
DOI:10.1016/j.bmcl.2013.02.072
The V600E BRAF kinase mutation, which activates the downstream MAPK signaling pathway, commonly occurs in about 8% of all human malignancies and about 50% of all melanomas. In this study, we employed virtual screening and chemical synthesis to identify a series of N-(thiophen-2-yl) benzamide derivatives as potent BRAFV600E inhibitors. Structure–activity relationship studies of these derivatives revealed that compounds b40 and b47 are the two most potent BRAFV600E inhibitors in this series.
Co-reporter:Kongkai Zhu;Junyan Lu;Zhongjie Liang
Journal of Computer-Aided Molecular Design 2013 Volume 27( Issue 3) pp:247-256
Publication Date(Web):2013 March
DOI:10.1007/s10822-012-9630-6
New Delhi metallo-β-lactamase-1 (NDM-1) has emerged as a major global threat to human health for its rapid rate of dissemination and ability to make pathogenic microbes resistant to almost all known β-lactam antibiotics. In addition, effective NDM-1 inhibitors have not been identified to date. In spite of the plethora of structural and kinetic data available, the accurate molecular characteristics of and details on the enzymatic reaction of NDM-1 hydrolyzing β-lactam antibiotics remain incompletely understood. In this study, a combined computational approach including molecular docking, molecular dynamics simulations and quantum mechanics/molecular mechanics calculations was performed to characterize the catalytic mechanism of meropenem catalyzed by NDM-1. The quantum mechanics/molecular mechanics results indicate that the ionized D124 is beneficial to the cleavage of the C–N bond within the β-lactam ring. Meanwhile, it is energetically favorable to form an intermediate if no water molecule coordinates to Zn2. Moreover, according to the molecular dynamics results, the conserved residue K211 plays a pivotal role in substrate binding and catalysis, which is quite consistent with previous mutagenesis data. Our study provides detailed insights into the catalytic mechanism of NDM-1 hydrolyzing meropenem β-lactam antibiotics and offers clues for the discovery of new antibiotics against NDM-1 positive strains in clinical studies.
Co-reporter:Baoen Chen ; Fei Ye ; Lu Yu ; Guifang Jia ; Xiaotian Huang ; Xueju Zhang ; Shuying Peng ; Kai Chen ; Meining Wang ; Shouze Gong ; Ruihan Zhang ; Jinya Yin ; Haiyan Li ; Yiming Yang ; Hong Liu ; Jiwen Zhang ; Haiyan Zhang ; Ao Zhang ; Hualiang Jiang ; Cheng Luo ;Cai-Guang Yang
Journal of the American Chemical Society 2012 Volume 134(Issue 43) pp:17963-17971
Publication Date(Web):October 9, 2012
DOI:10.1021/ja3064149
The direct nucleic acid repair dioxygenase FTO is an enzyme that demethylates N6-methyladenosine (m6A) residues in mRNA in vitro and inside cells. FTO is the first RNA demethylase discovered that also serves a major regulatory function in mammals. Together with structure-based virtual screening and biochemical analyses, we report the first identification of several small-molecule inhibitors of human FTO demethylase. The most potent compound, the natural product rhein, which is neither a structural mimic of 2-oxoglutarate nor a chelator of metal ion, competitively binds to the FTO active site in vitro. Rhein also exhibits good inhibitory activity on m6A demethylation inside cells. These studies shed light on the development of powerful probes and new therapies for use in RNA biology and drug discovery.
Co-reporter:Juxian Wang ; Limin Chen ; Sarmistha Halder Sinha ; Zhongjie Liang ; Huifang Chai ; Sakthivel Muniyan △; Yu-Wei Chou △; Chao Yang ; Leilei Yan ; You Feng ; Keqin Kathy Li ; Ming-Fong Lin △; Hualiang Jiang ; Yujun George Zheng
Journal of Medicinal Chemistry 2012 Volume 55(Issue 18) pp:7978-7987
Publication Date(Web):August 28, 2012
DOI:10.1021/jm300521m
Protein arginine methyltransferases (PRMTs) are proved to play vital roles in chromatin remodeling, RNA metabolism, and signal transduction. Aberrant regulation of PRMT activity is associated with various pathological states such as cancer and cardiovascular disorders. Development and application of small molecule PRMT inhibitors will provide new avenues for therapeutic discovery. The combination of pharmacophore-based virtual screening methods with radioactive methylation assays provided six hits identified as inhibitors against the predominant arginine methyltransferase PRMT1 within micromolar potency. Two potent compounds, A9 and A36, exhibited the inhibitory effect by directly targeting substrate H4 other than PRMT1 and displayed even higher inhibition activity than the well-known PRMT inhibitors AMI-1. A9 significantly inhibits proliferation of castrate-resistant prostate cancer cells. Together, A9 may be a potential inhibitor against advanced hormone-independent cancers, and the work will provide clues for the future development of specific compounds that block the interaction of PRMTs with their targets.
