Co-reporter:Pingyuan Wang, Zhiqing Liu, Haiying Chen, Na Ye, Xiaodong Cheng, Jia Zhou
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 8(Issue 8) pp:
Publication Date(Web):15 April 2017
DOI:10.1016/j.bmcl.2017.02.065
Exchange proteins directly activated by cAMP (EPACs) are critical cAMP-dependent signaling pathway mediators. The discovery of EPAC proteins has significantly facilitated understanding on cAMP-dependent signaling pathway and efforts along this line open new avenues for developing novel therapeutics for cancer, diabetes, heart failure, inflammation, infections, neurological disorders and other human diseases. Over the past decade, important progress has been made in the identification of EPAC agonists, antagonists and their biological and pharmacological applications. In this review, we briefly summarize recently reported novel functions of EPACs and the discovery of their small molecule modulators. The challenges and future perspectives are also discussed.Download high-res image (108KB)Download full-size image
Co-reporter:Zhiqing Liu, Yingmin Zhu, Haiying Chen, Pingyuan Wang, Fang C. Mei, Na Ye, Xiaodong Cheng, Jia Zhou
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 23(Issue 23) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.bmcl.2017.10.056
Exchange proteins directly activated by cAMP (EPACs) are critical cAMP-dependent signaling pathway mediators that play important roles in cancer, diabetes, heart failure, inflammations, infections, neurological disorders and other human diseases. EPAC specific modulators are urgently needed to explore EPAC’s physiological function, mechanism of action and therapeutic applications. On the basis of a previously identified EPAC specific inhibitor hit ESI-09, herein we have designed and synthesized a novel series of 2-substituted phenyl-N-phenyl-2-oxoacetohydrazonoyl cyanides as potent EPAC inhibitors. Compound 31 (ZL0524) has been discovered as the most potent EPAC inhibitor with IC50 values of 3.6 µM and 1.2 µM against EPAC1 and EPAC2, respectively. Molecular docking of 31 onto an active EPAC2 structure predicts that 31 occupies the hydrophobic pocket in cAMP binding domain (CBD) and also opens up new space leading to the solvent region. These findings provide inspirations for discovering next generation of EPAC inhibitors.Download high-res image (148KB)Download full-size image
Co-reporter:Na Ye, Yingmin Zhu, Zhiqing Liu, Fang C. Mei, Haiying Chen, Pingyuan Wang, Xiaodong Cheng, Jia Zhou
European Journal of Medicinal Chemistry 2017 Volume 134(Volume 134) pp:
Publication Date(Web):7 July 2017
DOI:10.1016/j.ejmech.2017.04.001
•Further structural modifications and SAR studies based on ESI-09 are presented.•Two series of novel diversified analogues have been designed and synthesized.•14, 32 and 33 identified as potent EPAC antagonists with low micromolar activities.•Molecular dockings on ligand-EPAC2 protein binding interactions are explored.•Benzo[d]isoxazol analogues offer new lead scaffolds for further optimization.Two series of novel EPAC antagonists are designed, synthesized and evaluated in an effort to develop diversified analogues based on the scaffold of the previously identified high-throughput (HTS) hit 1 (ESI-09). Further SAR studies reveal that the isoxazole ring A of 1 can tolerate chemical modifications with either introduction of flexible electron-donating substitutions or structurally restrictedly fusing with a phenyl ring, leading to identification of several more potent and diversified EPAC antagonists (e.g., 10 (NY0617), 14 (NY0460), 26 (NY0725), 32 (NY0561), and 33 (NY0562)) with low micromolar inhibitory activities. Molecular docking studies on compounds 10 and 33 indicate that these two series of compounds bind at a similar site with substantially different interactions with the EPAC proteins. The findings may serve as good starting points for the development of more potent EPAC antagonists as valuable pharmacological probes or potential drug candidates.Download high-res image (189KB)Download full-size image
Co-reporter:Ye Ding, Chunyong Ding, Na Ye, Zhiqing Liu, Eric A. Wold, Haiying Chen, Christopher Wild, Qiang Shen, Jia Zhou
European Journal of Medicinal Chemistry 2016 Volume 122() pp:102-117
Publication Date(Web):21 October 2016
DOI:10.1016/j.ejmech.2016.06.015
•Oridonin displays significant anticancer activities via multi-signaling pathways.•Recent advances in medicinal chemistry of oridonin-like compounds are presented.•The article summarizes the SAR and mechanism studies of relevant drug candidates.•The milestones and future direction of oridonin-based drug discovery are discussed.Natural products have historically been, and continue to be, an invaluable source for the discovery of various therapeutic agents. Oridonin, a natural diterpenoid widely applied in traditional Chinese medicines, exhibits a broad range of biological effects including anticancer and anti-inflammatory activities. To further improve its potency, aqueous solubility and bioavailability, the oridonin template serves as an exciting platform for drug discovery to yield better candidates with unique targets and enhanced drug properties. A number of oridonin derivatives (e.g. HAO472) have been designed and synthesized, and have contributed to substantial progress in the identification of new agents and relevant molecular mechanistic studies toward the treatment of human cancers and other diseases. This review summarizes the recent advances in medicinal chemistry on the explorations of novel oridonin analogues as potential anticancer therapeutics, and provides a detailed discussion of future directions for the development and progression of this class of molecules into the clinic.
