Co-reporter:Philipp Baer;Patrick Rabe;Christian A. Citron;Carina C. de Oliveira Mann;Norman Kaufmann; Dr. Michael Groll;Dr. Jeroen S. Dickschat
ChemBioChem 2014 Volume 15( Issue 2) pp:213-216
Publication Date(Web):
DOI:10.1002/cbic.201300708
Abstract
The biosynthesis of terpenes is catalysed by class I and II terpene cyclases. Here we present structural data from a class I hedycaryol synthase in complex with nerolidol, serving as a surrogate for the reaction intermediate nerolidyl diphosphate. This prefolded ligand allows mapping of the active site and hence the identification of a key carbonyl oxygen of Val179, a highly conserved helix break (G1/2) and its corresponding helix dipole. Stabilising the carbocation at the substrate's C1 position, these elements act in concert to catalyse the 1,10 ring closure, thereby exclusively generating the anti-Markovnikov product. The delineation of a general mechanistic scaffold was confirmed by site-specific mutations. This work serves as a basis for understanding carbocation chemistry in enzymatic reactions and should contribute to future application of these enzymes in organic synthesis.
Co-reporter:Martin L. Stein;Haissi Cui;Philipp Beck;Christian Dubiella;Constantin Voss;Dr. Achim Krüger;Dr. Boris Schmidt;Dr. Michael Groll
Angewandte Chemie 2014 Volume 126( Issue 6) pp:1705-1709
Publication Date(Web):
DOI:10.1002/ange.201308984
Abstract
Das Ubiquitin-Proteasom-System (UPS) wurde in akademischer wie pharmazeutischer Forschung erfolgreich für onkologische und immunologische Anwendungen adressiert. Typische Proteasominhibitoren basieren auf einem peptidischen Rückgrat, das mit einem elektrophilen C-Terminus zur Bindung an die aktiven proteolytischen Zentren gekuppelt ist. Obwohl der peptidische Anteil viel Interesse bezüglich der Untereinheitenselektivität hervorgerufen hat, wird die Wirkspezifität und biologische Stabilität der Substanzen entscheidend durch die reaktiven Kopfgruppen geprägt. In dieser Studie wurde daher eine systematische Untersuchung mit In-vitro-, In-vivo- und strukturbiologischen Methoden durchgeführt, um die Auswirkungen unterschiedlicher Funktionalitäten und chemischer Reaktivitäten aufzudecken. Dies ermöglichte die Einführung und Charakterisierung der Klasse der α-Ketoamide als potenteste reversible Inhibitoren mit möglichen Anwendungen für die Behandlung von soliden Tumoren und Autoimmunerkrankungen.
Co-reporter:Roger Müller;Melissa A. Gräwert;Thomas Kern;Tobias Madl;Jirka Peschek;Michael Sattler;Johannes Buchner
PNAS 2013 110 (25 ) pp:10183-10188
Publication Date(Web):2013-06-18
DOI:10.1073/pnas.1300547110
IgM is the first antibody produced during the humoral immune response. Despite its fundamental role in the immune system,
IgM is structurally only poorly described. In this work we used X-ray crystallography and NMR spectroscopy to determine the
atomic structures of the constant IgM Fc domains (Cµ2, Cµ3, and Cµ4) and to address their roles in IgM oligomerization. Although
the isolated domains share the typical Ig fold, they differ substantially in dimerization properties and quaternary contacts.
Unexpectedly, the Cµ4 domain and its C-terminal tail piece are responsible and sufficient for the specific polymerization
of Cµ4 dimers into covalently linked hexamers of dimers. Based on small angle X-ray scattering data, we present a model of
the ring-shaped Cµ4 structure, which reveals the principles of IgM oligomerization.
Co-reporter:Melissa Ann Gräwert and Michael Groll
Chemical Communications 2012 vol. 48(Issue 10) pp:1364-1378
Publication Date(Web):31 Oct 2011
DOI:10.1039/C1CC15273D
Cancer is the No. 2 cause of death in the Western world and one of the most expensive diseases to treat. Thus, it is not surprising, that every major pharmaceutical and biotechnology company has a blockbuster oncology product. In 2003, Millennium Pharmaceuticals entered the race with Velcade®, a first-in-class proteasome inhibitor that has been approved by the FDA for treatment of multiple myeloma and its sales have passed the billion dollar mark. Velcade®'s extremely toxic boronic acid pharmacophore, however, contributes to a number of severe side effects. Nevertheless, the launching of this product has validated the proteasome as a target in fighting cancer and further proteasome inhibitors have entered the market as anti-cancer drugs. Additionally, proteasome inhibitors have found application as crop protection agents, anti-parasitics, immunosuppressives, as well as in new therapies for muscular dystrophies and inflammation. Many of these compounds are based on microbial metabolites. In this review, we emphasize the important role of the structural elucidation of the various unique binding mechanisms of these compounds that have been optimized throughout evolution to target the proteasome. Based on this knowledge, medicinal chemists have further optimized these natural products, resulting in potential drugs with reduced off-target activities.
Co-reporter:M.Chem. Nerea Gallastegui;M.Sc. Philipp Beck;M.Sc. Marcelino Arciniega;Dr. Robert Huber;Stefan Hillebr;Dr. Michael Groll
Angewandte Chemie 2012 Volume 124( Issue 1) pp:251-254
Publication Date(Web):
DOI:10.1002/ange.201106010
Co-reporter:M.Sc. Felix Quitterer;M.Sc. Anja List;Priv.-Doz.Dr. Wolfgang Eisenreich;Dr.Dr. Adelbert Bacher ;Dr. Michael Groll
Angewandte Chemie 2012 Volume 124( Issue 6) pp:1367-1370
Publication Date(Web):
DOI:10.1002/ange.201106765
Co-reporter:M.Sc. Felix Quitterer;M.Sc. Anja List;Priv.-Doz.Dr. Wolfgang Eisenreich;Dr.Dr. Adelbert Bacher ;Dr. Michael Groll
Angewandte Chemie 2012 Volume 124( Issue 6) pp:
Publication Date(Web):
DOI:10.1002/ange.201107547
Co-reporter:Felix Quitterer;Anja List;Priv.-Doz.Dr. Wolfgang Eisenreich;Dr.Dr. Adelbert Bacher ;Dr. Michael Groll
Angewandte Chemie International Edition 2012 Volume 51( Issue 6) pp:1339-1342
Publication Date(Web):
DOI:10.1002/anie.201106765
Co-reporter:Markus Kaiser;Philipp Beck;Robert Dudler;Christian F. W. Becker;Martin L. Stein
PNAS 2012 Volume 109 (Issue 45 ) pp:18367-18371
Publication Date(Web):2012-11-06
DOI:10.1073/pnas.1211423109
Natural products represent valuable lead structures for drug discovery. However, for most bioactive compounds no cellular
target is yet identified and many substances predicted from genome analysis are inaccessible due to their life stage-dependent
biosynthesis, which is not reflected in common isolation procedures. In response to these issues, an NMR-based and target-directed
protease assay for inhibitor detection of the proteasome was developed. The methodology is suitable for one-shot identification
of inhibitors in conglomerates and crude culture broths. The technique was applied for analysis of the different life stages
of the bacterium Photorhabdus luminescens, which resulted in the isolation and characterization of cepafungin I (CepI), the strongest proteasome inhibitor described
to date. Its biosynthesis is strictly regulated and solely induced by the specific environmental conditions determined by
our methodology. The transferability of the developed technique to other drug targets may disclose an abundance of novel compounds
applicable for drug development.
Co-reporter:M.Chem. Nerea Gallastegui;M.Sc. Philipp Beck;M.Sc. Marcelino Arciniega;Dr. Robert Huber;Stefan Hillebr;Dr. Michael Groll
Angewandte Chemie International Edition 2012 Volume 51( Issue 1) pp:247-249
Publication Date(Web):
DOI:10.1002/anie.201106010
Co-reporter:Felix Quitterer;Anja List;Priv.-Doz.Dr. Wolfgang Eisenreich;Dr.Dr. Adelbert Bacher ;Dr. Michael Groll
Angewandte Chemie International Edition 2012 Volume 51( Issue 6) pp:
Publication Date(Web):
DOI:10.1002/anie.201107547
Co-reporter:Dr. Melissa Ann Gräwert;Nerea Gallastegui;Martin Stein;Dr. Boris Schmidt;Dr. Peter-Michael Kloetzel;Dr. Robert Huber;Dr. Michael Groll
Angewandte Chemie International Edition 2011 Volume 50( Issue 2) pp:542-544
Publication Date(Web):
DOI:10.1002/anie.201005488
Co-reporter:Dr. Melissa Ann Gräwert;Nerea Gallastegui;Martin Stein;Dr. Boris Schmidt;Dr. Peter-Michael Kloetzel;Dr. Robert Huber;Dr. Michael Groll
Angewandte Chemie 2011 Volume 123( Issue 2) pp:563-566
Publication Date(Web):
DOI:10.1002/ange.201005488
