Co-reporter:Feng-Wei Gao, Rong-Lin Zhong, Hong-Liang Xu, and Zhong-Min Su
The Journal of Physical Chemistry C November 16, 2017 Volume 121(Issue 45) pp:25472-25472
Publication Date(Web):October 30, 2017
DOI:10.1021/acs.jpcc.7b08172
Based on s-indaceno[1,2,3-cd;5,6,7-c′d′]diphenalene (1) consisting of two phenalenyl moieties, the monomer 2 and its dimer 22 are designed by boron and nitrogen atoms substituting the central carbon atoms of phenalenyl moieties. Calculated energy decompose analysis (EDA) shows that the orbital interaction for 22 possesses a large attractive contribution of −18.31 kcal mol–1, which is dominated by the π–π stacking interaction between the upper and the lower π-conjugated units. Interestingly, the natural population analysis (NPA) charge and the transition density matrix (TDM) show that both intramolecular charge transfer and intermolecular charge transfer (CT) exist in 22. Further, the first hyperpolarizability (βtot = 4.56 × 104 au) of 2 with intramolecular CT is greatly larger than that of reported molecule 3 (5.45 × 103 au) with intermolecular CT. Significantly, 22 exhibits the largest βtot value to be 1.42 × 105 au, which is caused by combining the intra- and intermolecular CT transitions (βx = 1.40 × 105 au and βz = 2.27 × 104 au). Correspondingly, highest occupied molecular orbital (HOMO) → lowest unoccupied molecular orbital (LUMO) (intramolecular CT) in the low-energy electronic transition of 22 is 68%, while HOMO → LUMO + 1 (intermolecular CT) is 18%, which demonstrates that the intramolecular CT effect on the βtot value is stronger than the case of the intermolecular CT effect. The present work might provide rich insight into designing and developing potential second-order optical nonlinearity materials with inter- and intramolecular CT characters.
Co-reporter:Linlin Sun, Ting Zhang, Bo Zhu, Caixia Wu, Likai Yan, Zhongmin Su
Dyes and Pigments 2017 Volume 137() pp:372-377
Publication Date(Web):February 2017
DOI:10.1016/j.dyepig.2016.10.035
•The introduction of POM leads to red shift and broadened absorption peak.•The energy gaps for systems 4–6 are smaller than those of systems 1–3.•Thiophene as π linker in system 5 has the best performance.A series of hexamolybdate-based organic-inorganic hybrids containing double donor-π-linker-acceptor chains were studied by using density functional theory and time-dependent density functional theory methods. The energy levels of frontier molecular orbitals, absorption spectra, electronic transition characters and photovoltaic parameters of dyes 1–6 were systematically evaluated. The results show that the absorption spectra are broadened and red-shifted by introducing hexamolybdate and different π-linkers. The absorption spectrum of dye 5 (thiophene as π-linker) has three strong and broad absorption bands in 300–750 nm. The hole injection energy, dye regeneration energy and charge recombination energy of dye 5 are smaller than others, which are advantageous to the hole injection and dye regeneration. The introduction of thiophene (dye 5) in this study is the most effective strategy to improve the absorption and increase the efficiency of dye in dye-sensitized solar cell (DSSC).A theoretical design and study on hexamolybdate-based organic-inorganic hybrids with double D-π-A chains for high performance p-type dye-sensitized solar cells.
Co-reporter:Minghao Qin, Dan Jin, Weilong Che, Yang Jiang, Liping Zhang, Dongxia Zhu, Zhongmin Su
Inorganic Chemistry Communications 2017 Volume 75() pp:25-28
Publication Date(Web):January 2017
DOI:10.1016/j.inoche.2016.11.013
•Simple, highly selective and sensitive fluorescence probe for Cu2 + in aqueous medium: 2-(2-hydroxyphenyl)benzimidazole (L1).•The probe can be applied to the monitoring of Cu2 + with wide pH range from 6 to 11.•It is also demonstrated that L1 can be used in biological imaging for the detection of Cu2 + in living cells.We present a simple but highly selective and sensitive fluorescence probe for Cu2 + in aqueous medium: 2-(2-hydroxyphenyl)benzimidazole (L1). The probe can be applied to the monitoring of Cu2 + with wide pH range from 6 to 11 and used in biological imaging for the detection of Cu2 + in living cells.
Co-reporter:Feng-Wei Gao, Rong-Lin Zhong, Hong-Liang XuZhong-Min Su
The Journal of Physical Chemistry C 2017 Volume 121(Issue 7) pp:
Publication Date(Web):February 7, 2017
DOI:10.1021/acs.jpcc.6b11732
Phenalenyl π-dimer (PLY2) has recently attracted intensive research interest due to its unique structure and binding characteristics (two-electron/12-center bonding). The directional transfer of electron or electron pair under the external electric field can produce a new structure with interesting properties. In the present work, we investigate for the first time the effect of the external electric field along the main molecule axis on PLY2. Two unpaired electrons between two layers are gradually shifted to the upper layer with increasing of the external electric field strength (Fext): the weaker the two-electron/12-center bonding, the stronger the electrostatic interaction between two layers. Significantly, a small increment of Fext makes a big difference: the interlayer distance in the PLY2 is sharply elongated from 3.241 Å (Fext = 203 × 10–4 au) to 3.485 Å (Fext = 204 × 10–4 au), which leads to the two-electron/12-center bonding breaking at 204 × 10–4 au. Therefore, the Fext = 204 × 10–4 au is regarded as the critical electric field. In this case, the interaction between two layers in PLY2 is exclusively governed by the electrostatic interaction. Besides this, the effect of the external electric field brings some distinctive changes in its diradical character (y0), the Wiberg bond index (WBI), the interaction energy (Eint), and the frontier molecular orbital (FMO) that can be used to explore the conversion between bonding and electrostatic interactions. This study can deepen the understanding for the effect of the external electric field on structures and electric properties for molecule and be an open a door for the discovery and development of new switching devices.
Co-reporter:Rui-Xin Bian, Xiao-Tong Wu, Fang Chai, Lu Li, Ling-Yu Zhang, Ting-Ting Wang, Chun-Gang Wang, Zhong-Min Su
Sensors and Actuators B: Chemical 2017 Volume 241() pp:592-600
Publication Date(Web):31 March 2017
DOI:10.1016/j.snb.2016.10.120
•Au nanoclusters-based test papers are prepared for Hg2+ and Pb2+ detection.•They are used based on aggregation-induced fluorescence quenching and enhancement.•Recycle of the portable test papers are also investigated.•Test paper was competent for a practical application in environmental water samples.A simple, portable and recyclable method has been developed for on-site and real-time detection of Hg2+ and Pb2+ by naked eyes by using Au nanoclusters-based test paper as the fluorescent sensor. The high specificity of Hg2+ and Pb2+ to Au nanoclusters provided excellent selectivity over 15 kinds of other ions. Interestingly, an apparent fluorescent quenching of the test paper was shown when exposed to Hg2+, while a significant fluorescent enhancement was observed in the presence of Pb2+, which could be attributed to aggregation-induced fluorescence quenching and enhancement. The limit of detection for Hg2+ and Pb2+ by naked eyes was 5 μM and 50 μM, respectively. More importantly, the cyclic utilization of the test paper was achieved by immersing the test papers into ethylenediamine tetraacetic acid to recover the fluorescence of the test paper, which contributed to reducing resource consumption. Otherwise, the GSH-AuNCs test paper was competent for a practical application in environmental water samples with strong anti-interference ability.Glutathione coated Au nanoclusters-based test papers were fabricated for visual detection of Hg2+ through aggregation-induced fluorescence quenching and Pb2+ through aggregation-induced fluorescence enhancement. The activity of test papers could be recovered by ethylenediamine tetraacetic acid after detection of Hg2+ and Pb2+ so that these can be reused for additional rounds.Figure optionsDownload full-size imageDownload high-quality image (89 K)Download as PowerPoint slide
Co-reporter:Yang Jiang;Guangfu Li;Weilong Che;Yingjie Liu;Bin Xu;Guogang Shan;Dongxia Zhu;Zhongmin Su;Martin R. Bryce
Chemical Communications 2017 vol. 53(Issue 21) pp:3022-3025
Publication Date(Web):2017/03/09
DOI:10.1039/C7CC00769H
A neutral dinuclear Ir(III) Schiff base complex PIBIP has been synthesized and shown to exhibit both piezochromic luminescence (PCL) and aggregation induced emission (AIE) behaviour. An efficient second-level anti-counterfeit trademark and a data encryption device were fabricated using PIBIP as the active material.
Co-reporter:Xiao-Xin Li;Feng-Cui Shen;Jiang Liu;Shun-Li Li;Long-Zhang Dong;Qiang Fu;Ya-Qian Lan
Chemical Communications 2017 vol. 53(Issue 72) pp:10054-10057
Publication Date(Web):2017/09/05
DOI:10.1039/C7CC05552H
A novel polyoxometalate-based metal–organic framework (POMOF) with an ABW network, NENU-601, was synthesized in situ. To the best of our knowledge, this is the first POMOF with a zeolite-like structure, which was designed by regulating the length and angle of mixed ligands and rationally choosing suitable Polyoxometalates (POMs) as nodes.
Co-reporter:Bai-Qiao Song;Da-Qin Chen;Zhenguo Ji;Junhong Tang;Xin-Long Wang;Hong-Ying Zang
Chemical Communications 2017 vol. 53(Issue 11) pp:1892-1895
Publication Date(Web):2017/02/02
DOI:10.1039/C6CC07729C
We have demonstrated for the first time that isostructural homochiral metal–organic frameworks (MOFs) can be synthesized directly from chiral ligands and indirectly from achiral ligands via spontaneous resolution combined with cooperative chirality induction, respectively. Moreover, the usage of different ligands leads to distinct proton conduction behaviors.
Co-reporter:Liu Yang;Li Cao;Xiao Li;Chao Qin;Liang Zhao;Kui-Zhan Shao
Dalton Transactions 2017 vol. 46(Issue 23) pp:7567-7576
Publication Date(Web):2017/06/13
DOI:10.1039/C7DT01306J
Four metal–organic frameworks, namely [Cd2(tib)(btb)(H2O)2]·NO3·2.5DMF (1), [Cd(tib)(H2dhbqdc)0.5(NO3)]·6H2O (2), [Co(tib)(1,4-ndc)]·2DMF (3), and [Cu3(tib)2(2,6-ndc)2(H2O)2]·2NO3·2H2O (4), were synthesized based on 1,3,5-tris(1-imidazolyl)benzene and diverse carboxylic acid ligands. They have been characterized by elemental analysis, infrared spectroscopy (IR), powder X-ray diffraction (PXRD), thermogravimetric analyses (TGA) and single crystal X-ray diffraction. Compound 1 is a 3D framework constructed from a binuclear Cd cluster with (3,3,6)-connected (63)2(69·86) topology. Compound 2 exhibits a 2D wavy layered structure with (3,4)-connected topology, and compound 3 displays a two-fold interpenetrating network with (3,5)-connected topology. Compound 4 can be regarded as a three-fold interpenetrating framework. Moreover, compounds 1 and 2 can be used as fluorescent sensors sensing small molecules with high selectivity. In this context, we selected the typical toxic explosives, TNP, namely 2,4,6-trinitrophenol and NB, namely nitrobenzene, as examples to investigate the properties of sensing. Furthermore, the magnetic properties of compounds 3 and 4 are investigated.
Co-reporter:Peng Huang;Xiao-Jun Wang;Jiao-Jiao Qi;Xin-Long Wang;Min Huang;Hai-Yang Wu;Chao Qin
Journal of Materials Chemistry A 2017 vol. 5(Issue 44) pp:22970-22974
Publication Date(Web):2017/11/14
DOI:10.1039/C7TA06822K
Two nanoscale polytantalotungstates (POTTs), Cs5K4{Cr3[Ta3P2W15O62]2(H2O)12}·15H2O (1) and Cs8.5K8Na2H5.5{Cr4[Ta3P2W15O62]4(H2O)12}·53H2O (2), based on unprecedented {Cr3Ta6} and {Cr4Ta12} clusters, respectively, were hydrothermally synthesized. A photocatalytic study revealed that 1 and 2 exhibit significant photocatalytic water splitting activities.
Co-reporter:Li-Li Wen;Xue-Gang Hou;Guo-Gang Shan;Wei-Lin Song;Shu-Ran Zhang;Hai-Zhu Sun
Journal of Materials Chemistry C 2017 vol. 5(Issue 41) pp:10847-10854
Publication Date(Web):2017/10/26
DOI:10.1039/C7TC03535G
Development of appropriate luminophores that can achieve effective sensing of nitroaromatic explosives is a crucial issue for our daily life safety and homeland security. Herein, supersensitive and highly selective detection of one nitroaromatic explosive, i.e. 2,4,6-trinitrophenol (TNP) in aqueous media, by taking advantage of rationally designed aggregation-induced emission (AIE) cationic Ir(III) phosphors with carbazole end-capped flexible ligands, is successfully realized. To decipher a detailed sensing mechanism and provide an alternative strategy for the molecular design in future, systematic experimental and theoretical investigations are performed. Comprehensive studies demonstrate that both electron and energy transfer, strong electrostatic interactions between TNP and cationic Ir(III) complexes, as well as specific intraligand charge transfer excited-state character are responsible for the high sensitivity and selectivity. We believe that the obtained results will pave a feasible avenue to construct novel phosphorescent materials that could be used for potentially efficient detection of TNP.
Co-reporter:Hang Yin, Yun Geng, Guang-Yan SunZhong-Min Su
The Journal of Physical Chemistry C 2017 Volume 121(Issue 4) pp:
Publication Date(Web):January 13, 2017
DOI:10.1021/acs.jpcc.6b12174
The intermolecular stacking and crystallization of perylene diimides (PDIs) has become research obstacles for small molecule acceptors (SMAs). For breaking molecular rigidity and planarity, it is an executable way to increase the distortion between two PDI units. A class of PDI dimers were designed via bridging different linkers in bay positions (1–1′ bridge) and headland positions (1–2′ bridge) to screen suitable acceptor materials for organic photovoltaic cells (OPVs). Density functional theory (DFT) and time-dependent density functional theory (TD-DFT) calculations were performed to investigate their electronic structures, open circuit voltage (VOC), driving forces (ΔEL-L), and some major parameters related to the short-circuit current density (JSC) such as absorption spectrum and carrier transport ability. Meanwhile, the intermolecular charge transfer (inter-CT) and charge recombination (inter-CR) rates were calculated for a further analysis on charge transfer properties at donor/acceptor (D/A) interface by employing the Marcus semiclassical model. The results manifest that the investigated 1–2′ bridged molecules possess low-lying LUMO energy levels, relatively bigger ΔEL-L, bathochromic-shifted absorption, as well as the strongest maximum absorption and more effective charge transport than 1–1′ bridged molecules. Surprisingly, compared with P3HT/(1–1′ bridged PDI dimers) interface, almost constant reorganization energy (λ), higher Gibbs free energy change of exciton dissociation (ΔGCT), and considerable inter-CT/inter-CR rates ratios (kinter-CT/kinter-CR) of P3HT/(1–2′ bridged PDI dimers) provides further evidence for that 1–2′ bridged PDI dimers as acceptors might perform higher efficiency in OPV device. Moreover, constructing NDT and DPPT as bridged linkers in PDI dimers as “push–pull” structures may rationally expect more favorable properties as acceptors in OPVs, which might provide theoretical guideline for the design and synthesis of new organic SMAs.
Co-reporter:Liu Yang;Xiao Li;Chun-Yi Sun;Han Wu;Chun-Gang Wang
New Journal of Chemistry (1998-Present) 2017 vol. 41(Issue 9) pp:3661-3666
Publication Date(Web):2017/05/02
DOI:10.1039/C7NJ00389G
By using a new N-donor ligand, a 3D stable pillared-layer metal–organic framework, namely [Cu4(1,4-ndc)4(L)2]·H2O·2DMF (1), has been synthesized under solvothermal conditions (1,4-H2ndc = 1,4-naphthalenedicarboxylate; L = 1-(1-(1H-imidazol-1-yl)naphthalen-4-yl)-1H-imidazole). X-ray single crystal diffraction analysis was utilized to obtain the crystal structure of 1 and 1 was further characterized by elemental analysis, powder X-ray diffraction (PXRD), thermogravimetric analysis (TGA) and infrared spectroscopy (IR). Based on dinuclear Cu(II) clusters as nodes, compound 1 is a 6-connected two-fold interpenetrating pcu network. Moreover, using rhodamine B (RB) and methylene blue (MB) as examples, 1 has the ability to separate dye molecules. Finally, the magnetic properties of 1 have been investigated.
Co-reporter:Liu Yang;Xiao Li;Chun-Yi Sun;Han Wu;Chun-Gang Wang
New Journal of Chemistry (1998-Present) 2017 vol. 41(Issue 9) pp:3661-3666
Publication Date(Web):2017/05/02
DOI:10.1039/C7NJ00389G
By using a new N-donor ligand, a 3D stable pillared-layer metal–organic framework, namely [Cu4(1,4-ndc)4(L)2]·H2O·2DMF (1), has been synthesized under solvothermal conditions (1,4-H2ndc = 1,4-naphthalenedicarboxylate; L = 1-(1-(1H-imidazol-1-yl)naphthalen-4-yl)-1H-imidazole). X-ray single crystal diffraction analysis was utilized to obtain the crystal structure of 1 and 1 was further characterized by elemental analysis, powder X-ray diffraction (PXRD), thermogravimetric analysis (TGA) and infrared spectroscopy (IR). Based on dinuclear Cu(II) clusters as nodes, compound 1 is a 6-connected two-fold interpenetrating pcu network. Moreover, using rhodamine B (RB) and methylene blue (MB) as examples, 1 has the ability to separate dye molecules. Finally, the magnetic properties of 1 have been investigated.
Co-reporter:Feng-Wei Gao;Hong-liang Xu
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 47) pp:31958-31964
Publication Date(Web):2017/12/06
DOI:10.1039/C7CP06412H
In this work, we applied an external electric field (F) to a biphenalenyl derivative (BN-PLY2) in the direction of the negative z-axis (F−z) and the positive z-axis (F+z), respectively. The influence of the two directions of F on the molecular structures and electronic properties is investigated, which gives interesting results. Density functional theory (DFT) calculations show that the application of F−z (F−z = 0 to −190 × 10−4) is an advantage toward improving π-dimer stability, which is attributed to an increase in bonding and attractive electrostatic interactions. Interestingly, a large amount of negative charge is induced by applying F−z to the upper layer, resulting in an increase in the electron density in the upper layer, which is the main factor for the formation of a symmetric highest occupied molecular orbital (HOMO) at F−z = −180 × 10−4 au (−9.26 × 109 V m−1). Moreover, when F+z is applied, the HOMO and HOMO−1 undergo orbital interchange in the π-dimer at F+z = 100/110 × 10−4 au. Significantly, the effect of the external electric field effectively regulates the first hyperpolarizabilities (βtot). When the F+z ranges from 0 to 140 × 10−4 au, the βtot values slightly decrease to 0 au. Note that, upon increasing F+z, the βtot values sharply increase to 6.67 × 103 au (F+z = 190 × 10−4 au). Furthermore, the evolutions of the absorption spectra under F might well explain the trend of βtot values. When the F+z ranges from 0 to 140 × 10−4 au, the broad absorption spikes with a low-energy are significantly blue-shifted, while only absorption spikes with a high-energy are significantly red-shifted (F+z = 140 to 190 × 10−4 au). The present work not only provides a deeper understanding of the relationships between the molecular structure and the electronic properties of a π-dimer system, but can also be developed for designing highly efficient nonlinear optical materials through the influence of an external electric field.
Co-reporter:Pingxiao Liu, Lingbao Xing, Hongtao Lin, Haining Wang, ... Zhongmin Su
Science Bulletin 2017 Volume 62, Issue 13(Volume 62, Issue 13) pp:
Publication Date(Web):15 July 2017
DOI:10.1016/j.scib.2017.05.031
In the present work, a novel porous, and chemically stable amine-based covalent organic polymer (POP-1) was designed and synthesized under solvothermal conditions. The porosity, crystallinity, chemical stability, electrochemical properties, and diffuse reflectance of POP-1 were investigated via N2 sorption experiment, power X-ray diffraction, thermogravimetric analysis, cyclic voltammetry, and ultraviolet visible near infrared spectrometry, respectively. POP-1 exhibits good chemical stability in both acidic and alkaline aqueous solutions, as well as in organic solvents. Undoped POP-1 can be directly used as a photocatalyst for rhodamine B irradiation degradation under light-emitting diode and natural light. The Ea of POP-1 for RhB degradation is 82.37 kJ/mol. Furthermore, POP-1 can be reused as a catalyst in RhB degradation without degraded catalytic activity.Download high-res image (153KB)Download full-size image
Co-reporter:Peng Huang;Xin-Long Wang;De-Qing He;Hai-Yang Wu;Chao Qin;Meng Du;Chao Lai
Dalton Transactions 2017 vol. 46(Issue 39) pp:13345-13348
Publication Date(Web):2017/10/10
DOI:10.1039/C7DT02737K
Two novel mixed-addenda Nb/W polyoxometalates are successfully fabricated, and investigated as anode materials for lithium-ion batteries. For the Cs5K4{Cr3(H2O)12[P2W15Nb3O62]2}·60H2O sample, a reversible capacity (466.6 mA h g−1) and stable cycle performance are obtained.
Co-reporter:Yuan-Yuan Wang;Mi Zhang;Shun-Li Li;Shu-Ran Zhang;Wei Xie;Jun-Sheng Qin;Ya-Qian Lan
Chemical Communications 2017 vol. 53(Issue 37) pp:5204-5207
Publication Date(Web):2017/05/04
DOI:10.1039/C6CC10208E
Two novel isostructural polyoxometalate (POM)-based metal–organic frameworks (MOFs) with diamond topology, NENU-506 and NENU-507, were hydrothermally synthesized. They not only combine the advantages of both POMs and MOFs, but also show excellent chemical and thermal stability. Notably, NENU-507 exhibited a high reversible capacity of 640 mA h g−1 after 100 cycles when applied as an anode material in lithium-ion batteries.
Co-reporter:Yan-Qing Jiao;Meng-Ting Li;Chao Qin
CrystEngComm (1999-Present) 2017 vol. 19(Issue 13) pp:1721-1724
Publication Date(Web):2017/03/27
DOI:10.1039/C6CE02420C
The first mixed-valence nickel-supported polyoxometalate-based metal–organic framework, (H2mim)[NiIII(Hmim)4][SiW11NiIIO37(OH)2(Hmim)]·3H2O (1) (Hmim = 2-methylimidazole), has been synthesized using a steam-assisted conversion method, and is constructed from noncovalent interactions (hydrogen bonds and π⋯π stacking interactions). 1 has a repeating honeycomb-shaped internal structure with one-dimensional channels along the direction of the c axis, where the diameter of the aperture is as large as 18.83 Å. Furthermore, the stability and the optical band gaps are investigated.
Co-reporter:Ying-Chen Duan;Yong Wu;Xin-Yao Ren;Liang Zhao;Yun Geng;Min Zhang;Guang-Yan Sun
Dalton Transactions 2017 vol. 46(Issue 34) pp:11491-11502
Publication Date(Web):2017/08/29
DOI:10.1039/C7DT02684F
Two reported Ir(III) complexes 1a and 1b containing oxazoline and imidazoline in ancillary ligand, respectively, were investigated by DFT/TD-DFT method. In order to obtain full-color display materials, we designed a group of Ir(III) complexes 2a–3d based on 1a, which exhibits higher quantum efficiency in phosphorescence, by introducing electron-donating/electron-withdrawing moieties to different positions of the ancillary ligand to adjust emission color. In addition to calculating the radiation rate and analyzing its determining factors, we also estimated nonradiative ability by evaluating the spin–orbit coupling matrix element between the ground state (S0) and the lowest triplet state (T1) as well as the reorganization energy from T1 to S0 to estimate quantum efficiency more accurately. In particular, an in-depth analysis on the contribution of each vibration mode to reorganization energy helped us to identify the effect of substituents on the nonradiative process. Besides, charge injection/transfer properties and energy relation of the states related to exciton quenching via the triplet metal-centered state were also examined, which provide an estimation on the OLED performance of our designed complexes. Overall, we expect 2b and 3c to be more efficient blue-emitting emitters than 1a and 3a and 3b to be efficient green and red emitters, respectively.
Co-reporter:Peng Huang;Hai-Yang Wu;Min Huang;Meng Du;Chao Qin;Xin-Long Wang
Dalton Transactions 2017 vol. 46(Issue 31) pp:10177-10180
Publication Date(Web):2017/08/08
DOI:10.1039/C7DT01890H
An unprecedented polytantalotungstate (POTT), Cs12.5K4.5H[Ta12Si4W37O158]·25H2O (1), based on the {SiW9Ta3O40}7− cluster was hydrothermally synthesized. A photocatalytic study revealed that 1 exhibits significant photocatalytic water splitting activity.
Co-reporter:Mei-Jie Wei;Jia-Qi Fu;Yi-Di Wang;Yi Zhang;Hong-Ying Zang;Kui-Zhan Shao;Yang-Guang Li
CrystEngComm (1999-Present) 2017 vol. 19(Issue 46) pp:7050-7056
Publication Date(Web):2017/11/27
DOI:10.1039/C7CE01589E
Developing a new type of high-performing proton-conducting electrolyte associated with proton exchange membrane fuel cells (PEMFCs) is one of the attractive and challenging topics in the modern energy field. Here, three coordination compounds have been synthesized via the HCl steaming-assisted conversion approach by using multiple functional groups including the sulfonate group and the Cl− or HPO42− group, namely, Cu2H2(Hspip)2Cl4·H2O (1), Cu(H2spip)Cl2·H2O (2) and CuH(Hspip)(HPO4)·H2O (3) (where H2spip is 2-sulfophenylimidazo(4,5-f)(1,10)-phenanthroline). We disclose the relationship between the structure characteristics and the nature of proton conductivity. The protonated sulfonate group together with the halide Cl− group plays a positive role in increasing proton conductivity; meanwhile, the packing mode of the structure is also an important factor influencing proton conduction. The compounds exhibit high proton conductivity values in the range of 10−4–10−2 S cm−1 at 95 °C and 97% relative humidity (RH), in which compound 2 exhibited the highest value of 1.09 × 10−2 S cm−1 at 97% RH and 368 K.
Co-reporter:Tian-Tian Zheng;Jiao Zhao;Zhou-Wen Fang;Meng-Ting Li;Chun-Yi Sun;Xiao Li;Xin-Long Wang
Dalton Transactions 2017 vol. 46(Issue 8) pp:2456-2461
Publication Date(Web):2017/02/21
DOI:10.1039/C6DT04630D
The crystal structure of Cd-MOF-74 was obtained for the first time that possesses high sensitivity for the detection of copper ions from water and simulated biological fluids based on changes in luminescent intensity. Furthermore, Cd-MOF-74 could selectively remove Cu2+ from simulated biological fluids that contain Mg2+, Co2+, Zn2+, Fe2+, Ni2+, Na+, and K+. The adsorption capacity of this adsorbent for copper ions reached 189.5 mg g−1 and it quickly adsorbed copper ions within 10 minutes under 10 ppm Cu2+ in the simulated biological system. XPS, PXRD, and gas adsorption measurements revealed that this high sensitivity and selectivity of Cd-MOF-74 resulted from the partial substitution of Cd2+ by Cu2+ in the framework. Although many MOF materials have been employed for sensor or selective adsorption of Cu2+, Cd-MOF-74 is the first example of MOFs showing both capabilities in simulated biological fluids, which represents a pioneering work that extends the applications of MOF materials in the biological field.
Co-reporter:Wei Xie;Jun-Sheng Qin;Wen-Wen He;Kui-Zhan Shao;Dong-Ying Du;Shun-Li Li;Ya-Qian Lan
Inorganic Chemistry Frontiers 2017 vol. 4(Issue 3) pp:547-552
Publication Date(Web):2017/03/14
DOI:10.1039/C6QI00528D
We successfully synthesized a novel anionic luminescent metal–organic framework (MOF) (NENU-524) with a lonsdaleite topology. NENU-524 contains a trigonal prismatic unit {Zn8(btca)6(2-NH2-bdc)3} that can be regarded as a double secondary building unit with an unusual triply bound triangular frustum geometry. The prepared NENU-524 had a permanent porosity and excellent stability in air. NENU-524 was used as a platform to encapsulate yellow-emitting [Ir(ppy)2(bpy)]+ cations in the nanotube channels of the blue-emitting MOF via an ion-exchange process. The [Ir(ppy)2(bpy)]+@NENU-524 MOF ([Ir(ppy)2(bpy)]+ concentration 3.86 wt%) emitted a pure white light with CIE coordinates of (0.300, 0.336) and a high quantum yield of up to 15.2%. The white light-emitting diodes assembled using [Ir(ppy)2(bpy)]+@NENU-524 as a white phosphor emitted a bright white light, suggesting that the composite is a promising material for use in lighting. The assembled white light-emitting diodes continued to emit a bright white light for up to one month. This simple and feasible approach could be used to develop luminescent luminophor@MOFs composites for practical applications.
Co-reporter:Xiao Li;Liu Yang;Tan Su;Xinlong Wang;Chunyi Sun;Zhongmin Su
Journal of Materials Chemistry A 2017 vol. 5(Issue 10) pp:5000-5006
Publication Date(Web):2017/03/07
DOI:10.1039/C6TA10405C
In this study, we synthesized a low-cost electrocatalyst, Ni/Mo2C nanoparticles coated with graphene shells (denoted as NiMo2C@C), via a facile carburization process of porous bimetallic metal–organic frameworks (NiMo-MOF). This is the first example of a Ni and Mo2C nanocomposite derived from a bimetallic MOF that demonstrates excellent electrocatalytic activity and remarkable durability as long as 10 h under acidic and basic conditions. The overpotentials are 169 mV and 181 mV to reach the current density of 10 mA cm−2, respectively. The favorable performance can be ascribed to the synergistic effect between Mo2C and Ni as well as the homogeneous distribution, graphene coating and mesoporous structure which is in favor of the charge transfer in the HER. This work may provide some guidelines for fabricating nanostructured hybrids composed of versatile transition metal carbides and graphene with high performance and stability in different media based on designed MOFs.
Co-reporter:Mei-Jie Wei;Jia-Qi Fu;Yi-Di Wang;Jing-Yang Gu;Bai-Ling Liu;Hong-Ying Zang;En-Long Zhou;Kui-Zhan Shao
Journal of Materials Chemistry A 2017 vol. 5(Issue 3) pp:1085-1093
Publication Date(Web):2017/01/17
DOI:10.1039/C6TA08581D
It is essential and vital to develop high-performance proton-conducting solid electrolyte materials for proton exchange membrane fuel cells (PEMFCs), but it remains challenging to design and synthesise such electrolytes with high proton conductivity which are also stable enough to be applied in PEMFCs. Herein, we employed the HCl steam-assisted conversion method to synthesize nonporous coordination complexes with a gradual increase of proton conductivity by stepwise protonation of sulfonated ligands and introduction of halide ions, including [Cu(Hsfpip)(H2O)2]·H2O (1), [CuH2(Hsfpip)2(H2O)] (2) and [CuH(Hsfpip)Cl(H2O)] (3) (where Hsfpip is 2-(2,4-disulfophenyl)imidazo(4,5-f)(1,10)-phenanthroline). We reveal the relationship between the nature of proton conduction and structural features. Three resulting coordination complexes showed high proton conductivity with a maximum value of 1.43 mS cm−1 for 1, 2.58 mS cm−1 for 2 and 15 mS cm−1 for 3 at 95 °C and 97% RH, and meanwhile, we proved their proton conduction nature and electron resistance using D2O-exchange experiments and the Hebb–Wagner polarization method. We believe that these nonporous solid electrolytes intrinsically possess proton carriers and may avoid fuel crossover, which makes them good candidates for PEMFCs in real-life applications.
Co-reporter:Yang Jiang;Guangfu Li;Dongxia Zhu;Zhongmin Su;Martin R. Bryce
Journal of Materials Chemistry C 2017 vol. 5(Issue 46) pp:12189-12193
Publication Date(Web):2017/11/30
DOI:10.1039/C7TC04066K
A neutral multifunctional dinuclear Ir(III) complex 1 with a Schiff base bridging ligand is shown to combine aggregation-induced emission (AIE), piezochromic luminescence (PCL) and vapochromism. The complex displays a reversible colour change in its phosphorescence in the solid state between faint red and bright orange with high contrast intensity, triggered by high polarity volatile organic compounds (VOCs) or by mechanical grinding within 10 s. Notably, unlike many known vapochromic systems, complex 1 exhibits ultrahigh stability, with the orange colour remaining unchanged in air for several months at room temperature. A simple and efficient monitoring device has been fabricated in which highly polar VOCs act as a switch to “turn on” the device by changing the aggregation state of complex 1.
Co-reporter:Jie Wu;Yuhe Kan;Zhenhua Xue;Jintian Huang;Peng Chen;Xiaofang Yu;Zeyu Guo;Zhongmin Su
Journal of Materials Chemistry C 2017 vol. 5(Issue 35) pp:9088-9097
Publication Date(Web):2017/09/14
DOI:10.1039/C7TC02336G
Cycloparaphenylenes (CPPs) have attracted the attention of researchers in various fields because of their unique properties, but studies and applications on host materials in the optoelectronics field are lacking. We undertook preliminary exploration and finally selected 1,4-azaborine and [6]CPP as the basic building blocks to construct a series of BN-[6]CPP hybrids. BN-[6]CPPs with various triplet energies (ET = 1.66–3.52 eV) could be the potential hosts in full-color phosphorescent organic light-emitting diodes (OLEDs). DFT/TDA-DFT calculations showed that introduction of 1,4-azaborine rings at different positions of the [6]CPP hoop could efficiently control delocalization (localization) of triplet excitons and T1-state aromaticity (non-aromaticity), and thus varied with the ET. This work provides not only an efficient strategy for realizing ET controllability for CPP-based materials by chemical modification of CPP frameworks, but also theoretical guidance for the design and prediction of novel hoop-shaped materials with high ET in the organic optoelectronics field.