Co-reporter:Jai S. Rudra, Ye Ding, Harshini Neelakantan, Chunyong Ding, Rajagopal Appavu, Sonja Stutz, Joshua D. Snook, Haiying Chen, Kathryn A. Cunningham, and Jia Zhou
ACS Chemical Neuroscience 2016 Volume 7(Issue 5) pp:546
Publication Date(Web):February 29, 2016
DOI:10.1021/acschemneuro.5b00345
The development of anti-cocaine vaccines that counteract the rewarding effects of the drug are currently being investigated as adjunct therapies for prevention of relapse in abstinent users. However, cocaine is weakly immunogenic and requires conjugation to carrier proteins and coadministration with strong adjuvants, which carry the risk of local reactogenicity and systemic toxicity. Here we report synthetic and multivalent self-assembling peptide nanofibers as adjuvant-free carriers for cocaine vaccines. A novel cocaine hapten modified at the P3 site was conjugated to the N-terminus of an amphipathic self-assembling domain KFE8. In aqueous buffers the cocaine-KFE8 conjugate assembled into β-sheet rich nanofibers, which raised anti-cocaine antibodies without the need for added adjuvants in mice. Vaccinated mice were treated with cocaine and a significant negative correlation was observed between antibody levels and cocaine-evoked hyperactivity. These totally synthetic and multivalent nanofibers with well-defined chemical composition represent the first generation of adjuvant-free cocaine vaccines.Keywords: Antibody; cocaine; hyperactivity; peptide-nanofiber; self-assembly; vaccine
Co-reporter:Christopher T. Wild, Yingmin Zhu, Ye Na, Fang Mei, Marcus A. Ynalvez, Haiying Chen, Xiaodong Cheng, and Jia Zhou
ACS Medicinal Chemistry Letters 2016 Volume 7(Issue 5) pp:460
Publication Date(Web):March 28, 2016
DOI:10.1021/acsmedchemlett.5b00477
N,N-Diphenylamines were discovered as potent and selective EPAC2 inhibitors. A study was conducted to determine the structure–activity relationships in a series of inhibitors of which several compounds displayed submicromolar potencies. Selectivity over the related EPAC1 protein was also demonstrated. Computational modeling reveals an allosteric site that is distinct from the cAMP binding domain shared by both EPAC isoforms, providing a theory with regards to subtype selectivity.Keywords: Buchwald−Hartwig amination; diphenylamines; Exchange proteins directly activated by cAMP (EPAC) inhibitors; structure−activity relationship
Co-reporter:Na Ye, Haiying Chen, Eric A. Wold, Pei-Yong Shi, and Jia Zhou
ACS Infectious Diseases 2016 Volume 2(Issue 6) pp:382
Publication Date(Web):April 26, 2016
DOI:10.1021/acsinfecdis.6b00041
Antiviral therapeutics with profiles of high potency, low resistance, panserotype, and low toxicity remain challenging, and obtaining such agents continues to be an active area of therapeutic development. Due to their unique three-dimensional structural features, spirooxindoles have been identified as privileged chemotypes for antiviral drug development. Among them, spiro-pyrazolopyridone oxindoles have been recently reported as potent inhibitors of dengue virus NS4B, leading to the discovery of an orally bioavailable preclinical candidate (R)-44 with excellent in vivo efficacy in a dengue viremia mouse model. This review highlights recent advances in the development of biologically active spirooxindoles for their antiviral potential, primarily focusing on the structure–activity relationships (SARs) and modes of action, as well as future directions to achieve more potent analogues toward a viable antiviral therapy.Keywords: antiviral agents; DENV; HIV; influenza virus; RSV; spirooxindoles
Co-reporter:Na Ye; Yingmin Zhu; Haijun Chen; Zhiqing Liu; Fang C. Mei; Christopher Wild; Haiying Chen; Xiaodong Cheng
Journal of Medicinal Chemistry 2015 Volume 58(Issue 15) pp:6033-6047
Publication Date(Web):July 7, 2015
DOI:10.1021/acs.jmedchem.5b00635
Exchange proteins directly activated by cAMP (EPAC) as guanine nucleotide exchange factors mediate the effects of the pivotal second messenger cAMP, thereby regulating a wide variety of intracellular physiological and pathophysiological processes. A series of novel 2-(isoxazol-3-yl)-2-oxo-N′-phenyl-acetohydrazonoyl cyanide EPAC antagonists was synthesized and evaluated in an effort to optimize properties of the previously identified high-throughput (HTS) hit 1 (ESI-09). Structure–activity relationship (SAR) analysis led to the discovery of several more active EPAC antagonists (e.g., 22 (HJC0726), 35 (NY0123), and 47 (NY0173)) with low micromolar inhibitory activity. These inhibitors may serve as valuable pharmacological probes to facilitate our efforts in elucidating the biological functions of EPAC and developing potential novel therapeutics against human diseases. Our SAR results have also revealed that further modification at the 3-, 4-, and 5-positions of the phenyl ring as well as the 5-position of the isoxazole moiety may allow for the development of more potent EPAC antagonists.
Co-reporter:Haijun Chen, Yu Gao, Ailan Wang, Xiaobin Zhou, Yunquan Zheng, Jia Zhou
European Journal of Medicinal Chemistry 2015 Volume 92() pp:648-655
Publication Date(Web):6 March 2015
DOI:10.1016/j.ejmech.2015.01.031
•Recent developments on medicinal aspects of UA analogues were discussed.•Significant bioactive compounds as anticancer agents were documented.•Relevant SAR studies and potential mechanisms were summarized.•Future direction for further UA-based drug discovery was elaborated.Currently, there is a renewed interest in common dietaries and plant-based traditional medicines for the prevention and treatment of cancer. In the search for potential anticancer agents from natural sources, ursolic acid (UA), a pentacyclic triterpenoid widely found in various medicinal herbs and fruits, exhibits powerful biological effects including its attractive anticancer activity against various types of cancer cells. However, the limited solubility, rapid metabolism and poor bioavailability of UA restricted its further clinical applications. In the past decade, with substantial progress toward the development of new chemical entities for the treatment of cancer, numerous UA derivatives have been designed and prepared to overcome its disadvantages. Despite extensive effort, discovery of effective UA derivatives has so far met with only limited success. This review summarizes the current status of the structural diversity and evolution in medicinal chemistry of UA analogues and provides a detailed discussion of future direction for further research in the chemical modifications of UA.