Co-reporter:Xiangqian Kong, Jie Qin, Zeng Li, Adina Vultur, Linjiang Tong, Enguang Feng, Geena Rajan, Shien Liu, Junyan Lu, Zhongjie Liang, Mingyue Zheng, Weiliang Zhu, Hualiang Jiang, Meenhard Herlyn, Hong Liu, Ronen Marmorstein and Cheng Luo
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 36) pp:7402-7417
Publication Date(Web):13 Jul 2012
DOI:10.1039/C2OB26081F
Oncogenic mutations in critical nodes of cellular signaling pathways have been associated with tumorigenesis and progression. The B-Raf protein kinase, a key hub in the canonical MAPK signaling cascade, is mutated in a broad range of human cancers and especially in malignant melanoma. The most prevalent B-RafV600E mutant exhibits elevated kinase activity and results in constitutive activation of the MAPK pathway, thus making it a promising drug target for cancer therapy. Herein, we describe the development of novel B-RafV600E selective inhibitors via multi-step virtual screening and hierarchical hit optimization. Nine hit compounds with low micromolar IC50 values were identified as B-RafV600E inhibitors through virtual screening. Subsequent scaffold-based analogue searching and medicinal chemistry efforts significantly improved both the inhibitor potency and oncogene selectivity. In particular, compounds 22f and 22q possess nanomolar IC50 values with selectivity for B-RafV600Ein vitro and exclusive cytotoxicity against B-RafV600E harboring cancer cells.
Co-reporter:Dengyou Zhang, Jing Ai, Zhongjie Liang, Chunpu Li, Xia Peng, YinChun Ji, Hualiang Jiang, Meiyu Geng, Cheng Luo, Hong Liu
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 17) pp:5169-5180
Publication Date(Web):1 September 2012
DOI:10.1016/j.bmc.2012.07.007
A series of 2-aminopyridine-3-carboxamide derivatives against c-Met were designed and synthesized by employing bioisosteric replacement of heterocyclic moieties with the amide bond. The structure–activity relationship (SAR) at various positions of the scaffold was explored. In this study, a promising compound (S)-24o with a c-Met IC50 of 0.022 μM was identified. The compound exhibited dose-dependent inhibition of the phosphorylation of c-Met as well as downstream signaling in EBC-1 cells. Furthermore, the interactive binding model of (S)-24o with c-Met was elucidated by virtue of a molecular modeling study.A series of 2-aminopyridine-3-carboxamide derivatives against c-Met were designed and synthesized by employing bioisosteric replacement of heterocyclic moieties with the amide bond. The SAR exploration led to the identification of a potent compound (S)-24o with a c-Met IC50 of 0.022 μM. This compound exhibited dose-dependent inhibition of the phosphorylation of c-Met as well as c-Met downstream signaling in EBC-1 cells. The interactive binding model of (S)-24o was elucidated using AutoDock4.2.
Co-reporter:Zhongjie Liang, Liying Zhang, Lianchun Li, Jun Liu, Hongling Li, Luyong Zhang, Limin Chen, Keguang Cheng, Mingyue Zheng, Xiaoan Wen, Pu Zhang, Jia Hao, Yanchun Gong, Xia Zhang, Xiaoyun Zhu, Jun Chen, Hong Liu, Hualiang Jiang, Cheng Luo, Hongbin Sun
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 6) pp:2011-2021
Publication Date(Web):June 2011
DOI:10.1016/j.ejmech.2011.02.053
Naturally occurring pentacyclic triterpenes (PT), a novel class of inhibitors against glycogen phosphorylase (GP), hold promise for the treatment of type-2 diabetes and other diseases with disorders in glycogen metabolism. To identify novel and more potent GP inhibitors, the receptor-based comparative molecular field analysis (CoMFA) and comparative molecular similarity analysis (CoMSIA) approaches were performed to investigate the quantitative structure–activity relationships (QSAR) among 106 PT analogues. The validated models demonstrated that the elongated or bulky substitutions in C17 position and/or C2, C3 positions are favorable. Then based on the structural information extracted from these models, 56 derivatives were synthesized and biochemically tested in this study. The IC50 value of the most potent compound P50 was found to be 1.1 μM.Quantitative structure–activity relationship (QSAR) models were setup from the receptor-based molecules alignment, and the predictive data were consistent with the experimental data.Highlights► Pentacyclic triterpenes were inhibitors against glycogen phosphorylase (GP). ► Receptor-based QSAR model leads to the identification of novel GP inhibitors. ► The dimer-like derivatives provide new scaffold for further inhibitor development.
