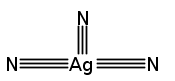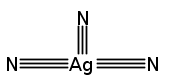Co-reporter:Denis A. Rychkov, Steven Hunter, Viktor Yu. Kovalskii, Alexander A. Lomzov, Colin R. Pulham, Elena V. Boldyreva
Computational and Theoretical Chemistry 2016 Volume 1088() pp:52-61
Publication Date(Web):15 July 2016
DOI:10.1016/j.comptc.2016.04.027
•A potential energy surface was calculated for the 5-HT molecule in aqueous media.•Minima and TS were located using fully relaxed optimization at different LOT.•5-HT conformational and lattice energies of serotonin salts were calculated.•Comparison of conformations in crystals and crystallisation media were preformed.•New approach for crystal structure prediction using conformational study is suggested.A potential energy surface (PES) has been calculated for the protonated serotonin (5-HT) molecule in aqueous media. Three pairs of symmetrically equivalent minima were located using fully relaxed optimization at B3LYP/6-31G(d,p) level of theory. Insensitivity to basis set was verified over a range of different functionals and levels of theory. All 9 transition structures were found using the QST3 method with ansatz structures taken directly from the result of the calculated PES. Energies associated with conformational changes of protonated 5-HT in aqueous media were calculated and found to vary between 6 and 23 kJ mol−1. Lattice energies of serotonin picrate monohydrate and serotonin adipate were calculated as −741.7 and −716.9 kJ mol−1, respectively, and compared to conformational energies to consider the relative importance of each interaction type. The study contributes to achieving better insight into the importance of molecular conformations in solution as a precursor for the formation of the final crystal structure.
Co-reporter:Steven Hunter, Tuuli Sutinen, Stewart F. Parker, Carole A. Morrison, David M. Williamson, Stephen Thompson, Peter J. Gould, and Colin R. Pulham
The Journal of Physical Chemistry C 2013 Volume 117(Issue 16) pp:8062-8071
Publication Date(Web):March 27, 2013
DOI:10.1021/jp4004664
We have performed simulations utilizing the dispersion-corrected density functional theory method (DFT-D) as parametrized by Grimme on selected polymorphs of RDX (cyclotrimethylenetrinitramine). Additionally, we present the first experimental determination of the enthalpy of fusion (ΔHfus) of the highly metastable β-form of RDX. The characteristics of fusion for β-RDX were determined to be 186.7 ± 0.8 °C, 188.5 ± 0.4 °C, and 12.63 ± 0.28 kJ mol–1 for the onset temperature, peak temperature (or melting point), and ΔHfus, respectively. The difference in experimental ΔHfus for the α- and β-forms of RDX is 20.46 ± 0.92 kJ mol–1. Ambient-pressure lattice energies (EL) of the α- and β-forms of RDX have been calculated and are in excellent agreement with experiment. In addition the computationally predicted difference in EL (20.35 kJ mol–1) between the α- and β-forms is in excellent agreement with the experimental difference in ΔHfus. The response of the lattice parameters and unit-cell volumes to pressure for the α- and γ-forms have been investigated, in addition to the first high-pressure computational study of the ε-form of RDX—these results are in very good agreement with experimental data. Phonon calculations provide good agreement for vibrational frequencies obtained from Raman spectroscopy, and a predicted inelastic neutron scattering (INS) spectrum of α-RDX shows excellent agreement with experimental INS data determined in this study. The transition energies and intensities are reproduced, confirming that both the eigenvalues and the eigenvectors of the vibrations are correctly described by the DFT-D method. The results of the high-pressure phonon calculations have been used to show that the heat capacities of the α-, γ-, and ε-forms of RDX are only weakly affected by pressure.
Co-reporter:Steven Hunter ; Alistair J. Davidson ; Carole A. Morrison ; Colin R. Pulham ; Patricia Richardson ; Matthew J. Farrow ; William G. Marshall ; Alistair R Lennie ;Peter J. Gould
The Journal of Physical Chemistry C 2011 Volume 115(Issue 38) pp:18782-18788
Publication Date(Web):August 25, 2011
DOI:10.1021/jp2012599
We have obtained detailed structural information for the energetic salt ammonium perchlorate (AP) at pressures up to ∼8 GPa through a combination of X-ray and neutron diffraction. Under hydrostatic conditions, AP undergoes a first-order phase transition at 3.98(5) GPa, broadly consistent with results from previous studies. We have successfully solved and refined the structure of the new orthorhombic phase (phase II, space group Pnma), which features a more close-packed structure with more extensive hydrogen bonding than the polymorph obtained at ambient pressure (phase I). Equations of state have been obtained for phase I from 0 to 3.5 GPa and for the new phase 4 to 8.1 GPa. To complement these experimental studies, we have also performed density functional theory (DFT) calculations of the hydrostatic compression of AP in the region of 0.0–3.5 GPa. A comparison of the performance of different pseudopotentials and DFT dispersion correction schemes in calculating crystal geometries at high pressure has been performed. The results highlight the fact that care must be taken when choosing pseudopotentials for high-pressure studies and that no significant improvements in the calculation of crystal geometries of AP are obtained by employing DFT-D corrections.