Co-reporter:Haijun Chen ; Christopher Wild ; Xiaobin Zhou ; Na Ye ; Xiaodong Cheng
Journal of Medicinal Chemistry 2014 Volume 57(Issue 9) pp:3651-3665
Publication Date(Web):November 20, 2013
DOI:10.1021/jm401425e
3′,5′-Cyclic adenosine monophosphate (cAMP) is a pivotal second messenger that regulates numerous biological processes under physiological and pathological conditions, including cancer, diabetes, heart failure, inflammation, and neurological disorders. In the past, all effects of cAMP were initially believed to be mediated by protein kinase A (PKA) and cyclic nucleotide-regulated ion channels. Since the discovery of exchange proteins directly activated by cyclic adenosine 5′-monophosphate (EPACs) in 1998, accumulating evidence has demonstrated that the net cellular effects of cAMP are also regulated by EPAC. The pursuit of the biological functions of EPAC has benefited from the development and applications of a growing number of pharmacological probes targeting EPACs. In this review, we seek to provide a concise update on recent advances in the development of chemical entities including various membrane-permeable analogues of cAMP and newly discovered EPAC-specific ligands from high throughput assays and hit-to-lead optimizations.
Co-reporter:Na Ye ; Ye Ding ; Christopher Wild ; Qiang Shen
Journal of Medicinal Chemistry 2014 Volume 57(Issue 16) pp:6930-6948
Publication Date(Web):May 15, 2014
DOI:10.1021/jm5004733
Activator protein 1 (AP-1) is a pivotal transcription factor that regulates a wide range of cellular processes including proliferation, apoptosis, differentiation, survival, cell migration, and transformation. Accumulating evidence supports that AP-1 plays an important role in several severe disorders including cancer, fibrosis, and organ injury, as well as inflammatory disorders such as asthma, psoriasis, and rheumatoid arthritis. AP-1 has emerged as an actively pursued drug discovery target over the past decade. Excitingly, a selective AP-1 inhibitor T-5224 (51) has been investigated in phase II human clinical trials. Nevertheless, no effective AP-1 inhibitors have yet been approved for clinical use. Despite significant advances achieved in understanding AP-1 biology and function, as well as the identification of small molecules modulating AP-1 associated signaling pathways, medicinal chemistry efforts remain an urgent need to yield selective and efficacious AP-1 inhibitors as a viable therapeutic strategy for human diseases.
Co-reporter:Chunyong Ding, Lili Wang, Haijun Chen, Christopher Wild, Na Ye, Ye Ding, Tianzhi Wang, Mark A. White, Qiang Shen and Jia Zhou
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 42) pp:8442-8452
Publication Date(Web):02 Sep 2014
DOI:10.1039/C4OB01040J
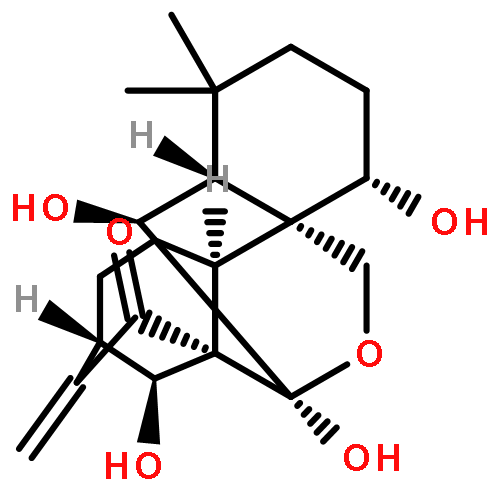
A mild and concise approach for the construction of a 3,4-dihydro-2H-pyran ring integrated into the A-ring of the natural product oridonin using an optimized inverse electron demand hetero-Diels–Alder (IED HDA) reaction is reported herein. A self-dimerization of the exocyclic enone installed in the A-ring through a homo-HDA reaction was identified to exclusively give a dimeric ent-kaurane diterpenoid with the spirochroman core. Moreover, efficient cross-HDA cycloadditions of this enone with various vinyl ethers or vinyl sulfides, instead of its own homo-HDA dimerization, were achieved in a regio- and stereoselective manner, thus providing access to novel dihydropyran-fused diterpenoids as potential anticancer agents to overcome chemoresistance.
Co-reporter:Haijun Chen, Zhengduo Yang, Chunyong Ding, Ailian Xiong, Christopher Wild, Lili Wang, Na Ye, Guoshuai Cai, Rudolfo M. Flores, Ye Ding, Qiang Shen, Jia Zhou
European Journal of Medicinal Chemistry 2014 Volume 82() pp:195-203
Publication Date(Web):23 July 2014
DOI:10.1016/j.ejmech.2014.05.049
•O-Alkylamino-tethered optimization strategy was utilized.•Novel diversified analogues based on HJC0149 have been designed and synthesized.•HJC0416 identified as a potent STAT3 inhibitor with an enhanced anticancer profile.•HJC0416 significantly suppressed breast cancer xenograft tumor growth in vivo.In a continuing effort to develop orally bioavailable small-molecule STAT3 inhibitors as potential therapeutic agents for human cancer, a series of novel diversified analogues based on our identified lead compound HJC0149 (1) (5-chloro-N-(1,1-dioxo-1H-1λ6-benzo[b]thiophen-6-yl)-2-hydroxybenzamide, Eur. J. Med. Chem. 2013, 62, 498–507) have been rationally designed, synthesized, and pharmacologically evaluated. Molecular docking studies and biological characterization supported our earlier findings that the O-alkylamino-tethered side chain on the hydroxyl group is an effective and essential structural determinant for improving biological activities and druglike properties of these molecules. Compounds with such modifications exhibited potent antiproliferative effects against breast and pancreatic cancer cell lines with IC50 values from low micromolar to nanomolar range. Among them, the newly discovered STAT3 inhibitor 12 (HJC0416) displayed an intriguing anticancer profile both in vitro and in vivo (i.p. & p.o.). More importantly, HJC0416 is an orally bioavailable anticancer agent as a promising candidate for further development.
Co-reporter:Haijun Chen, Guihua He, Cailong Li, Longrong Dong, Xiaobo Xie, Jianlei Wu, Yu Gao and Jia Zhou
RSC Advances 2014 vol. 4(Issue 85) pp:45151-45154
Publication Date(Web):10 Sep 2014
DOI:10.1039/C4RA08573F
A novel environment-friendly method to access bioactive oroxin A through a one-pot/two-step process from naturally abundant and inexpensive baicalin is described. The procedure presented here has several advantages including clean, one-pot, synthetic ease, and large-scale feasibility. This work also provides a model strategy for rapid and diverse access to natural molecules sharing the common skeleton of this family.
Co-reporter:Haijun Chen, Amy A. Mrazek, Xiaofu Wang, Chunyong Ding, Ye Ding, Laura J. Porro, Huiling Liu, Celia Chao, Mark R. Hellmich, Jia Zhou
Bioorganic & Medicinal Chemistry 2014 22(13) pp: 3393-3404
Publication Date(Web):
DOI:10.1016/j.bmc.2014.04.043
Co-reporter:Chunyong Ding, Yusong Zhang, Haijun Chen, Christopher Wild, Tianzhi Wang, Mark A. White, Qiang Shen, and Jia Zhou
Organic Letters 2013 Volume 15(Issue 14) pp:3718-3721
Publication Date(Web):July 8, 2013
DOI:10.1021/ol4015865
Efficient and concise synthetic approaches have been developed for the rapid and diverse installation of azide functionalities at the C-1, C-2, or C-3 positions of oridonin (1) with highly controlled regio- and stereoselectivity, while keeping key reactive pharmacophores intact by utilizing unique preactivation strategies based on the common synthon 4. Further functionalization of these azides through click chemistry yielding triazole derivatives successfully provides access to an expanded natural scaffold-based compound library for potential anticancer agents.
Co-reporter:Chunyong Ding ; Yusong Zhang ; Haijun Chen ; Zhengduo Yang ; Christopher Wild ; Lili Chu ; Huiling Liu ; Qiang Shen
Journal of Medicinal Chemistry 2013 Volume 56(Issue 12) pp:5048-5058
Publication Date(Web):June 7, 2013
DOI:10.1021/jm400367n
Oridonin (1), a complex ent-kaurane diterpenoid isolated from the traditional Chinese herb Isodon rubescens, has demonstrated great potential in the treatment of various human cancers due to its unique and safe anticancer pharmacological profile. Nevertheless, the clinical development of oridonin for cancer therapy has been hampered by its relatively moderate potency, limited aqueous solubility, and poor bioavailability. Herein, we report the concise synthesis of a series of novel nitrogen-enriched oridonin derivatives with thiazole-fused A-ring through an efficient protecting group-free synthetic strategy. Most of them, including compounds 7–11, 13, and 14, exhibited potent antiproliferative effects against breast, pancreatic, and prostate cancer cells with low micromolar to submicromolar IC50 values as well as markedly enhanced aqueous solubility. These new analogues obtained by rationally modifying the natural product have been demonstrated not only to significantly induce the apoptosis and suppress growth of triple-negative MDA-MB-231 breast cancer both in vitro and in vivo but also effective against drug-resistant ER-positive MCF-7 clones.
Co-reporter:Chunyong Ding ; Yusong Zhang ; Haijun Chen ; Zhengduo Yang ; Christopher Wild ; Na Ye ; Corbin D. Ester ; Ailian Xiong ; Mark A. White ; Qiang Shen
Journal of Medicinal Chemistry 2013 Volume 56(Issue 21) pp:8814-8825
Publication Date(Web):October 15, 2013
DOI:10.1021/jm401248x
Oridonin (1) has attracted considerable attention in recent years because of its unique and safe anticancer pharmacological profile. Nevertheless, it exhibits moderate to poor effects against highly aggressive cancers including triple-negative and drug-resistant breast cancer cells. Herein, we report the rational design and synthesis of novel dienone derivatives with an additional α,β-unsaturated ketone system diversely installed in the A-ring based on this class of natural scaffold that features dense functionalities and stereochemistry-rich frameworks. Efficient and regioselective enone construction strategies have been established. Meanwhile, a unique 3,7-rearrangement reaction was identified to furnish an unprecedented dienone scaffold. Intriguingly, these new analogues have been demonstrated to significantly induce apoptosis and inhibit colony formation with superior antitumor effects against aggressive and drug-resistant breast cancer cells in vitro and in vivo while also exhibiting comparable or lower toxicity to normal human mammary epithelial cells in comparison with 1.
Co-reporter:Haijun Chen ; Tamara Tsalkova ; Oleg G. Chepurny ; Fang C. Mei ; George G. Holz ; Xiaodong Cheng
Journal of Medicinal Chemistry 2013 Volume 56(Issue 3) pp:952-962
Publication Date(Web):January 3, 2013
DOI:10.1021/jm3014162
EPAC1 and EPAC2, two isoforms of exchange proteins directly activated by cAMP (EPAC), respond to the second messenger cAMP and regulate a wide variety of intracellular processes under physiological and pathophysiological circumstances. Herein, we report the chemical design, synthesis, and pharmacological characterization of three different scaffolds (diarylsulfones, N,N-diarylamines, and arylsulfonamides) as highly potent and selective antagonists of EPAC2. Several selective EPAC2 antagonists have been identified including 20i (HJC0350), which has an apparent IC50 of 0.3 μM for competing with 8-NBD-cAMP binding of EPAC2 and is about 133-fold more potent than cAMP. Compounds 1 (ESI-05), 14c (HJC0338), and 20i, selected from each series, have exhibited no inhibition of EPAC1-mediated Rap1-GDP exchange activity at 25 μM, indicating that they are EPAC2-specific antagonists. Moreover, live-cell imaging studies using EPAC1, EPAC2, or PKA FRET sensor also demonstrate that 20i functions as an EPAC2 specific antagonist.
Co-reporter:Haijun Chen, Zhengduo Yang, Chunyong Ding, Lili Chu, Yusong Zhang, Kristin Terry, Huiling Liu, Qiang Shen, Jia Zhou
European Journal of Medicinal Chemistry 2013 Volume 62() pp:498-507
Publication Date(Web):April 2013
DOI:10.1016/j.ejmech.2013.01.023
Fragment-based drug design (FBDD) is a promising approach for the generation of lead molecules with enhanced activity and especially drug-like properties against therapeutic targets. Herein, we report the fragment-based drug design, systematic chemical synthesis and pharmacological evaluation of novel scaffolds as potent anticancer agents by utilizing six privileged fragments from known STAT3 inhibitors. Several new molecules such as compounds 5, 12, and 19 that may act as advanced chemical leads have been identified. The most potent compound 5 (HJC0123) has demonstrated to inhibit STAT3 promoter activity, downregulate phosphorylation of STAT3, increase the expression of cleaved caspase-3, inhibit cell cycle progression and promote apoptosis in breast and pancreatic cancer cells with low micromolar to nanomolar IC50 values. Furthermore, compound 5 significantly suppressed estrogen receptor (ER)-negative breast cancer MDA-MB-231 xenograft tumor growth in vivo (p.o.), indicating its great potential as an efficacious and orally bioavailable drug candidate for human cancer therapy.Graphical abstractHighlights► First FBDD approach utilizing privileged fragments to target STAT3. ► Several novel scaffolds identified that may act as advanced chemical leads. ► Compound 5 (HJC0123) identified as a potent STAT3 inhibitor. ► The antiproliferative activity ranging from low micromolar to nanomolar IC50 values. ► HJC0123 significantly suppressed breast cancer xenograft tumor growth in vivo.
Co-reporter:Haijun Chen, Zhengduo Yang, Chunyong Ding, Lili Chu, Yusong Zhang, Kristin Terry, Huiling Liu, Qiang Shen, and Jia Zhou
ACS Medicinal Chemistry Letters 2013 Volume 4(Issue 2) pp:180
Publication Date(Web):January 15, 2013
DOI:10.1021/ml3003082
Niclosamide has been identified to potently inhibit the activation, nuclear translocation, and transactivation of STAT3. Nevertheless, the poor aqueous solubility and bioavailability of niclosamide have hindered its further clinical development for cancer therapy. To discover new molecules with enhanced druglike properties, a series of novel O-alkylamino-tethered derivatives of niclosamide have been designed, synthesized, and biologically evaluated. Among them, compound 11 (HJC0152) has been demonstrated to significantly suppress MDA-MB-231 xenograft tumor growth in vivo (ip and po), indicating its great potential as efficacious and orally bioavailable therapeutics for human cancer.Keywords: niclosamide derivatives; orally bioavailable; STAT3; water solubility
Co-reporter:Haijun Chen, Chunyong Ding, Christopher Wild, Huiling Liu, Tianzhi Wang, Mark A. White, Xiaodong Cheng, Jia Zhou
Tetrahedron Letters 2013 Volume 54(Issue 12) pp:1546-1549
Publication Date(Web):20 March 2013
DOI:10.1016/j.tetlet.2013.01.024
A concise and efficient synthetic approach to producing a novel non-cyclic nucleotide EPAC antagonist ESI-09 and its new analogs is reported. Key features of the synthesis include a mild and reliable one-pot procedure for an isoxazole synthon, as well as a modified one-pot protocol for the cyanomethyl ketone key intermediate. The synthesis requires inexpensive starting materials and only three linear steps for the completion in a total yield of 53%.
Co-reporter:Dr. Haijun Chen;Dr. Cheng Z. Wang;Dr. Chunyong Ding;Christopher Wild;Dr. Bryan Copits; Geoffrey T. Swanson; Kenneth M. Johnson; Jia Zhou
ChemMedChem 2013 Volume 8( Issue 2) pp:226-230
Publication Date(Web):
DOI:10.1002/cmdc.201200554
Co-reporter:Dr. Haijun Chen;Dr. Cheng Z. Wang;Dr. Chunyong Ding;Christopher Wild;Dr. Bryan Copits; Geoffrey T. Swanson; Kenneth M. Johnson; Jia Zhou
ChemMedChem 2013 Volume 8( Issue 2) pp:
Publication Date(Web):
DOI:10.1002/cmdc.201390004
Co-reporter:Chunyong Ding, Nicole M. Bremer, Thressa D. Smith, Patricia K. Seitz, Noelle C. Anastasio, Kathryn A. Cunningham, and Jia Zhou
ACS Chemical Neuroscience 2012 Volume 3(Issue 7) pp:538
Publication Date(Web):April 19, 2012
DOI:10.1021/cn300020x
Allosteric modulators of the serotonin (5-HT) 5-HT2C receptor (5-HT2CR) present a unique drug design strategy to augment the response to endogenous 5-HT in a site- and event-specific manner with great potential as novel central nervous system probes and therapeutics. To date, PNU-69176E is the only reported selective positive allosteric modulator for the 5-HT2CR. For the first time, an optimized synthetic route to readily access PNU-69176E (1) and its diastereomer 2 has been established in moderate to good overall yields over 10 steps starting from commercially available picolinic acid. This synthetic approach not only enables a feasible preparation of a sufficient amount of 1 for use as a reference compound for secondary pharmacological studies, but also provides an efficient synthesis of key intermediates to develop novel and simplified 5-HT2CR allosteric modulators. Compound 1 and its diastereomer 2 were functionally characterized in Chinese hamster ovary (CHO) cells stably transfected with the 5-HT2CR using an intracellular calcium (Cai2+) release assay. Compound 1 demonstrated efficacy and potency as an allosteric modulator for the 5-HT2CR with no intrinsic agonist activity. Compound 1 did not alter 5-HT-evoked Cai2+ in CHO cells stably transfected with the highly homologous 5-HT2AR. In contrast, the diastereomer 2 did not alter 5-HT-evoked Cai2+ release in 5-HT2AR-CHO or 5-HT2CR-CHO cells or exhibit intrinsic agonist activity.Keywords: 5-HT2C receptor; allosteric modulator; diastereomer; PNU-69176E; synthesis
Co-reporter:Haijun Chen, Tamara Tsalkova, Fang C. Mei, Yaohua Hu, Xiaodong Cheng, Jia Zhou
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 12) pp:4038-4043
Publication Date(Web):15 June 2012
DOI:10.1016/j.bmcl.2012.04.082
Exchange proteins directly activated by cAMP (Epac) are a family of guanine nucleotide exchange factors that regulate a wide variety of intracellular processes in response to second messenger cAMP. To explore the structural determinants for Epac antagonist properties of high throughput screening (HTS) hit ESI-08, pyrimidine 1, a series of 5-cyano-6-oxo-1,6-dihydro-pyrimidine analogues have been synthesized and evaluated for their activities for Epac inhibition. Structure–activity relationship (SAR) analysis led to the identification of three more potent Epac antagonists (6b, 6g, and 6h). These inhibitors may serve as valuable pharmacological probes for further elucidation of the physiological functions and mechanisms of Epac regulation. Our SAR results and molecular docking studies have also revealed that further optimization of the moieties at the C-6 position of pyrimidine scaffold may allow us to discover more potent Epac-specific antagonists.
Co-reporter:Zhiqing Liu, Christopher Wild, Ye Ding, Na Ye, ... Jia Zhou
Drug Discovery Today (June 2016) Volume 21(Issue 6) pp:989-996
Publication Date(Web):1 June 2016
DOI:10.1016/j.drudis.2015.11.008
•No Bcl-2 inhibitors targeting the BH3 domain approved for clinical use.•BH4 domain of Bcl-2 is identified as a promising novel target for cancer therapy.•Bcl-2 BH4 domain interacts with Bcl-2 family members as well as non-Bcl-2 proteins.•Interactions of BH4 domain with various proteins promote resistance to apoptosis.•Targeting the BH4 domain of Bcl-2 offers great potential to overcome drug resistance.Overexpression of B cell lymphoma 2 (Bcl-2) proteins is associated with therapy resistance in various human cancers. Traditional approaches target the Bcl-2 homology (BH)3 domain of Bcl-2; however, the BH4 domain represents a superior therapeutic target in light of its unique structure and crucial involvement in many cellular functions. In this critical review, we focus on the structural and functional basis of targeting the BH4 domain of Bcl-2, and highlight the recent advances in drug discovery efforts toward small-molecule BH4 domain inhibitors (e.g. BDA-366). The proof-of-concept studies support the hypothesis that targeting the BH4 domain of Bcl-2 holds promise to offer a novel anticancer therapy through the induction of apoptosis and an increased potential to overcome therapeutic resistance.
Co-reporter:Haijun Chen, Xiaobin Zhou, Ailan Wang, Yunquan Zheng, ... Jia Zhou
Drug Discovery Today (January 2015) Volume 20(Issue 1) pp:105-113
Publication Date(Web):1 January 2015
DOI:10.1016/j.drudis.2014.09.015
•The higher investment on HTS did not result in enhanced productivity of new drugs.•FBDD emerged as an attractive strategy in drug discovery over the past two decades.•Construction of fragment library is the critical step for FBDD.•The pros and cons of various fragment-based screening are discussed.•Deconstruction-reconstruction approach offers promise in current drug discovery.Recent advances in the understanding of molecular recognition and protein–ligand interactions have facilitated rapid development of potent and selective ligands for therapeutically relevant targets. Over the past two decades, a variety of useful approaches and emerging techniques have been developed to promote the identification and optimization of leads that have high potential for generating new therapeutic agents. Intriguingly, the innovation of a fragment-based drug design (FBDD) approach has enabled rapid and efficient progress in drug discovery. In this critical review, we focus on the construction of fragment libraries and the advantages and disadvantages of various fragment-based screening (FBS) for constructing such libraries. We also highlight the deconstruction–reconstruction strategy by utilizing privileged fragments of reported ligands.
Co-reporter:Chunyong Ding, Lili Wang, Haijun Chen, Christopher Wild, Na Ye, Ye Ding, Tianzhi Wang, Mark A. White, Qiang Shen and Jia Zhou
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 42) pp:NaN8452-8452
Publication Date(Web):2014/09/02
DOI:10.1039/C4OB01040J
A mild and concise approach for the construction of a 3,4-dihydro-2H-pyran ring integrated into the A-ring of the natural product oridonin using an optimized inverse electron demand hetero-Diels–Alder (IED HDA) reaction is reported herein. A self-dimerization of the exocyclic enone installed in the A-ring through a homo-HDA reaction was identified to exclusively give a dimeric ent-kaurane diterpenoid with the spirochroman core. Moreover, efficient cross-HDA cycloadditions of this enone with various vinyl ethers or vinyl sulfides, instead of its own homo-HDA dimerization, were achieved in a regio- and stereoselective manner, thus providing access to novel dihydropyran-fused diterpenoids as potential anticancer agents to overcome chemoresistance.









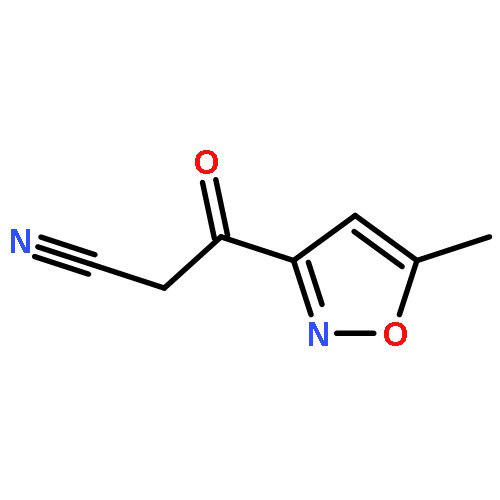
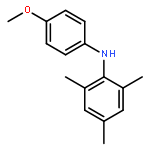
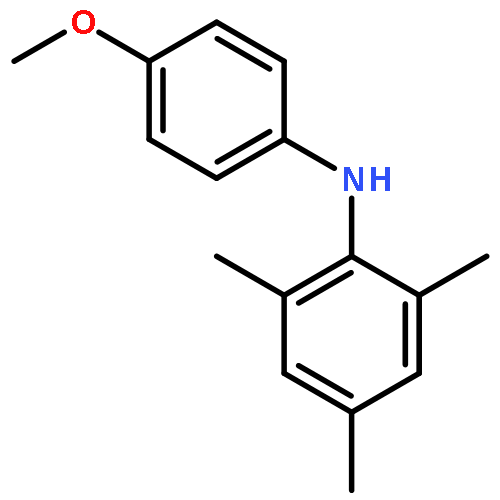


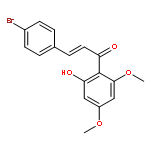





![2-Propen-1-one,1-(2-hydroxy-4,6-dimethoxyphenyl)-3-[4-(2-propenyloxy)phenyl]-](http://img.cochemist.com/ccimg/76600/76554-28-4.png)
![2-Propen-1-one,1-(2-hydroxy-4,6-dimethoxyphenyl)-3-[4-(2-propenyloxy)phenyl]-](http://img.cochemist.com/ccimg/76600/76554-28-4_b.png)
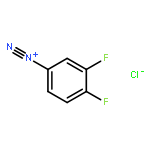
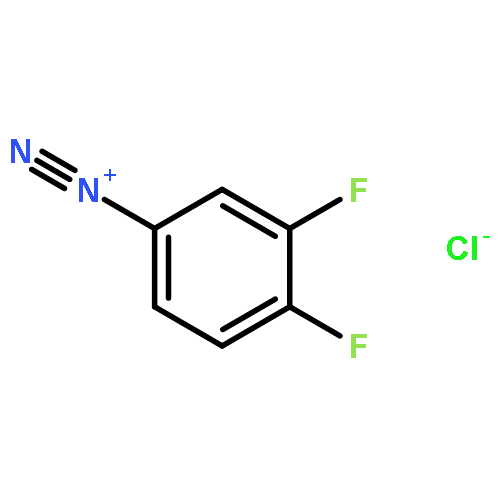

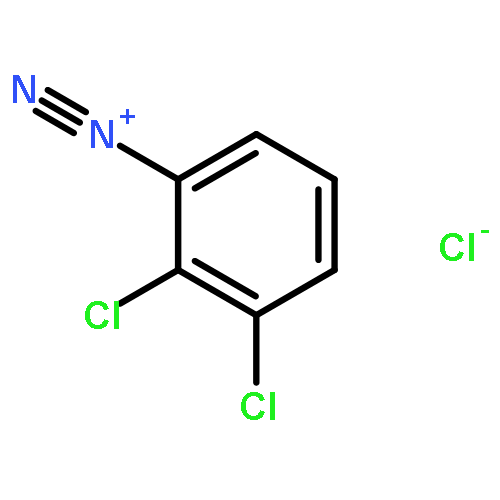
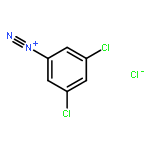



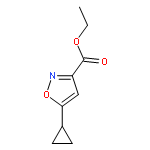
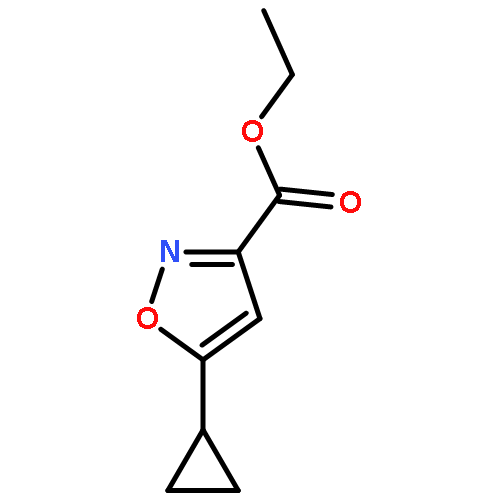
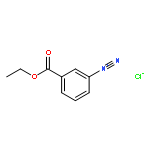
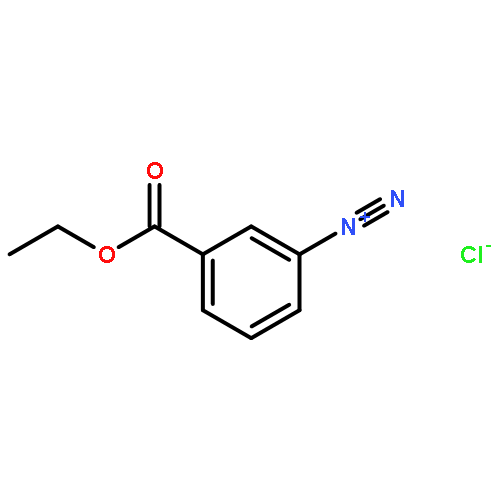


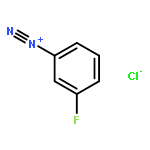
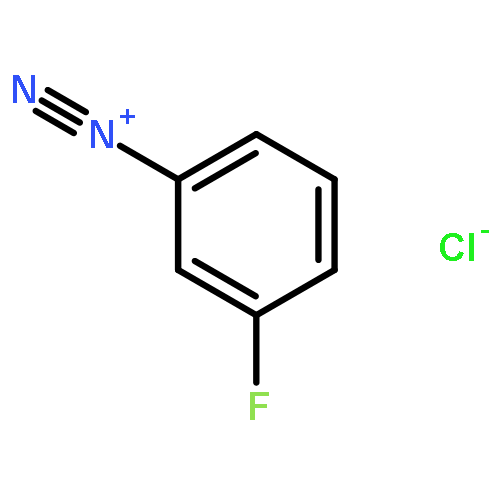
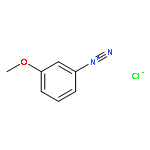

![8-Azabicyclo[3.2.1]octane-2-carboxylicacid, 3-(benzoyloxy)-, methyl ester, (1R,2R,3S,5S)-](http://img.cochemist.com/ccimg/18800/18717-72-1.png)
![8-Azabicyclo[3.2.1]octane-2-carboxylicacid, 3-(benzoyloxy)-, methyl ester, (1R,2R,3S,5S)-](http://img.cochemist.com/ccimg/18800/18717-72-1_b.png)


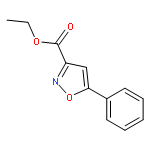