Co-reporter:Zhongjie Liang, Dengyou Zhang, Jing Ai, Limin Chen, Hengshuai Wang, Xiangqian Kong, Mingyue Zheng, Hong Liu, Cheng Luo, Meiyu Geng, Hualiang Jiang, Kaixian Chen
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 12) pp:3749-3754
Publication Date(Web):15 June 2011
DOI:10.1016/j.bmcl.2011.04.064
A series of N′-(2-oxoindolin-3-ylidene)hydrazide derivatives were identified as moderately potent inhibitors against c-Met kinase by pharmacophore-based virtual screening and chemical synthesis methods. The structure–activity relationship (SAR) at various positions of the scaffold was investigated and its binding mode with c-Met kinase was analyzed by molecular modeling studies. In this study, two potent compounds D2 and D25, with IC50 value at 1.3 μM and 2.2 μM against c-Met kinase respectively, were identified. Finally, based on the clues extracted from this study, future development for the optimization of this scaffold was discussed.Compound D2 was discovered by pharmacophore-based virtual screening against SPECS database. Based on the SAR and binding modes analysis, modifications on the N′-(2-oxoindolin-3-ylidene)hydrazide scaffold were proposed.
Co-reporter:Liang Chen, Xiangqian Kong, Zhongjie Liang, Fei Ye, Kunqian Yu, Weiyi Dai, Daocheng Wu, Cheng Luo, and Hualiang Jiang
The Journal of Physical Chemistry B 2011 Volume 115(Issue 44) pp:13019-13025
Publication Date(Web):September 12, 2011
DOI:10.1021/jp206297d
Esterase EstB from Burkholderia gladioli belongs to a novel class of esterases homologous to penicillin binding proteins, notably DD-peptidase and class C β-lactamases. It can cleave the side chain acetyl ester group from cephalosporins leaving the β-lactam ring intact, which is a feature of relevance to industrial biocatalytic applications in the production of semisynthetic cephalosporin derivatives. Due to its important role as a potential biocatalyst in industry, the significance of EstB has been greatly appreciated. However, the molecular basis for those residues involving catalysis of EstB remains elusive. By analyzing the crystal structure of EstB, we identified a conserved water molecule in active-site cavity which might mediate an intramolecular proton transfer (PT) from Lys78 to Asp186 via Tyr133. Then a combined computational approach including molecular dynamics (MD) simulations and quantum mechanics/molecular mechanics (QM/MM) calculations was employed to explore this presumable PT mode in the native enzyme. A 30 ns MD simulation of the enzyme highlights the conserved H-bond network involving Lys78, Tyr133, Asp186, and the conserved water molecule in the active site. In particular, the water molecule did not exchange with bulk solvent, indicating its structural and functional relevance. The energy profile calculated by QM/MM approach displayed a notably low PT barrier (2.2 kcal/mol) and a dramatic energy difference (14.1 kcal/mol) in reactants versus immediate products, which implies that the proposed proton shuttle is concerted and energetically favorable. Our studies offer a reasonable pathway to yield a free base by assisting Lys78 deprotonation, thereby paving the way for future studies on Ser75 activation that is a critical step in catalysis by EstB, as well as biocatalyst development by rational attempts. This PT mode would also afford clues for the forthcoming investigation on acyltransferase LovD that is homologous to EstB.