Co-reporter:Cai Xia Wu;Shi Zheng Wen;Li Kai Yan;Min Zhang;Teng Ying Ma;Yu He Kan;Zhong Min Su
Journal of Materials Chemistry C 2017 vol. 5(Issue 16) pp:4053-4062
Publication Date(Web):2017/04/20
DOI:10.1039/C6TC05545A
Using density functional theory (DFT) in combination with non-equilibrium Green's functions, we have investigated the electronic structures, magnetization, and quantum transport properties of zigzag graphene nanoribbons (ZGNRs) functionalized with conventional conductive metal adatoms (Al, Cu, Ag and Au). On the basis of the adsorption energies, our simulation demonstrates that Al and Cu adatoms are chemically bonded with ZGNRs, while the adsorptions for Ag and Au are between weak chemisorption and strong physisorption. The properties of charge transfer and magnetic moment are in reasonable agreement with the previous calculations. The adsorption of metal adatoms induce a net magnetic moment of −1 μB in 6ZGNR–metal systems. On the other hand, the transport studies of metal adatoms adsorbed ZGNRs suggest that the metal adatoms play an important role in the transport properties of devices and exhibit different effects on the transport properties of 6ZGNR-based and 7ZGNR-based devices. The 7ZGNR-based devices show the opposite conductive order in 6ZGNR-based devices. For 6ZGNR-based devices, the transport current in 6ZGNRs can be enhanced effectively by the adsorption of metal adatoms. However, the currents in 7ZGNR functionalized with conductive metal atoms are obviously smaller than that in pristine 7ZGNR, implying that metal adsorptions reduce the electrical conductivity of 7ZGNR-based devices. In contrast to the properties of the bulk materials, the conductivity of 6ZGNR–Al is highest among 6ZGNR–metal systems, which is in agreement with that of single atomic wires of Ag, Al, Au, and Cu.
Co-reporter:Cai Xia Wu;Shi Zheng Wen;Li Kai Yan;Min Zhang;Teng Ying Ma;Yu He Kan;Zhong Min Su
Journal of Materials Chemistry C 2017 vol. 5(Issue 16) pp:4053-4062
Publication Date(Web):2017/04/20
DOI:10.1039/C6TC05545A
Using density functional theory (DFT) in combination with non-equilibrium Green's functions, we have investigated the electronic structures, magnetization, and quantum transport properties of zigzag graphene nanoribbons (ZGNRs) functionalized with conventional conductive metal adatoms (Al, Cu, Ag and Au). On the basis of the adsorption energies, our simulation demonstrates that Al and Cu adatoms are chemically bonded with ZGNRs, while the adsorptions for Ag and Au are between weak chemisorption and strong physisorption. The properties of charge transfer and magnetic moment are in reasonable agreement with the previous calculations. The adsorption of metal adatoms induce a net magnetic moment of −1 μB in 6ZGNR–metal systems. On the other hand, the transport studies of metal adatoms adsorbed ZGNRs suggest that the metal adatoms play an important role in the transport properties of devices and exhibit different effects on the transport properties of 6ZGNR-based and 7ZGNR-based devices. The 7ZGNR-based devices show the opposite conductive order in 6ZGNR-based devices. For 6ZGNR-based devices, the transport current in 6ZGNRs can be enhanced effectively by the adsorption of metal adatoms. However, the currents in 7ZGNR functionalized with conductive metal atoms are obviously smaller than that in pristine 7ZGNR, implying that metal adsorptions reduce the electrical conductivity of 7ZGNR-based devices. In contrast to the properties of the bulk materials, the conductivity of 6ZGNR–Al is highest among 6ZGNR–metal systems, which is in agreement with that of single atomic wires of Ag, Al, Au, and Cu.
Co-reporter:Mei-Si Yang;Wei-Chao Chen;Chao Qin;Wei Yao;Ying-Xue Yin
New Journal of Chemistry (1998-Present) 2017 vol. 41(Issue 19) pp:10532-10536
Publication Date(Web):2017/09/25
DOI:10.1039/C7NJ02011B
For the first time, an unprecedented 3D framework based on lanthanide-containing Nb/W mixed-addendum polyoxometalate [H3La8(H2O)32(C6H5NO2)6][SiW9Nb3O40]3·8H2O (1) has been obtained by utilizing the steam-assisted conversion method. Meanwhile, the stability, electrochemical behavior, and photoluminescence properties of 1 have also been discussed.
Co-reporter:Jie Pan;Hang Yin;Yu-Zhong Xie;Guang-Yan Sun
RSC Advances (2011-Present) 2017 vol. 7(Issue 51) pp:31800-31806
Publication Date(Web):2017/06/21
DOI:10.1039/C7RA02360J
For the purpose of improving the power conversion efficiencies (PCEs) of bulk heterojunction (BHJ) devices, non-fullerene small acceptors have been presented by modifying desirable donors, such as a diketopyrrolopyrrole (DPP)-based acceptor with two thiazoles, namely poly{3,6-bis(5-hexyldecyl-2thenyl)-2,5-dihydro-2,5-di(alkyl)pyrrolo[3,4]pyrrolo-1,4-dionethiazole} (PDPP2TzT), synthesized by introducing two nitrogens onto the copolymer based on DPP and terthiophene (poly[{2,5-bis(2-hexyldecyl)-2,3,5,6-tetrahydro-3,6-dioxopyrrolo[3,4-c]pyrrole-1,4-diyl}]-alt-{[2,2′:5′,2′′-terthiophene-5,5′′-diyl}]) (PDPP3T). In order to clarify the transfer mechanism, we put forward a comparative theoretical study of PDPP3T and PDPP2TzT using density functional theory/time-dependent density functional theory (DFT/TD-DFT) calculations. Subsequently, we determined that the mechanism of the conversion from a donor to an acceptor was that the insertion of nitrogen could reduce the LUMO energy by changing the electron density and intramolecular interactions. Based on this mechanism, a series of acceptors were designed with the advantages of PDPP2TzT. The results illustrate that b4 showed the best performance when blended with PDPP5T as the donor for BHJ devices. Finally, we hope our investigations will provide guidelines for further rational design of acceptor materials for BHJ devices.
Co-reporter:Qing-Qing Pan;Shuang-Bao Li;Ying-Chen Duan;Yong Wu;Ji Zhang;Yun Geng;Liang Zhao
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 46) pp:31227-31235
Publication Date(Web):2017/11/29
DOI:10.1039/C7CP05938H
The interface characteristic is a crucial factor determining the power conversion efficiency of organic solar cells (OSCs). In this work, our aim is to conduct a comparative study on the interface characteristics between the very famous non-fullerene acceptor, ITIC, and a fullerene acceptor, PC71BM by combining molecular dynamics simulations with density functional theory. Based on some typical interface models of the acceptor ITIC or PC71BM and the donor PBDB-T selected from MD simulation, besides the evaluation of charge separation/recombination rates, the relative positions of Frenkel exciton (FE) states and the charge transfer states along with their oscillator strengths are also employed to estimate the charge separation abilities. The results show that, when compared with those for the PBDB-T/PC71BM interface, the CT states are more easily formed for the PBDB-T/ITIC interface by either the electron transfer from the FE state or direct excitation, indicating the better charge separation ability of the former. Moreover, the estimation of the charge separation efficiency manifests that although these two types of interfaces have similar charge recombination rates, the PBDB-T/ITIC interface possesses the larger charge separation rates than those of the PBDB-T/PC71BM interface. Therefore, the better match between PBDB-T and ITIC together with a larger charge separation efficiency at the interface are considered to be the reasons for the prominent performance of ITIC in OSCs.
Co-reporter:Shao-Fen Huang, Hai-Zhu Sun, Guo-Gang Shan, Fu-Shan Li, Qun-Ying Zeng, Kai-Yue Zhao, Zhong-Min Su
Dyes and Pigments 2017 Volume 139(Volume 139) pp:
Publication Date(Web):1 April 2017
DOI:10.1016/j.dyepig.2016.12.056
•Cationic Ir(III) dyes with triazole-type main and ancillary ligands were synthetized.•Bright emissions from deep-blue to red were achieved.•Theoretical calculations were performed to study their intrinsic photophysical behaviors.•Solution-processed blue-green device exhibited efficiencies of 2.19 cd A−1 and 0.97 lm W−1.Triazole-type ligand has been approved to be an excellent building block to construct efficient cationic Ir(III) dyes. Herein, a new family of Ir(III) complexes employing two different triazole fragments as cyclometalated and ancillary ligands, respectively, has been synthesized and their photophysical and electrochemical properties are investigated in detail. Through careful modification of the used ligands, efficiently tuning emission color from deep-blue (459 nm) to red (649 nm) is realized and the intrinsic structure-property relationship is also studied by the comprehensive theoretical calculations. Choosing a greenish blue emitting dye 3 that shows relatively high photoluminescence yield in neat film, the solution-processed single-layer device with peak current efficiency of 2.19 cd A−1 and power efficiency of 0.97 lm W−1, respectively, has been achieved.To construct efficient panchromatic cationic Ir(III) emitters and deeply understand the relationship between structures and properties, a series of Ir(III) dyes employing modified triazole-type cyclometalated and ancillary ligands are synthesized. Tuning the emission colors of resulting dyes from deep-blue to red is achieved.Download high-res image (274KB)Download full-size image
Co-reporter:Hanni Wu, Tengying Ma, Caixia Wu, Likai Yan, Zhongmin Su
Dyes and Pigments 2017 Volume 142(Volume 142) pp:
Publication Date(Web):1 July 2017
DOI:10.1016/j.dyepig.2017.03.062
•Charge transfer directions are all from donor side to POM, and injecting into TiO2 via COOH.•The introduction of POM results in stronger, longer, and tighter CT process.•The introduction of POM leads to a larger responsive range of spectraA series of donor-acceptor-π-bridge-acceptor (D-A-π-A) type hybrids were designed by introducing Lindqvist-type polyoxometalate (POM) into synthesized dyes aiming at revealing the impact of POMs on the performance of dye. The spectroscopic properties and charge transfer (CT) characters were studied using density functional theory (DFT) and time-dependent DFT methods. Compared with the synthesized dyes, designed dyes show larger responsive range of spectra. CT of the designed dyes are all from donor side to POM and COOH, suggesting strong electronic coupling with TiO2. Further analysis on CT parameters indicate that the introduction of POM results in stronger, longer, and tighter CT process. These designed dyes can be promising candidates for DSSCs.Four D-A-π-A hybrids were designed by introducing polyoxometalate for improving photovoltaic performance.Download high-res image (278KB)Download full-size image
Co-reporter:Lu Han, Jie Zhou, Xiao Li, Chun-Yi Sun, Liang Zhao, Yu-Teng Zhang, Man Zhu, Xin-Long Wang, Zhong-Min Su
Inorganic Chemistry Communications 2017 Volume 86(Volume 86) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.inoche.2017.10.015
•A novel MOF material, MOF1, containing hexagonal hydrophobic pores has been designed and synthesized;•Benefiting from appropriate porous size and condition, MOF1 shows enhanced luminescent intensity response to FAH molecules;•Different extent of luminescent enhancement was observed indicated MOF1 could be potential probe material to FAHs.A novel metal-organic framework (MOF) with a π-conjugated porous condition is reported to sensor fused aromatic hydrocarbons molecules. This MOF assembled by N-heterocyclic multi-carboxyl ligands and Cd ions shows hexagonal hydrophobic channels along [100] and [010] directions with size 18.4 × 15.5 Å2. Under the excitation of 338 nm, this MOF emits blue light peaking at 397 nm. Under immersing this MOF in the solutions containing aromatic hydrocarbon, including naphthalene, anthracene and phenanthrene within different time, the luminescent intensity of MOF enhanced different degree upon encapsulating aromatic hydrocarbons guest molecules. Therefore, this material shows favourable property of recognition for aromatic hydrocarbons molecules via solid-state photoluminescence.A novel metal-organic framework (MOF) with a π-conjugated porous condition is reported to sensor fused aromatic hydrocarbons molecules. This MOF assembled by N-heterocyclic multi-carboxyl ligand and Cd ions shows hexagonal hydrophobic channels along [100] and [010] directions with size 18.4 × 15.5 Å2. Under immersing this MOF in the solutions containing aromatic hydrocarbon, including anthracene, phenanthrene within different time, the luminescent intensity of MOF enhanced different degree upon encapsulating aromatic hydrocarbons guest molecules.Download high-res image (93KB)Download full-size image
Co-reporter:Lei Chen, Li Zhang, Ya-Hui Zhao, Kui-Zhan Shao, Xin-Long Wang, Ming-Xin Huo, Zhong-Min Su
Inorganic Chemistry Communications 2017 Volume 86(Volume 86) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.inoche.2017.09.028
•A new coordination polymer, compound 1, was synthesized under hydrothermal reaction.•The framework of 1 shows a 3D (3,8)-connected net with (53)2(58·64·78·84·94) topology.•By virtue of photoluminescent property, Compound 1 can be used to detect nitro explosives such as NB, NT and DNT.A novel 3D coordination polymer [Zn2.5(L)(trz)2(H2O)2]·2H2O (1) (H3L = 5-(4-carboxybenzyloxy)isophthalic acid, trz = 1,2,4-triazole) has been synthesized hydrothermally. Compound 1 displays a 3D (3,8)-connected net with (53)2(58·64·78·84·94) topology. The luminescent property of 1 dispersed in different solvents as well as nitro compounds have been investigated systematically, demonstrating high detection sensitivity via a fluorescence quenching mechanism.A novel 3D coordination polymer [Zn2.5(L)(trz)2(H2O)2]·2H2O (1) (H3L = 5-(4-carboxybenzyloxy)isophthalic acid, trz = triazole) has been synthesized hydrothermally. Compound 1 displays a 3D (3,8)-connected net with (53)2(58·64·78·84·94) topology. The luminescent property of 1 dispersed in different solvents as well as nitrobenzene have been investigated systematically, demonstrating high sensitivity for the detection of nitro compound via a fluorescence quenching mechanism.Download high-res image (509KB)Download full-size image
Co-reporter:Qing-Qing Wang, Xiao Li, Guang-zhe Li, Kui-Zhan Shao, Zhong-Min Su
Inorganic Chemistry Communications 2017 Volume 86(Volume 86) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.inoche.2017.09.002
•Two isostructural metal organic frameworks were synthesized with one-dimensional channels, showing 3-fold interpenetrated 6-connected pcu topologies based on bimetallic tetracarboxylate units.•By virtue of photoluminescent property, 1 can be used to detect nitro explosives.•Cupric ions also can be detected by 1 indicating it is worthy to be confirmed as an efficient fluorescent sensor.Two isostructural metal-organic frameworks, [M2(4-bpytm)(bdc)2] [M = Zn (1), Cu (2)] (4-bpytm = bis(4-pyridylthio)methane, bdc = terephthalic acid), were synthesized under hydrothermal reactions. Single-crystal X-ray diffraction analyses have been applied to determine their structures. Furthermore, they are characterized by IR, TG, PXRD, element analysis and PL spectra. Both 1 and 2 showed 3-fold interpenetrated 6-connected pcu topologies based on bimetallic tetracarboxylate units with one-dimensional channels. The luminescence properties of 1 are investigated systematically due to its one dimensional channels. The results show that 1 has the ability to sense explosives and inorganic ions.Two isostructural metal organic frameworks, [M2(4-bpytm)(bdc)2] [M = Zn (1), Cu(2)] was synthesized under hydrothermal reaction. 1 and 2 showed 3-fold interpenetrated 6-connected pcu topologies based on bimetallic tetracarboxylate units with one-dimensional channels. The luminescence properties of 1 are investigated systematically.Download high-res image (278KB)Download full-size image
Co-reporter:Man Zhu, Meng-Ting Li, Liang Zhao, Kui-Zhan Shao, Zhong-Min Su
Inorganic Chemistry Communications 2017 Volume 79(Volume 79) pp:
Publication Date(Web):1 May 2017
DOI:10.1016/j.inoche.2017.03.020
•Three new MOFs were obtained constructed on threefold symmetric ligand 1,3,5-tris(4-carbonylphenyloxy)benzene under solvothermal conditions.•Compound 1 and 2 are 2D grid layers, while compound 3 is a 3D interpenetated structure with dia topology.•Compound 1 and 3 exhibited strong photoluminescence. Comparing to the photoluminescent property of free tcpb and 4,4′-bipyridine ligands, compound 1 and 3 display remarkable red-shift.Three new metal–organic frameworks, namely, [Cd1.5(tcpb)(DMF)(H2O)] (1), [Co1.5(tcpb)(DMF)2]·3DMF (2) and [Zn(Htcpb)(4,4′-bpy)0.5]·1.5H2O (3) (tcpb = 1.3,5-tris(4-carbonylphenyloxy)benzene) have been synthesized by solvent-thermal method. The single-crystal X-ray diffraction analysis shows that compound 1 is 2D grid layers, with the trinuclear cadmium clusters acting as secondary building units (SBUs), and the tcpb ligand serveing as organic linkers. Compound 1 and Compound 2 are isomorphic structures containning similar connection modes of tcpb ligands with trinuclear metal clusters. Compound 3 is a 3D interpenetrated framework based on dinuclear Zn cluster with dia topology. The fluorescence properties for solid state compound 1 and 3 were examined at room temperature. Compared with free tcpb ligands, both of them display remarkable red-shift.Three new metal-organic frameworks constructed from d10 metal and 1,3,5-tris(4-carbonylphenyloxy)benzene (tcpb) were synthesis under solvothermal condition. Compound 1 and 2 are 2D grid layers, in which the trinuclear metal clusters act as secondary building units (SBUs), and the tcpb ligand serves as organic linkers. Compound 3 is a 3D interpenetated structure with dia topology based on dinuclear Zn cluster as SBUs. All these frameworks had good stability in the air and Compound 1 and 3 exhibited strong photoluminescence at room temperature.Download high-res image (239KB)Download full-size image
Co-reporter:Shu-Ran Zhang, Wei Wang, Guang-Juan Xu, Chan Yao, Yan-Hong Xu, Zhong-Min Su
Inorganic Chemistry Communications 2017 Volume 84(Volume 84) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.inoche.2017.07.008
•A luminescent coordination polymer, JLNU-1 has been prepared by hydrothermal method and characterized by analytical tools.•JLNU-1 exhibits excellent chemical stability in aqueous solution. The suspension of JLNU-1 in NB showed highest quenching.•JLNU-1 can be used as a fluorescent probe for sensitive and recyclable sensing of pollutant NB in aqueous solution.A luminescent coordination polymer, [Cd(H2biim)2SO4]·3H2O (JLNU-1, JLNU = Jilin Normal University); H2Biim = 2,2′-biimidazole, has been prepared by hydrothermal method and characterized by corresponding analytical tools. JLNU-1 exhibits outstanding chemical stability in aqueous solution and solvent-dependent luminescent behaviour. In particular, stable suspension of JLNU-1 in nitrobenzene showed highest quenching. The emission spectra were nearly completely quenched at a concentration of 100 ppm nitrobenzene in aqueous solution. JLNU-1 can be regenerative for 6 cycles by centrifugation of the solution after use and washing several times with H2O. Therefore, JLNU-1 can be used as a fluorescent probe for sensitive and recyclable sensing of small-molecule pollutant nitrobenzene in aqueous solution.JLNU-1 with 1D zigzag chain has been synthesized by hydrothermal reaction. JLNU-1 displays distinct PL emissions in different solvents. Especially, the dispersed solution of JLNU-1 in aqueous solution exhibits strong fluorescence emission, which could be quenched by trace amounts of nitrobenzene at ppm level. JLNU-1 can be utilized as a fluorescent sensor for the detection of nitrobenzene with high selectivity, sensitivity, and recyclability.Download high-res image (184KB)Download full-size image
Co-reporter:Zhi-Wen Zhao, Qing-Qing Pan, Shuang-Bao Li, Yu-Ai Duan, Yun Geng, Min Zhang, Zhong-Min Su
Journal of Molecular Graphics and Modelling 2017 Volume 77(Volume 77) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.jmgm.2017.07.027
•Explaining the effect of fluorine and cyano substitution in polymer donor materials from a theoretical perspective.•The substituent of cyano groups and fluorine in polymer 3 and 4 performs better balance between Voc and Jsc in OSCs.•The designed molecules 3 and 4 with higher ratios of rates kinter-CT/kinter-CR) are promising donor materials.A series of polymer donor materials 1-5 based on diketopyrrolopyrrole and thiophene unit which have been widely used in organic solar cells (OSCs) were investigated based on quantum chemical calculations. The effect of fluorine and cyano substitutions in polymer donor materials was focused on. Based on the investigation on electronic structures and optical properties of the reported molecules 1 and 2 and the analysis on some parameters relevant to charge dissociation ability at donor/acceptor interface constituted by 1 and 2 with PC61BM such as intermolecular charge transfer and recombination, driving force and Coulombic bound energy, we explained why fluorine substitution can improve OPV efficiency through strengthening eletron-withdrawing ability from a theoretical perspective. Then we designed cyano-substituted polymers 3-5 with the aim of obtaining better photovoltaic donor materials. The results reveal that our attempt to design donor materials which can balance large open-circuit voltage (Voc) and high short-circuit current (Jsc) in OSCs has worked out. It is worth noting that the substitutions of fluorine and cyano groups synergistically reduce energy gap and HOMO energy level of polymers 3 and 4. Moreover, 3/PC61BM and 4/PC61BM heterojunctions show over 107 and 104 times higher than 1/PC61BM on the ratios of intermolecular charge transfer and recombination rates (kinter-CT/kinter-CR). Thus, our work here may provide an efficient strategy to design promising donor materials in OPVs and we hope it could be useful in the future experimental synthesis.Download high-res image (140KB)Download full-size image
Co-reporter:Ying Gao, Tan Su, Liang Zhao, Yun Geng, Yong Wu, Min Zhang, Zhong-Min Su
Organic Electronics 2017 Volume 50(Volume 50) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.orgel.2017.07.024
•1 only exhibits a prompt emission whereas 2 has a prominent TADF characteristic.•HOMO and LUMO distributions of 1 and 2 are similar and kr of 1 is even greater than that of 2.•2 has the smaller ΔEST and larger SOC strength than 1.•2 has a smaller energy gap between S1/T1 MECP and S1 than 1.Density functional theory calculation was performed to analyze the critical factors determining whether molecule has a thermally activated delayed fluorescence (TADF) characteristic by comparing two similar molecules (1 and 2), which have a little difference in structure but opposite TADF performance. Some parameters relevant to TADF property were evaluated in this work. The results indicate that although the HOMO and LUMO distributions of 1 and 2 are similar and the radiative decay rate (kr) of 1 is even greater than that of 2, 2 has the smaller singlet-triplet energy splitting (ΔEST) and larger spin-orbital coupling (SOC) strength than 1. Besides, 2 has a smaller energy gap between S1/T1 minimum energy crossing point (MECP) and S1 than 1, indicating that it is easy for 2 to realize reverse intersystem crossing (RISC) from T1 to S1. Therefore, a comprehensive comparison between 1 and 2 suggests that the parameters including ΔEST, SOC and MECP between S1 and T1 are fairly important factors determining the TADF characteristic of 2.The results indicate that the HOMO and LUMO distributions of 1 and 2 are similar and the radiative decay rate (kr) of 1 is even greater than that of 2. However, 2 has smaller singlet-triplet energy splitting (ΔEST) and larger spin-orbital coupling (SOC) strength than 1. Besides, 2 has a smaller energy gap between S1/T1 minimum energy crossing point (MECP) and S1 than 1, indicating that it is easy for 2 to realize reverse intersystem crossing (RISC) from T1 to S1. Therefore, the parameters including ΔEST, SOC and MECP between S1 and T1 are fairly important factors determining the TADF characteristic of 2.Download high-res image (238KB)Download full-size image
Co-reporter:Dr. Lin Ding;Chi-Yuan Yang;Yu-Qing Zheng; Jie-Yu Wang; Jian Pei; Dr. Zhongmin Su
Asian Journal of Organic Chemistry 2017 Volume 6(Issue 9) pp:1231-1234
Publication Date(Web):2017/09/01
DOI:10.1002/ajoc.201700203
AbstractOrganic electron-acceptor materials play an important role in organic electronics, especially in organic bulk heterojunction (BHJ) solar cells. Recently, many non-fullerene small molecular acceptors have been developed, in which aromatic fused-imides were proven to be a promising family of excellent electron acceptors. We developed a series of acenaphtho[1,2-k]fluoranthene-fused diimides, AFIs (AFI1, AFI2 and AFI3), employing the Diels–Alder reaction as the crucial synthetic step. With electron-withdrawing diimide groups in their large conjugated backbone, AFIs display a LUMO level around −3.50 eV. These AFI compounds were used as the electron acceptors to fabricate organic photovoltaic devices. The power conversion efficiency (PCE) of up to 2.37 % was achieved with a high open-circuit voltage (Voc) up to 0.96 V.
Co-reporter:Bo Zhu, Wei Guan, Li-Kai Yan, and Zhong-Min Su
Journal of the American Chemical Society 2016 Volume 138(Issue 35) pp:11069-11072
Publication Date(Web):August 22, 2016
DOI:10.1021/jacs.6b02433
The cleavage of inert C–C bonds is a central challenge in modern chemistry. Multinuclear transition metal complexes would be a desirable alternative because of the synergetic effect of multiple metal centers. In this work, carbon–carbon bond cleavage and rearrangement of benzene by a trinuclear titanium hydride were investigated using density functional theory. The reaction occurs via a novel “two-state reactivity” mechanism. The important elementary steps consist of hydride transfer, benzene coordination, dehydrogenation, oxidative addition, hydride–proton exchange, and reductive elimination. Most importantly, the ground-state potential energy surface switches from nearly degenerate triplet and antiferromagnetic singlet states to a closed-shell singlet state in the dearomatization of benzene, which effectively decreases the activation barrier. Furthermore, the roles of the transition metal centers and hydrides were clarified.
Co-reporter:Ronglin Zhong, Min Zhang, Hongliang Xu and Zhongmin Su
Chemical Science 2016 vol. 7(Issue 2) pp:1028-1032
Publication Date(Web):27 Oct 2015
DOI:10.1039/C5SC03437J
Besides the classic double bond scheme, several novel schemes have been proposed to describe the nature of the chemical bond in dicarbon (C2), including a quadruple bond and a singlet diradical state. The results from a symmetry-broken CASSCF(8,8)/aug-cc-pVTZ study present a harmony between MO and VB theories, based on the orthogonal hybridization of the 3σg and 2σu orbitals together with the other six pristine valence orbitals. This scheme achieves the same bonding energy, RC–C, ωe and one electron density as that from the eight pristine valence orbitals. A quadruple bond scheme, identical to Prof. Shaik's result from VB theory, is achieved with the 4th bond energy in the range of 12.8–27.6 kcal mol−1. Meanwhile, the weight of a singlet open-shell configuration is the highest among all the possible configurations.
Co-reporter:Dashu Chen, Hongzhu Xing, Chungang Wang and Zhongmin Su
Journal of Materials Chemistry A 2016 vol. 4(Issue 7) pp:2657-2662
Publication Date(Web):01 Feb 2016
DOI:10.1039/C6TA00429F
A new microporous robust zirconium metal–organic framework (Zr-MOF), NNU-28, has been synthesized and employed as a visible-light photocatalyst for carbon dioxide (CO2) reduction to produce formate. NNU-28 is constructed by using a visible light responsive organic ligand derived from an anthracene group. Studies reveal that the as-prepared Zr-MOF shows desirable characteristics including excellent chemical and thermal stability, high CO2 uptake, broad-band visible light absorption and efficient photoinduced charge generation. Remarkably, NNU-28 is highly efficient for visible-light-driven CO2 reduction with a formate formation rate of 183.3 μmol h−1 mmolMOF−1, which is among the highest performances of Zr-MOFs. Both photocatalytic experiments and electron paramagnetic resonance (EPR) studies reveal that both the inorganic building unit Zr6 oxo cluster and the anthracene-based ligand contribute to the highly efficient photocatalysis of CO2 reduction. The dual photocatalytic routes are demonstrated here to be more efficient for visible-light-driven CO2 photoreduction than that typically relying on a ligand-to-metal charge transfer process, illustrating a new strategy to design and synthesize novel visible-light photocatalysts for CO2 reduction with high efficiency.
Co-reporter:Yu-Teng Zhang, Xin-Long Wang, Shuang-Bao Li, Ya-Ru Gong, Bai-Qiao Song, Kui-Zhan Shao and Zhong-Min Su
Chemical Communications 2016 vol. 52(Issue 62) pp:9632-9635
Publication Date(Web):24 Jun 2016
DOI:10.1039/C6CC04583A
Unprecedented Anderson-like alkoxo-polyoxovanadate [V6O6(OCH3)9(μ6-SO4)(COO)3]2− polyanions can serve as 3-connected second building units (SBUs) that assemble with dicarboxylate or tricarboxylate ligands to form a new family of metal organic tetrahedrons of V4E6 and V4F4 type (V = vertex, E = edge, and F = face). To our knowledge, this alkoxo-polyoxovanadate-based SBU is the first ever reported.
Co-reporter:Wei-Chao Chen, Xin-Long Wang, Chao Qin, Kui-Zhan Shao, Zhong-Min Su and En-Bo Wang
Chemical Communications 2016 vol. 52(Issue 61) pp:9514-9517
Publication Date(Web):21 Jun 2016
DOI:10.1039/C6CC03763A
A carbon-free, stable, homogeneous water oxidation catalyst based on the unique hepta-nuclear cobalt–arsenic core (“fused” double-quasi-cubane) and polyoxometalate ligands, Na12[{CoII7AsIII6O9(OH)6}(A-α-SiW9O34)2]·8H2O (1), was synthesized, thoroughly characterized and employed to catalyze water oxidation under visible-light-driven conditions.
Co-reporter:Wei Xie, Wen-Wen He, Dong-Ying Du, Shun-Li Li, Jun-Sheng Qin, Zhong-Min Su, Chun-Yi Sun and Ya-Qian Lan
Chemical Communications 2016 vol. 52(Issue 16) pp:3288-3291
Publication Date(Web):16 Dec 2015
DOI:10.1039/C5CC08703A
A stable mesoporous blue-emitting MOF NENU-521 was successfully constructed. NENU-521 can serve as a host for encapsulating Alq3 to obtain tunable and efficient white-light emission. The Alq3@NENU-521 composite possesses excellent stability and can be used as a promising white phosphor in WLEDs.
Co-reporter:Lifei He, Li Chen, Yue Zhao, Weilin Chen, Chunhui Shan, Zhongmin Su, Enbo Wang
Journal of Power Sources 2016 Volume 328() pp:1-7
Publication Date(Web):1 October 2016
DOI:10.1016/j.jpowsour.2016.07.085
•Highly dispersed POM nanoparticles were synthesized by micelle directed method.•TiO2 composited with POM nanoparticles was studied as photoanodes.•Both high dispersion and nanoscale increased the active site.•POM addition accelerated electron transition and retarded electron recombination.•The composite yielded an efficiency improvement from 5.9% to 8.4%.In this work, two kinds of polyoxometalate (POM) nanoparticles with controlled shapes and structures were synthesized by micelle directed method and then composited with TiO2 via calcination to remove the surfactants owing to the excellent electronic storage and transmission ability of POM, finally obtaining two kinds of TiO2 composites with highly dispersed and small-sized POM nanoparticles (∼1 nm). The TiO2 composites were then induced into the photoanodes of dye-sensitized (N719) solar cells (DSSCs). The separation of electron-holes becomes more favorable due to the nanostructure and high dispersion of POM which provide more active sites than pure POM tending to agglomeration. The TiO2 composite photoanodes finally yielded the power conversion efficiency (PCE) of 8.4% and 8.2%, respectively, which were 42% and 39% higher than the pristine TiO2 based anodes. In addition, the mechanisms of POM in DSSC are proposed.Two polyoxometalate (POM) nanoparticles with controlled shape and structure were synthesized by micelle directed method, and then composited with TiO2 via calcination to remove the surfactant to obtain TiO2 complexes with highly dispersed and small-sized polyoxometalate nanoparticles, which were firstly applied as photoanodes for dye-sensitized (N719) solar cells with enhanced photovoltaic performance.
Co-reporter:Yuteng Zhang, Xinlong Wang, Shuangbao Li, Baiqiao Song, Kuizhan Shao, and Zhongmin Su
Inorganic Chemistry 2016 Volume 55(Issue 17) pp:8770-8775
Publication Date(Web):August 12, 2016
DOI:10.1021/acs.inorgchem.6b01338
Three new polyoxovanadate-based metal–organic polyhedra (VMOPs) have been successfully synthesized and structurally characterized. Single crystals of three VMOPs were obtained by reaction of VCl3 and different carboxylate ligands (2,5-H2TDA = thiophene-2,5-dicarboxylic acid for VMOP-4, m-H2BDC = 1,3-benzenedicarboxylic acid for VMOP-5, 2,6-H2NDC = 2,6-naphthalenedicarboxylic aid for VMOP-6) under solvothermal conditions. Though all of the three hybrids feature the same {VVV4IV} units, their structures exhibit differences changing from truncated triangular prism to truncated quadrangular prism to octahedron, mainly depending on the nature of carboxylate ligands. Furthermore, the magnetic investigations reveal that VMOP-4–6 show similar ferromagnetic behaviors.
Co-reporter:Dong-Mei Gu, Jian-Zhao Zhang, Min Zhang, Yun Geng, Zhong-Min Su
Dyes and Pigments 2016 Volume 132() pp:136-141
Publication Date(Web):September 2016
DOI:10.1016/j.dyepig.2016.04.025
•two homologous organic dyes are calculated to reveal the regeneration mechanism.•three kinds of dye regeneration reaction processes by iodide have been investigated.•dye+ and I2- is the most favorite regeneration mechanism.•theoretically prove that S-substitution in donor unit further decreases ΔG barrier.In order to explore the mechanism of dye regeneration, three possible kinds of dye regeneration reaction processes for two homologous organic dyes by iodide have been investigated theoretically. The objective of this study is to interpret the significantly different regeneration rate constant between the organic dyes in the change from oxygen to sulfur in two triphenylamine donor unit. The corresponding surface electrostatic potential, the interaction energy and the Gibbs free energy differences in every reaction steps are calculated to reveal the different effect between iodide and O/S-containing organic dyes. The data indicate that the direct reaction between dye+ and I2- is the most favorite regeneration mechanism. Furthermore, the S-substitution in donor unit further decreases Gibbs free energy differences barrier and positively impacts the regeneration rate.The different effect between iodide and O/S-containing homologous organic dyes are calculated to reveal that the direct reaction between dye+ and I2- is the most favorite regeneration mechanism. Furthermore, the S-substitution in donor unit further decreases ΔG barrier and positively impact the regeneration rate.