![Benzene,1,3,5-trimethyl-2-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/5200/5184-64-5.png)
![Benzene,1,3,5-trimethyl-2-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/5200/5184-64-5_b.png)


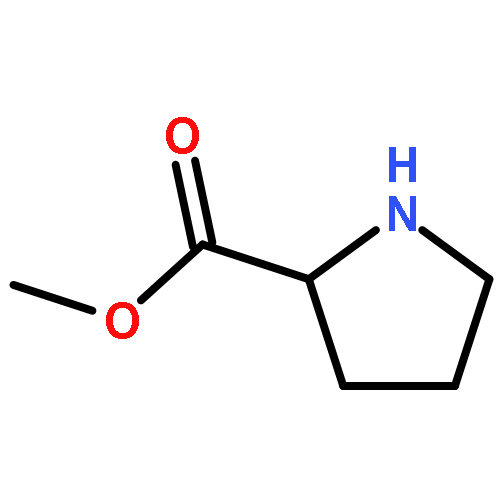

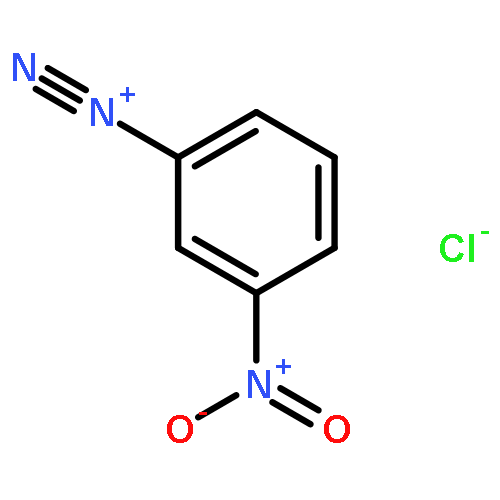


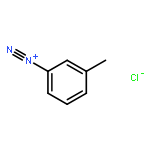
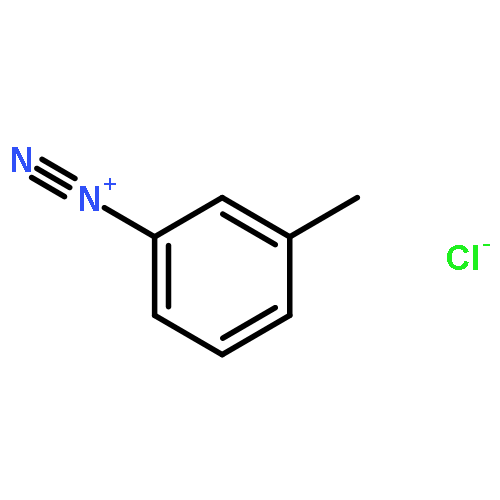
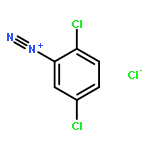
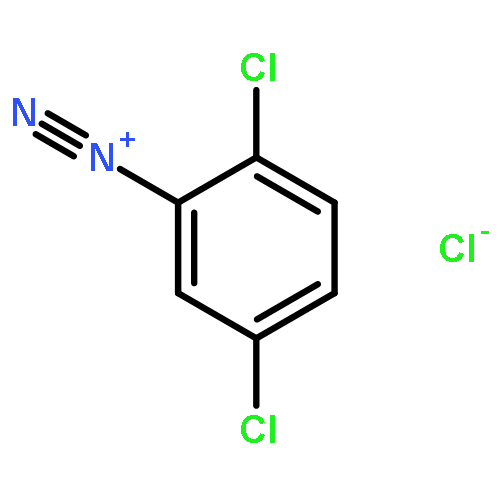



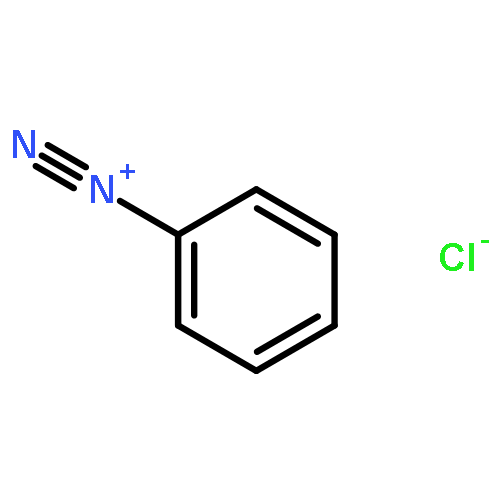
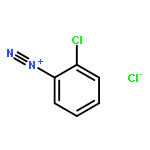


![8-Azabicyclo[3.2.1]octane-2-carboxylicacid, 3-(benzoyloxy)-8-methyl-, methyl ester, hydrochloride (1:1),(1R,2R,3S,5S)-](http://img.cochemist.com/ccimg/100/53-21-4.png)
![8-Azabicyclo[3.2.1]octane-2-carboxylicacid, 3-(benzoyloxy)-8-methyl-, methyl ester, hydrochloride (1:1),(1R,2R,3S,5S)-](http://img.cochemist.com/ccimg/100/53-21-4_b.png)
![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2.png)
![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2_b.png)
![3-?Isoxazolepropanenitr?ile, α-?[2-?(3-?chlorophenyl)?hydrazinylidene]?-?5-?(1,?1-?dimethylethyl)?-?β-?oxo-](http://img.cochemist.com/ccimg/263800/263707-16-0.png)
![3-?Isoxazolepropanenitr?ile, α-?[2-?(3-?chlorophenyl)?hydrazinylidene]?-?5-?(1,?1-?dimethylethyl)?-?β-?oxo-](http://img.cochemist.com/ccimg/263800/263707-16-0_b.png)