Co-reporter:JulieG. Geist;Susan Lauw Dr.;Victoria Illarionova Dr.;Boris Illarionov Dr.;Markus Fischer Dr.;Tobias Gräwert Dr.;Felix Rohdich Dr.;Wolfgang Eisenreich Dr.;Johannes Kaiser Dr. Dr.;Christian Scheurer;Sergio Wittlin Dr.;JoséL. Alonso-Gómez Dr.;W.Bernd Schweizer Dr.;Adelbert Bacher Dr.;François Diederich Dr.
ChemMedChem 2010 Volume 5( Issue 7) pp:1092-1101
Publication Date(Web):
DOI:10.1002/cmdc.201000083
Abstract
A library of 40 000 compounds was screened for inhibitors of 2-methylerythritol 2,4-cyclodiphosphate synthase (IspF) protein from Arabidopsis thaliana using a photometric assay. A thiazolopyrimidine derivative resulting from the high-throughput screen was found to inhibit the IspF proteins of Mycobacterium tuberculosis, Plasmodium falciparum, and A. thaliana with IC50 values in the micromolar range. Synthetic efforts afforded derivatives that inhibit IspF protein from M. tuberculosis and P. falciparum with IC50 values in the low micromolar range. Several compounds act as weak inhibitors in the P. falciparum red blood cell assay.
Co-reporter:Dr. Michael Groll;Nerea Gallastegui;Xavier Maréchal;Dr. Virginie LeRavalec;Dr. Nicolas Basse;Dr. Nicolas Richy;Dr. Emilie Genin;Dr. Robert Huber;Dr. Luis Moroder;Dr. Joëlle Vidal;Dr. Michèle Reboud-Ravaux
ChemMedChem 2010 Volume 5( Issue 10) pp:1701-1705
Publication Date(Web):
DOI:10.1002/cmdc.201000293
Co-reporter:Anna Stengel;Jürgen Soll;Ferdinand Alte;Bettina Bölter;J. Philipp Benz;Eike Petersen
PNAS 2010 Volume 107 (Issue 45 ) pp:19260-19265
Publication Date(Web):2010-11-09
DOI:10.1073/pnas.1009124107
Ferredoxin:NADPH oxidoreductase (FNR) is a key enzyme of photosynthetic electron transport required for generation of reduction
equivalents. Recently, two proteins were found to be involved in membrane-anchoring of FNR by specific interaction via a conserved
Ser/Pro-rich motif: Tic62 and Trol. Our crystallographic study reveals that the FNR-binding motif, which forms a polyproline
type II helix, induces self-assembly of two FNR monomers into a back-to-back dimer. Because binding occurs opposite to the
FNR active sites, its activity is not affected by the interaction. Surface plasmon resonance analyses disclose a high affinity
of FNR to the binding motif, which is strongly increased under acidic conditions. The pH of the chloroplast stroma changes
dependent on the light conditions from neutral to slightly acidic in complete darkness or to alkaline at saturating light
conditions. Recruiting of FNR to the thylakoids could therefore represent a regulatory mechanism to adapt FNR availability/activity
to photosynthetic electron flow.
Co-reporter:Dr. Tobias Gräwert;Ingrid Span;Dr.Dr. Adelbert Bacher ;Dr. Michael Groll
Angewandte Chemie International Edition 2010 Volume 49( Issue 47) pp:8802-8809
Publication Date(Web):
DOI:10.1002/anie.201000833
Abstract
The biosynthesis of natural products is a treasure trove of unusual reaction mechanisms. This Minireview summarizes recent work on the structure and mechanism of IspH protein, which catalyzes the reductive dehydroxylation of an allyl alcohol in a biosynthetic pathway leading to isoprenoid precursors.
Co-reporter:Tobias Gräwert;Ingrid Span;Wolfgang Eisenreich;Felix Rohdich;Jörg Eppinger;Adelbert Bacher
PNAS 2010 Volume 107 (Issue 3 ) pp:1077-1081
Publication Date(Web):2010-01-19
DOI:10.1073/pnas.0913045107
Isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP) represent the two central intermediates in the biosynthesis
of isoprenoids. The recently discovered deoxyxylulose 5-phosphate pathway generates a mixture of IPP and DMAPP in its final
step by reductive dehydroxylation of 1-hydroxy-2-methyl-2-butenyl 4-diphosphate. This conversion is catalyzed by IspH protein
comprising a central iron-sulfur cluster as electron transfer cofactor in the active site. The five crystal structures of
IspH in complex with substrate, converted substrate, products and PPi reported in this article provide unique insights into the mechanism of this enzyme. While IspH protein crystallizes with
substrate bound to a [4Fe-4S] cluster, crystals of IspH in complex with IPP, DMAPP or inorganic pyrophosphate feature [3Fe-4S]
clusters. The IspH:substrate complex reveals a hairpin conformation of the ligand with the C(1) hydroxyl group coordinated
to the unique site in a [4Fe-4S] cluster of aconitase type. The resulting alkoxide complex is coupled to a hydrogen-bonding
network, which serves as proton reservoir via a Thr167 proton relay. Prolonged x-ray irradiation leads to cleavage of the
C(1)-O bond (initiated by reducing photo electrons). The data suggest a reaction mechanism involving a combination of Lewis-acid activation and proton coupled electron transfer. The resulting allyl radical intermediate can acquire a second electron
via the iron-sulfur cluster. The reaction may be terminated by the transfer of a proton from the β-phosphate of the substrate
to C(1) (affording DMAPP) or C(3) (affording IPP).
Co-reporter:Michael Groll ; Katherine A. McArthur ; Venkat R. Macherla ; Rama Rao Manam ;Barbara C. Potts
Journal of Medicinal Chemistry 2009 Volume 52(Issue 17) pp:5420-5428
Publication Date(Web):August 13, 2009
DOI:10.1021/jm900559x
Many marketed drugs contain fluorine, reflecting its ability to modulate a variety of biological responses. The unique 20S proteasome inhibition profile of fluorosalinosporamide compared to chlorinated anticancer agent salinosporamide A (NPI-0052) is exemplary and relates to each halogen’s leaving group potential. Crystal structures of fluoro-, hydroxy-, and bromosalinosporamide in complex with the yeast 20S proteasome core particle (CP) provide mechanistic insights into ligand binding and leaving group elimination and the ability to fine-tune the duration of proteasome inhibition. Fluorosalinosporamide/CP crystal structures determined over time offer striking snapshots of the ligand trapped with an intact fluoroethyl group in anticipation of fluoride elimination, followed by complete nucleophilic displacement of fluoride to give the highly stabilized cyclic ether found for salinosporamide A and bromosalinosporamide. This two-step reaction pathway is consistent with a mechanism for partially reversible proteasome inhibition by fluorosalinosporamide. Proteasome catalyzed fluoride displacement provides preliminary insights into the active site Thr1N pKa.
Co-reporter:Tobias Gräwert Dr.;Felix Rohdich Dr.;Ingrid Span;Adelbert Bacher ;Wolfgang Eisenreich ;Jörg Eppinger Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 31) pp:5756-5759
Publication Date(Web):
DOI:10.1002/anie.200900548
Co-reporter:Patrick Schreiner,
Xiang Chen,
Koraljka Husnjak,
Leah Randles,
Naixia Zhang,
Suzanne Elsasser,
Daniel Finley,
Ivan Dikic,
Kylie J. Walters
&
Michael Groll
Nature 2008 453(7194) pp:548
Publication Date(Web):2008-05-22
DOI:10.1038/nature06924
Targeted protein degradation is largely performed by the ubiquitin–proteasome pathway, in which substrate proteins are marked by covalently attached ubiquitin chains that mediate recognition by the proteasome. It is currently unclear how the proteasome recognizes its substrates, as the only established ubiquitin receptor intrinsic to the proteasome is Rpn10/S5a (ref. 1), which is not essential for ubiquitin-mediated protein degradation in budding yeast2. In the accompanying manuscript we report that Rpn13 (refs 3–7), a component of the nine-subunit proteasome base, functions as a ubiquitin receptor8, complementing its known role in docking de-ubiquitinating enzyme Uch37/UCHL5 (refs 4–6) to the proteasome. Here we merge crystallography and NMR data to describe the ubiquitin-binding mechanism of Rpn13. We determine the structure of Rpn13 alone and complexed with ubiquitin. The co-complex reveals a novel ubiquitin-binding mode in which loops rather than secondary structural elements are used to capture ubiquitin. Further support for the role of Rpn13 as a proteasomal ubiquitin receptor is demonstrated by its ability to bind ubiquitin and proteasome subunit Rpn2/S1 simultaneously. Finally, we provide a model structure of Rpn13 complexed to diubiquitin, which provides insights into how Rpn13 as a ubiquitin receptor is coupled to substrate deubiquitination by Uch37.
Co-reporter:Eva M. Huber, Michael Groll
Structure (7 March 2012) Volume 20(Issue 3) pp:387-388
Publication Date(Web):7 March 2012
DOI:10.1016/j.str.2012.02.006
After elucidation of the atomic details of 20S proteasomes, current research focuses on the regulatory 19S particle. In this issue of Structure, He et al. present the crystal structure of Rpn2 and use electron microscopy to examine differences between Rpn2 and Rpn1.
Co-reporter:Nerea Gallastegui, Michael Groll
Structure (14 October 2009) Volume 17(Issue 10) pp:1279-1281
Publication Date(Web):14 October 2009
DOI:10.1016/j.str.2009.09.003
Principles of intracellular protein degradation remain among the most challenging questions in cell biology. Here, we discuss Wang and colleagues' crystal structure elucidation of the intermediate domain of Mpa, a regulatory particle of Mtb proteasome, the core proteolytic machinery of Mycobacterium tuberculosis.
Co-reporter:Felix Quitterer, Anja List, Philipp Beck, Adelbert Bacher, Michael Groll
Journal of Molecular Biology (14 December 2012) Volume 424(Issue 5) pp:270-282
Publication Date(Web):14 December 2012
DOI:10.1016/j.jmb.2012.09.007
The second step in the biosynthesis of the 22nd genetically encoded amino acid pyrrolysine (Pyl) is catalyzed by PylC that forms the pseudopeptide l-lysine-Nε-3R-methyl-d-ornithine. Here, we present six crystal structures of the monomeric active ligase in complex with substrates, reaction intermediates, and products including ATP, the non-hydrolyzable ATP analogue 5′‐adenylyl‐β‐γ‐imidodiphosphate, ADP, d-ornithine (d-Orn), l-lysine (Lys), phosphorylated d‐Orn, l-lysine-Nε-d-ornithine, inorganic phosphate, carbonate, and Mg2 +. The overall structure of PylC reveals similarities to the superfamily of ATP-grasp enzymes; however, there exist unique structural and functional features for a topological control of successive substrate entry and product release. Furthermore, the presented high‐resolution structures provide detailed insights into the reaction mechanism of isopeptide bond formation starting with phosphorylation of d‐Orn by transfer of a phosphate moiety from activated ATP. The binding of Lys to the enzyme complex is then followed by an SN2 reaction resulting in l‐lysine‐Nε‐d‐ornithine and inorganic phosphate. Surprisingly, PylC harbors two adenine nucleotides bound at the active site, what has not been observed in any ATP-grasp protein analyzed to date. Whereas one ATP molecule is involved in catalysis, the second adenine nucleotide functions as a selective anchor for the C- and N-terminus of the Lys substrate and is responsible for protein stability as shown by mutagenesis.Graphical AbstractDownload high-res image (210KB)Download full-size imageHighlights► PylC catalyzes the second step of the Pyl biosynthesis. ► PylC is an ATP-grasp enzyme with an unexpected extra adenine nucleotide binding site. ► PylC complex structures reveal detailed insights into the enzymatic reaction mechanism. ► Ordered substrate entry to the active site is necessary for isopeptide bond formation.
Co-reporter:Ingrid Span, Tobias Gräwert, Adelbert Bacher, Wolfgang Eisenreich, Michael Groll
Journal of Molecular Biology (10 February 2012) Volume 416(Issue 1) pp:1-9
Publication Date(Web):10 February 2012
DOI:10.1016/j.jmb.2011.11.033
Isoprenoids derive from two universal precursors, isopentenyl diphosphate and dimethylallyl diphosphate, which in most human pathogens are synthesized in the deoxyxylulose phosphate pathway. The last step of this pathway is the conversion of (E)-1-hydroxy-2-methylbut-2-enyl-4-diphosphate into a mixture of isopentenyl diphosphate and dimethylallyl diphosphate catalyzed by the iron–sulfur protein IspH. The crystal structures reported here of the IspH mutant proteins T167C, E126D and E126Q reveal an alternative substrate conformation compared to the wild-type structure. Thus, the previously observed alkoxide complex decomposes, and the substrate's hydroxymethyl group rotates to interact with Glu126. The carboxyl group of Glu126 then donates a proton to the hydroxyl group to enable water elimination. The structural and functional studies provide further knowledge of the IspH reaction mechanism, which opens up new routes to inhibitor design against malaria and tuberculosis.Download high-res image (185KB)Download full-size imageResearch Highlights► Characterization of the iron–sulfur protein IspH and three active-site mutants. ► Crystal structures of mutant IspH proteins in complex with the substrate (E)-1-hydroxy-2-methylbut-2-enyl-4-diphosphate. ► Mutant structures reveal an alternative substrate conformation during catalysis. ► Rotation of the substrate's hydroxymethyl group occurs prior to water elimination.
Co-reporter:Matthias Lee, Tobias Gräwert, Felix Quitterer, Felix Rohdich, ... Michael Groll
Journal of Molecular Biology (10 December 2010) Volume 404(Issue 4) pp:600-610
Publication Date(Web):10 December 2010
DOI:10.1016/j.jmb.2010.09.050
IspG protein serves as the penultimate enzyme of the recently discovered non-mevalonate pathway for the biosynthesis of the universal isoprenoid precursors, isopentenyl diphosphate and dimethylallyl diphosphate. The enzyme catalyzes the reductive ring opening of 2C-methyl-d-erythritol 2,4-cyclodiphosphate, which affords 1-hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate. The protein was crystallized under anaerobic conditions, and its three-dimensional structure was determined to a resolution of 2.7 Å. Each subunit of the c2 symmetric homodimer folds into two domains connected by a short linker sequence. The N-terminal domain (N domain) is an eight-stranded β barrel that belongs to the large TIM-barrel superfamily. The C-terminal domain (C domain) consists of a β sheet that is flanked on both sides by helices. One glutamate and three cysteine residues of the C domain coordinate a [4Fe–4S] cluster. Homodimer formation involves an extended contact area (about 1100 Å2) between helices 8 and 9 of each respective β barrel. Moreover, each C domain contacts the N domain of the partner subunit, but the interface regions are small (about 430 Å2). We propose that the enzyme substrate binds to the positively charged surface area at the C-terminal pole of the β barrel. The C domain carrying the iron–sulfur cluster could then move over to form a closed conformation where the substrate is sandwiched between the N domain and the C domain. This article completes the set of three-dimensional structures of the non-mevalonate pathway enzymes, which are of specific interest as potential targets for tuberculostatic and antimalarial drugs.Download high-res image (96KB)Download full-size imageResearch Highlights► [4Fe–4S] protein IspG (GcpE) of Aquifex aeolicus is a c2 symmetric functional dimer. ► IspG catalytic domain comprises a TIM-barrel fold with positively charged patch. ► Reduction of negatively charged substrate by FeS cluster upon proposed domain flip. ► [4Fe–4S] cluster coordinated by glutamate and three cysteines in C-terminal domain.
Co-reporter:Melissa Ann Gräwert and Michael Groll
Chemical Communications 2012 - vol. 48(Issue 10) pp:NaN1378-1378
Publication Date(Web):2011/10/31
DOI:10.1039/C1CC15273D
Cancer is the No. 2 cause of death in the Western world and one of the most expensive diseases to treat. Thus, it is not surprising, that every major pharmaceutical and biotechnology company has a blockbuster oncology product. In 2003, Millennium Pharmaceuticals entered the race with Velcade®, a first-in-class proteasome inhibitor that has been approved by the FDA for treatment of multiple myeloma and its sales have passed the billion dollar mark. Velcade®'s extremely toxic boronic acid pharmacophore, however, contributes to a number of severe side effects. Nevertheless, the launching of this product has validated the proteasome as a target in fighting cancer and further proteasome inhibitors have entered the market as anti-cancer drugs. Additionally, proteasome inhibitors have found application as crop protection agents, anti-parasitics, immunosuppressives, as well as in new therapies for muscular dystrophies and inflammation. Many of these compounds are based on microbial metabolites. In this review, we emphasize the important role of the structural elucidation of the various unique binding mechanisms of these compounds that have been optimized throughout evolution to target the proteasome. Based on this knowledge, medicinal chemists have further optimized these natural products, resulting in potential drugs with reduced off-target activities.