Co-reporter:Zhongjie Liang, Ting Shi, Sisheng Ouyang, Honglin Li, Kunqian Yu, Weiliang Zhu, Cheng Luo and Hualiang Jiang
The Journal of Physical Chemistry B 2010 Volume 114(Issue 36) pp:11927-11933
Publication Date(Web):August 20, 2010
DOI:10.1021/jp1054183
Sir2, the histone deacetylase III family, has been subjected to a wide range of studies because of their crucial roles in DNA repair, longevity, transcriptional silencing, genome stability, apoptosis, and fat mobilization. The enzyme binds NAD+ and acetyllysine as substrates and generates lysine, 2′-O-acetyl-ADP-ribose, and nicotinamide as products. However, the mechanism of the first step in Sir2 deacetylation reaction from various studies is controversial. To characterize this catalytic mechanism of acetyllysine deacetylation by Sir2, we employed a combined computational approach to carry out molecular modeling, molecular dynamics (MD) simulations, quantum mechanics/molecular mechanics (QM/MM) calculations on catalysis by both yeast Hst2 (homologue of SIR two 2) and bacterial Sir2TM (Sir2 homologue from Thermatoga maritima). Our three-dimensional (3D) model of the complex is composed of Sir2 protein, NAD+, and acetyllysine (ALY) substrate. A 15-ns MD simulation of the complex revealed that Gln115 and His135 play a determining role in deacetylation. These two residues can act as bases to facilitate the deprotonation of 2′-OH from N-ribose. The result is in great agreement with previous mutagenesis analysis data. QM/MM calculations were further performed to study the mechanism of the first step in deacetylation in the two systems. The predicted potential energy barriers for yHst2 and Sir2TM are 12.0 and 15.7 kcal/mol, respectively. The characteristics of the potential energy surface indicated this reaction belongs to a SN2-like mechanism. These results provide insights into the Sir2 mechanism of nicotinamide inhibition and have important implications for the discovery of effectors against Sir2 enzymes.
Co-reporter:Xiangqian Kong, Jie Qin, Zeng Li, Adina Vultur, Linjiang Tong, Enguang Feng, Geena Rajan, Shien Liu, Junyan Lu, Zhongjie Liang, Mingyue Zheng, Weiliang Zhu, Hualiang Jiang, Meenhard Herlyn, Hong Liu, Ronen Marmorstein and Cheng Luo
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 36) pp:NaN7417-7417
Publication Date(Web):2012/07/13
DOI:10.1039/C2OB26081F
Oncogenic mutations in critical nodes of cellular signaling pathways have been associated with tumorigenesis and progression. The B-Raf protein kinase, a key hub in the canonical MAPK signaling cascade, is mutated in a broad range of human cancers and especially in malignant melanoma. The most prevalent B-RafV600E mutant exhibits elevated kinase activity and results in constitutive activation of the MAPK pathway, thus making it a promising drug target for cancer therapy. Herein, we describe the development of novel B-RafV600E selective inhibitors via multi-step virtual screening and hierarchical hit optimization. Nine hit compounds with low micromolar IC50 values were identified as B-RafV600E inhibitors through virtual screening. Subsequent scaffold-based analogue searching and medicinal chemistry efforts significantly improved both the inhibitor potency and oncogene selectivity. In particular, compounds 22f and 22q possess nanomolar IC50 values with selectivity for B-RafV600Ein vitro and exclusive cytotoxicity against B-RafV600E harboring cancer cells.
Co-reporter:Yiqian Xie, Ran Zhou, Fulin Lian, Yan Liu, Limin Chen, Zhe Shi, Naixia Zhang, Mingyue Zheng, Bairong Shen, Hualiang Jiang, Zhongjie Liang and Cheng Luo
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 47) pp:NaN9673-9673
Publication Date(Web):2014/10/28
DOI:10.1039/C4OB01591F
Protein arginine methylation is a common post-translational modification which is crucial for a variety of biological processes. Dysregulation of protein arginine methyltransferases (PRMTs) activity has been implicated in cancer and other serious diseases. Thus, small molecule inhibitors against PRMT have great potential for therapeutic development. Herein, through the combination of virtual screening and bioassays, six small molecular compounds were identified as PRMT1 inhibitors. Amongst them, the binding affinity of compounds DCLX069 and DCLX078 with PRMT1 was further validated by T1ρ and saturation transfer difference (STD) NMR experiments. Most important of all, both compounds effectively blocked cell proliferation in breast cancer, liver cancer and acute myeloid leukemia cell lines. The binding mode analysis from molecular docking simulations theoretically indicated that both inhibitors occupied the SAM binding pocket to exert the inhibitory effect. Taken together, our compounds enriched the structural scaffolds as PRMT1 inhibitors and afforded clues for further optimization.