Co-reporter:David I. A. Millar, Iain D. H. Oswald, Christopher Barry, Duncan J. Francis, William G. Marshall, Colin R. Pulham and Adam S. Cumming
Chemical Communications 2010 vol. 46(Issue 31) pp:5662-5664
Publication Date(Web):09 Jul 2010
DOI:10.1039/C0CC00368A
The high-pressure, high-temperature ε-form of the widely used explosive RDX has been structurally characterised using a combination of diffraction techniques, and a sample of this form has been successfully recovered to ambient pressure.
Co-reporter:David I. A. Millar, Iain D. H. Oswald, Duncan J. Francis, William G. Marshall, Colin R. Pulham and Adam S. Cumming
Chemical Communications 2009 (Issue 5) pp:562-564
Publication Date(Web):05 Dec 2008
DOI:10.1039/B817966B
The crystal structure of the highly metastable β-form of RDX shows that the molecules adopt different conformations compared to the α-form and that, contrary to previous reports, the β-form obtained at ambient pressure is not the same form as that obtained at elevated temperatures and pressures.
Co-reporter:Iain D. H. Oswald, Isabelle Chataigner, Stephen Elphick, Francesca P. A. Fabbiani, Alistair R. Lennie, Jacques Maddaluno, William G. Marshall, Timothy J. Prior, Colin R. Pulham and Ronald I. Smith
CrystEngComm 2009 vol. 11(Issue 2) pp:359-366
Publication Date(Web):28 Nov 2008
DOI:10.1039/B814471K
The reproducible crystallisation of elusive polymorphs and solvates of molecular compounds at high pressure has been demonstrated through studies on maleic acid, malonamide, and paracetamol. These high-pressure methods can be scaled-up to produce ‘bulk’ quantities of metastable forms that can be recovered to ambient pressure for subsequent seeding experiments. This has been demonstrated for paracetamol form II and paracetamol monohydrate. The studies also show that the particular solid form can be tuned by both pressure and concentration.
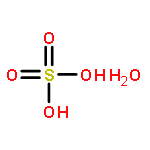
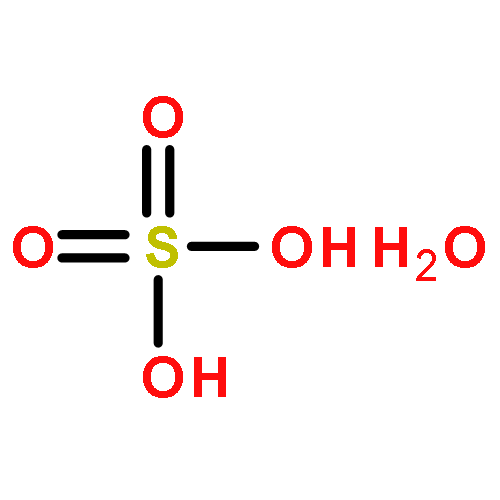
Co-reporter:Francesca P.A. Fabbiani, David R. Allan, Alice Dawson, Duncan J. Francis, William G. Marshall, Colin R. Pulham
Inorganica Chimica Acta 2008 Volume 361(Issue 2) pp:487-494
Publication Date(Web):15 January 2008
DOI:10.1016/j.ica.2007.05.006
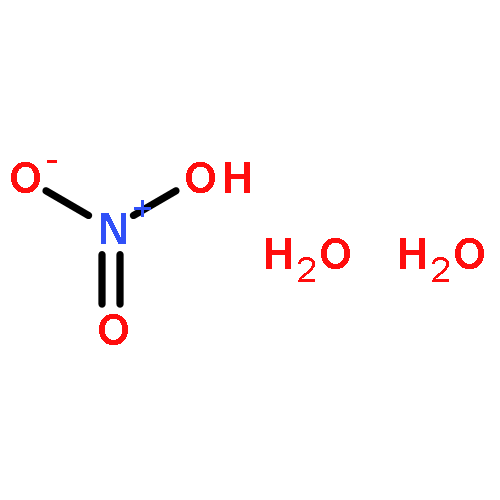
Two high-pressure polymorphs of sulfuric acid monohydrate (oxonium hydrogensulfate) have been obtained at ambient temperature by crystallisation at high pressure from the liquid at 1.3 GPa (form III) and by direct compression of the ambient-pressure form I first to 1.26 GPa (form II) and then to 1.72 GPa (form III). The structure of form III was solved by single crystal X-ray diffraction and this structure was used as the basis for the refinement of hydrogen positions using high-pressure neutron powder diffraction data. Form III crystallises in the orthorhombic crystal system at 1.97 GPa, and features parallel chains of hydrogensulfate ions linked by oxonium ions to form a three-dimensional hydrogen-bonded network. On further compression to 3.05 GPa, the direction of maximum compressibility is found to be along the a-axis and is associated with the shortening of a hydrogen bond between a hydrogensulfate ion and an oxonium ion. The structure of form II remains elusive although at ambient temperature it is stable (or metastable) at pressures as low as 0.42 GPa, perhaps indicating that it could be recoverable to ambient-pressure at low temperature.A high-pressure polymorph of sulfuric acid monohydrate has been obtained by crystallisation at high-pressure from the liquid at 1.3 GPa and by direct compression of the ambient-pressure form to 1.72 GPa. The structure was solved by single crystal X-ray diffraction and the hydrogen positions refined using neutron powder diffraction data.
Co-reporter:Francesca P. A. Fabbiani, David R. Allan, Alice Dawson, William I. F. David, Pamela A. McGregor, Iain D. H. Oswald, Simon Parsons and Colin R. Pulham
Chemical Communications 2003 (Issue 24) pp:3004-3005
Publication Date(Web):07 Nov 2003
DOI:10.1039/B310394C
Recrystallisation of paracetamol from a solution in methanol at a pressure of 0.62 GPa gives a new 1 ∶ 1 solvate that has been characterised by single crystal X-ray diffraction.
Co-reporter:Pamela A. McGregor;David R. Allan;Simon Parsons
Journal of Pharmaceutical Sciences 2002 Volume 91(Issue 5) pp:1308-1311
Publication Date(Web):28 MAR 2002
DOI:10.1002/jps.10131
This work reports the preparation and crystal structure of a trihydrate of paracetamol. Crystals were grown by slow cooling of an aqueous solution of paracetamol to 0°C. Single-crystal X-ray diffraction shows that the trihydrate crystallizes in the orthorhombic crystal system, space group Pbca, Z = 8, a = 7.3324(16), b = 12.590(3), c = 22.636(6) Å, V = 2089.7(9) Å3. The crystals of the trihydrate dehydrate rapidly at 20°C to give anhydrous paracetamol as its monoclinic form. © 2002 Wiley-Liss, Inc. and the American Pharmaceutical Association J Pharm Sci 91:1308–1311, 2002
Co-reporter:Lucy E. Griffiths, Martin R. Lee, Andrew R. Mount, Hiroshi Kondoh, Toshiaki Ohta and Colin R. Pulham
Chemical Communications 2001 (Issue 6) pp:579-580
Publication Date(Web):07 Mar 2001
DOI:10.1039/B100713K
Electrochemical oxidation of a titanium electrode in a
solution of potassium amide in liquid ammonia resulted in the deposition of
titanium nitride either as a thin film or as nanoparticles.
Co-reporter:David I. A. Millar, Iain D. H. Oswald, Duncan J. Francis, William G. Marshall, Colin R. Pulham and Adam S. Cumming
Chemical Communications 2009(Issue 5) pp:NaN564-564
Publication Date(Web):2008/12/05
DOI:10.1039/B817966B
The crystal structure of the highly metastable β-form of RDX shows that the molecules adopt different conformations compared to the α-form and that, contrary to previous reports, the β-form obtained at ambient pressure is not the same form as that obtained at elevated temperatures and pressures.
Co-reporter:David I. A. Millar, Iain D. H. Oswald, Christopher Barry, Duncan J. Francis, William G. Marshall, Colin R. Pulham and Adam S. Cumming
Chemical Communications 2010 - vol. 46(Issue 31) pp:NaN5664-5664
Publication Date(Web):2010/07/09
DOI:10.1039/C0CC00368A
The high-pressure, high-temperature ε-form of the widely used explosive RDX has been structurally characterised using a combination of diffraction techniques, and a sample of this form has been successfully recovered to ambient pressure.
















![5,2,6-(Iminomethenimino)-1H-imidazo[4,5-b]pyrazine,octahydro-1,3,4,7,8,10-hexanitro-](http://img.cochemist.com/ccimg/135300/135285-90-4.png)
![5,2,6-(Iminomethenimino)-1H-imidazo[4,5-b]pyrazine,octahydro-1,3,4,7,8,10-hexanitro-](http://img.cochemist.com/ccimg/135300/135285-90-4_b.png)