![6-Oxa-2-azabicyclo[3.2.0]heptane-3,7-dione,1-[(S)-(1S)-2-cyclohexen-1-ylhydroxymethyl]-4-ethyl-5-methyl-,(1R,4R,5S)-](http://img.cochemist.com/ccimg/863200/863126-95-8.png)
![6-Oxa-2-azabicyclo[3.2.0]heptane-3,7-dione,1-[(S)-(1S)-2-cyclohexen-1-ylhydroxymethyl]-4-ethyl-5-methyl-,(1R,4R,5S)-](http://img.cochemist.com/ccimg/863200/863126-95-8_b.png)
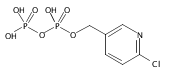
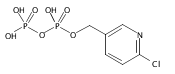
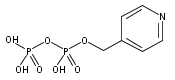
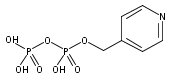
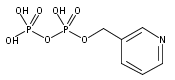
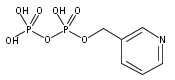
![Methyl 1H-pyrazolo[3,4-b]pyridine-5-carboxylate](http://img.cochemist.com/ccimg/1196200/1196156-42-9.png)
![Methyl 1H-pyrazolo[3,4-b]pyridine-5-carboxylate](http://img.cochemist.com/ccimg/1196200/1196156-42-9_b.png)


![1-isopropyl-1H-pyrazolo[3,4-b]pyridine-5-carboxylic acid](http://img.cochemist.com/ccimg/923700/923689-61-6.png)
![1-isopropyl-1H-pyrazolo[3,4-b]pyridine-5-carboxylic acid](http://img.cochemist.com/ccimg/923700/923689-61-6_b.png)






![Phosphonic acid, [(3,4-difluorophenyl)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/451500/451455-95-1.png)
![Phosphonic acid, [(3,4-difluorophenyl)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/451500/451455-95-1_b.png)



![(10S,11R,12S,15S,18S)-15-(Carbamoylmethyl)-10,11,23-trihydroxy-18-[3(S)-methyl-2-oxopentanamido]-9,14,17-trioxo-N-[1(Z)-propenyl]-8,13,16-triazatetracyclo[18.3.1.0(2,7).0(6,10)]tetracosa-1(24),2,4,6,20,22-hexaene-12-carboxamide](http://img.cochemist.com/ccimg/220700/220666-21-7.png)
![(10S,11R,12S,15S,18S)-15-(Carbamoylmethyl)-10,11,23-trihydroxy-18-[3(S)-methyl-2-oxopentanamido]-9,14,17-trioxo-N-[1(Z)-propenyl]-8,13,16-triazatetracyclo[18.3.1.0(2,7).0(6,10)]tetracosa-1(24),2,4,6,20,22-hexaene-12-carboxamide](http://img.cochemist.com/ccimg/220700/220666-21-7_b.png)




![2-[(2S,5R,8S,11S)-5-benzyl-11-[3-(diaminomethylideneamino)propyl]-7-methyl-3,6,9,12,15-pentaoxo-8-propan-2-yl-1,4,7,10,13-pentazacyclopentadec-2-yl]acetic acid](http://img.cochemist.com/ccimg/189000/188968-51-6.png)
![2-[(2S,5R,8S,11S)-5-benzyl-11-[3-(diaminomethylideneamino)propyl]-7-methyl-3,6,9,12,15-pentaoxo-8-propan-2-yl-1,4,7,10,13-pentazacyclopentadec-2-yl]acetic acid](http://img.cochemist.com/ccimg/189000/188968-51-6_b.png)




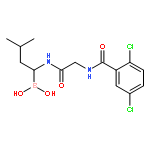
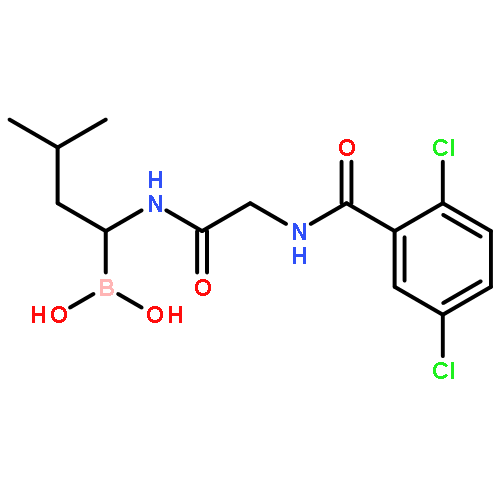
![L-Leucinamide,N-[(phenylmethoxy)carbonyl]-L-leucyl-N-[(1R)-1-borono-3-methylbutyl]-](http://img.cochemist.com/ccimg/179400/179324-22-2.png)
![L-Leucinamide,N-[(phenylmethoxy)carbonyl]-L-leucyl-N-[(1R)-1-borono-3-methylbutyl]-](http://img.cochemist.com/ccimg/179400/179324-22-2_b.png)
![N-[(Benzyloxy)carbonyl]-L-leucyl-N-{(1R)-3-methyl-1-[(1S,2R,6S,8S)-2,9,9-trimethyl-3,5-dioxa-4-boratricyclo[6.1.1.02,6]dec-4-yl]butyl}-D-leucinamide](http://img.cochemist.com/ccimg/179400/179324-21-1.png)
![N-[(Benzyloxy)carbonyl]-L-leucyl-N-{(1R)-3-methyl-1-[(1S,2R,6S,8S)-2,9,9-trimethyl-3,5-dioxa-4-boratricyclo[6.1.1.02,6]dec-4-yl]butyl}-D-leucinamide](http://img.cochemist.com/ccimg/179400/179324-21-1_b.png)
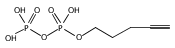
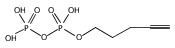
![Acetic acid, 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/1152400/172531-37-2.png)
![Acetic acid, 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/1152400/172531-37-2_b.png)
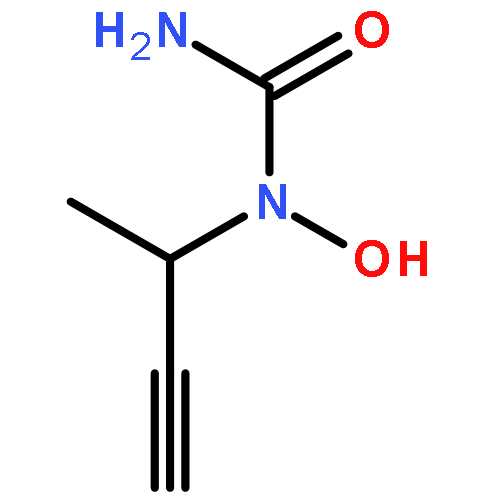
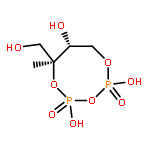
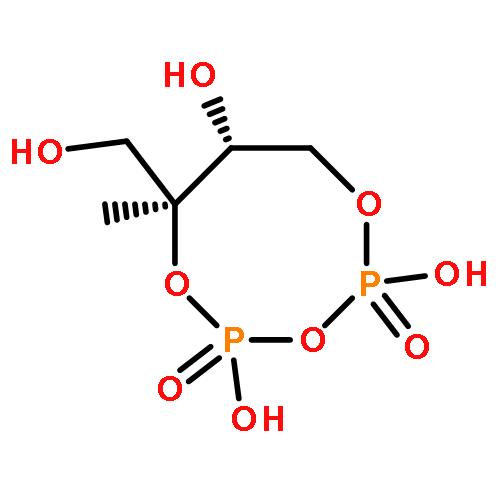
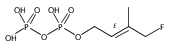
![L-Threoninamide,N-acetyl-N-methyl-L-isoleucyl-L-isoleucyl-N-[(1S)-3-methyl-1-[[(2R)-2-methyl-2-oxiranyl]carbonyl]butyl]-](http://img.cochemist.com/ccimg/134400/134381-21-8.png)
![L-Threoninamide,N-acetyl-N-methyl-L-isoleucyl-L-isoleucyl-N-[(1S)-3-methyl-1-[[(2R)-2-methyl-2-oxiranyl]carbonyl]butyl]-](http://img.cochemist.com/ccimg/134400/134381-21-8_b.png)
![L-Cysteine,N-acetyl-S-[(3S,4R)-3-hydroxy-2-[(1S)-1-hydroxy-2-methylpropyl]-4-methyl-5-oxo-D-prolyl]-](http://img.cochemist.com/ccimg/133400/133343-34-7.png)
![L-Cysteine,N-acetyl-S-[(3S,4R)-3-hydroxy-2-[(1S)-1-hydroxy-2-methylpropyl]-4-methyl-5-oxo-D-prolyl]-](http://img.cochemist.com/ccimg/133400/133343-34-7_b.png)
![(1s,4s,7z,10s,16e,21r)-7-ethylidene-4,21-di(propan-2-yl)-2-oxa-12,13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone](http://img.cochemist.com/ccimg/128600/128517-07-7.png)
![(1s,4s,7z,10s,16e,21r)-7-ethylidene-4,21-di(propan-2-yl)-2-oxa-12,13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone](http://img.cochemist.com/ccimg/128600/128517-07-7_b.png)
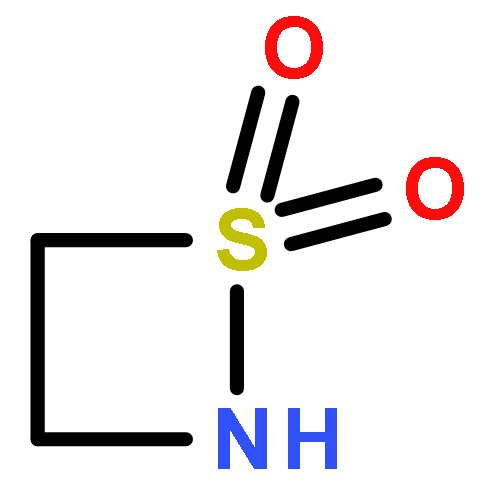
![1,2-Thiazetidine, 2-[(1,1-dimethylethyl)dimethylsilyl]-, 1,1-dioxide](http://img.cochemist.com/ccimg/121500/121444-55-1.png)
![1,2-Thiazetidine, 2-[(1,1-dimethylethyl)dimethylsilyl]-, 1,1-dioxide](http://img.cochemist.com/ccimg/121500/121444-55-1_b.png)
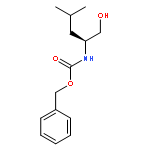
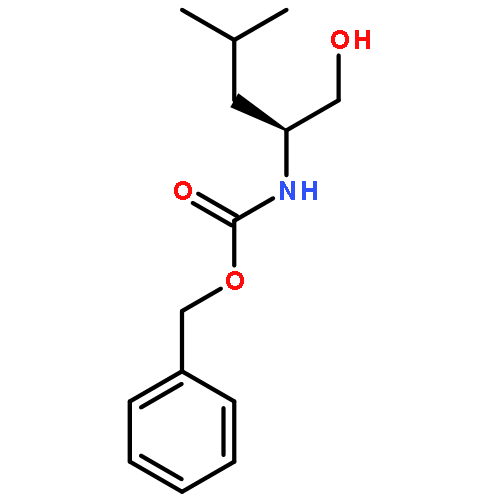
![Carbamic acid,N-[(1S)-2-cyclohexyl-1-(hydroxymethyl)ethyl]-, phenylmethyl ester](http://img.cochemist.com/ccimg/113900/113828-85-6.png)
![Carbamic acid,N-[(1S)-2-cyclohexyl-1-(hydroxymethyl)ethyl]-, phenylmethyl ester](http://img.cochemist.com/ccimg/113900/113828-85-6_b.png)
![2,4-Dodecadienamide,N-[(1S,2R)-2-hydroxy-1-[[[(3E,5S,8S,10S)-10-hydroxy-5-methyl-2,7-dioxo-1,6-diazacyclododec-3-en-8-yl]amino]carbonyl]propyl]-,(2E,4E)-](http://img.cochemist.com/ccimg/108400/108351-50-4.png)
![2,4-Dodecadienamide,N-[(1S,2R)-2-hydroxy-1-[[[(3E,5S,8S,10S)-10-hydroxy-5-methyl-2,7-dioxo-1,6-diazacyclododec-3-en-8-yl]amino]carbonyl]propyl]-,(2E,4E)-](http://img.cochemist.com/ccimg/108400/108351-50-4_b.png)


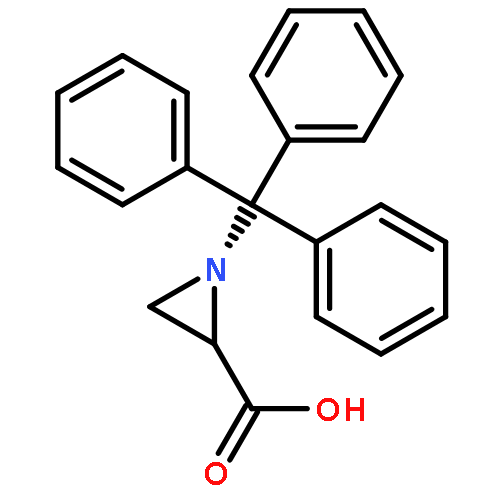
![1-{2-AMINO-5-CHLORO-4-[(TRIISOPROPYLSILYL)OXY]PHENYL}-2,2,2-TRIFL<WBR />UOROETHANONE](http://img.cochemist.com/ccimg/78400/78397-39-4.png)
![1-{2-AMINO-5-CHLORO-4-[(TRIISOPROPYLSILYL)OXY]PHENYL}-2,2,2-TRIFL<WBR />UOROETHANONE](http://img.cochemist.com/ccimg/78400/78397-39-4_b.png)





![N~2~-[(benzyloxy)carbonyl]-N-[(2S)-4-chloro-1-(4-hydroxyphenyl)-3-oxobutan-2-yl]-L-leucinamide](http://img.cochemist.com/ccimg/57000/56979-35-2.png)
![N~2~-[(benzyloxy)carbonyl]-N-[(2S)-4-chloro-1-(4-hydroxyphenyl)-3-oxobutan-2-yl]-L-leucinamide](http://img.cochemist.com/ccimg/57000/56979-35-2_b.png)





![ALANINE, N-[(1,1-DIMETHYLETHOXY)CARBONYL]LEUCYL-, METHYL ESTER](http://img.cochemist.com/ccimg/43200/43189-51-1.png)
![ALANINE, N-[(1,1-DIMETHYLETHOXY)CARBONYL]LEUCYL-, METHYL ESTER](http://img.cochemist.com/ccimg/43200/43189-51-1_b.png)
![L-Alanine, 1-[(1,1-dimethylethoxy)carbonyl]-L-prolyl-, methyl ester](/data/chemimg/1803600/36302-04-2.png)
![L-Alanine, 1-[(1,1-dimethylethoxy)carbonyl]-L-prolyl-, methyl ester](/data/chemimg/1803600/36302-04-2_b.png)











![L-Leucine, 1-[(1,1-dimethylethoxy)carbonyl]-L-prolyl-, methyl ester](http://img.cochemist.com/ccimg/15200/15136-16-0.png)
![L-Leucine, 1-[(1,1-dimethylethoxy)carbonyl]-L-prolyl-, methyl ester](http://img.cochemist.com/ccimg/15200/15136-16-0_b.png)










![2-benzyl-1-[(4-methoxyphenyl)amino]-3-methylpyrido[1,2-a]benzimidazole-4-carbonitrile](http://img.cochemist.com/ccimg/5900/5876-90-4.png)
![2-benzyl-1-[(4-methoxyphenyl)amino]-3-methylpyrido[1,2-a]benzimidazole-4-carbonitrile](http://img.cochemist.com/ccimg/5900/5876-90-4_b.png)


![N-(5-oxo-4,5-dihydro[1,2]dithiolo[4,3-b]pyrrol-6-yl)acetamide](http://img.cochemist.com/ccimg/500/488-04-0.png)
![N-(5-oxo-4,5-dihydro[1,2]dithiolo[4,3-b]pyrrol-6-yl)acetamide](http://img.cochemist.com/ccimg/500/488-04-0_b.png)














![Diphosphoric acid, mono[(2E)-4-hydroxy-3-methyl-2-butenyl] ester](http://img.cochemist.com/ccimg/396800/396726-03-7.png)
![Diphosphoric acid, mono[(2E)-4-hydroxy-3-methyl-2-butenyl] ester](http://img.cochemist.com/ccimg/396800/396726-03-7_b.png)






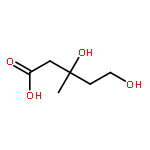

![6-Oxa-2-azabicyclo[3.2.0]heptane-3,7-dione,1-[(1S)-1-hydroxy-2-methylpropyl]-4-methyl-, (1R,4R,5S)-](http://img.cochemist.com/ccimg/154300/154226-60-5.png)
![6-Oxa-2-azabicyclo[3.2.0]heptane-3,7-dione,1-[(1S)-1-hydroxy-2-methylpropyl]-4-methyl-, (1R,4R,5S)-](http://img.cochemist.com/ccimg/154300/154226-60-5_b.png)


![10H-3,10a-Epidithiopyrazino[1,2-a]indole-1,4-dione,2,3,5a,6-tetrahydro-6-hydroxy-3-(hydroxymethyl)-2-methyl-, (3R,5aS,6S,10aR)-](http://img.cochemist.com/ccimg/100/67-99-2.png)
![10H-3,10a-Epidithiopyrazino[1,2-a]indole-1,4-dione,2,3,5a,6-tetrahydro-6-hydroxy-3-(hydroxymethyl)-2-methyl-, (3R,5aS,6S,10aR)-](http://img.cochemist.com/ccimg/100/67-99-2_b.png)
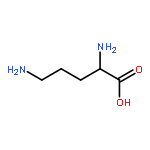
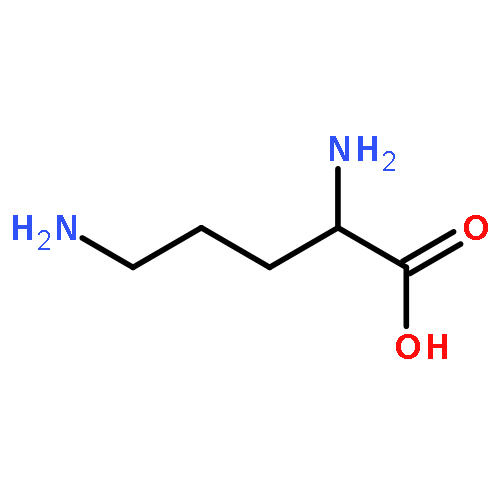
![L-Lysine, N6-[(3R)-1,5-didehydro-3-methyl-D-prolyl]-](/data/chemimg/106600/448235-52-7.png)
![L-Lysine, N6-[(3R)-1,5-didehydro-3-methyl-D-prolyl]-](/data/chemimg/106600/448235-52-7_b.png)
![L-Leucinamide,N-[(phenylmethoxy)carbonyl]-L-leucyl-N-[(1S)-1-formyl-3-methylbutyl]-](http://img.cochemist.com/ccimg/133500/133407-82-6.png)
![L-Leucinamide,N-[(phenylmethoxy)carbonyl]-L-leucyl-N-[(1S)-1-formyl-3-methylbutyl]-](http://img.cochemist.com/ccimg/133500/133407-82-6_b.png)