![4-(4-(5,5-Dimethyl-4,5-dihydrothiazol-2-yl)piperazin-1-yl)-6-propylthieno[2,3-d]pyrimidine](http://img.cochemist.com/ccimg/1271800/1271738-62-5.png)
![4-(4-(5,5-Dimethyl-4,5-dihydrothiazol-2-yl)piperazin-1-yl)-6-propylthieno[2,3-d]pyrimidine](http://img.cochemist.com/ccimg/1271800/1271738-62-5_b.png)
![<br>N,N'-(piperazine-1,4-diyldipropane-3,1-diyl)bis[2-(5-chlorothiophen-2-yl)qu inoline-4-carboxamide]](http://img.cochemist.com/ccimg/438500/438455-59-5.png)
![<br>N,N'-(piperazine-1,4-diyldipropane-3,1-diyl)bis[2-(5-chlorothiophen-2-yl)qu inoline-4-carboxamide]](http://img.cochemist.com/ccimg/438500/438455-59-5_b.png)
















![Spiro[3H-indole-3,6'(4'aH)-[1H]pyrano[3,4-f]indolizine]-4'-carboxylicacid, 1,2,5',5'a,7',8',10',10'a-octahydro-1'-methyl-2-oxo-, methyl ester,(1'S,3R,4'aS,5'aS,10'aR)-](http://img.cochemist.com/ccimg/600/509-80-8.png)
![Spiro[3H-indole-3,6'(4'aH)-[1H]pyrano[3,4-f]indolizine]-4'-carboxylicacid, 1,2,5',5'a,7',8',10',10'a-octahydro-1'-methyl-2-oxo-, methyl ester,(1'S,3R,4'aS,5'aS,10'aR)-](http://img.cochemist.com/ccimg/600/509-80-8_b.png)












![Thieno[3,2-d]pyrimidine, 2-(1H-indazol-4-yl)-6-[[4-(methylsulfonyl)-1-piperazinyl]methyl]-4-(4-morpholinyl)-](http://img.cochemist.com/ccimg/957100/957054-30-7.png)
![Thieno[3,2-d]pyrimidine, 2-(1H-indazol-4-yl)-6-[[4-(methylsulfonyl)-1-piperazinyl]methyl]-4-(4-morpholinyl)-](http://img.cochemist.com/ccimg/957100/957054-30-7_b.png)


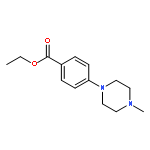
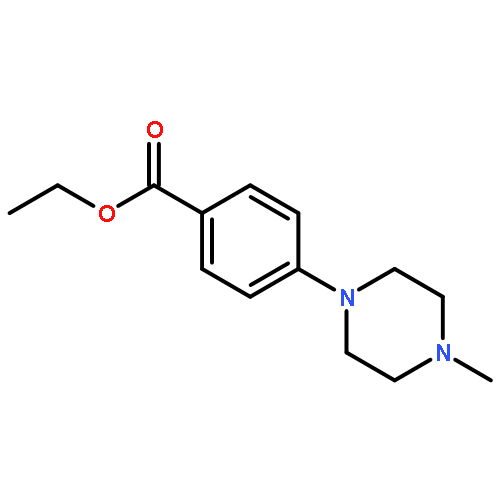
![<br>2-[4-(4-Fluoro-phenyl)-6-trifluoromethyl-pyrimidin-2-ylsulfanyl]-N-methyl-N -phenyl-acetamide](http://img.cochemist.com/ccimg/505100/505049-89-8.png)
![<br>2-[4-(4-Fluoro-phenyl)-6-trifluoromethyl-pyrimidin-2-ylsulfanyl]-N-methyl-N -phenyl-acetamide](http://img.cochemist.com/ccimg/505100/505049-89-8_b.png)


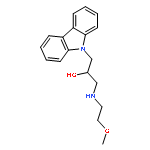
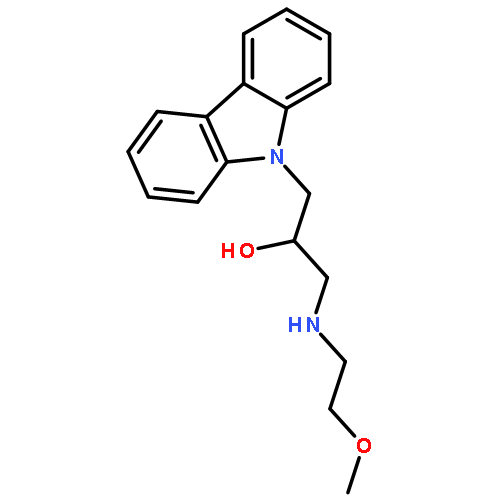




![9H-Carbazole-9-ethanol,a-[[(3-hydroxypropyl)amino]methyl]-](http://img.cochemist.com/ccimg/324800/324773-66-2.png)
![9H-Carbazole-9-ethanol,a-[[(3-hydroxypropyl)amino]methyl]-](http://img.cochemist.com/ccimg/324800/324773-66-2_b.png)
![(1-Adamantan-1-yl-ethyl)-benzo[1,3]dioxol-5-ylmethyl-amine](http://img.cochemist.com/ccimg/312600/312519-09-8.png)
![(1-Adamantan-1-yl-ethyl)-benzo[1,3]dioxol-5-ylmethyl-amine](http://img.cochemist.com/ccimg/312600/312519-09-8_b.png)
![N-[4-(acetylsulfamoyl)phenyl]-4-(4-chloro-2-methylphenoxy)butanamide](http://img.cochemist.com/ccimg/310500/310453-07-7.png)
![N-[4-(acetylsulfamoyl)phenyl]-4-(4-chloro-2-methylphenoxy)butanamide](http://img.cochemist.com/ccimg/310500/310453-07-7_b.png)




























![(2E)-1-{3-[(2E)-3,7-dimethylocta-2,6-dien-1-yl]-2,4-dihydroxyphenyl}-3-(4-hydroxyphenyl)prop-2-en-1-one](http://img.cochemist.com/ccimg/63000/62949-76-2.png)
![(2E)-1-{3-[(2E)-3,7-dimethylocta-2,6-dien-1-yl]-2,4-dihydroxyphenyl}-3-(4-hydroxyphenyl)prop-2-en-1-one](http://img.cochemist.com/ccimg/63000/62949-76-2_b.png)
![(E)-1-[2-HYDROXY-4-METHOXY-3-(3-METHYLBUT-2-ENYL)PHENYL]-3-(4-HYDROXYPHENYL)PROP-2-EN-1-ONE](http://img.cochemist.com/ccimg/56000/55912-03-3.png)
![(E)-1-[2-HYDROXY-4-METHOXY-3-(3-METHYLBUT-2-ENYL)PHENYL]-3-(4-HYDROXYPHENYL)PROP-2-EN-1-ONE](http://img.cochemist.com/ccimg/56000/55912-03-3_b.png)





![(9R)-8,8-dimethyl-2-oxo-9,10-dihydro-2H,8H-pyrano[2,3-f]chromen-9-yl (2Z)-2-methylbut-2-enoate](http://img.cochemist.com/ccimg/19500/19427-82-8.png)
![(9R)-8,8-dimethyl-2-oxo-9,10-dihydro-2H,8H-pyrano[2,3-f]chromen-9-yl (2Z)-2-methylbut-2-enoate](http://img.cochemist.com/ccimg/19500/19427-82-8_b.png)












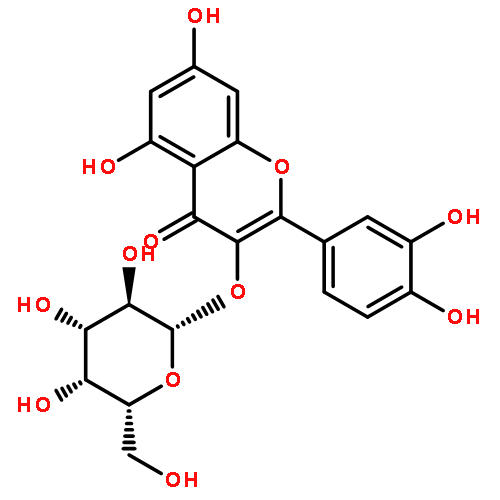
![2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-3-[(2R,3S,4R,5S,6S)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydropyran-2-yl]oxy-chromen-4-one](http://img.cochemist.com/ccimg/500/482-35-9.png)
![2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-3-[(2R,3S,4R,5S,6S)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydropyran-2-yl]oxy-chromen-4-one](http://img.cochemist.com/ccimg/500/482-35-9_b.png)


![1-(4-Fluorobenzyl)-N-(1-(4-methoxyphenethyl)piperidin-4-yl)-1H-benzo[d]imidazol-2-amine](http://img.cochemist.com/ccimg/68900/68844-77-9.png)
![1-(4-Fluorobenzyl)-N-(1-(4-methoxyphenethyl)piperidin-4-yl)-1H-benzo[d]imidazol-2-amine](http://img.cochemist.com/ccimg/68900/68844-77-9_b.png)