Co-reporter:Bai-Yu Li, Xiao-Lin Zhang, Ling-Yu Zhang, Ting-Ting Wang, Lu Li, Chun-Gang Wang, Zhong-Min Su
Dyes and Pigments 2016 Volume 134() pp:178-185
Publication Date(Web):November 2016
DOI:10.1016/j.dyepig.2016.07.014
•Upconversion nanorods are used for blood fingerprints detection.•They show high sensitivity, low background, high efficiency and low toxicity.•They are superior to Acid Yellow 7 (a blood-sensitive reagent) on all substrates.•The whole procedure is facile and convenient to use at the scene of the crime.In this paper, the NaYF4:Yb,Er,Gd fluorescent upconversion nanorods (UCNRs) were successfully employed as a promising fluorescent label for the detection of blood fingerprints with high sensitivity, low background, high efficiency, and low toxicity on a range of substrates including non-infiltrating materials (glass, ceramic tile, aluminium foil, stainless steel sheet, coin and marble), and infiltrating materials (magazine cover and train ticket). To assess the efficiency of this approach, comparisons were performed with Acid Yellow 7 (AY7, an effective blood reagent) on above-mentioned eight substrates. The fingerprints were cut in halves and the halves treated separately with UCNRs and AY7. The results manifested that UCNRs were superior to AY7 on all test substrates. This work shows that UCNRs are a versatile fluorescent label for the detection of blood fingerprints in criminal science for individual identification.NaYF4:Yb,Er,Gd upconversion nanorods were employed as a promising fluorescent label for the detection of blood fingerprints with high sensitivity, low background, high efficiency and low toxicity on a range of substrates including non-infiltrating materials (glass, ceramic tile, aluminium foil, stainless steel sheet, coin and marble), and infiltrating materials (magazine cover and train ticket).
Co-reporter:En-Long Zhou, Chao Qin, Xin-Long Wang, Kui-Zhan Shao and Zhong-Min Su
CrystEngComm 2016 vol. 18(Issue 34) pp:6370-6377
Publication Date(Web):26 Apr 2016
DOI:10.1039/C6CE00021E
Two hybrid materials based on Keggin-type polyoxometalate (POM), [Zn12.5(trz)17(H2O)7(SiW12O40)2]·2H2O (1) and (trz = 1,2,4-triazole) and [Pb2(dpdo)5(H2O)3(SiW12O40)]·H2O (2), (dpdo = 4,4′-bipyridine-N,N′-dioxide), have been hydrothermally synthesized and structurally characterized by elemental analysis, IR spectroscopy, thermal analysis, powder X-ray diffraction analysis and single crystal X-ray diffraction. Compound 1 contains 1D channels and giant cages occupied by Keggin-type POMs. Meanwhile, in compound 2, 2D coordination polymer sheets are constructed from Pb2+ and dpdo ligands which are further pillared by POM clusters into 3D porous frameworks with pcu topology. Interestingly, perpendicular to the 2D sheet, the dpdo ligands hang from both surfaces of the sheet; one end coordinates with the Pb2+ centre, while the other end is uncoordinated. Compounds 1 and 2 both show structural integrity in aqueous solutions in a wide pH range (from 2 to 12) and in common organic solvents (methanol and DMF). Moreover, the optical band gap, photocatalysis activity and electrochemical properties of both compounds have been investigated.
Co-reporter:Liu Yang, Xiao Li, Chao Qin, Kui-Zhan Shao and Zhong-Min Su
CrystEngComm 2016 vol. 18(Issue 25) pp:4765-4771
Publication Date(Web):25 Apr 2016
DOI:10.1039/C6CE00745G
A 3D porous layer metal–organic framework, namely [Cd(BPDC)0.5(L1)(NO3)]·3.4DMF (1), was synthesized under solvothermal conditions. The structure was characterized by single crystal X-ray diffraction analysis, powder X-ray diffraction (PXRD) analysis, thermogravimetric analysis (TGA), infrared spectrophotometry analysis and elemental analysis. The structure of 1 displays a 3D porous layer and can be classified as a (3,4)-connected topology with a point symbol of (42·6·38)2(42·6). The framework is stable to a certain degree. 1 can also be applied as a fluorescent sensor for small-molecule sensing, such as nitrobenzene (NB) and 2,4,6-trinitrophenol (TNP). In addition, 1 is a rare example of MOF-based fluorescent probes targeting environmentally relevant guest species, such as Hg(II) ions in aqueous solution, with high selectivity and sensitivity.
Co-reporter:Wei-Chao Chen, Chao Qin, Xin-Long Wang, Cai-Xia Wu, Yang-Guang Li, Hong-Ying Zang, Kui-Zhan Shao, Zhong-Min Su and En-Bo Wang
CrystEngComm 2016 vol. 18(Issue 16) pp:2820-2824
Publication Date(Web):15 Mar 2016
DOI:10.1039/C6CE00377J
The trimeric organic–inorganic hybrid selenotungstate(IV) [{Sn(CH3)2(CH3COO)}3{Sn(CH3)2}3{Se2W18O62(OH)(H2O)}3]18− is composed of three Wells–Dawson-type {α-Se2W18} fragments and six {(CH3)2Sn2+} groups involving the first reported carboxyl-modified dimethyltin moieties, exhibiting photocatalytic H2 evolution activity.
Co-reporter:Jun Liang, Xue-Song Wu, Xin-Long Wang, Chao Qin, Kui-Zhan Shao, Zhong-Min Su and Rong Cao
CrystEngComm 2016 vol. 18(Issue 13) pp:2327-2336
Publication Date(Web):17 Feb 2016
DOI:10.1039/C5CE02090E
Five new metal–organic rotaxane frameworks (MORFs) have been hydrothermally synthesized by systematically combining cucurbit[6]uril-based pseudorotaxanes, rigid carboxylate ligands and d10 metal ions. Compound 1 presents a three-periodic pillared structure; 2 and 3 are two-periodic layers with hcb topologies; 4 presents a 3D structure with snw topology and 5 is a three-periodic network, featuring from interpenetrating to non-interpenetrating 3D MORFs. The cucurbit [6]uril-based MORFs obtained in this work illustrate the importance of non-covalent interactions around CB[6]s in the assembly of resultant super architectures. Furthermore, their TGA analysis, solid photoluminescence properties and sensitization to lanthanide ions are investigated.
Co-reporter:Wei-Chao Chen, Chao Qin, Xin-Long Wang, Kui-Zhan Shao, Zhong-Min Su, and En-Bo Wang
Crystal Growth & Design 2016 Volume 16(Issue 5) pp:2481
Publication Date(Web):April 8, 2016
DOI:10.1021/acs.cgd.6b00285
The reaction of Na2WO4 and Na2SeO3 in the presence of MnCl2 under moderately acidic conditions yielded two unprecedented tungstoselenites: dimeric K2Na10[K2⊂{MnSe4W23O85(H2O)6}]·29H2O (1) and trimeric wheel-shaped K2Na10[K2⊂{Mn3Se7W39O131(OH)20(H2O)2}]·60H2O (2). The assemblies of 1 and 2 are based upon the structure directing effects of SeIV heteroatoms for generating diverse well-defined vacancy selenotungstate precursors during the formation. The polyoxoanion of 1 contains two novel Wells–Dawson-type-like {Se2W11} fragments, which are constructed from novel {SeW4} and {SeW7} species derived from Wells–Dawson-type {α-Se2W14} fragments and one disorder of Mn/W center. The polyoxoanion of 2 exhibits a crown-type structure composed of a [Se6W38O120(OH)18(H2O)2]6– “host” (abbreviated as {Se6W38}) encapsulating SeO32–-modified Mn/W and two K+ “guests”. Remarkably, the crown {Se6W38} shell remains a new type of {Se2W12}-based trimeric aggregate in the polyoxometalates chemistry. The two compounds were characterized by single-crystal X-ray structure analysis, IR spectroscopy, thermogravimetric, UV/vis spectroscopy, and ESI–MS. Moreover, their photocatalytic H2 evolution activity was also investigated.
Co-reporter:Rong-Lin Zhong, Hong-Liang Xu and Zhong-Min Su
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 20) pp:13954-13959
Publication Date(Web):20 Apr 2016
DOI:10.1039/C6CP00647G
Carbon–boron–nitride heteronanotubes (BNCNT) have attracted a lot of attention because of their adjustable properties and potential applications in many fields. In this work, a series of CA, PA and HA armchair BNCNT models were designed to explore their nonlinear optical (NLO) properties and provide physical insight into the structure–property relationships; CA, PA and HA represent the models that are obtained by doping the carbon segment into pristine boron nitride nanotube (BNNT) fragments circularly around the tube axis, parallel to the tube axis and helically to the tube axis, respectively. Results show that the first hyperpolarizability (β0) of an armchair BNCNT model is dramatically dependent on the connecting patterns of carbon with the boron nitride fragment. Significantly, the β0 value of PA-6 is 2.00 × 104 au, which is almost two orders of magnitude larger than those (6.07 × 102 and 1.55 × 102 au) of HA-6 and CA-6. In addition, the β0 values of PA and CA models increase with the increase in carbon proportion, whereas those of HA models show a different tendency. Further investigations on transition properties show that the curved charge transfer from N-connecting carbon atoms to B-connecting carbon atoms of PA models is essentially the origin of the big difference among these models. This new knowledge about armchair BNCNTs may provide important information for the design and preparation of advanced NLO nano-materials.
Co-reporter:Xiao Li, Liu Yang, Liang Zhao, Xin-Long Wang, Kui-Zhan Shao, and Zhong-Min Su
Crystal Growth & Design 2016 Volume 16(Issue 8) pp:4374-4382
Publication Date(Web):June 23, 2016
DOI:10.1021/acs.cgd.6b00482
Five new luminescent metal–organic frameworks (MOFs), [Cd(bdc)(dia)0.5(MeOH)](DMA)0.5 (1), [Cd2O(1,4-ndc)2(dia)2(H2O)]·DMA (2), [Cd(1,4-ndc)(dia)]·MeOH (3), [Cd(bpdc)(dia)0.5]·DMF (4), [Cd(bpdc) (dia)]·DEF (5) (dia = 9,10-bis(1H-imidazol-1-yl)anthracene), were obtained from solvothermal conditions and characterized by PXRD, IR, TG, and PL spectra. Single-crystal X-ray diffraction analyses showed that all compounds 1–5 display three-dimensional architectures with diverse topologies. Compound 1 was a 6-con pcu network based on dinuclear Cd cluster. Compound 2 features an eight-connected network constructed from a binuclear Cd cluster. Compound 3 presents a 3-fold interpenetrated four-connected dia topology. Compounds 4 and 5 exhibit a 3-fold interpenetrated six-connected pcu framework and a 3-fold interpenetrated four-connected dia net, respectively. Because compounds 3–5 have one-dimensional (1D) channels, the luminescence properties of 3–5 in different solvents were investigated systematically. Furthermore, compounds 3–5 display highly sensitive, selective, and well-recyclable properties in the detection of nitrobenzene (NB) and 2,4,6-trintrophenol (TNP) as fluorescent sensors.
Co-reporter:Yu-Teng Zhang, Xin-Long Wang, En-Long Zhou, Xue-Song Wu, Bai-Qiao Song, Kui-Zhan Shao and Zhong-Min Su
Dalton Transactions 2016 vol. 45(Issue 9) pp:3698-3701
Publication Date(Web):27 Jan 2016
DOI:10.1039/C5DT04764A
Three polyoxovanadate-based metal–organic polyhedra (denoted as VMOP-1, -2, and -3), adopting isostructural discrete octahedral cage geometries, were successfully synthesized under solvothermal conditions. These structures are all built up from the same pentavanadate {V5O9Cl} cluster connected by linear bidentate ligands (H2L1 = H2BDC, H2L2 = H2BDC-NH2, H2L3 = H2BDC-Br), respectively.
Co-reporter:Shao-Fen Huang, Hai-Zhu Sun, Guo-Gang Shan, Yong Wu, Min Zhang and Zhong-Min Su
New Journal of Chemistry 2016 vol. 40(Issue 5) pp:4635-4642
Publication Date(Web):08 Mar 2016
DOI:10.1039/C5NJ03045E
Pyridine-azole moieties have proved to be an attractive building block for multifunctional cationic Ir(III) complexes, however, few highly efficient blue materials have been demonstrated and the deep structure–property relationships need to be revealed. Herein, a series of cationic [Ir(dfppz)2(N∧N)][PF6] complexes (1–4) based on azole-type ancillary ligands, namely, 1,1′-diphenyl-1H,1′H-[2,2′]biimidazolyl (Phbid), 2-(1-phenyl-1H-imidazol-2-yl)-pyridine (Bpyim), 2-(1-methyl-1H-imidazol-2-yl)pyridine (Mpyim), and 2-(1,5-dimethyl-1H-[1,2,4]triazole-3-yl)pyridine (Mpytz), respectively, have been prepared, and their photophysical, electrochemical and charge transporting properties are investigated. The reported complexes exhibit strong perceived green to blue emission as well as excellent redox reversibility at room temperature. Comprehensive density functional theory calculations are performed to provide insight into the electronic structures of 1–4 and disclose the ancillary ligand effect on the emission behavior in detail. The simple methyl group modification endows the triazole-type ancillary with an exceedingly large π–π* energy gap, resulting in the blue emitting complex 4, namely [Ir(dfppz)2(Mpytz)][PF6] with a peak value at 462 nm. Meanwhile, despite the significant 3LC character, the high efficiency with a quantum yield of 31.6% of 4 is realized in neat film, which is higher than those of reported cationic Ir(III) complexes with similar emissions. Additionally, the calculation results also suggest that complexes 2–4 possess better electron-transporting abilities in comparison to those of 1.
Co-reporter:Hongzhu Xing, Dashu Chen, Xingyu Li, Yue Liu, Chungang Wang and Zhongmin Su
RSC Advances 2016 vol. 6(Issue 71) pp:66444-66450
Publication Date(Web):12 Jul 2016
DOI:10.1039/C6RA12134A
Visible-light-induced controlled/living radical polymerization (C/LRP) mediated by photoactive zirconium metal–organic framework (Zr–MOF) NNU-28 has been explored. The photopolymerization is carried out at room temperature under 520 nm monochromatic light. The results show that anthracene-based NNU-28 is a promising heterogeneous sensitizer to initiate the photopolymerization where the robust Zr–MOF exhibits enhanced visible light absorption and characteristics of photoinduced charge generation. The NNU-28 mediated CRP relies on the in situ photoreduction of a Cu(II) complex deactivator to Cu(I) complex activator through an electron transfer process. The photopolymerization of methacrylates shows typical characteristics of controlled radical polymerization, where the as-prepared polymers have controlled molecular weights, narrow molecular weight distribution and high retention of chain-end groups. Besides, the NNU-28 mediated photopolymerization can be temporally and spatially controlled by light. The visible-light-induced polymerization investigated here suggests the promising application of photoactive MOFs for well-designed polymer synthesis.
Co-reporter:Li-Li Wen, Jing Yu, Hai-Zhu Sun, Guo-Gang Shan, Kai-Yue Zhao, Wen-Fa Xie, Zhong-Min Su
Organic Electronics 2016 Volume 35() pp:142-150
Publication Date(Web):August 2016
DOI:10.1016/j.orgel.2016.05.002
•Three Ir(III) dyes with different substituents on 1,2-diphenyl-1H-benzoimidazole ligands are designed.•They exhibit bright lights with almost identical emissions in neat films.•The relationship between the chemical structures and EL performances were investigated.•Non-doped device N3 shows high efficiencies of 18.6 cd A−1 and 16.2 lm W−1.•Device N3 simultaneously displays low efficiency roll-off at high luminance.To construct efficient emitters suitable for non-doped devices and deeply understand the relationship between structures and performances, we designed and synthesized two heteroleptic iridium(III) complexes based on 1,2-diphenyl-1H-benzoimidazole (PBI) ligands whose substituents are varied simply from methyl (complex 2) to tert-butyl groups (complex 3). The parent complex 1 with non-substituent on PBI ligand has also been presented for a better comparison. Their photophysical, electrochemical and electroluminescent (EL) performances are investigated systematically. Despite their structural modification, all complexes exhibit almost identical emission and excited-state characters, which are rationalized by the quantum-chemical calculations. However, the obvious differences on device performances are found. Non-doped device employing 3 as emitting layer displays the highest EL performance with maximum current efficiency (ηc, max) of 18.6 cd A−1 and power efficiency (ηp, max) of 16.2 lm W−1 accompanied by low efficiency roll-off values, which is much higher than those of complexes 1 and 2. The obtained results herein suggest that introduction of the simple substituent into PBI ligand is an effective and feasible approach to develop highly efficient non-doped phosphors.To construct efficient emitters suitable for non-doped devices and deeply understand the relationship between structures and performances, three heteroleptic Ir(III) dyes employing modified 1,2-diphenyl-1H-benzoimidazole ligands whose substituents are varied simply from non-substituent (1) to tert-butyl groups (3) are synthesized. Non-doped device using 3 as emitting layer displays the highest EL performance with ηc, max of 18.6 cd A−1 and ηp, max of 16.2 lm W−1 accompanied by low efficiency roll-off values.
Co-reporter:Rong-Lin Zhong, Hong-Liang Xu, Zhong-Min Su
Chemical Physics Letters 2016 Volume 658() pp:230-233
Publication Date(Web):1 August 2016
DOI:10.1016/j.cplett.2016.06.054
•The physical properties of Li@BCN models are dependent on the different chemical environment of the tube termination.•The crucial electron population in the BCNs of Li@B-BCN and Li@N-BCN series is significantly different.•The first hyperpolarizabilities of Li@N-BCN series are dramatically larger than that of Li@B-BCN series.A series of Li@BCN models were systematically investigated to explore the physical origin of the interaction between lithium atoms and BCNs. Theoretical results show that the crucial electron population in the BCNs of Li@B-BCN and Li@N-BCN series is dramatically different. As results, the first hyperpolarizability of Li@B-BCN series increases with the increase of carbon proportion whereas that of Li@N-BCN series significantly decreases with the increase of carbon proportion. The results indicate that the physical properties of Li@BCN models are significantly dependent on the different chemical environment of the tube termination.
Co-reporter:Bao-Li Li, Hai-Ning Wang, Liang Zhao, Guang-Zhe Li, Zhong-Min Su
Inorganic Chemistry Communications 2016 Volume 66() pp:87-89
Publication Date(Web):April 2016
DOI:10.1016/j.inoche.2015.12.019
•A new metal–organic framework has been obtained and described.•It possesses a non-interpenetrating pillar-layer framework with 1D channels.•It can be used as a luminescent probe to detect small molecule acetone.•Its luminescent intensity decreases with the increase of amounts of acetone.The reactions of 4,4-bipyridine and 5-Hydroxyisophthalic acid with Zn(NO3)2·3H2O in N,N′-dimethylacetamide lead to the formation of [Zn(5-hip)(bpy)]·2DMA (1). Compound 1 possesses a non-interpenetrating pillar-layer framework with 1D rectangular-shape channels. The experimental results show that the luminescent intensity of 1 highly depends on small solvent molecules, particularly CH3OH and acetone. Compound 1 can be used as a luminescent probe to detect small molecule acetone.The reactions of 4,4-bipyridine and 5-Hydroxyisophthalic acid with Zn(NO3)2·3H2O in N,N′-dimethylacetamide lead to the formation of [Zn(5-hip)(bpy)]·2DMA (1). Compound 1 can be used as a luminescent probe to detect small molecule acetone. The results show that the luminescent intensity of 1 highly depends on small molecule acetone. The luminescent intensity decreases gradually with the increase of amounts of acetone.
Co-reporter:Weilong Che, Tiecheng Yu, Dan Jin, Xinyao Ren, Dongxia Zhu, Zhongmin Su, Martin R. Bryce
Inorganic Chemistry Communications 2016 Volume 69() pp:89-93
Publication Date(Web):July 2016
DOI:10.1016/j.inoche.2016.03.025
•2-(2′-Hydroxyphenyl)-2-oxazoline is shown to be a highly-selective Zn2 + sensor with very simple molecular structure. It demonstrates an excellent fluorescence “turn-on” response to Zn2 + in aqueous medium even in the presence of other competing anions and detection capability for studying the distribution of Zn2 + in living human HeLa cells as a proof-of-concept.2-(2′-Hydroxyphenyl)-2-oxazoline is shown to be a highly-selective Zn2 + sensor with very simple molecular structure. It demonstrates an excellent fluorescence “turn-on” response to Zn2 + in aqueous medium even in the presence of other competing anions and detection capability for studying the distribution of Zn2 + in living human HeLa cells as a proof-of-concept.2-(2′-Hydroxyphenyl)-2-oxazoline is shown to be a highly-selective Zn2 + sensor with very simple molecular structure.
Co-reporter:Ying Gao, Tan Su, Yong Wu, Yun Geng, Min Zhang, Zhong-Min Su
Chemical Physics Letters 2016 Volume 666() pp:7-12
Publication Date(Web):1 December 2016
DOI:10.1016/j.cplett.2016.10.069
•The ΔEST of compounds 2–5 is reduced largely, but the increase of kr is small.•The compound 6 not only reduces the ΔEST but also enhances kr.•The kr of compound 7 is comparable with that of compound 6, but the ΔEST is large.Based on a thermally activated delayed fluorescence (TADF) triptycene compound 1, compounds 2–7 were designed by varying electron-donating units (2–5) and increasing π-conjugation length (6 and 7). The results indicate that singlet-triplet energy splitting (ΔEST) of compounds 2–5 is reduced largely, but their improvement of radiative decay rate (kr) is slightly small. However, the compound 6 not only reduces the ΔEST but also enhances kr. For compound 7, the kr value is comparable with compound 6, but ΔEST value is quite large. Therefore, possessing an appropriate π-conjugation length might be helpful to improve TADF performances for the materials investigated here.Based on three-dimensional donor-acceptor triptycene activated delayed fluorescence (TADF) compound 1, we have designed six compounds 2–7 to investigate the effect of different electron-donating units and increasing the π-conjugation length on their a series of photoelectric properties related to the performance of these materials in OLEDs through using density functional methods. For a highly efficient TADF material, it is necessary to possess a small singlet-triplet splitting (ΔEST) and a reasonable radiative decay rate (kr). The calculated results indicate that: by varying the electron-donating units, the ΔEST values of compounds 2–5 are reduced by a great degree, but their improvement of kr values is slightly small. However, increasing the π-conjugation length for compound 6 not only reduces ΔEST value, but also enhances kr, indicating that compound 6 might be potential TADF materials.
Co-reporter:Han-Ni Wu, Jing Wang, Hong Li, Na-Na Ma, Ting Zhang, Shao-Qing Shi, Li-Kai Yan, Zhong-Min Su
Computational and Theoretical Chemistry 2016 Volume 1089() pp:28-34
Publication Date(Web):1 August 2016
DOI:10.1016/j.comptc.2016.05.003
•The effects of different functionals on the bond length of [PW12O40]3− were studied.•The effects of different functionals on the FMO energies of [PW12O40]3− were systematically investigated.•B3P86 with 13% HF exchange offers the reasonable results of M3PW12O40, M4SiW12O40 and M6P2W18O62 (M = Na, K).To obtain more reasonable Frontier Molecular Orbital (FMO) energies of α-Keggin anion [PW12O40]3−, the influence of different density functional theory (DFT) functionals were systematically investigated. Except LC-ωPBE, M06HF, LC-BLYP, HF and LC-BP86, the functionals we tested overestimate the bond length with mean unsigned error (MUE) values ranging from 0.02 Å to 0.03 Å. All the functionals used show the typical trend that the FMO energy levels are overestimated. The hybrid generalized gradient approximation (hGGA), such as B3P86 (MUE 0.465 eV), performs better than others. The results on Na3PW12O40 are more reasonable than those of [PW12O40]3− as the errors caused by the negative charge were compensated. B3P86 is superior to B3PW91 and B3LYP in the description on Na3PW12O40 with MUE 0.461 eV and mean signed error (MSE) 0.043 eV. B3P86 with 13% HF exchange offers the reasonable MUEs (≈0.12 eV) for M3PW12O40, M4SiW12O40 and M6P2W18O62 (M = Na, K).
Co-reporter:Hui-Ting Mao, Chun-Xiu Zang, Li−Li Wen, Guo-Gang Shan, Hai-Zhu Sun, Wen-Fa Xie, and Zhong-Min Su
Organometallics 2016 Volume 35(Issue 22) pp:3870-3877
Publication Date(Web):November 15, 2016
DOI:10.1021/acs.organomet.6b00753
Five neutral heteroleptic Ir(III) complexes 1–5 using the same cyclometalated ligand and different pyridine-1,2,4-triazolyl derivatives as ancillary ligands with fluorine substituents attached, were rationally designed and prepared. Their photophysical, electrochemical, and thermal properties were studied, and theoretical calculations were performed to understand the emission behaviors as well. Introducing fluorine atoms has little effect on the photophysical and thermal properties, but the performances of the resulting devices can be fine-tuned. Among them, a heavy doping level device employing a phosphor with five fluorine atoms delivers superior device efficiencies with ηc = 32.6 cd A–1 and ηp = 27.6 lm W–1, respectively, which is higher than those of other counterparts. Importantly, such a device exhibits almost negligible roll-off in luminance efficiency. Despite nondoped devices achieving good EL performance, more fluorine atoms lead to a relatively higher efficiency roll-off. The results suggest that rational incorporation of fluorine atoms into the ancillary ligands can significantly improve the performance of devices with features of high efficiency and small roll-off.
Co-reporter:Kai-Yue Zhao, Guo-Gang Shan, Qiang Fu, and Zhong-Min Su
Organometallics 2016 Volume 35(Issue 23) pp:3996-4001
Publication Date(Web):November 23, 2016
DOI:10.1021/acs.organomet.6b00788
A new design strategy to tune emission color of aggregation-induced emission (AIE) Ir(III) complexes, by simply adjusting the strength of donor/acceptor on ancillary ligands, is reported. Herein, all studied Ir(III) complexes employ 1-(2,4-difluorophenyl)-1H-pyrazole as cyclometalated ligands but different pyridine-1,2,4-triazolyl moieties as ancillary ligands that were modified with carbazole end-capped alkyl groups, in which pyridine-1,2,4-triazolyl moieties and functionalized carbazole act as donor and acceptor units, respectively. The experimental and theoretical results clearly reveal the intrinsic relationship between structures and emission behaviors. Enhancing the ability of donor and/or acceptor leads to red-shifted emission and vice versa. In addition, one of the designed dyes exhibits the reversible and significant mechanochromic luminescent behavior thanks to its efficient AIE characteristic and structural modification, providing a feasible way to design multifunctional materials.
Co-reporter:Xianchun Liu;Dashu Chen;Lin Chen;Renxi Jin;Dr. Shuangxi Xing;Dr. Hongzhu Xing; Yan Xing; Zhongmin Su
Chemistry - A European Journal 2016 Volume 22( Issue 27) pp:9293-9298
Publication Date(Web):
DOI:10.1002/chem.201600894
Abstract
In this paper, a facile strategy is reported for the preparation of well-dispersed Pt nanoparticles in ordered mesoporous silica (Pt@OMS) by using a hybrid mesoporous phenolic resin-silica nanocomposite as the parent material. The phenolic resin polymer is proposed herein to be the key in preventing the aggregation of Pt nanoparticles during their formation process and making contributions both to enhance the surface area and enlarge the pore size of the support. The Pt@OMS proves to be a highly active and stable catalyst for both gas-phase oxidation of CO and liquid-phase hydrogenation of 4-nitrophenol. This work might open new avenues for the preparation of noble metal nanoparticles in mesoporous silica with unique structures for catalytic applications.
Co-reporter:Jun-Sheng Qin; Dong-Ying Du; Wei Guan; Xiang-Jie Bo; Ya-Fei Li; Li-Ping Guo; Zhong-Min Su; Yuan-Yuan Wang; Ya-Qian Lan;Hong-Cai Zhou
Journal of the American Chemical Society 2015 Volume 137(Issue 22) pp:7169-7177
Publication Date(Web):May 1, 2015
DOI:10.1021/jacs.5b02688
Two novel polyoxometalate (POM)-based metal–organic frameworks (MOFs), [TBA]3[ε-PMoV8MoVI4O36(OH)4Zn4][BTB]4/3·xGuest (NENU-500, BTB = benzene tribenzoate, TBA+ = tetrabutylammonium ion) and [TBA]3[ε-PMoV8MoVI4O37(OH)3Zn4][BPT] (NENU-501, BPT = [1,1′-biphenyl]-3,4′,5-tricarboxylate), were isolated. In these compounds, the POM fragments serving as nodes were directly connected with organic ligands giving rise to three-dimensional (3D) open frameworks. The two anionic frameworks were balanced by TBA+ ions residing inside the open channels. They exhibit not only good stability in air but also tolerance to acidic and basic media. Furthermore, they were employed as electrocatalysts for the hydrogen evolution reaction (HER) owing to the combination of the redox activity of a POM unit and the porosity of a MOF. Meanwhile, the HER activities of ε(trim)4/3, NENU-5, and HKUST-1 were also studied for comparison. Remarkably, as a 3D hydrogen-evolving cathode operating in acidic electrolytes, NENU-500 exhibits the highest activity among all MOF materials. It shows an onset overpotential of 180 mV and a Tafel slope of 96 mV·dec–1, and the catalytic current density can approach 10 mA·cm–2 at an overpotential of 237 mV. Moreover, NENU-500 and NENU-501 maintain their electrocatalytic activities after 2000 cycles.
Co-reporter:Tengying Ma, Shizheng Wen, Caixia Wu, Likai Yan, Min Zhang, Yuhe Kan and Zhongmin Su
Journal of Materials Chemistry A 2015 vol. 3(Issue 39) pp:10085-10090
Publication Date(Web):29 Jul 2015
DOI:10.1039/C5TC00792E
The electronic and transport properties of a series of 11-ASiNRs (armchair silicene nanoribbons) at different torsion angles were studied by using density functional theory combined with nonequilibrium Green's function method. Several key factors determining the transport properties, such as the electron transmission coefficient and band structure, have been discussed. The interesting results suggest that the transport properties of ASiNRs are insensitive to the torsional silicene nanoribbon configuration in the scattering region. With the increase of the torsion angle, the transmission coefficient is still well maintained within the limits of the torsion angle. Although the torsion angle is increased to 120°, the current dropped by just 22% compared to the initial 11-ASiNRs at a torsion angle of 0°. Furthermore, all the configurations of 11-ASiNRs in this study behave as conventional conductors with nearly linear current–voltage dependence. On the basis of these distinctive transport properties with metabolic structure, ASiNRs present potential promising applications in silicon-based electronic nanodevices.
Co-reporter:Shu-Ran Zhang, Jing Li, Dong-Ying Du, Jun-Sheng Qin, Shun-Li Li, Wen-Wen He, Zhong-Min Su and Ya-Qian Lan
Journal of Materials Chemistry A 2015 vol. 3(Issue 46) pp:23426-23434
Publication Date(Web):12 Oct 2015
DOI:10.1039/C5TA07427D
In this work, a novel microporous anionic metal–organic framework (MOF), [Zn(ABTC)0.5(NO3)][(CH3)2NH2]·DMA·3H2O (NENU-505; NENU = Northeast Normal University; H4ABTC = 3,3′,5,5′-azobenzenetetracarboxylic acid; DMA = N,N-dimethylacetamide), has been rationally synthesized under solvothermal conditions. Single-crystal X-ray analysis reveals that NENU-505 is a (4,4)-connected 3D network with pts topology. Charge neutrality is achieved by [(CH3)2NH2]+ ions. It is noteworthy that NENU-505 displays high stability in air for more than two months. In particular, the adsorption ability of NENU-505 toward ionic dyes has been also investigated. According to the UV/vis spectroscopy analysis and the colour variance of NENU-505, we found that the cationic dyes could be efficiently adsorbed over a period of time, while the neutral and anionic dyes could not be adsorbed. Therefore, NENU-505 exhibits selective adsorption toward cationic dyes and can potentially serve as a column-chromatographic filler for the separation of dye molecules. Furthermore, the cationic dyes can be gradually released in the presence of NaCl. More interestingly, when NENU-505 was immersed in different metal ion DMA solutions, it performs as a rare example of a highly selective and sensitive sensor for Cr3+ ions. In connection to this, the probable sensing mechanism was also further investigated in detail in this paper. Remarkably, this is the first MOF to exhibit an excellent ability for the detection and adsorption of Cr3+ ions in a convenient, economical, and environmentally friendly manner.
Co-reporter:En-Long Zhou, Peng Huang, Chao Qin, Kui-Zhan Shao and Zhong-Min Su
Journal of Materials Chemistry A 2015 vol. 3(Issue 14) pp:7224-7228
Publication Date(Web):02 Mar 2015
DOI:10.1039/C5TA00231A
A stable luminescent anionic metal–organic framework with unprecedented topological structure was constructed, which showed a certain degree of framework stability in water and acid/base solutions. Furthermore, the compound showed moderate CO2 adsorption and extremely selective detection of 2,4,6-trinitrophenol (TNP) in both an organic solvent and aqueous solution.
Co-reporter:Xianchun Liu, Renxi Jin, Dashu Chen, Lin Chen, Shuangxi Xing, Hongzhu Xing, Yan Xing and Zhongmin Su
Journal of Materials Chemistry A 2015 vol. 3(Issue 8) pp:4307-4313
Publication Date(Web):09 Dec 2014
DOI:10.1039/C4TA06049K
In this study, we report a simple in situ auto-reduction strategy for the fabrication of Ag@FDU-15 nanocomposites, in which the small sized Ag nanoparticles are monodispersed in the channels of FDU-15 ordered mesopolymers. The as-prepared Ag@FDU-15 nanocomposites were characterized by small and wide angle X-ray diffraction (XRD), nitrogen sorption, thermo-gravimetric analysis (TGA), energy dispersive X-ray spectroscopy (EDS), X-ray photoelectron spectroscopy (XPS), transmission electron microscopy (TEM) and scanning transmission electron microscopy (STEM). TEM results show that the silver nanoparticles are uniformly dispersed with a mean diameter of 5.6 ± 0.5 nm. When used as a hydrogenation catalyst, the Ag@FDU-15 nanocomposites exhibit excellent catalytic performance and reusability for the reduction of 4-nitrophenol (4-NP) in the presence of NaBH4.
Co-reporter:Shao-Juan Bao, Rajamani Krishna, Ya-Bing He, Jun-Sheng Qin, Zhong-Min Su, Shun-Li Li, Wei Xie, Dong-Ying Du, Wen-Wen He, Shu-Ran Zhang and Ya-Qian Lan
Journal of Materials Chemistry A 2015 vol. 3(Issue 14) pp:7361-7367
Publication Date(Web):30 Jan 2015
DOI:10.1039/C5TA00256G
An air-stable tetrazolate-containing framework, [Zn2L2]·2DMF (NENU-520, H2L = 4-(1H-tetrazole-5-yl)biphenyl-4-carboxylic acid), with uncoordinated N atoms on its internal surface was solvothermally synthesized and structurally characterized. This metal–organic framework (MOF) exhibited high CO2 uptake of 79.9 cm3 cm−3 at 298 K and 100 kPa, as well as excellent adsorption selectivity for CO2 over CH4 and N2. Particularly, its exceptionally high selectivity of CO2 over N2 at 298 K has ranked NENU-520 among the highest MOFs for selective CO2 separation. Furthermore, the potential application of NENU-520 for the fixed bed pressure swing adsorption (PSA) separation of CO2 from CH4 and N2 has been validated via simulated breakthrough experiments. The small channel with the size of 3.6 Å, combined with CO2-accessible free nitrogen atoms directed toward the inner surface, is believed to contribute to its high CO2 uptake capacity and selectivity. Thus, this work represents a unique way to target MOF materials for highly selective CO2 separation by incorporating CO2-philic functional sites on pore surfaces, and at the same time optimizing pore sizes.
Co-reporter:Xiao-Li Hu, Chao Qin, Xin-Long Wang, Kui-Zhan Shao and Zhong-Min Su
Chemical Communications 2015 vol. 51(Issue 99) pp:17521-17524
Publication Date(Web):23 Sep 2015
DOI:10.1039/C5CC07004J
An anionic metal–organic framework (MOF) with 1D nanotube channels has been constructed. The charge and size dependent ion-exchange of cationic dyes was investigated. Rho@1 could be used as a dual-emitting fluorescent sensor for sensing explosives by self-referencing energy transfer behaviors.
Co-reporter:Bai-Qiao Song, Xin-Long Wang, Yu-Teng Zhang, Xue-Song Wu, Hong-Sheng Liu, Kui-Zhan Shao and Zhong-Min Su
Chemical Communications 2015 vol. 51(Issue 46) pp:9515-9518
Publication Date(Web):30 Apr 2015
DOI:10.1039/C5CC02639C
A unique cationic metal–organic framework was synthesized by connecting the neutral rod-shaped secondary building unit with a cationic dicarboxylate ligand. This framework showed a rare snub square tessellation pattern by the periodic tiling of triangular and square nanotubes. The charge- and size-dependent ion-exchange of anion dyes was investigated.
Co-reporter:Yan-Qing Jiao, Hong-Ying Zang, Xin-Long Wang, En-Long Zhou, Bai-Qiao Song, Chun-Gang Wang, Kui-Zhan Shao and Zhong-Min Su
Chemical Communications 2015 vol. 51(Issue 56) pp:11313-11316
Publication Date(Web):01 Jun 2015
DOI:10.1039/C5CC03357H
The first polyoxometalate-based metal–organic nanotube constructed via covalent bonds has been synthesized. POM anions stick the metal–organic nanotubes to build 3D nanotubular arrays. The stability, magnetic and proton conducting properties are investigated.
Co-reporter:Jun-Sheng Qin;Ji-Chuan Zhang;Min Zhang;Dong-Ying Du;Jing Li;Yuan-Yuan Wang;Si-Ping Pang;Sheng-Hua Li;Ya-Qian Lan
Advanced Science 2015 Volume 2( Issue 12) pp:
Publication Date(Web):
DOI:10.1002/advs.201500150
Co-reporter:Wei Xie; Shu-Ran Zhang; Dong-Ying Du; Jun-Sheng Qin; Shao-Juan Bao; Jing Li; Zhong-Min Su; Wen-Wen He; Qiang Fu;Ya-Qian Lan
Inorganic Chemistry 2015 Volume 54(Issue 7) pp:3290-3296
Publication Date(Web):March 13, 2015
DOI:10.1021/ic5029383
A stable porous carbazole-based luminescent metal–organic framework, NENU-522, was successfully constructed. It is extremely stable in air and acidic/basic aqueous solutions, which provides the strategy for luminescent material encapsulation of Ln3+ ions with tunable luminescence for application in light emission. More importantly, Ln3+@NENU-522 can emit white light by encapsulating different molar ratios of Eu3+ and Tb3+ ions. Additionally, Tb3+@NENU-522 is found to be useful as a fluorescent indicator for the qualitative and quantitative detection of nitroaromatic explosives with different numbers of −NO2 groups, and the concentrations of complete quenching are about 2000, 1000, and 80 ppm for nitrobenzene, 1,3-dinitrobenzene, and 2,4,6-trinitrophenol, respectively. Meanwhile, Tb3+@NENU-522 displays high selectivity and recyclability in the detection of nitroaromatic explosives.
Co-reporter:Yu-Teng Zhang, Chao Qin, Xin-Long Wang, Peng Huang, Bai-Qiao Song, Kui-Zhan Shao, and Zhong-Min Su
Inorganic Chemistry 2015 Volume 54(Issue 23) pp:11083-11087
Publication Date(Web):November 23, 2015
DOI:10.1021/acs.inorgchem.5b01936
An unprecedented octavanadium-substituted polyoxoniobate Na18[Nb48V8(OH)30O130]·33H2O (1), with a multiple-strand wheel structure, was successfully synthesized via a conventional aqueous method, which represents the largest vanadoniobate cluster reported to date. Single-crystal X-ray diffraction, ESI-MS spectrum, IR spectra, and UV–vis spectra were investigated. In addition, photocatalytic H2 evolution activity for 1 under UV light was observed with TEA as a sacrificial electron donor.
Co-reporter:Lihao Wang, Lin Xu, Ya Wang, Zhongmin Su, Ran Liu
Electrochimica Acta 2015 Volume 155() pp:1-7
Publication Date(Web):10 February 2015
DOI:10.1016/j.electacta.2014.11.114
In this work, we fabricate a ternary nanocomposite photoelectrode consisting of TiO2 nanoparticles, polyoxometalate (P2W18) and copper quantum dots (Cu QDs) so as to create a synergistic effect of the ternary nanocomposite on photovoltaic and photoelectrocatalytic performances. Both the high electron mobility from low cost Cu QDs and the high electro-hole separation efficiency from P2W18 could evidently improve the photoelectrochemical performance of TiO2. Transient photocurrent and current-voltage curves measurements demonstrate that both the photocurrent response and power conversion efficiency of the P2W18/Cu/TiO2 film were markedly enhanced in comparison with that of the only TiO2 film and the P2W18/TiO2 film, respectively. Furthermore, the photoelectrode also exhibited significant photoelectrocatalytic activity for formic acid oxidation. The substitute of copper quantum dots for noble metals in electrode is a favorable innovation for the practical application of photoelectrochemical devices.
Co-reporter:Hong-Tao Cao, Guo-Gang Shan, Yong-Ming Yin, Hai-Zhu Sun, Yong Wu, Wen-Fa Xie, Zhong-Min Su
Dyes and Pigments 2015 Volume 112() pp:8-16
Publication Date(Web):January 2015
DOI:10.1016/j.dyepig.2014.06.014
•Four Ir(III)-based dyes with 1,2-diphenyl-1H-benzoimidazole ligands were prepared.•They displayed green emissions around 492 nm with high quantum yields in CH2Cl2.•Their electroluminescence properties were successfully investigated.•A non-doped device displayed high efficiencies of 19.8 cd A−1 and 20.4 lm W−1.•A device simultaneously displayed low efficiency roll-off at high luminance.Four novel iridium(III) complexes containing 1,2-diphenyl-1H-benzoimidazole as cyclometalated ligands were successfully synthesized and characterized. The complexes displayed strong emissions around 492 nm with high photoluminescence quantum yields of 70–92% in dichloromethane solution at 298 K. Doped OLEDs based on the complexes were prepared, which showed a peak current efficiency of 34.5 cd A−1, power efficiency of 40.1 lm W−1. Non-doped OLEDs using the complexes as emitters were then fabricated for further investigation of their electroluminescence properties. Encouragingly, one non-doped device possessed outstanding performance with a maximum current efficiency of 19.8 cd A−1 and power efficiency of 20.4 lm W−1 whilst simultaneously displaying low efficiency roll-off at high luminance.Novel kinds of iridium(III) complexes with 1,2-diphenyl-1H-benzoimidazole ligands were prepared to fabricate high-performance non-doped OLEDs, which showed favorable performance with a maximum ηc of 19.8 cd A−1 and ηp of 20.4 lm W−1 accompanied by low efficiency roll-off at high luminance.
Co-reporter:Yang Cui, Yong-Min Yin, Hong-Tao Cao, Min Zhang, Guo-Gang Shan, Hai-Zhu Sun, Yong Wu, Zhong-Min Su, Wen-Fa Xie
Dyes and Pigments 2015 Volume 119() pp:62-69
Publication Date(Web):August 2015
DOI:10.1016/j.dyepig.2015.03.024
•Three pyridine-azole-based dyes modified by tetraphenylethene unit were prepared.•The dyes show intrinsic aggregated-induced emission features.•They dyes also exhibit the piezochromism with a high contrast ratio.•The emission color between blue and green can be reversibly and quickly switched.•Non-doped electroluminescent devices with ηc of 2.3 cd A−1 and ηp of 2.0 lm W−1 are achieved.In this work, three tetraphenylethene-functionalized pyridine-azole derivatives were successfully synthesized and characterized. Their photophysical properties in both solution and solid-state were investigated systematically. All luminogens are almost non-emissive in solution but highly emissive in the aggregated states, showing aggregated-induced emission. Importantly, their crystalline aggregates exhibit effective piezochromism with high contrast in both emission color and intensity. The emission color between blue and green can be reversibly and quickly switched by a grinding-heating process several times without any deterioration. The experimental data clearly demonstrate that the interconversion between crystalline and amorphous states is response for the present piezochromism. Non-doped electroluminescence devices using the dyes as light-emitting layers were fabricated. The devices display a peak current efficiency of 2.3 cd A−1 and power efficiency of 2.0 lm W−1, respectively. The obtained results will be useful in designing new efficient multifunctional materials and enriching piezochromic luminescent systems as well.Pyridine-azole based dyes functionalized with a tetraphenylethene moiety were synthesized and showed intrinsic AIE features and piezochromism. Efficient non-doped OLEDs based on the dyes were characterized.
Co-reporter:Hong-Tao Cao, Guo-Gang Shan, Yong-Ming Yin, Hai-Zhu Sun, Yong Wu, Wen-Fa Xie, Zhong-Min Su
Dyes and Pigments 2015 Volume 113() pp:655-663
Publication Date(Web):February 2015
DOI:10.1016/j.dyepig.2014.10.003
•Three Ir(III)-based dyes with N-heterocyclic phenyltriazole ligands are prepared.•They display blue emissions at 460–466 nm in CH2Cl2 at 298 K.•Their photoluminescence and electroluminescence properties are investigated.•They possess increased photoluminescence efficiencies with 14% and 18%.•They exhibit improved electroluminescence efficiencies with 4.1 and 4.6 lm W−1.Novel kinds of blue-emitting heteroleptic iridium(III) complexes (fpdmtz)2Ir(mpypz) (1), (fpmptz)2Ir(mpypz) (2) and (fpmptz)2Ir(pypz) (3) with N-heterocyclic phenyltriazole ligands were designed and synthesized under the guidance of theoretical calculations. Especially, the effect of substituent groups on their emission properties was systematically studied through introducing methyl and propyl moieties into the cyclometalated and ancillary ligands. The experimental results showed that 2 and 3 modified by propyl groups exhibited higher photoluminescence and electroluminescence efficiencies than 1 modified by methyl moiety in its cyclometalated ligands. Such greatly improved emission efficiencies were tentatively attributed to the suppressed intermolecular interactions in solid state caused by the introduction of propyl group, indicating that the photoluminescence and electroluminescence properties of iridium(III) complexes can be manipulated just by modifying simple small molecule groups into the cyclometalated ligands.Novel kinds of blue-emitting iridium(III) complexes adopting N-heterocyclic phenyltriazole ligands were designed and synthesized, which showed improved efficiencies through introducing propyl group into the cyclometalated ligands.
Co-reporter:Kun Zhou, Chao Qin, Li-Kai Yan, Wen-E Li, Xin-Long Wang, Hai-Ning Wang, Kui-Zhan Shao, Zhong-Min Su
Dyes and Pigments 2015 Volume 113() pp:299-306
Publication Date(Web):February 2015
DOI:10.1016/j.dyepig.2014.08.028
Co-reporter:Ting Zhang, Wei Guan, Likai Yan, Tengying Ma, Jing Wang and Zhongmin Su
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 7) pp:5459-5465
Publication Date(Web):22 Dec 2014
DOI:10.1039/C4CP04890C
Polyoxometalate (POM)-based organic–inorganic hybrid systems II1–II7 are designed as p-type dyes containing double D–A1–π–A2 chains. The A1 spacers are thiophene, 1,2,3-triazole, 1,3,4-oxadlazole, thienothiadiazole units or their combinations and the A2 spacer is hexamolybdate. The electronic structures, absorption spectra, and electronic transition characteristics of systems were systematically studied on the basis of density functional theory (DFT) and time-dependent DFT (TDDFT). The highest occupied molecular orbital (HOMO) levels of systems II1–II7 were below the valence bond (VB) of NiO and the lowest unoccupied molecular orbital (LUMO) levels of studied systems were higher than the I2/I3− redox level, which benefit hole injection and dye regeneration. The HOMOs of systems II1–II4 were predominantly delocalized over the organic groups and MoN, which are more helpful to hole injection than systems II5–II7. Introduction of thienothiadiazole units is an effective way to improve the light absorption ability of dyes, and inserting thiophene and 1,2,3-triazole as A1 spacers can increase the efficiency of dye in dye-sensitized solar cells (DSSC).
Co-reporter:Yong Wu, Guo-Gang Shan, Hai-Bin Li, Shui-Xing Wu, Xin-Yao Ren, Yun Geng and Zhong-Min Su
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 4) pp:2438-2446
Publication Date(Web):01 Dec 2014
DOI:10.1039/C4CP04919E
The geometries, electronic structures, photophysical properties and spin–orbit coupling (SOC) effects in the radiative process for the recently synthesized complexes (Bppy)Pt(acac) (1) and (BNppy)Pt(acac) (2) as well as the designed complexes 3–6 were investigated by DFT and TD-DFT calculations, to reveal the influences of the functional ligands on charge injection ability and phosphorescence efficiency of emitters. It is found that compared with electron acceptor complex 1, complexes 2–6 have lower ionization potentials and comparable high electronic affinities, which are suited for bipolar luminescent materials. The results also demonstrated that Bppy complexes 1, 5 and 6 have more 3MLCT compositions in T1 emitting states compared with BNppy complexes 2–4, which results in strong SOC and fast kr. Thus, the phosphorescence efficiency of 1 is higher than that of 2. In addition, 5 and 6 have the balanced charge transport and better hole injection ability when the hole-transporting ligand is incorporated to 1. Therefore, 5 and 6 can server as promising candidates for efficient multifunctional phosphorescent OLED emitters owing to their ambipolar characters, balanced charge carrier injection/transport features and high phosphorescence quantum efficiency.
Co-reporter:Caixia Wu, Tengying Ma, Likai Yan, Ting Zhang and Zhongmin Su
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 17) pp:11521-11526
Publication Date(Web):30 Mar 2015
DOI:10.1039/C4CP06042C
Quantum chemical calculations were performed to explore the structural and electronic properties of the two polyoxoaurates, [AuIII4AsV4O20]8− (Au4As4) and [AuIII4SeIV4O16]4− (Au4Se4), known to date, and a number of hypothetical polyoxoaurate derivatives comprising heteroatoms different from arsenic and selenium (namely, Si, Ge and P). In addition, the interactions of [AuIII4X4Om]n− (X = As, Se) with alkali-metal cations (Li+, Na+, K+ and Rb+) are also analysed. The studies suggest that the geometry structure, electronic properties and nucleophilicity of oxygen atoms of these polyoxoaurates are tuned by the size or electronegativity of the heteroatoms (Si, Ge, P, As and Se). Then, the geometry of [AuIII4X4Om]n− (X = As and Se) coordinating with alkali cations from Li+ to Rb+ and the complexation energy between [AuIII4X4Om]n− and alkali cations were compared. The results show that the stability and electronic structure of heteropolyoxoaurates depend on the entrapped cations. On the basis of the complexation energy, it can be concluded that the ion-pairing effect in arsenate-capped oxoaurate is stronger than that in selenite-capped oxoaurate. These heteropolyoxoaurates are expected to play a role in aqueous behaviour, self-assembly characteristics of polyoxoaurates, ion recognition, selectivity studies and may exhibit potential guest-switchable redox properties.
Co-reporter:Yan-Qing Jiao, Chao Qin, Hong-Ying Zang, Wei-Chao Chen, Chun-Gang Wang, Tian-Tian Zheng, Kui-Zhan Shao and Zhong-Min Su
CrystEngComm 2015 vol. 17(Issue 10) pp:2176-2189
Publication Date(Web):14 Jan 2015
DOI:10.1039/C4CE02007C
A series of organic–inorganic hybrid compounds built from Keggin and Wells–Dawson polyoxometalates and TM-trz complexes (Htrz = 1,2,4-triazole) were obtained at different pH values under hydrothermal conditions, namely, [Co2(H2O)2(Htrz)5][SiW12O40]·2.5H2O (1), [Zn5(Htrz)8(trz)2(H2O)8][SiW12O40]2·10H2O (2), [CuI2CuII4(Htrz)4(trz)4Cl2][GeW12O40]·15H2O (3), [CuI2CuII4(Htrz)4(trz)4Cl2][SiW12O40]·15H2O (4), K2Na2(H2O)2[Co11(trz)14(H2O)14][P2W18O62]2·29H2O (5) and [Cu12(trz)10(Htrz)2(OH)4(SO4)2(H2O)6][P2W18O62]·28H2O (6). All of the compounds were characterized using single-crystal X-ray diffraction, TG analysis and powder X-ray diffraction. Compound 1 contains a kind of bimetallic–organic segment [Co2(Htrz)5]4+, which could be regarded as a quasi-sinusoid-like chain. 2 is a dimer constructed from two [SiW12O40]4− units linked by a [Zn5(trz)2(Htrz)8]8+ motif. In 3 and 4, the 2D wall-like layers constructed by the Cu(II) ions and Htrz ligands are extended into 3D frameworks by polyanions. 5 shows a complicated three-dimensional (3D) framework with a {412·612·84}{46}2 topology. Compound 6 is also a complicated 3D structure based on a triangular trinuclear Cu-trz unit and [P2W18O62] polyanions. To the best of our knowledge, 6 represents the largest copper cluster linked by amines in a POM system. Structural diversities reveal that the pH value of the reaction system plays a crucial role in the assembly of POM-based hybrids. Moreover, the optical band gaps, electrochemical properties, magnetic properties and the photocatalytic properties of the compounds have been investigated.
Co-reporter:Liu Yang, Chao Qin, Bai-Qiao Song, Xiao Li, Chun-Gang Wang and Zhong-Min Su
CrystEngComm 2015 vol. 17(Issue 24) pp:4517-4524
Publication Date(Web):07 May 2015
DOI:10.1039/C5CE00539F
Five new coordination polymers constructed from 1,4-naphthalenedicarboxylate and N-donor ligands including [Cd2(1,4-ndc)2(bpy)2]·0.5H2O (1), [Cd(1,4-ndc)(phen)]·0.5H2O (2), [Cu2(1,4-ndc)2(L1)]·H2O (3), [Co3(H2O)2(1,4-ndc)4(HL2)2] (4), and [Cd2(NO3)2(1,4-ndc)(L3)2]·H2O (5) have been synthesized under hydrothermal conditions and characterized by elemental analysis, infrared spectroscopy, powder X-ray diffraction and single crystal X-ray diffraction. Compounds 1 and 2 are 2D layers with 4-connected (44·62) topology. Compound 3 is a 3D two-fold interpenetrating pcu network based on a binuclear cluster. Compound 4 exhibits an eight-connected bcu network constructed by a trinuclear cluster. Compound 5 is a 3D two-fold interpenetrating network with uninodal 4-connected cds (CdSO4) topology. Furthermore, the magnetic properties of compounds 3 and 4 were investigated and the photoluminescence properties of compounds 1, 2 and 5 show that the three compounds have strong emission under λex = 320 nm at room temperature.
Co-reporter:Bai-Qiao Song, Chao Qin, Yu-Teng Zhang, Xue-Song Wu, Hong-Sheng Liu, Kui-Zhan Shao and Zhong-Min Su
CrystEngComm 2015 vol. 17(Issue 16) pp:3129-3138
Publication Date(Web):11 Mar 2015
DOI:10.1039/C4CE02473G
Seven new coordination polymers, namely, [Zn(L)(bdc)]·2H2O (1), [Zn(L)(2-NH2-bdc)]·2H2O (2), [Zn(L)(bdc)]·1.75DMF·0.5H2O (3), [Zn(L)(oba)]·H2O (4), [Co(L)(oba)]·H2O (5), [Cd2(L)2(bdc)(NO3)2]·2DMF (6) and [Cd(L)(ndc)(H2O)] (7) (L = 4-amino-3,5-bis(4-imidazol-1-ylphenyl)-1,2,4-triazole, H2bdc = 1,4-benzenedicarboxylic acid, 2-NH2-H2bdc = 2-amino-1,4-benzenedicarboxylic acid, H2oba = 4,4′-oxydibenzoic acid and H2ndc = 1,4-naphthalenedicarboxylic acid), have been synthesized. The structures were determined by single-crystal X-ray diffraction analysis and further characterized by elemental analysis, IR, and thermogravimetric (TG) analysis. 1 and 2 feature a similar three-fold interpenetrating network combined with interdigitation which extends the structure into a three-dimensional framework. 3 is a two-fold interpenetrating framework showing intralayer porosity and interdigitation. Compounds 1 and 3 represent a pair of supramolecular isomers where structural motifs and different interpenetration modes exist; compounds 4 and 5 are isostructural and display the same three-fold interpenetrating framework where two types of interlaced triple-stranded helices A and B can be found. The triple-stranded helices with opposite chirality for each type are distributed in adjacent layers. Compound 6 exhibiting a unique simultaneous threading fashion of adjacent polymeric motifs gives the novel (2D → 3D) polythreaded array where coordinated NO3− anions act as the dangling ligands. Compound 7 is a 6-connected 3D self-penetrating network where right- and left-handed single-stranded helical chains (AR and AL) and interlaced triple-stranded helices (BR and BL) coexist. The luminescence properties of compounds 1–7 were also measured and discussed.
Co-reporter:Xue-Song Wu, Jun Liang, Xiao-Li Hu, Xin-Long Wang, Bai-Qiao Song, Yan-Qing Jiao, and Zhong-Min Su
Crystal Growth & Design 2015 Volume 15(Issue 9) pp:4311-4317
Publication Date(Web):August 18, 2015
DOI:10.1021/acs.cgd.5b00612
Four novel high-dimensional metal–organic rotaxane frameworks (MORFs) were successfully fabricated by cucurbit[6]uril-based pseudorotaxanes ([PR43]) and rigid carboxylate ligands by adjusting the synthesis temperature and the length of ligands. Compound 1 represents a three periodic 2-fold interpenetrating dia network based on a binuclear {Cd2Cl4O4} second building unit (SBU). 2 and 3 are isostructural pcu networks constructed by binuclear and trinuclear cadmium clusters, respectively, while compound 4 is a two-dimensional layered network with a large grid 44.3 × 19.8 Å2. Furthermore, their fluorescent properties in different solvents are investigated.
Co-reporter:Si-Quan Jiang, Zi-Yan Zhou, Shu-Ping Zhuo, Guo-Gang Shan, Ling-Bao Xing, Hai-Ning Wang and Zhong-Min Su
Dalton Transactions 2015 vol. 44(Issue 48) pp:20830-20833
Publication Date(Web):09 Nov 2015
DOI:10.1039/C5DT03814F
An efficient turn-on fluorescent sensor for PO43− has been developed by rationally designing an in situ-generated iron(III) complex with a 1,8-naphthalene-based Schiff base unit. The sensor exhibits high sensitivity and selectivity in both solution and solid-state film, even in the presence of other phosphate anions such as HPO42− and H2PO4−.
Co-reporter:Hong-Tao Cao, Lei Ding, Guo-Gang Shan, Hai-Zhu Sun, Yong Wu and Zhong-Min Su
Dalton Transactions 2015 vol. 44(Issue 46) pp:19997-20003
Publication Date(Web):19 Oct 2015
DOI:10.1039/C5DT03129J
A sulfur-free iridium(III) complex (pbi)2Ir(mtpy) (1) was successfully prepared and adopted as a Hg(II)-chemosensor with high selectivity and sensitivity. Multi-signaling responses towards Hg(II) ions were observed by UV−vis absorption, phosphorescence and electrochemistry measurements. With addition of Hg(II) ions, complex 1 presented quenched emission in its phosphorescence spectrum and the detection limit was as low as 2.5 × 10−7 M. Additionally, its redox peak currents showed a broad linear relationship with the concentration of Hg(II) ions ranging from 0 to 500 μM, which was beneficial for the quantitative detection. Based on the 1H NMR and ESI-MS analyses, the probing mechanism was tentatively supposed to be the Hg2+-induced changes in the local environment of complex 1. Such a response process was useful for achieving simple and effective detection of Hg(II) ions as well as developing more chemosensors.
Co-reporter:Bai-Qiao Song, Chao Qin, Yu-Teng Zhang, Xue-Song Wu, Liu Yang, Kui-Zhan Shao and Zhong-Min Su
Dalton Transactions 2015 vol. 44(Issue 42) pp:18386-18394
Publication Date(Web):09 Sep 2015
DOI:10.1039/C5DT03218K
Two unprecedented homochiral enantiomers based on two different kinds of rigid ligands, namely [Cd(NDC)L]2·H2O (1R and 1L), have been synthesized under hydrothermal conditions through spontaneous resolution. Their structures were determined by single-crystal X-ray diffraction analysis and further characterized by elemental analysis, IR, and thermogravimetric (TG) analysis. The resulting framework 1, constructed by four kinds of homo-handed helical chains represents the first 3D self-penetrating framework formed by decoration of single (10,3)-a net with helical chains. The single (10,3)-a net in 1 formed by three kinds of different homo-handed helical chains is different from the standard one, which should be ascribed to the usage of V-shaped ligand L. A unique self-penetration motif can be discovered in 1 where one helical chain alternately passes through 10-membered shortest circuits linked to each other and in contrary, the corresponding circuits are bound to the helical chain. Interestingly, 1 exhibits fluorescent emission in both the solid and solution phase. The uncoordinated nitrogen atom and amino group from the triazole core on the crystal surface make it suitable to detect picric acid in water. The luminescence intensity of 1 in water can be efficiently quenched by the addition of picric acid (PA). The sensitive detection of PA can be continuously performed for at least five cycles without diminishing the fluorescence intensity and destroying the framework structure of 1. The possible quenching mechanisms for PA are also investigated.
Co-reporter:Bai-Qiao Song, Xin-Long Wang, Chun-Yi Sun, Yu-Teng Zhang, Xue-Song Wu, Liu Yang, Kui-Zhan Shao, Liang Zhao and Zhong-Min Su
Dalton Transactions 2015 vol. 44(Issue 31) pp:13818-13822
Publication Date(Web):06 Jul 2015
DOI:10.1039/C5DT01560J
A novel 3D organic–inorganic hybrid framework constructed from tetra-CoII-substituted sandwich-type phosphotungstates with a rare 8-connected bcu topology is reported, which exhibited highly efficient photocatalysis activity under visible light and could be used for 5 cycles without any obvious decrease in activity.
Co-reporter:Wei-Chao Chen, Chao Qin, Xin-Long Wang, Yang-Guang Li, Hong-Ying Zang, Kui-Zhan Shao, Zhong-Min Su and En-Bo Wang
Dalton Transactions 2015 vol. 44(Issue 25) pp:11290-11293
Publication Date(Web):20 May 2015
DOI:10.1039/C5DT01711D
A large lanthanide-containing tungstotellurites(IV) nanocluster Na18[Ce10Te8W88O298(OH)12(H2O)40]·54H2O (1) was synthesized by combining cerium linkers and TeO32− heteroanion templates. The macroanion in 1 consists of two identical [Te4W42O144(OH)6Ce4(H2O)13]14− subunits and two triangle-shaped {W2O5Ce(H2O)7} linkers.
Co-reporter:Bo Zhu, Li-Kai Yan, Wei Guan and Zhong-Min Su
Dalton Transactions 2015 vol. 44(Issue 19) pp:9063-9070
Publication Date(Web):10 Apr 2015
DOI:10.1039/C5DT00318K
A thorough theoretical analysis was carried out on the sulfoxidation with H2O2 catalyzed by a tetranuclear peroxotungstate [SiO4{WO(O2)2}4]4−. The active species is the [SiO4{WO(O2)2}4(H2O2)]4− (SiW4(H2O2)) complex rather than [SiO4{WO(O2)2}4]4− (SiW4). The catalytic cycle consists of three elementary processes: oxygen transfer, sulfoxide dissociation, and catalyst regeneration. The oxygen transfer occurs from the peroxo oxygen atom O1 of SiW4(H2O2) to the sulfur center of dimethyl sulfide with a moderate Gibbs activation energy (ΔG°‡) of 17.1 kcal mol−1. By comparing potential energy surfaces and condensed Fukui functions (ƒ+), the electrophilicity of the outer peroxo atoms in SiW4(H2O2) determines which oxygen transfers to the dimethyl sulfide. Then, the sulfoxide dissociation proceeds with a small ΔG°‡ value of 2.3 kcal mol−1 by elongation of the peroxo O1–O4 distance and elimination of the product dimethylsulfoxide. Finally, the catalyst regeneration is found to occur via two successive proton transfers from H2O2 to the oxygen atoms of peroxotungstates with the ΔG°‡ values of 15.9 and 15.3 kcal mol−1, which has been firstly examined in the present study. All of these steps occur easily with moderate ΔG°‡ values, but the oxygen transfer is the rate-determining step of this catalytic reaction. In addition, the catalytic activity of peroxotungstates can be effectively tuned by changing the heteroatom X of [XO4{WO(O2)2}4(H2O2)]n− in the order: SeVI ≈ SVI > AsV ≈ PV > SiIV.
Co-reporter:Xiao-Li Hu, Fu-Hong Liu, Chao Qin, Kui-Zhan Shao and Zhong-Min Su
Dalton Transactions 2015 vol. 44(Issue 17) pp:7822-7827
Publication Date(Web):19 Mar 2015
DOI:10.1039/C5DT00515A
A novel Cd-MOF (metal organic framework) [Cd3(NTB)2(DMA)3]·2DMA (H3NTB = 4,4′,4′′-nitrilotrisbenzoic acid; DMA = N,N-dimethylacetamide) (1) was obtained under solvothermal conditions. The resulting MOF exhibits a novel (2D→3D) interdigitated architecture that is obtained from a bilayered motif with hexagonal grids. Luminescence properties of the activated phase of 1a well dispersed in different solvents have also been investigated systematically, which demonstrate distinct solvent-dependent luminescence spectra with emission intensities significantly quenched toward nitrobenzene (NB) and 2,4,6-trinitrophenol (TNP). The results reveal that 1 can be applied as a fluorescent sensor for the detection of TNP with high sensitivity, selectivity, and recyclability.
Co-reporter:Bai-Qiao Song, Chao Qin, Yu-Teng Zhang, Xue-Song Wu, Kui-Zhan Shao and Zhong-Min Su
Dalton Transactions 2015 vol. 44(Issue 9) pp:3954-3958
Publication Date(Web):26 Jan 2015
DOI:10.1039/C4DT03933E
A unique three-dimensional metal azolate framework containing a tetranuclear copper cluster constructed by six 1,2,4-triazole units was synthesized in which the 1,2,4-triazole units show unusual bridging “crevice” coordination mode with their 1- and 2-positioned sp2 N-atoms as symmetrically bridging centers. The photocatalytic activities of as-prepared compound were tested by degradation of rhodamine-B (RB) under different light irradiation.
Co-reporter:Bai-Qiao Song, Chao Qin, Yu-Teng Zhang, Li-Tao An, Kui-Zhan Shao and Zhong-Min Su
Dalton Transactions 2015 vol. 44(Issue 6) pp:2844-2851
Publication Date(Web):22 Oct 2014
DOI:10.1039/C4DT02808B
A novel interpenetrating metal–organic framework, namely [Zn3L2(oba)3(H2O)2]·4H2O (1), has been synthesized under hydrothermal conditions. Its structure was determined by single-crystal X-ray diffraction analysis and further characterized by elemental analysis, IR, and thermogravimetric (TG) analysis. In the structure of 1, the rigid and flexible V-shaped ligands link Zn(II) to form a 3D structure where two types of helices and four types of pseudo-helical chains containing three pairs of enantiomers and two pairs of conformational isomers have been characterized. One such 3D framework incorporates six identical networks to form a 7-fold interpenetrated 3D framework. From the topological analysis, the Zn(II) ions act as three- and four-connected nodes, and oba as well as L are linkers. The framework of compound 1 can be classified as a new (63)2(65·8) topology, which is a novel (3,3,4)-connected [4 + 3] 7-fold interpenetrating net showing 7-fold interlocking pseudo-helical chains and a unique catenane-like motif with Hopf links. In addition, the luminescence properties of the compound are discussed.
Co-reporter:Fu-Hong Liu, Chao Qin, Yan Ding, Han Wu, Kui-Zhan Shao and Zhong-Min Su
Dalton Transactions 2015 vol. 44(Issue 4) pp:1754-1760
Publication Date(Web):17 Nov 2014
DOI:10.1039/C4DT02961E
Two novel pillared MOFs (metal organic frameworks) [Zn2(trz)2(tda)]·DMA CH3OH (1) and [Zn2(trz)2(bpdc)]·DMA (2) were obtained under solvothermal conditions. The resulting MOFs show similar structures but with different interlayer distances based on the different carboxylate ligands. 1 and 2 display a certain degree of framework stability in both acid/base solutions and water. The luminescence intensities of the activated phases 1a and 2a are sensitive to metal ions, particularly Fe3+ and Cd2+ ions. Furthermore, the luminescent properties of 1a and 2a well dispersed in different solvents have also been investigated systematically, which demonstrate distinct solvent-dependent luminescent spectra with emission intensities that are significantly quenched by acetone, nitrobenzene and trinitrotoluene.
Co-reporter:Wei Du, Hai-Bin Li, Dong-Mei Gu, Yong Wu, Guang-Yan Sun, Yun Geng and Zhong-Min Su
RSC Advances 2015 vol. 5(Issue 121) pp:100169-100175
Publication Date(Web):10 Nov 2015
DOI:10.1039/C5RA17237C
In this work, we designed a series of ruthenium sensitizers DX2–DX5 derived from a phosphine-coordinated ruthenium sensitizer DX1 with a surprisingly high short-circuit photocurrent density (Jsc) of 26.8 mA cm−2 for dye sensitized solar cells (DSSCs), with the aim of enhancing the light harvesting ability in the near-infrared (NIR) region and further increasing the Jsc. Density functional theory (DFT) and relativistic time-dependent DFT calculations have been performed to evaluate the optical and photovoltaic properties of these Ru dyes, taking the effect of spin–orbit coupling (SOC) into consideration. The intrinsic causes for varied Jsc and open-circuit photovoltage (Voc) have been systematically discussed through investigating the light harvesting efficiency, electron injection driving force, dye regeneration driving force, electronic coupling and conduction band energy shift. The calculated results reveal that the designed DX5 has increased light harvesting efficiency in the NIR region and a higher conduction band energy shift compared with other sensitizers. That is, DX5 may have improved Jsc and Voc, which makes DX5 serve as a promising sensitizer for future DSSC applications.
Co-reporter:Hanni Wu, Ting Zhang, Likai Yan and Zhongmin Su
RSC Advances 2015 vol. 5(Issue 113) pp:93659-93665
Publication Date(Web):16 Oct 2015
DOI:10.1039/C5RA17343D
Based on a porphyrin derivative (system 1), Lindqvist-, Keggin-, and Anderson-type polyoxometalate (POM) organic–inorganic hybrids (systems 2–4) were designed with the aim of investigating their charge transfer character and screening them as high performance p-type sensitizers. The electronic structures and absorption spectra of systems 1–4 were systematically investigated by means of density functional theory (DFT) and time-dependent DFT (TD-DFT) methods. The results indicate that Lindqvist- and Keggin-type POMs affect the lowest unoccupied molecular orbital (LUMO) energy levels, while the Anderson-type POM does not contribute to the frontier molecular orbitals (FMOs). Furthermore, the absorption spectrum of the Lindqvist-type POM porphyrin derivative (system 2) exhibits strong and broad absorption in the visible region and is red shifted about 100 nm in comparison with system 1. Further studies point out that system 2 can balance the photovoltaic parameters, LHE, HJE, CRE and DRE, indicating that it will be a promising high performance dye sensitizer in p-type dye-sensitized solar cells (DSSCs).
Co-reporter:Xue Zhang, Shi-Ling Sun, Hong-Liang Xu and Zhong-Min Su
RSC Advances 2015 vol. 5(Issue 81) pp:65991-65997
Publication Date(Web):24 Jul 2015
DOI:10.1039/C5RA10156E
It is well known that ammonia borane (BH3NH3) is one of the simplest donor–acceptor complexes. The donor–acceptor bond (B–N bond) is formed by sharing lone pair electrons between BH3 and NH3 groups. In the present work, different strengths of external electric fields (Eext) are applied along the Z-axis direction to investigate the electric field induced effect on the BH3NH3 structure and properties. Interestingly, we have found that the lone pair electrons can be gradually moved in the “channel” between the BH3 and NH3 groups by modulating Eext. The donor–acceptor bond (B–N bond) is gradually elongated until it breaks when the Eext ranges from 0.0000 to 0.0321 au. The B–N bond is the shortest at Eext = −0.0519 au. Interestingly, the negative charge on BH3 groups sharply decreases from 0.161 to 0.005 (for NH3 groups decreases from 0.161 to 0.005) and the electron cloud of HOMO−2 exhibits an obvious transformation at “broking bonding” Eext ranging from 0.0320 to 0.0321 au, indicating the electron movement induced by the electric-field is the main reason to change the structure and stability of BH3NH3. Further, atoms in molecules (AIM) analysis shows that the B⋯N interactions are similar to that of hydrogen bonds at Eext = −0.0767 au and the iconicity of B–N bond in BH3NH3 is confirmed by its low electron density for B–N ρ(B–N) (0.103–0.073) in the region of Eext (0–0.025 au).
Co-reporter:Xiao Li, Liu Yang, Chao Qin, Fu-Hong Liu, Liang Zhao, Kui-Zhan Shao and Zhong-Min Su
RSC Advances 2015 vol. 5(Issue 73) pp:59093-59098
Publication Date(Web):17 Jun 2015
DOI:10.1039/C5RA09339B
Four new polyoxovanadate-based organic–inorganic hybrid materials [MnV2(bpp)2O6] (1), [Ag4V4(bpp)4O12]·2H2O (2) and [M3V6(bpp)4O18·4H2O]·2H2O [M = Ni (3), Zn (4), bpp = 1,3-bis(4-pyridyl)propane] have been synthesized under hydrothermal conditions through the self-assembly of transition metal salts, bpp ligands and ammonium metavanadate. Single-crystal X-ray diffraction analyses show that 1 possesses a three-dimensional framework, constructed from arrays of {V4O12} rings covalently linked through metal–organic units, {Mn(C13H4N2)2}. Compound 2 is an eight-connected self-catenated metal–organic framework, based on bimetallic {Ag4V4O12} clusters as nodes. Compounds 3 and 4 are isostructural, both of them exhibit an intriguing 6,10-connected network. The structure of 4, as an example, is based on single zinc atoms as six-connected nodes, as well as bimetallic {Zn2V6O18} clusters as ten-connected nodes, defining not only a new topology for three-periodic nets but also the first 6,10-connected framework using heterometallic clusters as nodes. Furthermore, the thermal stabilities and photocatalytic activities of 1–4 have been discussed in detail.
Co-reporter:Xiao-Li Hu, Chao Qin, Liang Zhao, Fu-Hong Liu, Kui-Zhan Shao and Zhong-Min Su
RSC Advances 2015 vol. 5(Issue 61) pp:49606-49613
Publication Date(Web):22 May 2015
DOI:10.1039/C5RA05945C
Two novel Zn(II) coordination polymer compounds, namely, [Zn3(HL)2(fma)2]·DMA·H2O (1) and [Zn2(μ3-OH)(HL)(Br-bdc)]·0.5DMA·CH3OH·H2O (2) have been successfully obtained with 1-(5-tetrazolyl)-4-(1-imidazolyl) benzene ligand and dicarboxylic acids (fma = fumaric acid; Br-bdc = 2-bromoterephthalic acid). Compound 1 exhibits a 3D framework with a square aperture diameter for the channel is 8.8 × 8.8 Å2, and the framework can be simplified as a (3,4)-connected tfj network with the point symbol of {4.82}4{42.84}2{84.122} topology. In compound 2, the HL ligand forms a 2D sheet of Zn(HL) by linking Zn ions along the c axis, which are further connected by double Br-bdc pillars resulting in unique bi-pillared-layer type 3D frameworks. The result, 1a, exhibits a certain degree of CO2 uptake and selective CO2/N2 adsorption capacity. Furthermore, luminescent properties of 1a well dispersed in different solvents have also been investigated systematically, which demonstrate distinct solvent-dependent luminescent spectra with emission intensities significantly quenched toward acetone and nitrobenzene.
Co-reporter:Hai-Ning Wang, Si-Quan Jiang, Qing-Yun Lu, Zi-Yan Zhou, Shu-Ping Zhuo, Guo-Gang Shan and Zhong-Min Su
RSC Advances 2015 vol. 5(Issue 60) pp:48881-48884
Publication Date(Web):20 May 2015
DOI:10.1039/C5RA08347H
A pillar-layer MOFs Cd(5-aip)L·3DMA (1) has been obtained under solvothermal conditions. 1 possesses non-interpenetrating pillar-layer framework with 1D rectangular-shape channels. The experimental results show that the luminescent intensity of 1 is highly dependent on metal ions and small solvent molecules, particularly Ni2+/Cd2+ ions and acetone.
Co-reporter:Lin Lin Sun, Ting Zhang, Jing Wang, Hong Li, Li Kai Yan and Zhong Min Su
RSC Advances 2015 vol. 5(Issue 50) pp:39821-39827
Publication Date(Web):24 Apr 2015
DOI:10.1039/C5RA05164A
A series of polyoxometalates (POMs)-based dyes with electron donating/withdrawing groups were investigated by using density functional theory (DFT) and time-dependent DFT (TDDFT) calculations. The light harvesting efficiency (LHE), dye regeneration efficiency (DRE), charge recombination efficiency (CRE), holes injecting efficiency (HJE) and reorganization energy (Ereorg) were systematically evaluated. The maximum absorptions of the designed dyes with electron donating groups (systems 1–3) are red shifted comparing with those containing the electron withdrawing groups (systems 4 and 5). Different electron donating groups have significant differences on the binding energy of dye–(NiO)4 system. In the dye regeneration, the I2 interacts with the terminal oxygen of POM cluster. System 1 (–NH2) balances all parameters, and it may perform better than others. Therefore, the introduction of –NH2 into POM-based organic–inorganic hybrids may improve the performance of dye in dye-sensitized solar cells (DSSCs).
Co-reporter:Ying Gao, Rong-Lin Zhong, Hong-Liang Xu, Shi-Ling Sun and Zhong-Min Su
RSC Advances 2015 vol. 5(Issue 38) pp:30107-30119
Publication Date(Web):23 Mar 2015
DOI:10.1039/C5RA01145K
In 2003, a novel compound 2 containing the benzo-15-crown-5 moiety was synthesized and described. In the present work, we have designed two compounds 1 (benzo-12-crown-4) and 3 (benzo-18-crown-6) on the basis of compound 2. Further, nine configurations N*M (N = 1, 2 and 3; M = Li+, Na+ and K+) are designed by the compounds 1, 2 and 3 complexing alkali metal cations. Density functional calculation is performed to investigate the effect of ring size and the nature of the alkali metal cations on the interaction energy, charge transfer and nonlinear optical properties. The results indicate that the interaction energy of N*M depends on both the ring size and the nature of the alkali metal cations. Moreover, the amount of net charge transfer is related to the diameters of the alkali metal cations. In addition, the calculated nonlinear optical properties reveal that compound 2 has the largest first hyperpolarizability among the three compounds 1, 2 and 3. However, the alkali metal cations give rise to different effects on the nonlinear optical properties. Significantly, the order of the first hyperpolarizability can be explained by the transition energy and the dipole moment variation within the two-state approximation.
Co-reporter:Shuang-Bao Li, Yu-Ai Duan, Yun Geng, Hong-Ze Gao, Yong-Qing Qiu and Zhong-Min Su
RSC Advances 2015 vol. 5(Issue 37) pp:29401-29411
Publication Date(Web):17 Mar 2015
DOI:10.1039/C5RA00785B
A class of D1–A–D2–A–D1-type small molecule (SM) donors (2–5) was engineered via modifying or replacing the core donor moiety in three building blocks based on the reported DTS(PTTh2)2 (1) to screen suitable donor materials for organic photovoltaics (OPV). Density functional theory calculation was performed to investigate the electronic structures, open circuit voltage (Voc) and key parameters closely relevant to the short-circuit current density (Jsc), including (i) absorption spectrum, (ii) electron–hole coherence, (iii) energy driving force, (iv) charge transfer dynamics, and (v) carrier transport efficiency. The results manifest that the designed 2–5 show good performance with large Voc, stable charge transfer and effective charge transport. Surprisingly, the ratios kinter-CT/kinter-CR of 2/PCBM, 3/PCBM, and 5/PCBM heterojunctions present over 104 times higher than that of 1/PCBM. Our conclusions indicate that designed PT-based SMs can better the performance of OPVs, which will provide theoretical guideline for the design and synthesis of new organic SM donors.
Co-reporter:Xiao-Li Hu, Chao Qin, Xin-Long Wang, Kui-Zhan Shao and Zhong-Min Su
New Journal of Chemistry 2015 vol. 39(Issue 10) pp:7858-7862
Publication Date(Web):20 Jul 2015
DOI:10.1039/C5NJ01237F
One new cluster-based metal–organic framework, namely, [Cd2.5Na(NTB)2(DMF)4]·3DMF (1) (NTB = 4,4′,4′′-nitrilotrisbenzoic acid; DMF = N,N-dimethylformamide) has been successfully obtained by employing a C3 symmetric ligand. In 1, the NTB links two different metal centers (single metal and Cd3 cluster) to form two kinds of 1D chains, which linked through the carboxylate O atom of the NTB ligand to give rise to a 3D framework. Moreover, desolved 1 can be well dispersed in different solvents, which demonstrate distinct solvent-dependent luminescence spectra with emission intensities significantly quenched toward nitrobenzene (NB) and 2,4,6-trinitrophenol (TNP). The results reveal that 1 could be applied as a fluorescence sensor for TNP with high sensitivity and selectivity.
Co-reporter:Chen Shao, Zhong-Min Su
Inorganic Chemistry Communications 2015 Volume 57() pp:4-7
Publication Date(Web):July 2015
DOI:10.1016/j.inoche.2015.04.011
•A new metal–organic framework was synthesized and structurally characterized.•This compound displays high thermal stability and excellent water resistance.•This compound exhibits highly selective and sensitive quenching effect toward Cu2 + in aqueous solutions.A new water-stable 3D metal–organic framework (MOF), [Cd2L2]·NMP·MeOH (1, H2L = 2-(3,5-dimethyl-1H-pyrazol-4-yl)-1,3-dioxoisoindoline-5,6-dicarboxylic acid, NMP = 1-methyl-2-pyrrolidinone) has been solvothermally synthesized and structurally characterized. 1 exhibits a 3D open-framework with a 2D metallic plane pillared by L2 − fragments. Meanwhile, luminescent studies indicate that the luminescence intensity of 1 was strongly dependent on different metal ions. Most interestingly, 1 exhibited significantly quenching effect toward Cu2 +, which implies that it may be used as a luminescent probe for the detection of Cu2 +.A new water-stable metal–organic framework (1) was solvothermally synthesized and structurally characterized. It exhibits a 3D open-framework with a 2D metallic plane pillared by L2 − fragments. Luminescent studies indicate that 1 exhibits significantly high specific, selective and sensitive quenching effect toward Cu2 +.
Co-reporter:Qian Li, Yuai Duan, Hong-Ze Gao, Zhong-Мin Su, Yun Geng
Journal of Molecular Graphics and Modelling 2015 Volume 59() pp:50-58
Publication Date(Web):June 2015
DOI:10.1016/j.jmgm.2015.04.001
•Fluorination and carbonylation effect on electron transport properties of thiophene-based materials were investigated.•Introducing F atom and CO group would effectively increase EA, reduce λele and manipulate packing manner of the material.•System 3d is a promising electron transporting material due to the biggest EA value and small λele.In this work, we concentrate on systematic investigation on the fluorination and carbonylation effect on electron transport properties of thiophene-based materials with the aim of seeking and designing electron transport materials. Some relative factors, namely, frontier molecular orbital (FMO), vertical electron affinity (VEA), electron reorganization energy (λele), electron transfer integral (tele), electron drift mobility (μele) and band structures have been calculated and discussed based on density functional theory. The results show that the introduction of fluorine atoms and carbonyl group especially for the latter could effectively increase EA and reduce λele, which is beneficial to the improvement of electron transport performance. Furthermore, these introductions could also affect the tele by changing molecular packing manner and distribution of FMO. Finally, according to our calculation, the 3d system is considered to be a promising electron transport material with small λele, high electron transport ability and good ambient stability.In this work, systematic investigations on the fluorination and carbonylation effect on electron transport properties of thiophene-based materials were focused on with the aim of seeking and designing electron transport materials.
Co-reporter:Shui-Xing Wu; Yu-He Kan; Hai-Bin Li; Liang Zhao; Yong Wu
The Journal of Physical Chemistry Letters 2015 Volume 6(Issue 15) pp:2950-2958
Publication Date(Web):July 14, 2015
DOI:10.1021/acs.jpclett.5b01182
It is well known that the aluminum cathode performs dramatically better when a thin lithium fluoride (LiF) layer inserted in organic electronic devices. The doping effect induced by the librated Li atom via the chemical reactions producing AlF3 as byproduct was previously proposed as one of possible mechanisms. However, the underlying mechanism discussion is quite complicated and not fully understood so far, although the LiF interlayer is widely used. In this paper, we perform theoretical calculations to consider the reactions between an aluminum atom and distinct LiF clusters. The reaction pathways of the Al–(LiF)n (n = 2, 4, 16) systems were discovered and the energetics were theoretically evaluated. The release of Li atom and the formation of AlF3 were found in two different chemical reaction routes. The undissociated Al–(LiF)n systems have chances to change to some structures with loosely bound electrons. Our findings about the interacted Al–(LiF)n systems reveal new insights into the LiF interlayer effects in organic electronics applications.
Co-reporter:Feng-Wei Gao;Rong-Lin Zhong;Shi-Ling Sun;Hong-Liang Xu
Journal of Molecular Modeling 2015 Volume 21( Issue 10) pp:
Publication Date(Web):2015 October
DOI:10.1007/s00894-015-2808-9
Very recently, two new cage-like radicals (C59B and C59N) formed by a boron or nitrogen atom substituting one carbon atom of C60 were synthesized and characterized. In order to explore the structure–property relationships of combination the cage-like radical and alkali metal, the endohedral Li@C59B and Li@C59N are designed by lithium (Li) atom encapsulated into the cage-like radicals C59B and C59N. Further, the structures, natural bond orbital (NBO) charges, and nonlinear optical (NLO) responses of C59B, C59N, Li@C59B, and Li@C59N were investigated by quantum chemical method. Three density functional methods (BHandHLYP, CAM-B3LYP, and M05-2X) were employed to estimate their first hyperpolarizabilities (βtot) and obtained the same trend in the βtot value. The βtot values by BHandHLYP functional of the pure cage-like radicals C59B (1.30 × 103 au) and C59N (1.70 × 103 au) are close to each other. Interestingly, when one Li atom encapsulated into the electron-rich radical C59N, the βtot value of the Li@C59N increases to 2.46 × 103 au. However, when one Li atom encapsulated into the electron-deficient radical C59B, the βtot value of the Li@C59B sharply decreases to 1.54 × 102 au. The natural bond orbital analysis indicates that the encapsulated Li atom leads to an obvious charge transfer and valence electrons distribution plays a significant role in the βtot value. Further, frontier molecular orbital explains that the interesting charge transfer between the encapsulated Li atom and cage-like radicals (C59B and C59N) leads to differences in the βtot value. It is our expectation that this work will provide useful information for the design of high-performance NLO materials.
Co-reporter:Rong-Lin Zhong; Hong-Liang Xu; Zhi-Ru Li
The Journal of Physical Chemistry Letters 2015 Volume 6(Issue 4) pp:612-619
Publication Date(Web):January 28, 2015
DOI:10.1021/jz502588x
The excess electron is a kind of special anion with dispersivity, loosely bounding and with other fascinating features, which plays a pivotal role (promote to about 106 times in (H2O)3{e}) in the large first hyperpolarizabilities (β0) of dipole-bound electron clusters. This discovery opens a new perspective on the design of novel nonlinear optical (NLO) molecular materials for electro-optic device application. Significantly, doping alkali metal atoms in suitable complexants was proposed as an effective approach to obtain electride and alkalide molecules with excess electron and large NLO responses. The first hyperpolarizability is related to the characteristics of complexants and the excess electron binding states. Subsequently, a series of new strategies for enhancing NLO response and electronic stability of electride and alkalide molecules are exhibited by using various complexants. These strategies include not only the behaviors of pushed and pulled electron, size, shape, and number of coordination sites of complexants but also the number and spin state of excess electrons in these unusual NLO molecules.
Co-reporter:Dr. Wen-Wen He;Dr. Guang-Sheng Yang;Dr. Yu-Jia Tang; Shun-Li Li;Dr. Shu-Ran Zhang; Zhong-Min Su; Ya-Qian Lan
Chemistry - A European Journal 2015 Volume 21( Issue 27) pp:9784-9789
Publication Date(Web):
DOI:10.1002/chem.201500815
Abstract
A series of isoreticular metal–organic frameworks (MOFs; NENU-511–NENU-514), which all have high surface areas and strong adsorption capacities, have been successfully constructed by using mixed ligands. NENU-513 has the highest benzene capacity of 1687 mg g−1at 298 K, which ranks as the top MOF material among those reported up to now. This NENU series has been used for adsorptive desulfurization because of its permanent porosity. The results indicate that this series has a higher adsorptive efficiency in the removal of organosulfur compounds than other MOF materials, especially NENU-511, which has the highest adsorptive efficiency in the ambient atmosphere. This study proves that the design and synthesis of targeted MOFs with higher surface areas and with functional groups present is an efficient method to enhance benzene-storage capacity and the adsorption of organosulfur compounds.
Co-reporter:Wei Du, Hai-Bin Li, Yun Geng, Yong Wu, Min Zhang, Zhong-Min Su
Journal of Photochemistry and Photobiology A: Chemistry 2015 Volume 301() pp:40-46
Publication Date(Web):15 March 2015
DOI:10.1016/j.jphotochem.2015.01.004
•Properties of dyes related to the Jsc and Voc of DSSCs were investigated based on DFT calculation.•The designed HIS-2a shows red-shifted absorption spectra and enhanced transition intensity in NIR region.•HIS-2a is a promising candidate to further increase efficiency.To overcome the shortcomings of traditional Ru dyes in low molar extinction coefficient (ϵ) of the metal-to-ligand charge transfer band in the near-infrared region, we designed and studied two Ru dyes, HIS-2a and HIS-2b derived from HIS-2. The intrinsic causes for varying short-circuit photocurrent density (Jsc) and open-circuit photovoltage (Voc) have been systematically investigated on the dyes and dye/(TiO2)38 systems including the conduction band energy shift, electronic structures, light harvesting efficiency, electron injection driving force, electronic coupling, dye regeneration driving force and so on. Those results suggest that, compared with HIS-2 and HIS-2b, HIS-2a has red-shifted absorption spectrum and increased transition intensity in NIR region, which would lead to a larger Jsc. At the same time, the conduction band energy of HIS-2a shifts little compared with other dyes, which will not have much effect on Voc. Hence, we could speculate that HIS-2a is a promising candidate with enhanced Jsc and a more efficient Ru(II) π-expanded terpyridyl dye. We also hope that this study will provide theoretical guidelines for the synthesis of new Ru dyes.To overcome the shortcomings of traditional Ru dyes in low molar extinction coefficient (ϵ) in the near-infrared region, two Ru dyes, HIS-2a and HIS-2b derived from HIS-2, were designed and studied in depth based on density functional theory calculations.
Co-reporter:Dong-Ying Du, Jun-Sheng Qin, Shun-Li Li, Zhong-Min Su and Ya-Qian Lan
Chemical Society Reviews 2014 vol. 43(Issue 13) pp:4615-4632
Publication Date(Web):27 Mar 2014
DOI:10.1039/C3CS60404G
Polyoxometalate (POM)-based metal–organic framework (MOF) materials contain POM units and generally generate MOF materials with open networks. POM-based MOF materials, which utilize the advantages of both POMs and MOFs, have received increasing attention, and much effort has been devoted to their preparation and relevant applications over the past few decades. They have good prospects in catalysis owing to the electronic and physical properties of POMs that are tunable by varying constituent elements. In this review, we present recent developments in porous POM-based MOF materials, including their classification, synthesis strategies, and applications, especially in the field of catalysis.
Co-reporter:Wen-Wen He, Shun-Li Li, Hong-Ying Zang, Guang-Sheng Yang, Shu-Ran Zhang, Zhong-Min Su, Ya-Qian Lan
Coordination Chemistry Reviews 2014 Volume 279() pp:141-160
Publication Date(Web):1 November 2014
DOI:10.1016/j.ccr.2014.03.022
•We have reviewed a good number of POM-based coordination polymers with entangled structures.•Different topological concepts are employed to describe the various types of these polymers.•Further classification is based on the dimensionalities of the individual motifs and the roles of the POMs.•The unique characteristics of the POMs make them perfect templates and linkers in the formation of these structures.Polyoxometalates (POMs) have attracted a lot of interest due to their novel structure characteristics and various connection modes. POM-based coordination polymers with entangled structures, an indispensable branch of entangled networks, take advantage of the features of POMs, and have received increasing attention. Much effort has been devoted over the past few decades toward their preparation and the analysis of their unusual entangled topology. In this review, we will summarize a number of examples of POM-based coordination polymer that have been described according to their different entangled characteristics. Different concepts, such as interpenetration, polycatenation, polyrotaxane, polypseudo-rotaxane and self-penetration, are employed to describe the various types of POM-based coordination polymers with entangled structures. In addition, we further classify POM-based coordination polymers with entangled structures based on the dimensionalities of the individual motifs and the roles of the POMs. Combining the advantages of the attractive potential applications of the POMs and structure diversities of the entangled frameworks, the investigation of POM-based coordination polymers with entangled structures will be a sustainable research field in coordination chemistry.
Co-reporter:Ping Shen, Wen-Wen He, Dong-Ying Du, Hai-Long Jiang, Shun-Li Li, Zhong-Ling Lang, Zhong-Min Su, Qiang Fu and Ya-Qian Lan
Chemical Science 2014 vol. 5(Issue 4) pp:1368-1374
Publication Date(Web):02 Dec 2013
DOI:10.1039/C3SC52666F
In this work, we have demonstrated an unprecedented single-crystal-to-single-crystal (SCSC) transformation between two 3D metal-organic frameworks (MOFs). The centrosymmetric IFMC-68 ([(Zn4O)2(L)3]·10H2O·46DMA) transforms into a chiral IFMC-69 ([(Zn4O)2(L)3H2O]·H2O·4DMA) doubly triggered by reaction temperature and time simultaneously in the presence or absence of solvent. To our knowledge, this is the first representative that the non-interpenetrated structure transforms into self-penetrated structure in MOFs. For the first time, we have studied the influence of reaction temperature and time on SCSC transformation, simultaneously, and get the transformation relationship among IFMC-68, IFMC-69 and the intermediate coming from the direct synthesis method and stepwise synthesis method at different temperatures and for different times. Meanwhile, we have achieved the conversion from an air-unstable to air-stable structure. Air-stable IFMC-69 exhibits the selective CO2 uptake over N2 and more excellent gas adsorption ability than IFMC-68. In addition, IFMC-69 shows an efficient capability in reversible adsorption of iodine. The electrical conductivity value (σ) of I2@IFMC-69 is much higher than the pristine MOF and thus is promising for potential semiconductor materials in the future.
Co-reporter:Li Wang, Yong Wu, Guo-Gang Shan, Yun Geng, Jian-Zhao Zhang, Dong-Mei Wang, Guo-Chun Yang and Zhong-Min Su
Journal of Materials Chemistry A 2014 vol. 2(Issue 16) pp:2859-2868
Publication Date(Web):14 Jan 2014
DOI:10.1039/C3TC31831A
A series of heteroleptic iridium(III) complexes were investigated by using the density functional theory/time-dependent density functional theory (DFT/TD-DFT) approach to determine the influence of the diphenylphosphoryl (Ph2PO) moiety on the electronic structures, phosphorescent properties and the organic light-emitting diode (OLED) performance. The results reveal that the introduction of the Ph2PO group could not only dramatically change the electron density distributions of the LUMO and cause red shifts of the emission wavelengths, but also increase the oscillator strengths and the metal character, thus leading to larger radiative decay rates. Additionally, compared with FIrpic (a widely used kind of blue guest material in OLED devices), those complexes with Ph2PO substituents could improve the electron injection/balance ability, increase the Förster energy transfer rate and confine the triplet excitons to the guest phosphors, hence resulting in better OLED performance. Interestingly, further analysis indicates that, compared to IrpicPO with the Ph2PO group sited at the phenyl ring of the phenylpyridine (ppy) ligands, IrpicPOpy with the Ph2PO group sited at the pyridine ring of the ppy ligands performs better in the hole-trapping and hole-injection ability. Finally, we hope our investigations will facilitate the future design of high efficient phosphorescent materials.
Co-reporter:Yan-Qing Jiao, Chao Qin, Xin-Long Wang, Fu-Hong Liu, Peng Huang, Chun-Gang Wang, Kui-Zhan Shao and Zhong-Min Su
Chemical Communications 2014 vol. 50(Issue 45) pp:5961-5963
Publication Date(Web):12 Dec 2013
DOI:10.1039/C3CC47703G
A novel δ-Dawson [(WO5)3W14Mn2IIIO44Cl2]12− compound induced by the Jahn–Teller distortion of MnIII has been synthesized through utilizing {Mn12} as a reactant, which exhibits photocatalytic H2 evolution activity. Its electrochemical behavior and magnetic properties were investigated.
Co-reporter:Hong Ren, Lingyu Zhang, Jiping An, Tingting Wang, Lu Li, Xiaoyan Si, Liu He, Xiaotong Wu, Chungang Wang and Zhongmin Su
Chemical Communications 2014 vol. 50(Issue 8) pp:1000-1002
Publication Date(Web):11 Nov 2013
DOI:10.1039/C3CC47666A
The polyacrylic acid@zeolitic imidazolate framework-8 (PAA@ZIF-8) nanoparticles (NPs) were first fabricated using a facile and simple route. It is worthwhile noting that the as-fabricated PAA@ZIF-8 NPs possessed ultrahigh doxorubicin (DOX) loading capability (1.9 g DOX g−1 NPs), which were employed as pH-dependent drug delivery vehicles.
Co-reporter:Xue-Gang Hou, Yong Wu, Hong-Tao Cao, Hai-Zhu Sun, Hai-Bin Li, Guo-Gang Shan and Zhong-Min Su
Chemical Communications 2014 vol. 50(Issue 45) pp:6031-6034
Publication Date(Web):22 Apr 2014
DOI:10.1039/C3CC49395D
A new cationic Ir(III) complex with AIE characteristics was designed and synthesized with the help of density functional theory calculations, which exhibits highly sensitive and selective detection of explosives (2,4,6-trinitrophenol, TNP).
Co-reporter:Kun Zhou, Yun Geng, Li-Kai Yan, Xin-Long Wang, Xian-Chun Liu, Guo-Gang Shan, Kui-Zhan Shao, Zhong-Min Su and Ying-Ning Yu
Chemical Communications 2014 vol. 50(Issue 80) pp:11934-11937
Publication Date(Web):13 Aug 2014
DOI:10.1039/C4CC05893C
An ultrastable [Ag55(MoO4)6]43+ ({Ag55Mo6} for short) nanocluster with a Ag-centered multishell structure in compound [Ag55(MoO4)6(CCtBu)24(CH3COO)18](OAc)·2H2O (1) has been obtained. The ultrastability of 1 was demonstrated by Mulliken population analysis. In addition, the potential wide gap semiconductor property and electrochemical properties of 1 were investigated.
Co-reporter:Wei-Chao Chen, Chao Qin, Xin-Long Wang, Yang-Guang Li, Hong-Ying Zang, Yan-Qing Jiao, Peng Huang, Kui-Zhan Shao, Zhong-Min Su and En-Bo Wang
Chemical Communications 2014 vol. 50(Issue 87) pp:13265-13267
Publication Date(Web):10 Sep 2014
DOI:10.1039/C4CC06447J
Two Fe-substituted Dawson-type nanoscale selenotungstate clusters, {Fe6Se6W34} and {Fe10Se8W62} involving {α-Se2W14} and {γ-Se2W14} building blocks, have been isolated, which exhibit photocatalytic H2 evolution activity. Their electrochemical behaviours and magnetic properties were also investigated.
Co-reporter:Jian-Zhao Zhang, Ji Zhang, Hai-Bin Li, Yong Wu, Hong-Liang Xu, Min Zhang, Yun Geng, Zhong-Min Su
Journal of Power Sources 2014 Volume 267() pp:300-308
Publication Date(Web):1 December 2014
DOI:10.1016/j.jpowsour.2014.05.085
•The different performance of reported dyes was rationalized by DFT calculations.•Present a comprehensive strategy to design high-efficiency dye.•Designed dyes 6 and 7 are promising candidates to further improve efficiency.Factors associated with short circuit current density (Jsc) and open circuit photovoltage (Voc) of dye sensitized solar cells (DSSCs) have been analyzed through DFT and TDDFT calculations to explore the origin of the significant performance differences with only tiny structure difference (1.24% for 1 and 8.21% for 2) (Advanced Functional Materials 2012, 22, 1291–1302). Our results reveal that the insertion of phenyl ring in 2 enlarges the distance between the dye cation hole and the semiconductor surface and makes the benzothiadiazole (BTDA) unit, which has strong interaction with the electrolyte, far away from the semiconductor, resulting in a decreased charge recombination rate compared with that of 1. However, the insertion of phenyl ring also results in a distortion of the molecular structure, leading to a decreased light harvesting ability. Hence, two dyes (6 and 7) derived from 2 with better conjugation degree, farther position of BTDA unit and longer molecular length have been designed to keep the advantages and overcome the disadvantages of 2 simultaneously. The results demonstrate that we get the desired properties of dyes through reasonable molecular design, and these two dyes could be promising candidates in DSSC field and further improve the performance of the cell.
Co-reporter:Wei-Chao Chen, Xin-Long Wang, Yan-Qing Jiao, Peng Huang, En-Long Zhou, Zhong-Min Su, and Kui-Zhan Shao
Inorganic Chemistry 2014 Volume 53(Issue 18) pp:9486-9497
Publication Date(Web):August 25, 2014
DOI:10.1021/ic500442k
A versatile one-pot strategy was employed to synthesize five cerium(III)-containing polyoxotungstate nanoclusters through pH-controlled and sulfite anion-directed assembly: [C2H8N]3Na7[Ce2(H2O)6W22O72(OH)4]·20H2O (1) at pH 5.0; [C2H8N]8Na16[Ce4(H2O)12W44O144(OH)12]·23H2O (2) at pH 4.5; [C2H8N]2Na4Ce2[Ce2(H2O)10W28O92(OH)2]·27H2O (3) at pH 2.8–3.3; [C2H8N]2Na7[{α-SW7O28}{Ce2(H2O)6}(W3O6){α-SW9O32}{α-SW9O31(OH)}]·18H2O (4) at pH 2.5; [C2H8N]2Na18[Ce2(H2O)9W36O110(OH)12]2·30H2O (5) at pH 1.5. These compounds were characterized by single-crystal X-ray structure analysis, IR spectroscopy, thermogravimetric (TG) analysis, X-ray photoelectron spectroscopy (XPS), and electrospray ionization mass spectrometry (ESI-MS). Moreover, their electrochemical properties were investigated. Single-crystal X-ray structure analysis revealed that 1 and 2 were di- and tetra-cerium(III)-bridged polyoxotungstates, constructed from two different types of lacunary {W11} units. 3 composed of the well-known cerium(III)-stabilized {W28} unit and organic amine–sodium–cerium cations, was isolated in the pH range 2.8–3.3. In this reaction system, the SO32– anion acted as a heteroanion template at a lower pH 2.5. 4 was isolated by the combination of cerium(III) centers and SO32– heteroanion template, which is the first lanthanide-containing polyoxotungstates with sulfur heteroatoms, and the 4f metal cerium(III) centers in 4 both have eight-coordinated modes and the SO32– heteroanion templates display μ7 and μ9 coordination modes. At a much lower pH 1.5, the polyanion of 5 was obtained, two triangular-shaped {W36} subunits were bridged by the cerium(III) ions, resulting in the largest lanthanide-containing iso-polyoxotungstates known to date.
Co-reporter:Shu-Ran Zhang, Dong-Ying Du, Jun-Sheng Qin, Shun-Li Li, Wen-Wen He, Ya-Qian Lan, and Zhong-Min Su
Inorganic Chemistry 2014 Volume 53(Issue 15) pp:8105-8113
Publication Date(Web):July 23, 2014
DOI:10.1021/ic5011083
In this work, five novel 2D isostructural Cd(II)–lanthanide(III) heterometallic–organic frameworks [CdCl(L)EuxTby(H2O)(DMA)](NO3)·3DMA (IFMC-36-EuxTby: x = 1, y = 0, IFMC-36-Eu; x = 0.6, y = 0.4, IFMC-36-Eu0.6Tb0.4; x = 0.5, y = 0.5, IFMC-36-Eu0.5Tb0.5; x = 0.4, y = 0.6, IFMC-36-Eu0.4Tb0.6; x = 0, y = 1, IFMC-36-Tb; H3L is 4,4′,4″-((2,2′,2″-(nitrilotris(methylene))tris(1H-benzo[d]imidazole-2,1-diyl))tris(methylene))tribenzoic acid; IFMC = Institute of Functional Material Chemistry) have been successfully synthesized by taking advantage of different molar ratios of lanthanide(III) (Ln(III)) and metalloligands under solvothermal conditions. Further luminescent measurements indicate that IFMC-36-EuxTby exhibits characteristic sharp emission bands of Eu(III) and Tb(III), and the intensities of red and green can be modulated correspondingly by tuning the ratios of Eu(III) and Tb(III). Particularly, the solvent-dependent luminescent behavior of IFMC-36-Eu shows a potential application in detection of small-molecule pollutant nitrobenzene by significant fluorescence quenching. Furthermore, IFMC-36-Eu displays preeminent anti-interference ability and could be used for sensing in the systems with complicated components. This is the first time that a d–f heterometallic–organic framework can be investigated as a chemical sensor for selective, sensitive, and recyclable detection of nitrobenzene.
Co-reporter:Yonghuai Wei, Ting Zhang, Zhongling Lang, Likai Yan, Zhongmin Su
Dyes and Pigments 2014 Volume 102() pp:6-12
Publication Date(Web):March 2014
DOI:10.1016/j.dyepig.2013.10.034
•Novel hexamolybdates dyes were designed with different electron donors.•The absorption spectra of substituted hexamolybdates dyes remarkably red shift.•The theoretical examination was performed on the key parameters.•Dye 6 containing a biTT unit has the largest λmax at 733 nm and shows a higher Jsc.•These dyes are promising candidates to obtain high performance solar cell materials.Novel organoimido-substituted hexamolybdates dyes were designed by introducing 3,4-ethylenedioxythiophene (EDOT) or thienothiophene (TT) unit as electron donor based on [Mo6O18(MBTH)]2−. The electronic structures, absorption spectra and transition natures of designed systems have been theoretically investigated according to density functional theory (DFT) and time-dependent DFT (TDDFT) calculations. Compared with dye 1, the absorption spectra of these designed organoimido-substituted hexamolybdates dyes exhibit both strong and broad absorptions from 400 to 800 nm, as well as remarkably red shift owing to the long π-conjugated bridge and high delocalization. Especially for dye 6, which contains a biTT unit, it has the largest maximum absorption wavelength (λmax) at 733 nm and may show a higher short-circuit current density (Jsc) as it possesses higher light harvesting efficiency (LHE) and reasonable driving force (ΔERP). Our work reveals that the designed molecule 6 is promising candidate for high performance solar cell materials.
Co-reporter:Hai-Bin Li, Ji Zhang, Yong Wu, Jun-Ling Jin, Yu-Ai Duan, Zhong-Min Su, Yun Geng
Dyes and Pigments 2014 Volume 108() pp:106-114
Publication Date(Web):September 2014
DOI:10.1016/j.dyepig.2014.04.029
•The π-linker effect on p-type DSSC performance was evaluated theoretically in detail.•The calculated charge transfer index is correlated well with the electron lifetime of dyes.•The high IPCE of O7 is due to the efficient regeneration and improved LHE.•The designed dyes all show the red-shifted absorption and larger LHE than O7.•Dye 4 is a promising candidate for the p-type sensitizer.The geometries, electronic structures, absorption spectra and the character of excited state for the reported p-type sensitizers O2, O6 and O7 and designed 1–5 with different π-linkers were investigated by DFT and TDDFT calculations, to reveal the influences of π-linkers on the performance of DSSCs. It is found that the higher IPCE of O7 as compared to O6 stems from the improved light harvesting efficiency (LHE) and the efficient regeneration. And, for O2 and O6 with comparable IPCE, the slightly increased Jsc for O2 can be mainly ascribed to the better spectrum matching with solar spectrum. The results also revealed that the calculated charge transfer index (DCT, the metric of photoinduced electron–hole separation) for reported sensitizers is well correlated with the measured electron lifetime pertaining to Voc. Importantly, dye 4 may be a promising candidate for the p-type sensitizer due to the red-shifted absorption, larger LHE and longer DCT in comparison with prototype O7.The performance of p-type sensitizers used in DSSCs was evaluated and predicted by DFT and TDDFT calculations.
Co-reporter:Ting Zhang, Nana Ma, Likai Yan, Tengying Ma, Zhongmin Su
Dyes and Pigments 2014 Volume 106() pp:105-110
Publication Date(Web):July 2014
DOI:10.1016/j.dyepig.2014.03.007
•The second-order NLO behaviors can be switched by reversible redox.•The absorption bands possess remarkably shift with the manipulable redox states.•The redox processes have influence on intramolecular donor or acceptor character.•The complex with reversible redox states might be excellent switchable NLO materials.•The β0 value of system, SP2−, is largest and is 525.94*10−30 esu.In this paper, the relationship between reversible redox properties and second-order nonlinear optical (NLO) responses for photochromic unsymmetrical Mn-Anderson type polyoxometalate covalently linked to one spiropyran has been systematically investigated by using density functional theory (DFT) method. It is noticeable that the second-order NLO behaviors can be switched by reversible redox for this complex. The gaining and losing of electrons cause significant change on NLO response. Among these studied systems, the β0 value of two-electron-oxidized system, SP2−, is largest and ∼48 times as large as that of system SP. The charge-transfer transition corresponding to the dominant contributions to β0 values indicates that redox processes affect on intramolecular donor and acceptor character. Therefore, this kind of complex with the facile and reversible redox states might potentially be used for switching purposes in NLO molecular devices.The unsymmetrical spiropyran–polyoxometalate complex with the facile and reversible redox states might become excellent switchable NLO material.
Co-reporter:Yong Li, Hong-Liang Xu, Heng-Qing Wu, Rong-Lin Zhong, Shi-Ling Sun and Zhong-Min Su
Dalton Transactions 2014 vol. 43(Issue 6) pp:2656-2660
Publication Date(Web):08 Nov 2013
DOI:10.1039/C3DT52923A
Recently, two isomeric thiophene-fused benzocarborane derivatives Tb1 and Tb2 with different locations of sulfur atoms, labeled as 1, 4 and 6, 9 of the thiophene were synthesized by Morisaki (Chem.–Eur. J., 2012, 18, 11251–11257) and Barrere (Macromolecules, 2009, 42, 2981–2987), respectively. In the present work, natural bond orbital (NBO) analysis shows that after doping one lithium atom into the isomeric structures Tb1 and Tb2, the electrons transfer to different regions in Tb1 and Tb2. For Tb1-Li, the transferred electrons mainly locate at S1, C2, C3, and S4, but for Tb2-Li, the transferred electrons mainly locate at C2, C3, C7, and C8. Significantly, the charge distribution is a crucial factor influencing the static first hyperpolarizabilities for Tb1-Li and Tb2-Li. Furthermore, the βtot value of Tb1-Li is 6222 au, which is about 160 times larger than that of Tb1 (39 au). However, the βtot value of Tb2-Li (498 au) is only about 5 times larger than that of the corresponding Tb2 (91 au). It is our expectation that this work could provide useful information for the development of nonlinear optical materials based on carboranes.
Co-reporter:Chun-Chen Yuan, Shi-Ming Wang, Wei-Lin Chen, Lin Liu, Chao Qin, Zhong-Min Su and En-Bo Wang
Dalton Transactions 2014 vol. 43(Issue 4) pp:1493-1497
Publication Date(Web):12 Nov 2013
DOI:10.1039/C3DT52676C
Two Anderson-type heteropolyanion-supported copper phenanthroline redox couples have been successfully introduced into dye-sensitized solar cells, which can significantly increase the short-circuit photocurrent, open-circuit voltage and the conversion efficiency by 2.2 times, 26.8% and 3.93 times respectively, compared to the pristine copper phenanthroline redox couple.
Co-reporter:Sa Chen, Shi-Ling Sun, Heng-Qing Wu, Hong-Liang Xu, Liang Zhao and Zhong-Min Su
Dalton Transactions 2014 vol. 43(Issue 33) pp:12657-12662
Publication Date(Web):02 Jun 2014
DOI:10.1039/C4DT01240B
Recently, the well-known phenalenyl radical π-dimer with its fascinating 2-electron/12-center (2e/12c) bond has attracted our attention. In this work, we designed two molecules, Li3O⋯C13H9 (1a) and BeF3⋯C13H9 (1b). Interestingly, owing to the inductive effect of superatoms, an electron is transferred from Li3O to phenalenyl in 1a, while an electron is transferred from phenalenyl to BeF3 in 1b. Further, we employed 1a and 1b as building blocks to assemble two novel molecules with 2e/12c bonds: Li3O⋯(C13H9)2⋯BeF3 (2a) and Li3O⋯(C13H9)2⋯BeF3 (2b). Remarkably, 2a and 2b with novel 2e/12c bonds exhibit a dramatic interlayer charge-transfer character, which results in a significant difference of dipole moments (Δμ: 2.6804 for 2a and 3.8019 Debye for 2b) between the ground state and the crucial excited state. As a result, the static first hyperpolarizabilities (β0: 5154 for 2a and 12500 au for 2b) are considerably larger than the values of 347 for 1a and 328 au for 1b. It is our expectation that the results of the present work might provide beneficial information for further theoretical and experimental studies on the fascinating properties of molecules with interlayer charge-transfer character.
Co-reporter:Kun Zhou, Chao Qin, Xin-Long Wang, Kui-Zhan Shao, Li-Kai Yan and Zhong-Min Su
Dalton Transactions 2014 vol. 43(Issue 28) pp:10695-10699
Publication Date(Web):17 Apr 2014
DOI:10.1039/C4DT00762J
We report a rare all-thiol-stabilized [Ag28(StBu)23]5+ ({Ag28S23} for short) nanocluster with a “crab-like” shape in compound [Ag28(StBu)23](CF3COO)5·8CH3OH (1), which has been synthesized by the self-assembly of AgStBu with CF3COOH, Et3N and KBr/KI in methanol. The diffuse reflection spectrum and luminescence spectra of 1 were investigated.
Co-reporter:Yu-Teng Zhang, Peng Huang, Chao Qin, Li-Kai Yan, Bai-Qiao Song, Zhao-Xia Yang, Kui-Zhan Shao and Zhong-Min Su
Dalton Transactions 2014 vol. 43(Issue 26) pp:9847-9850
Publication Date(Web):14 Apr 2014
DOI:10.1039/C4DT00507D
A new organic–inorganic hybrid titaniobate compound, [Cu(en)2][Cu(en)2(H2O)2]3[Ti2Nb8O28]·8H2O (1) (en = ethylenediamine), was successfully synthesized, characterized by single-crystal X-ray diffraction, IR spectroscopy, thermogravimetric analysis (TGA) and UV-Vis diffuse reflectance spectroscopy and its photoluminescence studied.
Co-reporter:Li-Jie Wang, Rong-Lin Zhong, Shi-Ling Sun, Hong-Liang Xu, Xiu-Mei Pan and Zhong-Min Su
Dalton Transactions 2014 vol. 43(Issue 25) pp:9655-9660
Publication Date(Web):10 Mar 2014
DOI:10.1039/C3DT53329H
Recently, a new sulfide cluster fullerene, Sc2S@Cs (10528)-C72 containing two pairs of fused pentagons has been isolated and characterized (Chen et al., J. Am. Chem. Soc., 2012, 134, 7851). Inspired by this investigation, we propose a question: what properties will be influenced by the interaction between the encapsulated V-shaped polar molecule and C72? To answer this question, four encapsulated metallic fullerenes (EMFs) M2N@C72 (M = Sc or Y, N = S or O) along with pristine Cs-C72 (10528) were investigated by quantum chemistry methods. The results show that the Egap (3.01–3.14 eV) of M2N@C72 are significantly greater than that of pristine Cs-C72 (10528) (2.34 eV). This indicates that the stabilities of these EMFs increase by encapsulating the V-shaped polar molecule into the fullerene. Furthermore, the natural bond orbital (NBO) charge analysis indicates electron transfer from M2N to C72 cage, which plays a crucial role in enhancing first hyperpolarizability (βtot). The βtot follows the order of 1174 au (Y2O@C72) ≈ 1179 au (Sc2O@C72) > 886 au (Y2S@C72) ≈ 864 au (Sc2S@C72) > 355 au (C72). This indicates that the βtot of M2N@C72 is more remarkable than that of pristine Cs-C72 (10528) due to the induction effect of the encapsulated molecule. Compared with sulfide cluster fullerenes (Y2S@C72 and Sc2S@C72), oxide cluster fullerenes (Sc2O@C72 and Y2O@C72) show much larger βtot due to the small ionic radius and the large electronegativity of oxygen. In contrast, the metal element (scandium and yttrium) has a slight influence on the βtot. Thus, oxide cluster fullerenes are candidates to become promising nonlinear optical materials with higher performance.
Co-reporter:Na-Na Ma, Shu-Jun Li, Li-Kai Yan, Yong-Qing Qiu and Zhong-Min Su
Dalton Transactions 2014 vol. 43(Issue 13) pp:5069-5075
Publication Date(Web):13 Dec 2013
DOI:10.1039/C3DT53298D
The rotary motion based on metallacarboranes around a molecular axis can be controlled by simple electron transfer processes, which provides a basis for the structure–property relationship for the nonlinear optical (NLO) switching. However, this phenomenon has not been previously reported in the development of NLO properties of metallacarboranes. In this work, the metallacarboranes [NiIII/IV(C2B9H11)2]−/0 and their C-,B-functionalized derivatives are studied by the density functional theory (DFT) method. By calculating relative energies, we obtained the stable states before and after rotation controlled by simple electron transfer. Then, the static and frequency-dependent second-order NLO properties were calculated by several DFT functionals. According to the TDDFT results, the large NLO responses of the studied compounds are mainly caused by substituent group-to-carborane cage charge transfer (L′LCT) and substituent group-to-metal charge transfer (L′MCT) processes. The order of first hyperpolarizabilities (β values) illustrates that the NLO response can be enhanced by introducing a strong electron-donating group. Significantly, the geometric interconversions resulting from the redox reaction of 1C/1T–6C/6T allow the NLO responses to be switched “ON” or “OFF”. The B(9,9′)-methoxyphenyl-functionalized derivative of nickelacarborane, having low energetic cost and large different NLO responses between two states (from 0 to 20998 a.u.), can be an excellent switchable NLO material.
Co-reporter:Kun Zhou, Chao Qin, Xin-Long Wang, Kui-Zhan Shao, Li-Kai Yan and Zhong-Min Su
CrystEngComm 2014 vol. 16(Issue 34) pp:7860-7864
Publication Date(Web):12 Jun 2014
DOI:10.1039/C4CE00867G
The first example of 1D self-assembly of carbonate-templated sandwich-like macrocycle-based Ag20S10 nanoclusters, {[(CO32−)@Ag20(StBu)10(CH3COO)8(DMF)2]·2H2O}n (1), is reported. Compound 1 is stable both in the solid state and in solution. It exhibits potential wide gap semiconductor properties, reversible thermochromic luminescence behaviour and excellent electrochemical properties.
Co-reporter:Wei-Li Zhou, Jun Liang, Chao Qin, Kui-Zhan Shao, Fang-Ming Wang and Zhong-Min Su
CrystEngComm 2014 vol. 16(Issue 32) pp:7410-7418
Publication Date(Web):09 Jun 2014
DOI:10.1039/C4CE00633J
Four inorganic–organic hybrids based on polyoxometalates (POMs), [Ag4(btz)2(Hbtz)2(H2O)Na(PMo12O40)·H2O] (1), [Ag2(4,4′-bpy)2Ag2(4,4′-bpy)(H2O)2(PMoVMoVI11O40)] (2), [Ag5(btz)2(4,4′-bpy)2(PMo12O40)·3H2O] (3), and [Ag5(btz)2(4,4′-bpy)2(PW12O40)·3H2O] (4) (Hbtz = 1H-1,2,3-benzotriazole, 4,4′-bpy = 4,4′-bipyridine), have been hydrothermally synthesized and structurally characterized by conventional techniques. Compound 1 is a 2D layer composed of AgI ions, btz ligands and [PMo12O40]3− anions. Compound 2 represents a rare 3D poly-threading framework based on infinite 2D layers made up of {POM–Ag2} inorganic building blocks and bpy ligands, which were threaded by silver–organic linear double chains. Compounds 3 and 4 are isostructural and exhibit three-fold interpenetrating diamondoid frameworks containing tri-nuclear silver centers as nodes and hexa-nuclear silver–organic macrocycles and [PMo12O40]3− anions, both as bi-connected units. Furthermore, their electrochemical properties, fluorescence properties and optical band gaps have been investigated in detail.
Co-reporter:Hai-Ning Wang, Guo-Gang Shan, Hai-Bin Li, Xin-Long Wang, Hong-Tao Cao and Zhong-Min Su
CrystEngComm 2014 vol. 16(Issue 13) pp:2754-2759
Publication Date(Web):20 Feb 2014
DOI:10.1039/C3CE41841C
A series of coordination compounds Htpim (1), {[Zn(Htpim)2(H2O)2]·(NO3)2·7.5H2O}n (2), {[Zn(Htpim)2(H2O)2]·(CH3COO)2·5H2O}n (3), {[Cd(Htpim)2(NO3)2]·9H2O}n (4), {[Cd(Htpim)2(CH3COO)(H2O)]·(CH3COO)·3H2O}n (5) were assembled from different metal(II) ions (ZnII for compounds 2 and 3, and CdII for compounds 4 and 5) and a building block of 2,4,5-tri(4-pyridyl)-imidazole (Htpim). All the compounds were fully characterized by single crystal X-ray diffraction analysis. Compounds 2, 3, 4 and 5 possess a 1D chain structure. The compounds herein exhibit interesting piezochromic properties. Their emission colors are blue-shifted by grinding. On the basis of the PXRD patterns, the piezochromic behavior is due to a conversion from the crystalline to the amorphous state by changes in the molecular arrangement and in the intermolecular π–π interactions. Moreover, compound 1 exhibits positive solvatochromism, and could behave as a luminescent sensor for the detection of the Cu2+ ion.
Co-reporter:Bai-Qiao Song, Xin-Long Wang, Guang-Sheng Yang, Hai-Ning Wang, Jun Liang, Kui-Zhan Shao and Zhong-Min Su
CrystEngComm 2014 vol. 16(Issue 30) pp:6882-6888
Publication Date(Web):23 May 2014
DOI:10.1039/C4CE00546E
A novel microporous metal–organic framework (MOF), namely {[Cd2L3(DMF)(NO3)]·2DMF·3H2O}n (1), where L = 1,3-bis(4-carboxyphenyl)imidazolium chloride, was prepared via a hydrothermal process. The structure of 1 shows the first example of 6-fold polyrotaxane-like interpenetration combined with interdigitation which extends the structure into a three-dimensional framework and exhibits reversible and irreversible guest-dependent structural conversion; because of the 1D channels running along the b axis, 1 can serve as a host to encapsulate and sensitize Eu3+ cations in water. Moreover, the framework also exhibits a proton conductivity of over 10−5 S cm−1 at 298 K and 98% relative humidity because the methylene hydrogens of the imidazolium moiety are highly acidic and aligned inside the channels enhancing the proton conductivity.
Co-reporter:Kun Zhou, Chao Qin, Xin-Long Wang, Li-Kai Yan, Kui-Zhan Shao and Zhong-Min Su
CrystEngComm 2014 vol. 16(Issue 45) pp:10376-10379
Publication Date(Web):30 Sep 2014
DOI:10.1039/C4CE01707B
An intriguing 2D cyano-bridged multinuclear silver(I) alkynyl network [Ag15(CCtBu)10(CF3COO)2(CN)3]n (1) is obtained by self-assembly. The existence of CN− in 1 is assumed to have originated from the Ag(I)-mediated C–C cleavage reaction of acetonitrile under mild solvothermal conditions. Furthermore, the photoluminescence property of 1 is investigated.
Co-reporter:Peng Huang, En-Long Zhou, Xin-Long Wang, Chun-Yi Sun, Hai-Ning Wang, Yan Xing, Kui-Zhan Shao and Zhong-Min Su
CrystEngComm 2014 vol. 16(Issue 41) pp:9582-9585
Publication Date(Web):20 Jun 2014
DOI:10.1039/C4CE00960F
New heteropolyniobates based on a bicapped Keggin-type {VNb14O42(NO3)2} (abbreviated as {VNb14}) cluster have been successfully synthesized by conventional aqueous methods. These clusters are a type of multifunctional material, which exhibit selective adsorption for methanol, ethanol and water and photocatalytic H2 evolution activity.
Co-reporter:Bai-Qiao Song, Xin-Long Wang, Jun Liang, Yu-Teng Zhang, Kui-Zhan Shao and Zhong-Min Su
CrystEngComm 2014 vol. 16(Issue 39) pp:9163-9167
Publication Date(Web):29 Jul 2014
DOI:10.1039/C4CE01148A
Two 2D isostructural organic–inorganic hybrid solid materials based on transition metal mono-substituted Keggin polyoxotungstate with an antenna molecule were synthesized under hydrothermal conditions. Compound 2 with the substituted Co(II) exhibits good photocatalytic activity in the degradation of rhodamine-B (RB) under visible light irradiation.
Co-reporter:Shuang-Bao Li, Yu-Ai Duan, Yun Geng, Hai-Bin Li, Jian-Zhao Zhang, Hong-Liang Xu, Min Zhang and Zhong-Min Su
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 47) pp:25799-25808
Publication Date(Web):03 Sep 2014
DOI:10.1039/C4CP03022B
In the current work, a series of bithiopheneimide (BTI)-based D–A copolymers were investigated based on the reported PDTSBTI (1) to screen excellent molecules toward organic photovoltaic (OPV) donor materials. It is found that the PCE based on the proposed derivative 4, where the silicon atom is replaced with vinyl and cyano groups on the DTS unit, shows a 70 percent improvement by Scharber diagrams compared with its prototype 1. Then, the charge transfer dynamics of 1/PC71BM and 4/PC71BM were investigated, including the intermolecular charge transfer (inter-CT) and recombination (inter-CR) rates. The theoretical data demonstrate that the ratio kinter-CT/kinter-CR of 4/PC71BM heterojunction is about 1 × 105 times higher than that of 1/PC71BM. These results clearly reveal that the designed donor molecule 4 will be a promising candidate for high-performance OPV device. We expect that this work from electron processing at the D/A interface may provide a theoretical guideline for further optimization and design of organic copolymer donor materials.
Co-reporter:Ji Zhang, Jian-Zhao Zhang, Hai-Bin Li, Yong Wu, Yun Geng and Zhong-Min Su
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 45) pp:24994-25003
Publication Date(Web):30 Sep 2014
DOI:10.1039/C4CP03355H
Ten porphyrin sensitizers with different electron-withdrawing groups derived from the best sensitizer SM315 were investigated by means of the density functional theory (DFT) and time-dependent DFT calculations. To this end, major factors affecting the performance of the cell, including light harvesting, electron injection, dye regeneration, and conduction band energy shift are taken into consideration. Especially, the calculated distance (r) from the electron recapture center to the semiconductor surface is used to probe the charge recombination process. In addition, considering the complexity of the porphyrin sensitizers' absorption, the maximum short circuit current density (Jmaxsc) is determined for investigating the light harvesting ability quantitatively. We find that when compared to SM315 with 2,1,3-benzothiadiazole, 1 with naphtho[1,2-c:5,6-c]bis[1,2,5]thiadiazole shows better performance due to both larger Jmaxsc and r, and 7 with diketopyrrolopyrrole could also be a promising candidate due to the much larger Jmaxsc and comparable r.
Co-reporter:Bo Zhu, Zhong-Ling Lang, Na-Na Ma, Li-Kai Yan and Zhong-Min Su
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 33) pp:18017-18022
Publication Date(Web):14 Jul 2014
DOI:10.1039/C4CP01389A
Density functional theory (DFT) calculations and natural bond orbital (NBO) analysis were carried out to investigate the electronic structures and bonding features between the ruthenium(II) atom and the SO2 molecule in two ruthenium–sulfur dioxide (SO2) adducts, trans-Ru(NH3)4(SO2)Cl+ and [{SiW11O39}RuII(SO2)]6−. In addition, the bonding interactions between SO2 and the metal-ruthenium fragment were determined by binding energy (ΔEabs) calculation and electronic structures. The results indicate that the η1-S-planar model in both trans-Ru(NH3)4(SO2)Cl+ and [{SiW11O39}RuII(SO2)]6− are more favorable. NBO analysis of the bonding interaction between ruthenium and sulfur centers in the [{SiW11O39}RuII(SO2)]6− complex shows that it possesses a σ and a π bond. It predicts that the polyoxometalate [SiW11O39Ru]6− can serve as a potential adsorbent for the SO2 molecule because of the strong Ru–S bond relative to Ru(NH3)4Cl+.
Co-reporter:Wei-Chao Chen, Li-Kai Yan, Cai-Xia Wu, Xin-Long Wang, Kui-Zhan Shao, Zhong-Min Su, and En-Bo Wang
Crystal Growth & Design 2014 Volume 14(Issue 10) pp:5099-5110
Publication Date(Web):July 8, 2014
DOI:10.1021/cg500719q
Using a pH-dependent synthetic approach, the combination of different simple metal salts or metal coordination complexes with SeO32– heteroanion templates was employed to synthesize five distinct assemblies of Keggin-/Dawson-type tungstoselenites: (C2H8N)10KNa[(α-SeW9O34){Zr(H2O)}{WO(H2O)}(WO2)(SeO3){α-SeW8O31Zr(H2O)}]2·14H2O (1) at pH = 1.3; (C2H8N)10KNa5[(Se2W18O60)2(μ2-O)4]·12H2O (2) at pH = 2.5; (C2H8N)4Na4[Se2W18O62(H2O)2]·13H2O (3) at pH = 3.6; (C2H8N)4K3Na10[(α-SeW9O33)2{Ce2(CH3COO)(H2O)3W3O6}(α-Se2W14O52)]·26H2O (4) at pH = 4.5; K10Na5[(α-SeW9O33)2{Ce2(H2O)4W3O6}{α-Se2W14O51(OH)}]·24H2O (5) at pH = 4.5. All five compounds were characterized by single-crystal X-ray structure analysis, IR spectroscopy, thermogravimetric, UV/vis spectroscopy, and ESI-MS. Moreover, their electrochemical properties were investigated. Keggin-type polyoxoanion of 1 remains the first reported Zr-containing tungstoselenites based on {α-SeW9} building blocks. X-ray analysis revealed that the 4d metal Zr centers have seven- and eight-coordinated modes, and SeO32– acts as the templates as well as the linkers. With the increasing of the pH, Dawson-type polyoxoanions of 2 and 3 based on the first reported basic lacunary {α-Se2W14} building blocks are obtained by using 3d-4f metal coordination complexes. Polyoxoanions of 4 and 5 remain similar structures stabilized by the 4f metal Ce centers at pH = 4.5 and that contain the basic Keggin-type {α-SeW9} and Dawson-type {α-Se2W14} building blocks in 1–3 at the same time, presenting the mixed multiple lacunary building blocks being combined into the single polyoxoanion architecture. Furthermore, the density functional theory calculations have been performed on polyoxoanions of 1 and 5 as the representatives to investigate their electronic properties.
Co-reporter:Yan-Chun Liu, Shui-Xing Wu, Zhong-Min Su and Hou-Yu Zhang
New Journal of Chemistry 2014 vol. 38(Issue 3) pp:1092-1099
Publication Date(Web):22 Nov 2013
DOI:10.1039/C3NJ00857F
The 14-π cross-linked annulene dicyclopenta[a,e]pentalene (dcpp) is suggested for the first time to function as a sandwich ligand. According to density functional theory (DFT) calculations, upon being sandwiched between two dcpp ligands, the tetravanadium chain, without the support of auxiliary ligands, has two unpaired electrons and has a tendency for deformation to gain extra stability through multicenter bondings in the ground state. The one-electron ligand chlorine can lead to two types of structures, one with two chlorine atoms connecting to the two central vanadium atoms and the other one with two chlorine atoms connecting to the two terminal vanadium atoms, both of which are energetically more favorable in the triplet state. In the carbonyl end-capping complex, the tetravanadium is stable as a linear metal–metal bonded array with alternating single and double bonds and all electrons paired for bonding, although a triplet state conformer exists with two extra carbonyl ligands bound to the two central vanadium atoms which is more energy favorable by 15.1 kcal mol−1. Bonding features within the tetravanadium moiety were obtained based on electron density analyses. We hope these discussions are helpful for the design of new extended metal atom chain (EMAC) systems and for the pursuit of effective catalysts based on the dcpp sandwich complexes.
Co-reporter:Peng Li;Guo-Gang Shan;Hong-Tao Cao;Dong-Xia Zhu;Rukkiat Jitchati;Martin R. Bryce
European Journal of Inorganic Chemistry 2014 Volume 2014( Issue 14) pp:2376-2382
Publication Date(Web):
DOI:10.1002/ejic.201400007
Abstract
The syntheses of two new heteroleptic cationic iridium complexes containing 2,6-diphenylpyridine (Hdppy) and 2,4,6-triphenylpyridine (Htppy) as the cyclometalated ligands, namely, [Ir(dppy)2phen]PF6 (1, phen = 1,10-phenanthroline) and [Ir(tppy)2phen]PF6 (2), are described. The X-ray crystal structure of 2 reveals a distorted octahedral geometry around the Ir center and close intramolecular face-to-face π–π stacking interactions between the pendant phenyl rings at the 2-position of the cyclometalated ligands and the NN ancillary ligand. This represents a new π–π stacking mode in charged Ir complexes. Complexes 1 and 2 are green photoemitters: their photophysical and electrochemical properties are interpreted with the assistance of density functional theory (DFT) calculations. These calculations also establish that the observed intramolecular interactions cannot effectively prevent the lengthening of the Ir–N bonds of the complexes in their metal-centered (3MC) states. Complexes 1 and 2 do not emit light in light-emitting electrochemical cells (LECs) under conditions in which the model compound [Ir(ppy)2phen]PF6 (3) emits strongly. This is explained by degradation reactions of the 3MC state of 1 and 2 under the applied bias during LEC operation facilitated by the enhanced distortions in the geometry of the complexes. These observations have important implications for the future design of complexes for LEC applications.
Co-reporter:Ying Gao, Shi-Ling Sun, Hong-Liang Xu, Liang Zhao and Zhong-Min Su
RSC Advances 2014 vol. 4(Issue 47) pp:24433-24438
Publication Date(Web):13 May 2014
DOI:10.1039/C4RA02238F
In this work, the 1-Li+, 1-Na+ and 1-K+ complexes formed by N-methylbenzoaza-18-crown-6-ether derivatives (1) with one alkali metal cation (Li+, Na+ and K+) were investigated. Significantly, the dipole moments of 1-Li+, 1-Na+ and 1-K+ enhance with increasing atomic number. However, their first hyperpolarizabilities (β0) decrease with increasing the atomic number. Further results show that the interaction energies increase in the order of 1-Li+ > 1-Na+ > 1-K+. Moreover, the transition energies of 1-Li+, 1-Na+ and 1-K+ are inversely proportional to the β0 values. Therefore, the interaction energy and transition energy are the major factors determining the β0 values of 1-Li+, 1-Na+ and 1-K+. The combining of large variations of the dipole moment and the first hyperpolarizability can be used as a detection sensor for alkali metal cations. We hope that this work will provide valuable knowledge for designing alkali metal cation sensors by electro-optical properties.
Co-reporter:Xin-Yao Ren, Yong Wu, Guo-Gang Shan, Li Wang, Yun Geng and Zhong-Min Su
RSC Advances 2014 vol. 4(Issue 107) pp:62197-62208
Publication Date(Web):24 Oct 2014
DOI:10.1039/C4RA07041K
A density functional theory/time-depended density functional theory was used to investigate a series of heteroleptic Ir(III) complexes (1–4) employing azadipyrromethene and closely related dipyrromethene derivatives as N^N ancillary ligands, in an effort to explore the underlying reasons of non-radiative behaviour of 1 and further adjust the photophysical properties by the modification of N^N ancillary ligands. The results reveal that the non-emissive phenomenon of 1 can be attributed to the weak 3ILCT character of the emissive excited state and large structure distortion, as well as the small Topt1–Sopt0 energy gap. Upon tailoring the N^N ancillary ligands, the geometry distortion of 2–4 becomes obviously smaller in comparison with 1, accordingly, the spectrum properties are also markedly affected. For instance, the enlargement of frontier molecular orbital energy gaps from 1 to 4 results in the blue-shift of their absorption and emission spectra, which is considered to be dominated by the ancillary ligand, while there is a little contribution from the Ir(III) center. Importantly, further analysis on the quantum yield (ΦPL) of these complexes also indicates a vital role of N^N ancillary ligands. It is intriguing to note that the designed complex 4 without pendant phenyl rings substituent in the ancillary ligand, possesses an efficient indirect spin-orbital coupling route, larger transition electric dipole moment (μS3), higher Topt1–Sopt0 energy gap and smaller S3–T1 splitting energy (ΔE(S3–T1)), which ensure its higher ΦPL compared to other complexes.
Co-reporter:Shi-Ming Wang, Lin Liu, Wei-Lin Chen, Zhong-Min Su, En-Bo Wang, and Chao Li
Industrial & Engineering Chemistry Research 2014 Volume 53(Issue 1) pp:150-156
Publication Date(Web):2017-2-22
DOI:10.1021/ie402074c
A new H3PW12O40 (PW12)-based interfacial layer for dye-sensitized solar cells (DSSCs) has been fabricated by the LBL method. The features of the interfacial layer were tested by IR and UV–vis spectroscopies and AFM. The cells were systemically tested by the photocurrent–voltage (J–V) curve, dark-current measurement, open-circuit voltage decay (OCVD), and monochromatic incident photon-to-photocurrent conversion efficiency (IPCE) techniques. The PW12-based interfacial layer was found to accelerate electron transfer and retard recombination, eventually leading to an efficient increase in energy conversion efficiency. The investigations indicate that the energy conversion efficiency of a (PW12/TiO2)3-based DSSC is significantly enhanced by 54% at 100 mW cm–2 compared with a DSSC with no treatment and by 20% compared with a TiCl4-treatment DSSC. Polyoxometalate was first introduced into the interfacial layer in modifying the photoanode to accelerate electron transfer and retard recombination to improve DSSC efficiency in this work.
Co-reporter:Yu-Ai Duan, Hai-Bin Li, Yun Geng, Yong Wu, Guang-Yu Wang, Zhong-Min Su
Organic Electronics 2014 Volume 15(Issue 2) pp:602-613
Publication Date(Web):February 2014
DOI:10.1016/j.orgel.2013.12.011
•Changing the position of heteroatom helps to control the transport channel.•The introduction of hexylthienyl results in a typical one-dimensional transport.•Hirshfeld surface analysis is useful for exploring the intermolecular interaction.•The intermolecular packing motifs can be rationalized by electrostatic potential analysis.•Distinct and anisotropic mobility is due to different intermolecular interaction.Chemical modifications such as changing the position of heteroatom, introducing different substituents and π-conjugated cores are powerful molecular design tools to modulate their optical and electrochemical performance. In this context, in-depth density functional theory investigations on the geometries, frontier molecular orbitals, reorganization energies, transfer integrals, anisotropic mobilities and band structures of tetrathienoarene derivatives were carried out to provide insights into the effects of these chemical modifications on their hole transport properties. The electrostatic potential, Hirshfeld surface analysis, energy decomposition analysis (EDA) and anisotropic mobility were also employed to shed light on the intricate interplays among molecular packings, intermolecular interactions and transport properties. It is found that compared with 2, 1a with sulfur atom inside has lower frontier orbital energy level, smaller reorganization energy and larger transfer integral and hole mobility. However, more S⋯S interactions in 2 could provide more effective transport channels for charge carrier transport. The introduction of hexylthienyl groups (1c) results in an enhancement of π–π interaction and leads to an increase in the highest occupied molecular orbital (HOMO) and transfer integrals. Meanwhile, the strongest intermolecular interaction energy of pathway A in 1c renders its transport behavior with typical one-dimensional (1D) transport. Moreover, anthracene as the π-conjugated core seemed to possess better transport properties in comparison with dibenzothiophene or chrysene acting as core. In addition, the dispersion energy of all investigated compounds plays a leading role in determining the energetically accessible stacking motifs. We hope that our speculation would facilitate the future design and preparation of high-performance charge-transport materials.
Co-reporter:Wei-Li Zhou, Jun Liang, Liang Zhao, Xin-Long Wang, Kui-Zhan Shao, Zhong-Min Su
Inorganic Chemistry Communications 2014 Volume 47() pp:48-51
Publication Date(Web):September 2014
DOI:10.1016/j.inoche.2014.07.014
•An unprecedented Ag-Pyrazine-Polyoxometalate framework has been synthesized.•Compound 1 exhibits a three-dimensional entangled architecture.•Compound 1 shows outstanding photocatalytic activity for the degradation of RhB.A new 3D inorganic–organic hybrid based on polyoxometalates (POMs), [Ag4(pz)6(SiW12O40)·H2O] (1), (pz = pyrazine)1 has been hydrothermally synthesized and structurally characterized by single-crystal X-ray diffraction, IR, TG, and UV–vis absorption spectra. In compound 1, there are two identical interpenetrating 3D frameworks containing deca-nuclear silver circles and pz ligands, which are further linked by [SiW12O40]4 − anions to exhibit an entangled architecture. Additionally, its optical band gap, photocatalytic performance and electrochemical behavior at room temperature have been investigated in detail.A novel three-dimensional AgI/POM-based hybrid has been synthesized and its optical band gaps, electrochemical and photocatalytic performance have been investigated.
Co-reporter:Lian-Jie Li, Xin-Long Wang, Kui-Zhan Shao, Zhong-Min Su, Hai-Ming Xie
Solid State Sciences 2014 Volume 34() pp:8-11
Publication Date(Web):August 2014
DOI:10.1016/j.solidstatesciences.2014.04.004
•Compound 1 presents a two-dimensional layers structure with point symbol of (36·46·53)-hxl topology.•Compound 2 exhibits the 2-fold interpenetrating 3D structure.•Compound 2 shows the point symbol of the (34·47·54)-pcu topology based on the hexanuclear zinc clusters as the six-connected node.•Compound 2 shows fluorescence property in the solid state at room temperature.Two metal–organic frameworks, namely, [Ni2(BIMB)2(ndd)2·H2O]n (1) and [Zn3(ndd)2.5(μ3-OH)(1,3-dpp)]n (2) (H2ndd = 2,2′-(naphthalene-1,5-diylbis(oxy))diacetic acid, BIMB = 1,4-bis[(1H-imidazol-1-ly)methyl]benzene, 1,3-dpp = 1,3-di(pyridin-4-yl)propane) have been synthesized under hydrothermal conditions and characterized by single-crystal X-ray diffraction and thermogravimetric analysis. Compound 1 presents a two-dimensional network with point symbol of (36·46·53)-hxl topology. Moreover, compound 2 displays a novel 2-fold interpenetrated structure with the point symbol of (412·63)-pcu topology based on the hexanuclear [Zn6(CO2)10(N)4] unit as a six-connected node. Meanwhile, compound 2 shows good fluorescence property in the solid state at room temperature.
Co-reporter:Gang Yuan, Kui-Zhan Shao, Xiang-Rong Hao, Ya-Ru Pan, Zhong-Min Su
Inorganic Chemistry Communications 2014 Volume 42() pp:15-19
Publication Date(Web):April 2014
DOI:10.1016/j.inoche.2014.01.004
•One new CdII complex was constructed from the designed semi-rigid bis-IDC ligand.•The H4L ligands displayed two different coordination modes in complex 1.•The 3D framework of 1 was built by H2L2 − anions linking chain-like SBUs.•Both HL ligand and complex displayed good solid state luminescent properties.A new 3D complex, {[Cd3(HL)2(H2L)(H2O)4]·2H2O}n (1) (H4L = 1,1′-(1,4-phenylenebis(methylene))bis(1H-imidazole-4,5-dicarboxylic acid), has been hydrothermally synthesized and characterized by elemental analyses, IR, TG, and X-ray single-crystal diffraction. Complex 1 is the first framework based on CdII ion and semi-rigid bis-IDC ligand. The (6,6)-net architecture of 1 is built from H2L2 − ligands linking 1D chain-like [Cd3(HL)2]n SBUs, which formed by HL3 − anions bridging two kinds of metal centers. In addition, complex 1 was demonstrated to display strong blue-violet fluorescence emission in the solid state at room temperature.Graphical abstractA new complex based on the designed H3IDC derivative ligand with two IDC groups has been successfully synthesized under hydrothermal condition. Complex 1 exhibits a 3D 6-connected framework constructed from H2L2 – ligands linking chain-like SBUs formed by HL3− ligands bridging two different metal nodes. Moreover, the thermostability and luminescent properties complex 1 have been investigated.
Co-reporter:Hong Li, Yong-Ming Yin, Hong-Tao Cao, Hai-Zhu Sun, Li Wang, Guo-Gang Shan, Dong-Xia Zhu, Zhong-Min Su, Wen-Fa Xie
Journal of Organometallic Chemistry 2014 Volume 753() pp:55-62
Publication Date(Web):1 March 2014
DOI:10.1016/j.jorganchem.2013.11.036
•Two Ir(III) complexes with carbene ligands are designed and synthesized.•Both complexes exhibit greenish-blue emission with high efficiency of ∼0.5.•The experimental results are rationalized by the theoretical calculations.•Greenish-blue OLEDs show good efficiencies of 11.78 cd A−1 and 11.43 lm W−1.•A white OLED using one of them as dopant is successfully fabricated.Two heteroleptic iridium(III) complexes using carbene as cyclometalated ligands and pyridine-triazole as ancillary ligand, namely (fpmi)2Ir(mtzpy) (1) and (fpmi)2Ir(phtzpy) (2) [fpmi = 1-(4-fluorophenyl)-3-methylimdazolin-2-ylidene-C,C2′, mtzpy = 2-(5-methyl-2H-1,2,4-triazol-3-yl)pyridine, phtzpy = 2-(5-phenyl-2H-1,2,4-triazol-3-yl)pyridine], were synthesized and their structural, photophysical and electrochemical properties investigated systematically. Both complexes exhibit bright greenish-blue phosphorescence (λmax ∼490 nm) with quantum yields of about 0.50. Comprehensive density functional theory (DFT) approach was then performed to gain insights into their photophysical and electrochemical characters. The fabrication of organic light-emitting diodes (OLEDs), employing complexes 1 and 2 as phosphorescent dopants, was successfully achieved. Among them, the device based on 1 exhibited considerable power efficiency (ηp) of 11.43 lm W−1 and current efficiency (ηc) of 11.78 cd A−1. With the merit of intrinsic characteristic of complex 1, a white OLED comprised of 1 and one orange phosphor (pbi)2Ir(biq) achieved a peak ηp of 9.95 lm W−1 and ηc of 10.81 cd A−1, together with Commission Internationale de l'Eclairage (CIE) coordinates of (0.34, 0.40). The results indicate that two iridium(III) complexes reported here are promising phosphorescent dyes for OLEDs.Two iridium(III) complexes containing carbene-based cyclometalated ligands and pyridine-triazole ancillary ligand exhibit intense greenish-blue phosphorescent emission. The greenish-blue and white OLEDs using the titled complexes as the dopant emitters show effective electroluminescence efficiencies.
Co-reporter:Zhao-Xia Yang, Peng Huang, Liang Zhao, Min Zhang, Yu-Teng Zhang, Zhong-Min Su
Inorganic Chemistry Communications 2014 Volume 44() pp:195-197
Publication Date(Web):June 2014
DOI:10.1016/j.inoche.2014.03.022
•Compound 1 was hydrothermally synthesized by the aggregation of niobium.•1 was depicted as four {SiW9O34} units linked together by a {Nb16O30} cluster.•The photocatalytic activity of 1 for H2 evolution was observed.A tetra-Keggin polyoxometalate (POM), Cs19K2[Nb4O6(SiW9Nb3O40)4]Cl·27H2O (1) with a photocatalytic property has been synthesized by utilizing the self-assembly of W/Nb mixed-addendum POM [SiW9(NbO2)3O37]7 − under hydrothermal conditions. Its structure was determined by single-crystal X-ray diffraction, and further characterized by IR spectroscopy, thermogravimetric analysis, and cyclic voltammetry. Moreover, the photocatalytic activity of 1 for H2 evolution from water was observed under UV irradiation, suggesting that 1 might be a promising photocatalytic catalyst.Compound 1 was obtained by the aggregation of niobium under hydrothermal conditions. 1 may be depicted as four {SiW9O34} units linked together by a {Nb16O30} cluster. Moreover, the photocatalytic activity of 1 for H2 evolution from water was observed under UV irradiation.
Co-reporter:Ya-Juan Xiao, Chun-Yi Sun, Guo-Chun Yang, Liang Zhao, Zhong-Min Su
Inorganic Chemistry Communications 2014 Volume 46() pp:248-250
Publication Date(Web):August 2014
DOI:10.1016/j.inoche.2014.05.016
•A fourfold interpenetrated three-dimensional microporous framework was synthesized.•This compound displays high adsorption of CO2 without any functional sites.•This compound exhibits good capability to tune luminescence properties of Alq3.A versatile three-dimensional four-fold interpenetrated metal-organic framework [Zn5(btz)6(bdc)2(H2O)2]·7DMA (1) has been synthesized under solvothermal condition, which has two types of channels running along a and b axes with dimension of approximately 7.5 × 9 Å2 and 8.5 × 11 Å2. With the appropriate and permanent porosity, compound 1 exhibits high adsorption of CO2 and good capability to encapsulate Alq3 (tris(8-hydro-xyquinoline)aluminum) chromophores to realize tunable fluorescence at solid state. To the best of our knowledge, this material is a rare example of MOFs that exhibit good capability to tune luminescence properties of Alq3.A three-dimensional fourfold interpenetrated MOF exhibits high adsorption of CO2 and good capability to encapsulate Alq3 chromophores to realize tunable fluorescence.
Co-reporter:Chun-Chen Yuan, Shi-Ming Wang, Wei-Lin Chen, Lin Liu, Zhi-Ming Zhang, Ying Lu, Zhong-Min Su, Si-Wen Zhang, En-Bo Wang
Inorganic Chemistry Communications 2014 Volume 46() pp:89-93
Publication Date(Web):August 2014
DOI:10.1016/j.inoche.2014.05.023
•This is the first report of a pure-inorganic-cobalt-compound redox couple for DSSCs.•Two Keggin-type POMs have been employed as a redox couple in DSSCs.•The redox potential of the {CoII/IIIW12} couple matches with dye and TiO2.•The {CoII/IIIW12} redox couple shows weak absorption in the visible light region.Two Keggin-type polyoxometalates [K6CoIIW12O40 (denoted as {CoIIW12}) and K5CoIIIW12O40 (denoted as {CoIIIW12})] were firstly employed as the redox couple in dye-sensitized solar cells. Photocurrent density–photovoltage curves, cyclic voltammetry curves, photocurrent action spectra and UV–vis spectra demonstrated that {CoIIW12} and {CoIIIW12} displayed weak absorption in the visible light region, potential matching with dye and TiO2 conducting band, and significant conversion efficiency.Two Keggin-type polyoxometalates have been successfully employed as the redox couple in dye-sensitized solar cells for the first time, which achieved relatively high values of open circuit voltage (651 mV) and fill factor (0.549).
Co-reporter:Dr. Jun-Sheng Qin;Dr. Shu-Ran Zhang;Dr. Dong-Ying Du;Ping Shen;Shao-Juan Bao; Ya-Qian Lan; Zhong-Min Su
Chemistry - A European Journal 2014 Volume 20( Issue 19) pp:5625-5630
Publication Date(Web):
DOI:10.1002/chem.201304480
Abstract
Herein, a novel anionic framework with primitive centered cubic (pcu) topology, [(CH3)2NH2]4[(Zn4dttz6)Zn3]⋅15 DMF⋅4.5 H2O, (IFMC-2; H3dttz=4,5-di(1H-tetrazol-5-yl)-2H-1,2,3-triazole) was solvothermally isolated. A new example of a tetranuclear zinc cluster {Zn4dttz6} served as a secondary building unit in IFMC-2. Furthermore, the metal cluster was connected by ZnII ions to give rise to a 3D open microporous structure. The lanthanide(III)-loaded metal–organic framework (MOF) materials Ln3+@IFMC-2, were successfully prepared by using ion-exchange experiments owing to the anionic framework of IFMC-2. Moreover, the emission spectra of the as-prepared Ln3+@IFMC-2 were investigated, and the results suggested that IFMC-2 could be utilized as a potential luminescent probe toward different Ln3+ ions. Additionally, the absorption ability of IFMC-2 toward ionic dyes was also performed. Cationic dyes can be absorbed, but not neutral and anionic dyes, thus indicating that IFMC-2 exhibits selective absorption toward cationic dyes. Furthermore, the cationic dyes can be gradually released in the presence of NaCl.
Co-reporter:Yang-Yang Hu;Dr. Shi-Ling Sun;Wen-Tao Tian; Wei Quan Tian;Dr. Hong-Liang Xu; Zhong-Min Su
ChemPhysChem 2014 Volume 15( Issue 5) pp:
Publication Date(Web):
DOI:10.1002/cphc.201490021
Co-reporter:Yang-Yang Hu;Dr. Shi-Ling Sun;Wen-Tao Tian; Wei Quan Tian;Dr. Hong-Liang Xu; Zhong-Min Su
ChemPhysChem 2014 Volume 15( Issue 5) pp:929-934
Publication Date(Web):
DOI:10.1002/cphc.201301149
Abstract
A series of spiral donor–π–acceptor frameworks (i.e. 2–2, 3–3, 4–4, and 5–5) based on 4-nitrophenyldiphenylamine with π-conjugated linear acenes (naphthalenes, anthracenes, tetracenes, and pentacenes) serving as the electron donor and nitro (NO2) groups serving as the electron acceptor were designed to investigate the relationships between the nonlinear optical (NLO) responses and the spirality in the frameworks. A parameter denoted as D was defined to describe the extent of the spiral framework. The D value reached its maximum if the number of NO2 groups was equal to the number of fused benzene rings contained in the linear acene. A longer 4-nitrophenyldiphenylamine chain led to a larger D value and, further, to a larger first hyperpolarizability. Different from traditional NLO materials with charge transfer occurring in the one-dimensional direction, charge transfer in 2–2, 3–3, 4–4, and 5–5 occur in three-dimensional directions due to the attractive spiral frameworks, and this is of great importance in the design of NLO materials. The origin of such an enhancement in the NLO properties of these spiral frameworks was explained with the aid of molecular orbital analysis.
Co-reporter:Wen-E Li, Fu-Hong Liu, Xiao-Ming Liu, Kun Zhou, Liang Zhao, Xiao-Li Hu, Zhong-Min Su
Inorganic Chemistry Communications 2014 40() pp: 8-10
Publication Date(Web):
DOI:10.1016/j.inoche.2013.11.005
Co-reporter:Yin Lin, Yan-Fei Zhu, Zhi-Hui Chen, Fu-Hong Liu, Liang Zhao, Zhong-Min Su
Inorganic Chemistry Communications 2014 40() pp: 22-25
Publication Date(Web):
DOI:10.1016/j.inoche.2013.11.023
Co-reporter:Rong-Lin Zhong;Dr. Shi-Ling Sun;Dr. Hong-Liang Xu; Yong-Qing Qiu ; Zhong-Min Su
ChemPlusChem 2014 Volume 79( Issue 5) pp:732-736
Publication Date(Web):
DOI:10.1002/cplu.201300381
Abstract
Carbon–boron–nitride heteronanotubes have attracted much interest owing to their adjustable properties and wide applications in many fields. In this work, the structures and first hyperpolarizabilities (β0) of a series of HA-n and isoelectronic PA-n heteronanotubes are investigated, in which HA-n is composed of a helical C segment and BN segment, and PA-n is composed of an arc C segment and BN segment. Interestingly, the helix C segment in HA-9 leads to a fascinating long-range charge-transfer character. As a result, the β0 value of HA-9 is 1.80×104 a.u., which is much larger than that of PA-9 (4.99×103 a.u.). Significantly, the β0 value of the C segment in HA-9 is 1.72×104 a.u., whereas that of the BN segment in HA-9 is much smaller (7.48×102 a.u.). In this context, the larger β0 value of HA-9 is essentially induced by the helix topological effect of the C segment. It is our expectation that this new knowledge about heteronanotubes might provide more ideas for further exploration of sp2-hybridized carbon segments with special topologies.
Co-reporter:Dr. Shu-Ran Zhang;Dr. Dong-Ying Du;Dr. Jun-Sheng Qin;Dr. Shao-Juan Bao; Shun-Li Li;Dr. Wen-Wen He; Ya-Qian Lan;Dr. Ping Shen; Zhong-Min Su
Chemistry - A European Journal 2014 Volume 20( Issue 13) pp:3589-3594
Publication Date(Web):
DOI:10.1002/chem.201304692
Abstract
A 2D, extremely stable, metal–organic framework (MOF), NENU-503, was successfully constructed. It displays highly selective and recyclable properties in detection of nitroaromatic explosives as a fluorescent sensor. This is the first MOF that can distinguish between nitroaromatic molecules with different numbers of NO2 groups.
Co-reporter:Li Wang, Yong Wu, Yun Geng, Jie Wu, Dong-Xia Zhu, and Zhong-Min Su
The Journal of Physical Chemistry A 2014 Volume 118(Issue 27) pp:5058-5067
Publication Date(Web):May 20, 2014
DOI:10.1021/jp4099649
The phosphorescent efficiencies of the Ir(III) carbene complexes 1–3 with wide-range color tuning were focused on in this work. A DFT/TDDFT (density functional theory/time-dependent density functional theory) investigation on the geometries in the ground and lowest triplet excited states, the frontier molecular orbitals, the absorption spectra, and d-orbital splittings of 1–3 were provided to get a better understanding of structure–property relationships. Importantly, to shed light on the difference in phosphorescent quantum yields for 1–3, radiative decay constants as well as zero-field-splitting parameters were calculated based on the estimation of spin–orbit coupling (SOC) matrix elements denoted as ⟨T1α|HSOC|Sn⟩. The results show that, for any complex, the radiative decay rates in the three substates (namely, Tx, Ty, and Tz) are not equal, and the largest radiative rates of 1–3 are all located in x substates with values of 1.0764 × 104, 0.8231 × 104, and 1.9596 × 104 s–1, respectively. Moreover, for 3 with the highest quantum efficiency, we make efforts to modify it through varying substituents and substituent positions not only to achieve blue shift in the emission but also to obtain improved triplet energy.
Co-reporter:Ying Gao, Shi-Ling Sun, Hong-Liang Xu, Zhong-Min Su
Chemical Physics Letters 2014 600() pp: 123-127
Publication Date(Web):
DOI:10.1016/j.cplett.2014.03.037
Co-reporter:Ting Zhang ; Wei Guan ; Shizheng Wen ; Tengying Ma ; Likai Yan ;Zhongmin Su
The Journal of Physical Chemistry C 2014 Volume 118(Issue 51) pp:29623-29628
Publication Date(Web):December 3, 2014
DOI:10.1021/jp509765a
We use time-dependent density functional theory methods to discuss the absorption spectra, electronic transition properties, and photovoltaic performance of metalloporphyrin–polyoxometalates (POM) complexes for p-type dye-sensitized solar cells (DSSCs). The results show that the energy levels of the frontier molecular orbitals for dyes 2–6 match the requirements of p-type DSSCs. The absorption spectra of dyes 2–6 exhibit larger and broader absorptions compared to that of dye 1 by the introduction of POM. In addition, the photovoltaic performances of dyes 2–6 are suitable for high-efficiency DSSCs. This paper is expected to advance the design of metalloporphyrin–POM hybrid dyes with excellent performance in DSSCs.
Co-reporter:Heng-Qing Wu ; Rong-Lin Zhong ; Shi-Ling Sun ; Hong-Liang Xu
The Journal of Physical Chemistry C 2014 Volume 118(Issue 13) pp:6952-6958
Publication Date(Web):March 17, 2014
DOI:10.1021/jp410560j
Adamantane (Ad) is the parent molecule of diamondoids, which has been the subject of much chemical interest because of its highly symmetric cage structure. Many valuable papers demonstrated that the H atom of the methine position in Ad is highly reactive. Compared with highly symmetric carbon structures such as graphene and single-walled carbon nanotubes (SWCNTs), Ad only possesses σ bonds. In this work, we report a quantum chemical calculation on three complexes (Ad-Li, Ad-Na, and Ad-K), which were obtained by substituting the H atom of methine position in Ad with alkali metal (Li, Na, and K). Interestingly, the substitution by alkali metals leads to absorption within the visible region. The maximum absorption wavelengths of the complexes show a red shift trend from the Li to the K complex. This trend indicates that the crucial transition energy becomes smaller, which might lead to a larger nonlinear optical response. Among the three structures, the largest first static hyperpolarizability (βtot) of the K complex was 76626 au, which is about 45 times larger than a prototypical second-order nonlinear optical (NLO) molecule of p-nitroaniline (βtot = 1679 au). Therefore, our results show that alkali metal substituted Ads may be novel potential candidates for high-performance NLO materials.
Co-reporter:Xin-Yao Ren, Yong Wu, Li Wang, Liang Zhao, Min Zhang, Yun Geng, Zhong-Min Su
Journal of Molecular Graphics and Modelling 2014 Volume 51() pp:149-157
Publication Date(Web):June 2014
DOI:10.1016/j.jmgm.2014.05.005
•One synthesized and two designed guanidinate-based iridium (III) complexes are investigated.•Phosphorescence quantum efficiency is evaluated with the aid of kr and knr values.•Different cyclometalated ligands can greatly affect the photophysical properties of complexes.•Changing the pyridine ring with diazole groups can significantly enhance the SOC effects.•The designed complex 3 is expected to be a promising candidate for highly efficient guanidinate-based phosphorescence emitter.A density functional theory/time-depended density functional theory was used to investigate the synthesized guanidinate-based iridium(III) complex [(ppy)2Ir{(NiPr)2C(NPh2)}] (1) and two designed derivatives (2 and 3) to determine the influences of different cyclometalated ligands on photophysical properties. Except the conventional discussions on geometric relaxations, absorption and emission properties, many relevant parameters, including spin-orbital coupling (SOC) matrix elements, zero-field-splitting parameters, radiative rate constants (kr) and so on were quantitatively evaluated. The results reveal that the replacement of the pyridine ring in the 2-phenylpyridine ligand with different diazole rings cannot only enlarge the frontier molecular orbital energy gaps, resulting in a blue-shift of the absorption spectra for 2 and 3, but also enhance the absorption intensity of 3 in the lower-energy region. Furthermore, it is intriguing to note that the photoluminescence quantum efficiency (ΦPL) of 3 is significantly higher than that of 1. This can be explained by its large SOC value (n = 3–4) and large transition electric dipole moment (μS3), which could significantly contribute to a larger kr. Besides, compared with 1, the higher emitting energy (ET1) and smaller 2 value for 3 may lead to a smaller non-radiative decay rate. Additionally, the detailed results also indicate that compared to 1 with pyridine ring, 3 with imidazole ring performs a better hole injection ability. Therefore, the designed complex 3 can be expected as a promising candidate for highly efficient guanidinate-based phosphorescence emitter for OLEDs applications.Calculation results indicate that upon changing the main ligand of guanidinate-based complex 1 from 2-phenylpyridine to different phenyl-diazoles, significant SOC effects can be observed, leading to higher kr values compared to 1. The designed complex 3 would be considered as a promising candidate for highly efficient guanidinate-based phosphorescence emitter in OLEDs.
Co-reporter:Dong-Ying Du, Li-Kai Yan, Zhong-Min Su, Shun-Li Li, Ya-Qian Lan, En-Bo Wang
Coordination Chemistry Reviews 2013 Volume 257(3–4) pp:702-717
Publication Date(Web):February 2013
DOI:10.1016/j.ccr.2012.10.004
Owing to the potential applications in catalysis, analytical chemistry, ion exchange, magnetism, biological chemistry and medicine, tremendous effort has been dedicated to exploring polyoxometalate (POM) chemistry. Chiral POM-based materials are particularly attractive due to the combination of the advantage of POMs with the importance of chirality. Nearly 100 chiral POM-based compounds were reported, which were mainly used as asymmetric catalysts, molecular recognition and nonlinear optical materials. In addition, the chirality within POM systems has attracted the attention of theoretical chemists and research was carried out to explore the origin of chirality by density functional theoretical methods. In this review, we summarize the developments of chiral POM-based materials, including their synthetic strategies, calculations on the origin of chirality and the relevant applications.Graphical abstractChiral POM-based materials are particularly attractive due to the combination of the advantage of POMs with the importance of chirality. In this review, we summarize the developments of chiral POM-based materials, including their synthetic strategies, the calculations on the origin of chirality and the relevant applications.Highlights► We review the synthesis strategies and recent results of chiral polyoxometalates (POMs). ► Theoretical calculations on the origin of chirality within chiral POMs were discussed. ► The applications of chiral POMs in asymmetric catalysis, molecular recognition and NLO materials were summarized. ► The challenges of preparation and applications of chiral POMs were discussed.
Co-reporter:Jing Zhang;Li Zhou;Hameed A. Al-Attar;Kuizhan Shao;Li Wang;Dongxia Zhu;Zhongmin Su;Martin R. Bryce;Andrew P. Monkman
Advanced Functional Materials 2013 Volume 23( Issue 37) pp:4667-4677
Publication Date(Web):
DOI:10.1002/adfm.201300344
Abstract
Eight new iridium(III) complexes 1-8, with 1,3,4-oxadiazole (OXD) derivatives as the cyclometalated C^N ligand and/or the ancillary N^N ligands are synthesized and their electrochemical, photophysical, and solid-state light-emitting electrochemical cell (LEC) properties are investigated. Complexes 1, 2, 7 and 8 are additionally characterized by single crystal X-ray diffraction. LECs based on complexes 1-8 are fabricated with a structure indium tin oxide (ITO)/poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS)/cationic iridium complex:ionic liquid/Al. LECs of complexes 1–6 with OXD derivatives as the cyclometalated ligands and as the ancillary ligand show yellow luminescence (λmax = 552–564 nm). LECs of complexes 7 and 8 with cyclometalated C^N phenylpyridine ligands and an ancillary N^N OXD ligand show red emission (λmax 616–624 nm). Using complex 7 external quantum efficiency (EQE) values of >10% are obtained for devices (210 nm emission layer) at 3.5 V. For thinner devices (70 nm) high brightness is achieved: red emission for 7 (8528 cd m−2 at 10 V) and yellow emission for 1 (3125 cd m−2 at 14 V).
Co-reporter:Shabbir Muhammad, Hong-Liang Xu, Rong-Lin Zhong, Zhong-Min Su, Abdullah G. Al-Sehemi and Ahmad Irfan
Journal of Materials Chemistry A 2013 vol. 1(Issue 35) pp:5439-5449
Publication Date(Web):06 Aug 2013
DOI:10.1039/C3TC31183J
Nonlinear optical (NLO) materials are the smartest materials of the era, and have the ability to generate new electromagnetic fields with changed frequencies, phases, and other physical properties. Recently, many cutting edge research reports have been focused on NLO materials especially on those which are composed of sp2 hybridized carbon nanostructures. As the carbon nanostructures are composed of abundant π-electrons and have significant delocalization, these are potential candidates for modern NLO materials. Generally, sp2 hybridized carbon nanostructures can be divided into zero-dimensional fullerenes, one-dimensional nanotubes and two-dimensional graphene nanoribbons and quantum dots etc. These dimensionally different carbon nanomaterials are promising candidates for a wide range of applications in next-generation nanotechnologies. In present feature article, we first briefly explain a theoretical structure–NLO property relationship based on perturbation theory and then elucidate the crucial factors to control the NLO responses. We put together the different random investigations of sp2 hybridized carbon nanostructures for NLO application by highlighting the importance of their several structural designs to tune NLO amplitudes. Furthermore, we make a comparative and updated analysis of the NLO properties of dimensionally different sp2 hybridized carbon nanomaterials i.e. fullerenes, carbon nanotubes, and graphene nanoribbons and quantum dots. Finally, we make a brief discussion about different aspects and opportunities to use the sp2 hybridized carbon nanomaterials as high performance NLO materials of the future. This review is a focused perspective based on different updated quantum chemical investigations about fullerenes, nanotubes and graphene nanoribbons and quantum dots for their possible use in nonlinear optical applications.
Co-reporter:Guo-Gang Shan, Hai-Bin Li, Hai-Zhu Sun, Dong-Xia Zhu, Hong-Tao Cao and Zhong-Min Su
Journal of Materials Chemistry A 2013 vol. 1(Issue 7) pp:1440-1449
Publication Date(Web):14 Dec 2012
DOI:10.1039/C2TC00558A
Three multifunctional cationic iridium(III)-based materials with aggregation-induced emission (AIE) and piezochromic luminescence (PCL) behavior have been rationally designed with the help of the theoretical calculations and successfully synthesized. All complexes contain the same cyclometalated ligand, 1-(2,4-difluorophenyl)-1H-pyrazole (dfppz), while functionalized ancillary ligands with different substitution are used to control their photophysical properties. Complex 1 and 2 with ancillary ligands modified with aliphatic chains and carbazole end-capped alkyl groups, respectively, undergo remarkable and reversible changes in emission color in the solid state upon grinding or heating. In addition, 2, characterized as having the 3ILCT excited-state feature, simultaneously exhibits an interesting AIE behavior, showing almost non-emission in good solution but enhanced emission in its solid state. Further modification of 2 by attachment of a tert-butyl group on the ligand obtains complex 3, an amorphous material, which only displays AIE activity. More importantly, with the merits of reversible PCL and AIE properties of 2, the rare multi-channel color change and temperature-dependent emission behavior of the iridium(III) complex have been observed. Furthermore, the emissive nanoaggregates of 2 can be efficiently quenched by picric acid, making it a highly sensitive chemosensor for explosives, which is demonstrated in iridium(III)-based luminescent materials for the first time.
Co-reporter:Yanling Si, Guochun Yang and Zhongmin Su
Journal of Materials Chemistry A 2013 vol. 1(Issue 7) pp:1399-1406
Publication Date(Web):13 Dec 2012
DOI:10.1039/C2TC00413E
Time-dependent density functional theory (TDDFT) calculations have been used to investigate chiroptical, linear, and second-order nonlinear optical (NLO) properties of the novel tetrathiafulvalenylallene in both neutral and two cationic states for the first time. The calculated UV-Vis/ECD spectra of the studied compound are in good agreement with the experimental ones, which can be used to assign its absolute configuration (AC) with high confidence. From neutral state to the two cationic states, the studied compound exhibits pronounced different chiroptical effects and second-order NLO response values. For example, the calculated β0 value of 12+ is 10.36 times as large as that of 1, while the β0 value of 14+ is 46.51 times as large as that of 1. These effects mainly result from the structural modifications of TTF units in the redox process. It is found that charge transfer between the tetrathiafulvalene (TTF) unit and the allene framework plays a key role in determining the chiroptical properties and electronic transition properties. It is interesting to find that the two benzene rings have vanishingly small effects on the chiroptical properties. The studied compound could act as both a chiroptical switch and NLO switch material from the standpoint of different chiroptical and NLO responses, reversible redox processes, and high stability. The effects of different functionals and basis sets, including solvent effects on the UV-Vis/ECD spectra were also considered.
Co-reporter:Hai-Ning Wang, Fu-Hong Liu, Xin-Long Wang, Kui-Zhan Shao and Zhong-Min Su
Journal of Materials Chemistry A 2013 vol. 1(Issue 42) pp:13060-13063
Publication Date(Web):30 Sep 2013
DOI:10.1039/C3TA13242K
By means of a stepwise synthesis strategy, three 12-connected MOFs with fcu topology have been obtained, which are rare examples of 3D frameworks constructed from a MOP precursor. In addition, the adsorption and separation of dye molecules have been investigated.
Co-reporter:Wen-Wen He, Shun-Li Li, Wen-Liang Li, Ji-Sen Li, Guang-Sheng Yang, Shu-Ran Zhang, Ya-Qian Lan, Ping Shen and Zhong-Min Su
Journal of Materials Chemistry A 2013 vol. 1(Issue 37) pp:11111-11116
Publication Date(Web):25 Jul 2013
DOI:10.1039/C3TA12662E
A novel microporous metal–organic framework, IFMC-16, has been successfully constructed by using mixed ligands. IFMC-16 has multipoint interaction sites and exhibits high hydrogen storage capacity not only at ambient pressure, but also at lower pressure in a high pressure region, and displays high adsorptive efficiency in the removal of organosulfur compounds.
Co-reporter:Guang-Sheng Yang, Zhong-Ling Lang, Hong-Ying Zang, Ya-Qian Lan, Wen-Wen He, Xiao-Liang Zhao, Li-Kai Yan, Xin-Long Wang and Zhong-Min Su
Chemical Communications 2013 vol. 49(Issue 11) pp:1088-1090
Publication Date(Web):10 Dec 2012
DOI:10.1039/C2CC36894C
Two S-containing MOFs, interpenetrating IFMC-27 and non-interpenetrating IFMC-28, were synthesized by altering solvent size. The nanoporous IFMC-28 reveals high selective adsorption for Cu2+ ions and has been applied as a chromatographic column for separating transition metal ions for the first time.
Co-reporter:Xiao-Li Hu, Chun-Yi Sun, Chao Qin, Xin-Long Wang, Hai-Ning Wang, En-Long Zhou, Wen-E Li and Zhong-Min Su
Chemical Communications 2013 vol. 49(Issue 34) pp:3564-3566
Publication Date(Web):14 Mar 2013
DOI:10.1039/C3CC39173F
Unprecedented 3d–4f MOFs encapsulating infinite linear polyiodide chains were firstly reported using iodine molecules as a versatile precursor template. They possess high framework stability in acid/base aqueous solutions. The kinetics of iodine molecule release/recovery and UV-light photocatalytic H2 evolution activities were investigated.
Co-reporter:Hong Ren, Lingyu Zhang, Tingting Wang, Lu Li, Zhongmin Su and Chungang Wang
Chemical Communications 2013 vol. 49(Issue 54) pp:6036-6038
Publication Date(Web):30 Apr 2013
DOI:10.1039/C3CC41284A
A novel, fast and simple method was developed to fabricate poly(acrylic acid sodium salt) microspheres (PAAS MSs). The resulting PAAS MSs were utilized as active templates to universally synthesize the mesoporous lanthanide-doped gadolinium oxide hollow nanospheres with multicolored upconversion emissions under mild conditions.
Co-reporter:Jun Liang, Xin-Long Wang, Yan-Qing Jiao, Chao Qin, Kui-Zhan Shao, Zhong-Min Su and Qing-Yin Wu
Chemical Communications 2013 vol. 49(Issue 76) pp:8555-8557
Publication Date(Web):31 Jul 2013
DOI:10.1039/C3CC43990A
Three novel self-catenated 4-connected uninodal (65·8)-mok metal–organic rotaxane frameworks (MORFs) containing cucurbit[6]uril were constructed from the in situ trans/cis-configuration (1:1) of rotaxanes by taking advantage of a d10 metal ion directed synthesis. It was revealed that the effect of hydrogen bonds and π–π stacking interactions play significant roles in the self-assembly process.
Co-reporter:Dong-Hua Hu, Chun-Yi Sun, Fu-Hong Liu, Chao Qin, Xin-Long Wang and Zhong-Min Su
CrystEngComm 2013 vol. 15(Issue 34) pp:6769-6775
Publication Date(Web):05 Jul 2013
DOI:10.1039/C3CE40782A
Six new coordination polymers, namely, [Zn2(L1)2phen2(H2O)2] (1), [M3(L1)2(OH)2 (4,4′-bpy)(H2O)2]·2H2O (M = Cd (2), Co(3)), [Cd5(L1)2(OH)2(trz)4(L3)]·2H2O (4), [Zn(HL2)(4,4′-bpy)] (5) and [Cd3(L2)2(L3)2(H2O)5]·H2O (6) (H2L1 = 4-(1-carboxy-ethoxy)-benzoic acid, H3L2 = 5-(1-carboxy-ethoxy)-isophthalic acid, L3 = 1,2-bis(1,2,4-triazole-1-yl)ethane, phen = 1,10-phenanthroline, 4,4′-bpy = 4,4′-bipyridine, Htrz = 1,2,4-triazole), have been hydrothermally synthesized and characterized by single-crystal X-ray diffraction. Compound 1 is a molecular square composed by two Zn ions, two L1 and two phen ligands. Compounds 2 and 3 are isostructural and display 3D frameworks constructed by 1D M–O–M chains. Compound 4 exhibits a 3D 8-connected network with hex topology based on the pentanuclear Cd cluster and the point symbol is (36·418·53·6). Compound 5 shows a 2D wavelike layer with sql topology. Compound 6 reveals a 3D three-fold interpenetrating architecture having (3,4)-connected dmd topology with the point symbol of (4·102)2(42·104). These compounds have been characterized by infrared spectroscopy (IR) and thermal gravimetric analyses (TGA). In addition, the photoluminescent behaviour of 1, 2, and 4–6 has been investigated in detail.
Co-reporter:Ji Zhang, Hai-Bin Li, Yun Geng, Shi-Zheng Wen, Rong-Lin Zhong, Yong Wu, Qiang Fu, Zhong-Min Su
Dyes and Pigments 2013 Volume 99(Issue 1) pp:127-135
Publication Date(Web):October 2013
DOI:10.1016/j.dyepig.2013.04.026
•Three dyes with different electron donors derived from C219 for DSSC were designed.•A set of key parameters affecting the efficiency of the cell have been discussed.•Dye 3 with Coumarin donor shows higher Voc and Jsc than that of champion dye C219.Dye sensitized solar cell sensitized by organic dye C219 (Chem. Mater, 2010, 22, 1915–1925) has reached the record efficiency (η) of 10.1% with comparable short-circuit current density (Jsc) but relatively lower open-circuit photovoltage (Voc) as compared to that of ruthenium-based cells. With the motivation of further enhancing Voc, we designed three dyes 1–3 with different electron donors, i.e. carbazole, indoline and coumarin. Based on the theoretical analysis of the conduction band energy shift of TiO2, electron injection rate and electron lifetime, the three designed dyes all show larger Voc than that of C219. On the other hand, 3 should also have larger Jsc compared with that of C219 due to its red-shifted spectrum and higher light harvesting efficiency. Thus we expect that the cell sensitized by 3 could exhibit both larger Voc and Jsc in comparison to the champion C219-based cells.A theoretically designed coumarin dye shows both higher Voc and Jsc potential compared with champion dye C219 through the reliable DFT/TDDFT calculations on semiconductor/sensitizer/electrolyte interfacial interaction.
Co-reporter:Guo-Gang Shan, Hai-Bin Li, Hai-Zhu Sun, Hong-Tao Cao, Dong-Xia Zhu and Zhong-Min Su
Dalton Transactions 2013 vol. 42(Issue 31) pp:11056-11065
Publication Date(Web):19 Mar 2013
DOI:10.1039/C3DT50358E
Herein we designed and synthesized a series of cationic iridium(III) complexes with a phenylbenzoimidazole-based cyclometalated ligand, containing different numbers of carbazole moieties from zero to three (complexes 1–4). The photophysical and electrochemical properties of this series have been systematically investigated. The complexes exhibit strong luminescence in both solution and in neat films, as well as excellent redox reversibility. Introducing carbazole groups into the complexes is found to lead to substantially enhanced photoluminescence quantum efficiency in the neat film, but has little effect on the emitting color and excited-state characteristics as supported by density functional theory (DFT) results. DFT calculations also suggest that functionalized complexes 2–4 reveal better hole-transporting properties than 1. More importantly, all complexes effectively reduce the degradation reaction to some extent in metal-centered (3MC) excited-states, demonstrating their stability. Further studies indicate that restriction of opening of the structures in the 3MC state is caused by the unique molecular conformation of the phenylbenzoimidazole ligand, which is first demonstrated here in cationic iridium(III) complexes without intramolecular π–π stacking. These results presented here would provide valuable information for designing and synthesizing highly efficient and stable cationic iridium(III) complexes suitable for the optical devices.
Co-reporter:Zhong-Ling Lang, Guo-Chun Yang, Na-Na Ma, Shi-Zheng Wen, Li-Kai Yan, Wei Guan and Zhong-Min Su
Dalton Transactions 2013 vol. 42(Issue 29) pp:10617-10625
Publication Date(Web):14 May 2013
DOI:10.1039/C3DT50666E
Water oxidation is a key half reaction in the energy conversion scheme. The reaction mechanism for the oxidation of H2O to O2 catalyzed by single-Ru-substituted polyoxometalates, [RuIII(H2O)XW11O39]n− (X = SiIV, GeIV), was investigated by means of density functional calculations. The electronic structure of the pre-activation intermediates indicates that the aqua ligand is prone to accommodate the proton coupled electron transfer (PCET) process to achieve the active group [RuVOa], and the high valent oxo-ruthenium(V) species are responsible for the O–O forming event. Three possible proton acceptors were designed for the rate-determining step (Ob, Oa, and H2O), the calculated results support that the bridge Ob atom of the polytungstate ligand will act as the most favorable proton acceptor in the O–O bond formation, with an energy barrier of 28.43 kcal mol−1. A detailed information of the peroxidic intermediates in the oxidation process was also characterized, both the peroxo-species [RuIV(OO)SiW11O39]6− and [RuV(OO)SiW11O39]5− show the six-coordinate isomer with an open terminal geometry is more favorable than the close seven-coordinate ones. In addition, the replacement of the heteroatom in XO4n− can effectively tune the catalytic activity of polyoxometalates, in the order of GeIV > SiIV.
Co-reporter:Hai-Ning Wang, Guang-Sheng Yang, Xin-Long Wang and Zhong-Min Su
Dalton Transactions 2013 vol. 42(Issue 18) pp:6294-6297
Publication Date(Web):07 Mar 2013
DOI:10.1039/C3DT32958E
pH-induced different crystalline behaviors based on the same reactants and reaction conditions are illustrated by our present study. Compound 2 has been used for the adsorption and delivery of 5-FU.
Co-reporter:Kun Zhou, Xin-Long Wang, Chao Qin, Hai-Ning Wang, Guang-Sheng Yang, Yan-Qing Jiao, Peng Huang, Kui-Zhan Shao and Zhong-Min Su
Dalton Transactions 2013 vol. 42(Issue 5) pp:1352-1355
Publication Date(Web):15 Nov 2012
DOI:10.1039/C2DT32145A
The first macrocycle-based high-nuclearity nanocluster [Ag20(StBu)10(CF3COO)2]8+ in complex 1, with a sandwich-like structure constructed by bilevel inversion Ag5S5 pentagrams and an interlayer Ag10 ring, has been obtained. Structure analysis indicates that Cl− acts as an anion template to direct the formation of 1. In addition, the photoluminescent property of complex 1 is also investigated.
Co-reporter:Da-Shu Chen, Li-Bo Sun, Zhi-Qiang Liang, Kui-Zhan Shao, Chun-Gang Wang, Zhong-Min Su, and Hong-Zhu Xing
Crystal Growth & Design 2013 Volume 13(Issue 9) pp:4092-4099
Publication Date(Web):August 19, 2013
DOI:10.1021/cg400913f
Six new coordination polymers, (Me2NH2)[In(L)]·DMF (1), (Me2NH2)2[In2(L)2]·(DMF)3·(H2O)2 (2), (Me2NH2)4[Cd2(L)2]·(DMF)2 (3), (Et2NH2)2[Cd(L)]·H2O (4), (Et2NH2)3[Cd2(L)2]·(DEF)2·(H2O)4 (5), (Et2NH2)3[Cd2(L)2]·(EtNH2)2·(H2O)2 (6) have been prepared based on a newly designed flexible tetracarboxylate ligand of H4L {H4L = 5-[bis(3-carboxybenzyl)amino]isophthalic acid; DMF = N,N-dimethylformamide; DEF = N,N-diethylformamide}. Compounds 1–6 possess very similar two-dimensional framework structures with (4,4)-net topology. These compounds comprise two groups of conformational supramolecular isomers, owing to identical compositions of the frameworks but different conformations of ligands. It is worth noting that as much as seven pairs of stereoisomers of ligands have been found in these compounds, which demonstrated that the introduction of multiple single bonds has greatly expanded the flexibility of the ligand. The photoluminescent properties of compounds 1–6 have also been investigated where the emission of these supramolecular isomers are from blue to green.
Co-reporter:Lei Chen, Li Zhang, Shun-Li Li, Yong-Qing Qiu, Kui-Zhan Shao, Xin-Long Wang and Zhong-Min Su
CrystEngComm 2013 vol. 15(Issue 40) pp:8214-8221
Publication Date(Web):31 Jul 2013
DOI:10.1039/C3CE40910D
Five metal–organic frameworks (MOFs), namely, [Zn2(L1)(BIMB)(μ2-OH)] (1), [Zn3(L1)2(BIMB)3]·4H2O (2), [Zn(HL2)(BIMB)0.5]·H2O (3), [Zn3(L2)2(BIMB)3]·8H2O (4) and [Zn3(L2)2(BIMB)3]·4H2O (5) (H3L1 = 5-(3-carboxybenzyloxy)isophthalic acid, H3L2 = 5-(4-carboxybenzyloxy)isophthalic acid and BIMB = 1,4-di(1H-imidazol-1-yl)benzene), have been synthesized under hydrothermal conditions. Their structures have been determined by single-crystal X-ray diffraction analyses and further characterized by elemental analyses, IR spectra, and thermogravimetric (TG) analyses. Complex 1 is a (3,4)-connected 3D interlocking polycatenated framework with the (63)(66) topology by 2D parallel interpenetration. Complex 2 adopts a self-penetrating 3D (3,4)-connected network with (6·82)(4·6·8)(6·84·9)(4·62·82·10) topology, which contains rotaxane- and catenane-like motifs together. Complex 3 is a 3D supramolecular structure formed by hydrogen bonds between adjacent 2D (4,4) sheets. For complex 4, it shows a three-folded 3D framework by a (3,4)-connected net with (62·8)(4·6·8)(63·7·8·9)(4·62·72·8)(62·72·92) topology. Complex 5 is a (3,4)-connected 3D framework. We have discussed the influence of synthetic conditions and different carboxylate ligands on the final structures. In addition, powder X-ray diffraction and photoluminescent properties for 1–5 have been studied in detail at room temperature.
Co-reporter:Hai-Ning Wang, Fu-Hong Liu, Yan-Fei Zhu, Xin-Long Wang, Zhi-Hui Chen and Zhong-Min Su
CrystEngComm 2013 vol. 15(Issue 37) pp:7402-7405
Publication Date(Web):13 Aug 2013
DOI:10.1039/C3CE41044G
Two Co6(μ3-OH)2 cluster based porous coordination polymers have been synthesized, in which different channels exist. The reported compounds represent the first porous examples based on this cluster.
Co-reporter:Shizheng Wen, Wei Guan, Yuhe Kan, Guochun Yang, Nana Ma, Likai Yan, Zhongmin Su and Guanhua Chen
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 23) pp:9177-9185
Publication Date(Web):11 Apr 2013
DOI:10.1039/C3CP51380G
Nano-hybrid materials based on a combination of polyoxometalate (POM) clusters and single-walled carbon nanotubes (SWNT) exhibit a great interesting application in molecular cluster batteries. The interactions between POM and SWNT and their detailed electronic properties have been investigated by employing first-principles calculations. Various models were constructed to study the geometries, interactions (binding sites and energies), and charge transfer behavior. Analysis of charge distributions reveals two different charge transfer characteristic depending on the type of POM interaction with SWNT. The simulation provides insight into the optimal structures in lieu of interfacial stability. Finally, the implications of these results for understanding the properties of molecular cluster batteries are discussed.
Co-reporter:Yu-Zhong Xie, Guo-Gang Shan, Peng Li, Zi-Yan Zhou, Zhong-Min Su
Dyes and Pigments 2013 Volume 96(Issue 2) pp:467-474
Publication Date(Web):February 2013
DOI:10.1016/j.dyepig.2012.09.020
A series of novel Zn(II) Schiff base complexes with diphenylamino and carbazole groups that exhibited aggregation-induced emission enhancement were designed and synthesized. The properties of four complexes were investigated by UV–Vis absorption and fluorescence emission spectroscopy, cyclic voltammetry and density functional theory calculations. The fluorescence intensities of the four dyes are weak in tetrahydrofuran, but become strong in a mixture of water/tetrahydrofuran (v/v = 9/1). This work constitutes the first observation of this phenomenon for Zn(II) Schiff base complexes. A simple model complex, without the possibility of intramolecular rotational motion, was prepared in order to determine the mechanism of the AIEE. The present aggregation-induced emission enhancement was attributed to restricted intramolecular rotational motions in the solid by carefully analyzing the difference in molecular structure and photophysical properties amongst the new complexes.Graphical abstractA series of Zn(II) Schiff base complexes with an intramolecular rotational motion group were prepared. An investigation of their photophysical properties showed that these complexes exhibited aggregation-induced emission enhancement (AIEE) properties.Highlights► A series of Zn(II) Schiff base complexes have been designed and synthesized. ► The complexes exhibit aggregation-induced emission enhancement property. ► The aggregation-induced emission enhancement phenomenon is related to restriction of intramolecular rotation. ► This is the first time aggregation-induced emission enhancement phenomenon has been observed for Zn(II) Schiff base complexes.
Co-reporter:Guo-Gang Shan, Hai-Bin Li, Hong-Tao Cao, Hai-Zhu Sun, Dong-Xia Zhu, Zhong-Min Su
Dyes and Pigments 2013 Volume 99(Issue 3) pp:1082-1090
Publication Date(Web):December 2013
DOI:10.1016/j.dyepig.2013.08.015
•Synthesis of Ir(III)-based dyes with different N-alkyl chains is described.•They show naked-eye visible and reversible piezochromism.•The chain length-dependent emissions are presented and investigated.•The dyes exhibit high quantum yields, redox reversibility and good stability.•The experimental results are rationalized by the theoretical calculations.To investigate the relationship between structures and piezochromic luminescence behavior and to develop excellent materials for efficient light-emitting electrochemical cells, a series of iridium(III) complexes with different length of alkyl chains have been designed and synthesized. The results show that all complexes exhibit not only naked-eye visible and reversible piezochromic behavior, but chain length-dependent emission properties: the shorter the alkyl chain is, the more remarkable mechanochromic behavior and higher recrystallization temperature will be. In light of powder X-ray diffractometry and differential scanning calorimetric data, the interconversion between crystalline and amorphous states upon external stimuli is response for the present piezochromism. Additionally, these complexes show high quantum yields of 55–65% in neat films as well as excellent redox reversibility. Despite changed alkyl chains are introduced into these complexes, the negligible effect on emitting colors and excited-state characters of them in both solutions and neat films have been observed. Such photophysical properties are also interpreted with the help of theoretical calculations. Moreover, the theoretical results suggest that the intrinsic intramolecular π–π stacking confirmed by the crystal structures can effectively restrict the opening of the structures in metal-centered excited-state, demonstrating their stability. The structure–property relationships and the results demonstrated here will help researchers develop and design more promising iridium(III)-based piezochromic materials and attractive phosphors for optical devices.A series of iridium(III)-based phosphorescent dyes showing naked-eye visible and reversible piezochromic luminescent behavior are successfully synthesized. Moreover, they are promising luminescent materials with high quantum yields of 55–65%, excellent redox reversibility and relative stability.
Co-reporter:Jing Wang, Hong Li, Na-Na Ma, Li-Kai Yan, Zhong-Min Su
Dyes and Pigments 2013 Volume 99(Issue 2) pp:440-446
Publication Date(Web):November 2013
DOI:10.1016/j.dyepig.2013.05.027
•The novel dyes of POM-based organic–inorganic hybrids were designed for DSSCs.•The absorption spectra of systems were systematically studied.•The electron injection efficiency (Φinj) from dyes to TiO2 was evaluated.•The light harvesting efficiency (LHE) of dyes was assessed.•The incident photon-to-electron conversion efficiency (IPCE) of dyes was assessed.A series of POM-based organic–inorganic hybrids with different π-conjugated bridges are investigated as sensitizers for application in dye-sensitized solar cells (DSSCs). A combination of density functional theory (DFT) and time-dependent DFT (TDDFT) approaches is employed. The effects of π-conjugated spacer size and π-conjugation length on the spectra of designed systems are demonstrated. The results show that the absorption spectra are systematically broadened and red-shifted with increasing sizes of the π-conjugated spacer and the length of π-conjugation. The theoretical examination was performed on the key parameters of incident photon-to-electron conversion efficiency (IPCE), light harvesting efficiency (LHE) and electron injection efficiency (Φinj). The result suggests that system 2 with thieno[3,2-b]thiophene demonstrates a balance of the two crucial factors and may result in the highest IPCE of DSSCs. This study is expected to deepen our understanding of POM-based organic–inorganic hybrid dyes and assist the molecular design of new dyes for the further optimization of DSSCs.
Co-reporter:Bing Zhang, Yu-He Kan, Yun Geng, Yu-Ai Duan, Hai-Bin Li, Jian Hua, Zhong-Min Su
Organic Electronics 2013 Volume 14(Issue 5) pp:1359-1369
Publication Date(Web):May 2013
DOI:10.1016/j.orgel.2013.02.017
Why does the fluorination of just one phenyl in 5,11-diphenyltetracene (PPT) bring about so tremendous change of the charge carrier mobility? Herein, we carried out density functional theory (DFT) to provide insight into this remarkable difference by investigating their geometries, electronic structures, reorganization energies, transfer integrals, intermolecular interactions and band structures. The improved charge mobility from PPT to FPPT (5-(perfluorophenyl)-11-phenyltetracene) can be attributed to favorable molecular packing due to the increase of π–π interaction which is confirmed by Hirshfeld surfaces analysis. Furthermore, we calculated charge mobilities of novel compound 4,11-diphenyltetracene (PPT′) and its fluorinated derivative 4-(perfluorophenyl)-11-phenyltetracene (FPPT′), on the basis of the predicted packing motifs. The largest charge mobility of FPPT′ (2.49 cm2/V s) exhibits one-fold higher than PPT′ (1.07 cm2/V s) due to dense packing structures, which further confirms our finding that fluorination may be an effective means to improve the carrier mobility. This work paves the way towards the development of a computational protocol that could be implemented not only for rationalizing synthetic efforts but also for design of high-performance organic transport materials.Graphical abstractThe impact of fluorination on the charge-transport parameters of organic filed-effect transistors was unambiguously proclaimed by theoretical calculations.Highlights► The impact of fluorination on the charge-transport parameters of OFET was unambiguously proclaimed. ► Mobility differences in rubrene derivatives were ascribed to the change of packing motifs. ► The novel fluorinated rubrene derivative FPPT′ possesses high theoretical mobility.
Co-reporter:Yan-Chun Liu;Shui-Xing Wu;Yu-He Kan;Hou-Yu Zhang
European Journal of Inorganic Chemistry 2013 Volume 2013( Issue 12) pp:2220-2230
Publication Date(Web):
DOI:10.1002/ejic.201201416
Abstract
A systematic density functional study was performed on the structures and bonding features in a homologous metal–metal (M–M) host–guest series M2@C50X10 (M = Zn, Cd, Hg; X = CH, N, B). Calculations indicated that D5d conformers are energy minima for Zn2@C50X10 (X = CH, N) and Cd2@C50X10 (X = CH, N, B), whereas Zn2@C50B10 has no imaginary frequency with the lowered C2h symmetry. Among these hybrid fullerene–metal complexes, those with dizinc and dicadmium guests adopt η5/η5 coordination, except for an η2/η2 case in C2h-Zn2@C50B10, to meet appropriate interactions between the metal atom and cage; and the η1/η1 coordination mode can only be satisfied in just one (Hg2@C50N10) of the complexes with Hg–Hg guests, regardless of the enforced coordination surroundings. The nature of metal–cage interactions between M22+ and C50X102– moieties and M–M bonds have been analyzed by means of the topological properties of electron densities and energy-partitioning methods, and results suggest that the two types of interactions are almost similar to those in their M2(C5H5)2 counterparts.
Co-reporter:Da-Shu Chen, Jia-Ming Cheng, Li-Bo Sun, Zhi-Qiang Liang, Kui-Zhan Shao, Chun-Gang Wang, Hong-Zhu Xing, Zhong-Min Su
Inorganic Chemistry Communications 2013 Volume 38() pp:104-107
Publication Date(Web):December 2013
DOI:10.1016/j.inoche.2013.10.022
Co-reporter:Jian-Sheng Li, Xiao-Jing Sang, Wei-Lin Chen, Lan-Cui Zhang, Zhong-Min Su, Chao Qin, En-Bo Wang
Inorganic Chemistry Communications 2013 Volume 38() pp:78-82
Publication Date(Web):December 2013
DOI:10.1016/j.inoche.2013.10.027
•The polyoxometalates (POMs) sensitized solar cell (PSSC) was studied firstly.•It is breakthrough to put forward that POMs export electrons under irradiation.•The energy level and band gap were investigated for the POMs base sensitizer.Keggin-type polyoxometalates (POMs) based photosensitizer [(CH3)4N]5[PW11O39RhCH2COOH]∙6H2O (PW11Rh-COOH) was firstly explored for assembling the POMs sensitized solar cells (PSSC). Electrochemical measurement, UV–vis diffuse reflectance spectrum, Surface photovoltage spectrum, and X-ray photoelectron spectroscopy demonstrated that PW11Rh-COOH displayed higher photovoltaic response than that of other POMs because of the better visible-light response, energy level matching and higher carrier separation efficiency.Keggin-type polyoxometalates (POMs) based photosensitizer [(CH3)4N]5[PW11O39RhCH2COOH]∙6H2O (PW11Rh-COOH) was firstly explored for assembling the POMs sensitized solar cells (PSSC), which displayed higher photovoltaic response than that of other POMs because of the better visible-light response, energy level matching and higher carrier separation efficiency.
Co-reporter:Xiang-Rong Hao, Guang-sheng Yang, Kui-Zhan Shao, Zhong-Min Su, Gang Yuan, Xin-Long Wang
Journal of Solid State Chemistry 2013 Volume 198() pp:143-148
Publication Date(Web):February 2013
DOI:10.1016/j.jssc.2012.09.008
Three metal–organic frameworks (MOFs), [Co2(BPDC)2(4-BPH)·3DMF]n (1), [Cd2(BPDC)2(4-BPH)2·2DMF]n (2) and [Ni2(BDC)2(3-BPH)2 (H2O)·4DMF]n (3) (H2BPDC=biphenyl-4,4′-dicarboxylic acid, H2BDC=terephthalic acid, BPH=bis(pyridinylethylidene)hydrazine and DMF=N,N′-dimethylformamide), have been solvothermally synthesized based on the insertion of heterogeneous BPH pillars. Framework 1 has “single-pillared” MOF-5-like motif with inner cage diameters of up to 18.6 Å. Framework 2 has “double pillared” MOF-5-like motif with cage diameters of 19.2 Å while 3 has “double pillared” 8-connected framework with channel diameters of 11.0 Å. Powder X-ray diffraction (PXRD) shows that 3 is a dynamic porous framework.Graphical abstractBy insertion of flexible BPH pillars based on “pillaring” strategy, three metal–organic frameworks are obtained showing that the porous frameworks can be constructed in a much greater variety.Highlights► Frameworks 1 and 2 have MOF-5 like motif. ► The cube-like cages in 1 and 2 are quite large, comparable to the IRMOF-10. ► Framework 1 is “single-pillared” mode while 2 is “double-pillared” mode. ► PXRD and gas adsorption analysis show that 3 is a dynamic porous framework.
Co-reporter:Ting Zhang, Nana Ma, Likai Yan, Shizheng Wen, Zhongmin Su
Chemical Physics Letters 2013 Volume 557() pp:123-128
Publication Date(Web):5 February 2013
DOI:10.1016/j.cplett.2012.12.024
The static second-order nonlinear optical (NLO) responses of a series of lacunary γ-Keggin polyanion derivatives, [XM2W10O38(μ-OH)2]n− (X = SiIV, GeIV, PV, AsV, M = AlIII, TiIV, VV; X = ZnII, VV, M = AlIII), were investigated by using density functional theory (DFT) methods. The results show that the molecular NLO activity of lacunary Keggin POM derivatives can be modified by replacing the central heteroatom (X) and the substituted addenda metal atom (M), which reveals the general rules to design the system with large optical nonlinearity.Graphical abstractHighlights► These inorganic complexes have evidently large static second-order polarizability. ► The molecular NLO activity of lacunary Keggin POM derivatives can be modified. ► These POMs seem to be promising inorganic candidates for application in nonlinear optics.
Co-reporter:Yan Liu, Guochun Yang, Shiling Sun, Yanling Si, Zhongmin Su
Journal of Molecular Graphics and Modelling 2013 Volume 44() pp:311-317
Publication Date(Web):July 2013
DOI:10.1016/j.jmgm.2013.07.006
•The MLCT transition is mainly responsible for the low-energy absorption band with relative smaller oscillator.•The first hyperpolarizability values of the v shaped complexes are larger than that of the linear shape complex.•These complexes exhibit two-dimensional second-order nonlinear optical (NLO) character.•The variation of first hyperpolarizabilities of the studied complexes can be explained by the two-level model.The photophysical properties of the linear and v shaped Pt(II) triarylborons with a 2,2′-bpy core derivatives have been investigated by density functional theory (DFT) method. The calculated electronic absorption wavelengths are in agreement with experimental ones, which can be described as a mixed transition of intra-ligand charge transfer (ILCT), ligand to ligand charge-transfer (LLCT), and metal-to-ligand charge transfer (MLCT). It is found that the MLCT transition is mainly responsible for the low-energy absorption band with relative smaller oscillator strength, while the high-energy absorption band mainly derives from ILCT and LLCT transition. Moreover, the electron absorption wavelengths are not only dependent on the position of the Ph-BMes2 but also on the electron-accepting ability of the acceptor groups. The first hyperpolarizability values of the v shaped complexes are larger than that of the linear shape complex, which indicates that larger intramolecular charge transfer for the v shaped complexes will come into being under the external electric field. