Co-reporter:Man Li, Xiao-Song Xue, and Jin-Pei Cheng
ACS Catalysis November 3, 2017 Volume 7(Issue 11) pp:7977-7977
Publication Date(Web):October 11, 2017
DOI:10.1021/acscatal.7b03007
The trifluoromethylthio (SCF3) group enjoys a privileged role in the field of drug discovery because its incorporation into a drug molecule often leads to significantly improved pharmacokinetics and efficacy. In spite of its prime importance in drug discovery, the stereospecific introduction of the SCF3 group into target molecules has remained an unmet challenge. A major breakthrough was made in 2013 when Rueping and Shen simultaneously and independently disclosed natural Cinchona alkaloid catalyzed asymmetric electrophilic trifluoromethylthiolation of β-keto esters. However, two key issues remain obscure. (a) What is the preferred mode of catalysis? (b) How is asymmetric induction accomplished? Here we report an in-depth computational exploration into the mechanism and origin of stereoinduction in Cinchona alkaloid catalyzed trifluoromethylthiolation of β-keto esters with N-trifluoromethylthiophthalimide as electrophilic SCF3 source. Three mechanistic possibilities, i.e., (a) the transfer-trifluoromethylthiolation, (b) the Wynberg ion pair-hydrogen bonding model, and (c) the Houk–Grayson bifunctional Brønsted acid-hydrogen bonding model, were evaluated with density functional theory (B3LYP-D3 and M06-2X functionals). Our calculations suggest that, in contrast to Cinchona alkaloid catalyzed conjugate additions, the most preferred mode for the title reaction is not the Houk–Grayson bifunctional Brønsted acid-hydrogen bonding model but instead the Wynberg ion pair-hydrogen bonding model, wherein the SCF3 transfer proceeds via an SN2-like mechanism. Consequently, although the Houk–Grayson bifunctional Brønsted acid–hydrogen bonding model has recently been demonstrated to be a general mechanistic model for Cinchona alkaloid catalyzed asymmetric Michael additions, this catalysis mode cannot be simply extended to an asymmetric SN2-type of reaction. The predicted enantioselectivities based on the Wynberg ion pair-hydrogen bonding model are in good agreement with experimental data, lending strong support to the plausibility of this mode of catalysis. Noncovalent interaction (NCI) analysis of the stereocontrolling transition state structures reveals that the enantioselectivity is mainly induced by the concerted action of multiple weak noncovalent substrate–catalyst interactions, such as C–H···O, C–H···S, C–H···π, and π···π interactions. Not only has this contribution provided insights into the mechanistic model and principles of stereocontrol by Cinchona alkaloids but also it should offer help in the future design of catalysts and asymmetric electrophilic trifluoromethylthiolation reactions.Keywords: asymmetric catalysis; Cinchona alkaloid; density functional calculation; noncovalent interaction; trifluoromethylthiolation;
Co-reporter:Shan Jiang, Tai-Shan Yan, Yong-Chao Han, Li-Qian Cui, Xiao-Song Xue, and Chi Zhang
The Journal of Organic Chemistry November 17, 2017 Volume 82(Issue 22) pp:11691-11691
Publication Date(Web):June 3, 2017
DOI:10.1021/acs.joc.7b00883
We have developed an efficient method for direct formation of epoxide groups from carbon(sp2)–carbon(sp3) single bonds of β-keto esters; the reaction is mediated by the water-soluble hypervalent iodine(V) reagent AIBX (5-trimethylammonio-1,3-dioxo-1,3-dihydro-1λ5-benzo[d][1,2]iodoxol-1-ol anion). On the basis of the results of density functional theory calculations and experimental studies, we propose that the reaction proceeds by a two-stage mechanism involving dehydrogenation of the β-keto ester substrates and epoxidation of the resulting enone intermediates. The rate-limiting step is abstraction of the β′-C–H (calculated free energy of activation, 24.5 kcal/mol).
Co-reporter:Man Li, Biying Zhou, Xiao-Song Xue, and Jin-Pei Cheng
The Journal of Organic Chemistry August 18, 2017 Volume 82(Issue 16) pp:8697-8697
Publication Date(Web):July 24, 2017
DOI:10.1021/acs.joc.7b01771
The recent recognition of the novel application of a few traditional electrophilic trifluoromethylthiolating reagents as SCF3 radical sources offers a remarkable new opportunity for the development of radical trifluoromethylthiolation reactions. Herein, the first trifluoromethylthio radical donating ability (Tt•DA) scale of electrophilic SCF3-transfer reagents has been developed. This scale is based on Y-SCF3 bond dissociation energies, which were obtained by density functional calculations (M06-2X). Single electron transfer is revealed to exhibit a substantial Y-SCF3 bond-weakening effect, thus significantly facilitating the SCF3 radical (•SCF3) release. The results may aid in future novel radical SCF3-transfer reagent design and new reaction development.
Co-reporter:Jin-Dong Yang, Ya Wang, Xiao-Song Xue, and Jin-Pei Cheng
The Journal of Organic Chemistry April 21, 2017 Volume 82(Issue 8) pp:4129-4129
Publication Date(Web):March 27, 2017
DOI:10.1021/acs.joc.7b00036
The recent discovery of the radical reactivity of a few traditionally electrophilic N–F reagents has sparked a renaissance of radical fluorination. A knowledge of the N–F bond dissociation enthalpies (BDE) of electrophilic N–F reagents is essential for understanding of their reactivities. However, a thorough literature survey revealed that such information is extremely sparse. This prompted us to carry out the first systematic computation on the N–F BDEs of electrophilic N–F reagents. The calculated N–F BDE scale of 88 electrophilic N–F reagents ranges from 49.3 to 80.0 kcal mol–1 in acetonitrile. The large variety of N–F reagents and wide span of N–F BDEs make the scale a useful tool not only for the future rational design of novel reagents but also for judicious selection of appropriate ones to explore new radical fluorinations.
Co-reporter:Biying Zhou, Taishan Yan, Xiao-Song Xue, and Jin-Pei Cheng
Organic Letters 2016 Volume 18(Issue 23) pp:6128-6131
Publication Date(Web):November 17, 2016
DOI:10.1021/acs.orglett.6b03134
Fluorination mediated by the cyclic hypervalent fluoroiodane reagent (1) often requires an exogenous Lewis acid. The widely accepted Lewis-acid-activation model is that a given Lewis acid binds to the oxygen atom of 1 (O-coordination) to polarize the I–O bond. Computational studies of silver-mediated geminal difluorination of styrenes with 1 reveal a new “F-coordination” model that is energetically much preferred over the commonly accepted “O-coordination” model. The calculations rationalize the regioselective formation of the geminal difluorination product.
Co-reporter:Man Li; Jinping Guo; Xiao-Song Xue;Jin-Pei Cheng
Organic Letters 2016 Volume 18(Issue 2) pp:264-267
Publication Date(Web):January 7, 2016
DOI:10.1021/acs.orglett.5b03433
A new parameter, trifluoromethylthio cation-donating ability (Tt+DA), is introduced as a quantitative descriptor for the propensity of electrophilic trifluoromethylthiolating reagents to transfer a CF3S moiety in organic synthesis. The first Tt+DA scale of popular reagents has been established through DFT calculations. Excellent correlation has been identified between the Tt+DAs of N-SCF3-type reagents and the pKa of the corresponding acids, offering a powerful avenue for the rational design of novel reagents.
Co-reporter:Panpan Zhang, Man Li, Xiao-Song Xue, Chunfa Xu, Qunchao Zhao, Yafei Liu, Haoyang Wang, Yinlong Guo, Long Lu, and Qilong Shen
The Journal of Organic Chemistry 2016 Volume 81(Issue 17) pp:7486-7509
Publication Date(Web):July 21, 2016
DOI:10.1021/acs.joc.6b01178
The super electrophilicity of a shelf-stable, easily prepared trifluoromethylthiolating reagent N-trifluoromethylthio-dibenzenesulfonimide 7 was demonstrated. Consistent with the theoretical prediction, 7 exhibits reactivity remarkably higher than that of other known electrophilic trifluoromethylthiolating reagents. In the absence of any additive, 7 reacted with a wide range of electron-rich arenes and activated heteroarenes under mild conditions. Likewise, reactions of 7 with styrene derivatives can be fine-tuned by simply changing the reaction solvents to generate trifluoromethylthiolated styrenes or oxo-trifluoromethylthio or amino-trifluoromethylthio difunctionalized compounds in high yields.
Co-reporter:Taishan Yan, Biying Zhou, Xiao-Song Xue, and Jin-Pei Cheng
The Journal of Organic Chemistry 2016 Volume 81(Issue 19) pp:9006-9011
Publication Date(Web):September 7, 2016
DOI:10.1021/acs.joc.6b01642
The mechanism and origin of the unexpected chemoselectivity in fluorocyclization of o-styryl benzamide with a cyclic hypervalent fluoroiodane reagent were explored with DFT calculations. The calculations suggested an alternative mechanism that is broadly similar to, but also critically different from, the previously proposed mechanism for the formation of an unexpected structurally novel seven-membered 4-fluoro-1,3-benzoxazepine. The amide group of o-styryl benzamide was revealed to be crucial for activating the fluoroiodane reagent and facilitating C–F bond formation. In contrast to the popular electrophilic N–F reagent Selectfluor, the F atom in the fluoroiodane reagent is nucleophilic, and the I(III) atom is the most electrophilic site, thus inducing a completely different reactivity pattern. The insights reported here will be valuable for the further development of new reactions based on the hypervalent fluoroiodane reagent.
Co-reporter:Xiao-Song Xue, Ya Wang, Man Li, and Jin-Pei Cheng
The Journal of Organic Chemistry 2016 Volume 81(Issue 10) pp:4280-4289
Publication Date(Web):April 27, 2016
DOI:10.1021/acs.joc.6b00683
Quantitative knowledge of the fluorinating strength of electrophilic N–F reagents is of crucial importance for rational design and optimization of novel reagents and new reactions. Herein, we report the first systematic computation of fluorinating potentials of 130 electrophilic N–F reagents in two commonly used solvents dichloromethane and acetonitrile in terms of the N–F bond heterolysis energies as expressed by the fluorine plus detachment (FPD) values. The calculated FPD scales of 130 N–F reagents cover a range from 112.3 to 290.4 kcal mol–1 and 110.9 to 278.4 kcal mol–1 in dichloromethane and acetonitrile, respectively. This comprehensive FPD database provides a valuable quantitative guide for studying the influence of structural variation on the fluorinating strength of the N–F reagents, opening a door to the rational design of novel reagents with appropriate fluorinating strength for specific purposes. It is demonstrated that the FPD values can reproduce the reactivity order for electrophilic N–F reagents better than other parameters.
Co-reporter:Man Li, Xiao-Song Xue, Jinping Guo, Ya Wang, and Jin-Pei Cheng
The Journal of Organic Chemistry 2016 Volume 81(Issue 8) pp:3119-3126
Publication Date(Web):March 21, 2016
DOI:10.1021/acs.joc.5b02821
This work established an energetic guide for estimating the trifluoromethyl cation-donating abilities (TC+DA) of electrophilic trifluoromethylating reagents through computing X–CF3 bond (X = O, S, Se, Te, and I) heterolytic dissociation enthalpies. TC+DA values for a wide range of popular reagents were derived on the basis of density functional calculations (M06-2X). A good correspondence has been identified between the computed TC+DA values and the experimentally observed relative trifluoromethylating capabilities of the reagents. Substituent effects hold good linear free energy relationships on the TC+DAs of the most widely used reagents including Umemoto reagent, Yagupolskii–Umemoto reagent, and Togni reagents, which allow their trifluoromethylating capabilities to be rationally tuned by substituents and thus extend their synthetic utility. All the information disclosed in this work would contribute to future rational exploration of the electrophilic trifluoromethylation chemistry.
Co-reporter:Xiao-Song Xue, Ya Wang, Chen Yang, Pengju Ji, and Jin-Pei Cheng
The Journal of Organic Chemistry 2015 Volume 80(Issue 18) pp:8997-9006
Publication Date(Web):August 24, 2015
DOI:10.1021/acs.joc.5b00693
In contrast to the great success of computational methodologies in molecular solvents, effective and accurate calculation of the fundamental bond energetic properties in ionic liquids (ILs) is essentially absent. This is largely due to the unusual complexity of handling solvation quantities of ILs and the lack of precisely determined bond parameters to serve as the authentic benchmark to calibrate the modeling methodology. Herein, we report the first accurate calculations of absolute pKa values in a commonly used IL, [Bmim][NTf2], with the carefully developed “ion-biased” cluster-continuum model. Experimental pKa values of benzoic acids and benzenethiols in [Bmim][NTf2] were reproduced with mean unsigned errors of only 0.3 and 0.5 pKa units, respectively, which enables theoretical approaches with a suitable strategy as a powerful tool to handle complicated problems in ILs and to eventually realize the rational design of the IL chemistry.
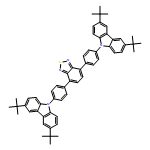
Co-reporter:Biying Zhou, Xiao-song Xue, Jin-pei Cheng
Tetrahedron Letters (29 March 2017) Volume 58(Issue 13) pp:1287-1291
Publication Date(Web):29 March 2017
DOI:10.1016/j.tetlet.2017.02.040

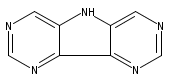



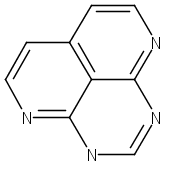

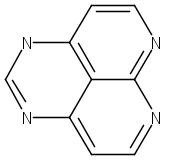
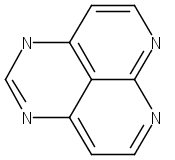







![9H-Carbazole, 9,9'-[2,1,3-benzothiadiazole-4,7-diylbis(2,1-ethynediyl-4,1-phenylene)]bis[3,6-bis(1,1-dimethylethyl)-](/data/chemimg/139700/1226860-84-9.png)
![9H-Carbazole, 9,9'-[2,1,3-benzothiadiazole-4,7-diylbis(2,1-ethynediyl-4,1-phenylene)]bis[3,6-bis(1,1-dimethylethyl)-](/data/chemimg/139700/1226860-84-9_b.png)

![9H-Carbazole, 9,9'-[9,10-anthracenediylbis(2,1-ethynediyl-4,1-phenylene)]bis[3,6-bis(1,1-dimethylethyl)-](/data/chemimg/139700/1226860-82-7.png)
![9H-Carbazole, 9,9'-[9,10-anthracenediylbis(2,1-ethynediyl-4,1-phenylene)]bis[3,6-bis(1,1-dimethylethyl)-](/data/chemimg/139700/1226860-82-7_b.png)

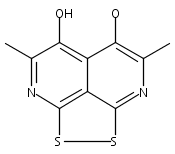
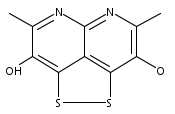

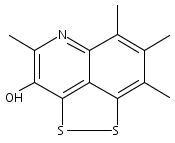
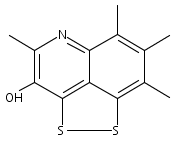

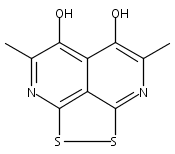

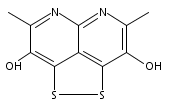
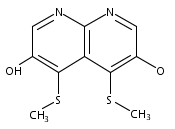


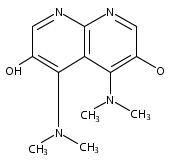


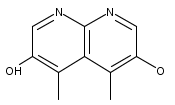

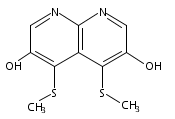




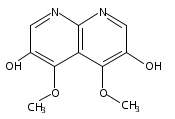
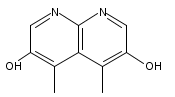

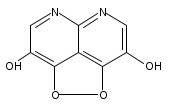
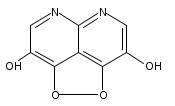

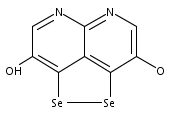
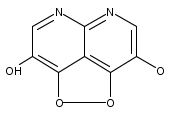

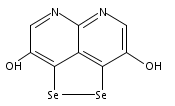
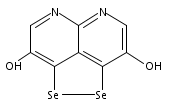
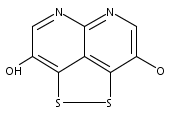
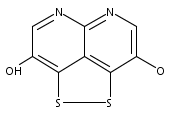
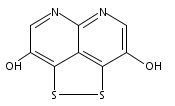
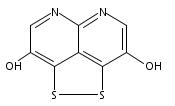
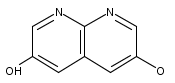

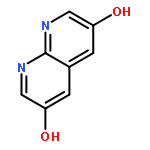
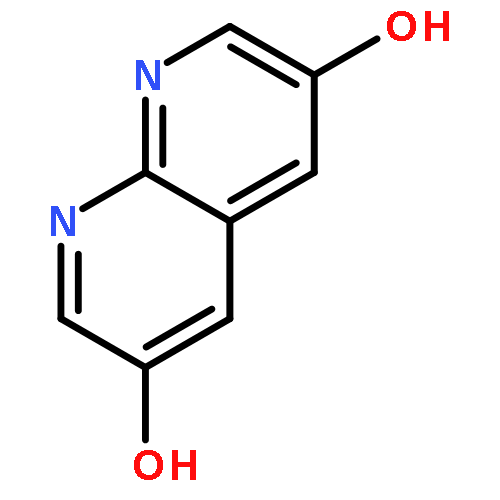
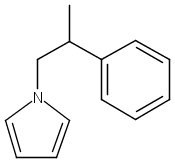
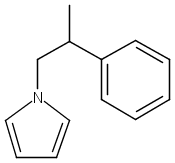

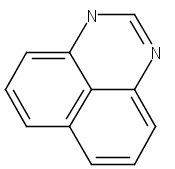

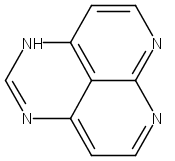

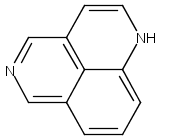
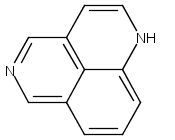


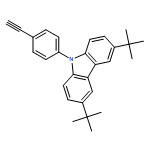
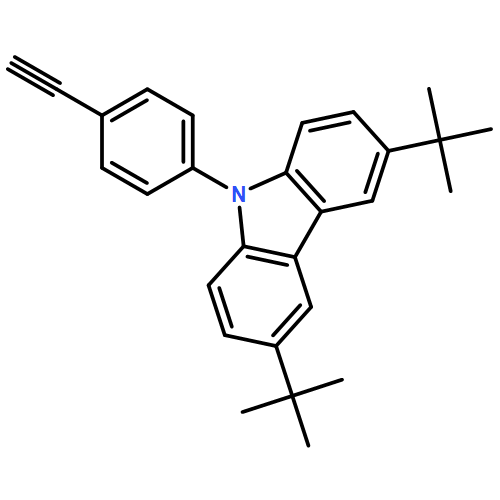

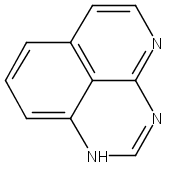
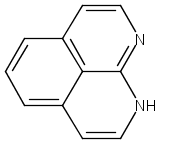

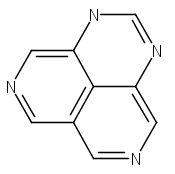
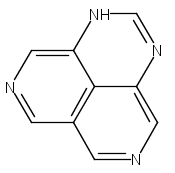
![3H-Pyrido[4,3,2-de]quinazoline](http://img.cochemist.com/ccimg/111500/111493-32-4.png)
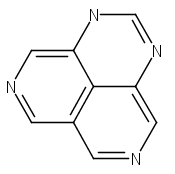
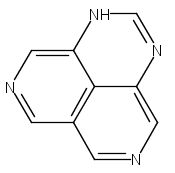
![3H-Pyrido[4,3,2-de]quinazoline](http://img.cochemist.com/ccimg/111500/111493-32-4_b.png)
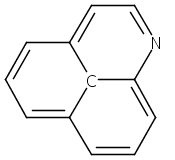
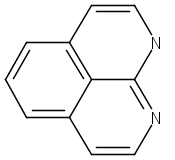
![3H-Pyrido[3,4,5-de]quinazoline](http://img.cochemist.com/ccimg/110800/110748-36-2.png)
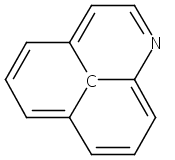
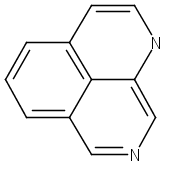
![3H-Pyrido[3,4,5-de]quinazoline](http://img.cochemist.com/ccimg/110800/110748-36-2_b.png)
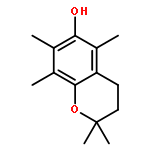
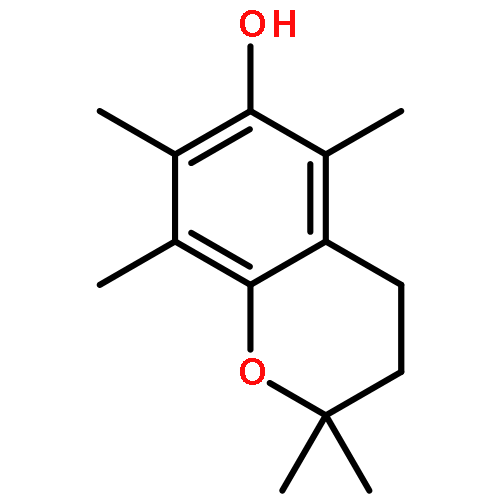
![9H-Pyrrolo[2,3-c:5,4-c']dipyridine](http://img.cochemist.com/ccimg/77300/77200-36-3.png)
![9H-Pyrrolo[2,3-c:5,4-c']dipyridine](http://img.cochemist.com/ccimg/77300/77200-36-3_b.png)
![1H-Benzo[de][1,6]naphthyridine](http://img.cochemist.com/ccimg/37000/36917-96-1.png)
![1H-Benzo[de][1,6]naphthyridine](http://img.cochemist.com/ccimg/37000/36917-96-1_b.png)
![1H-Benzo[de]cinnoline](http://img.cochemist.com/ccimg/28100/28022-68-6.png)
![1H-Benzo[de]cinnoline](http://img.cochemist.com/ccimg/28100/28022-68-6_b.png)
![1H-Benzo[ij][2,7]naphthyridine(8CI,9CI)](http://img.cochemist.com/ccimg/27300/27212-02-8.png)
![1H-Benzo[ij][2,7]naphthyridine(8CI,9CI)](http://img.cochemist.com/ccimg/27300/27212-02-8_b.png)
![1H-Dipyrido[2,3-b:3',2'-d]pyrrole](http://img.cochemist.com/ccimg/18000/17966-00-6.png)
![1H-Dipyrido[2,3-b:3',2'-d]pyrrole](http://img.cochemist.com/ccimg/18000/17966-00-6_b.png)
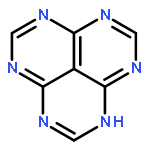
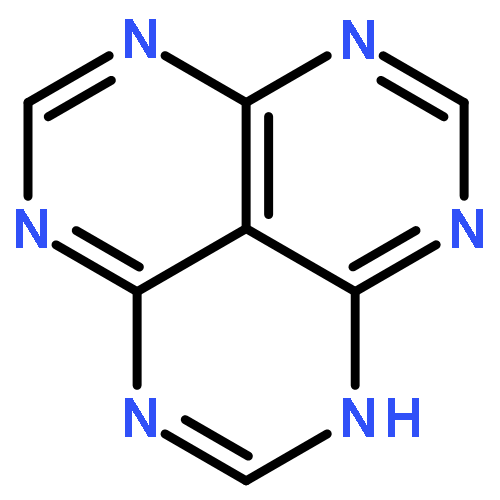




![1H-Benzo[de]quinoline](http://img.cochemist.com/ccimg/300/203-86-1.png)
![1H-Benzo[de]quinoline](http://img.cochemist.com/ccimg/300/203-86-1_b.png)

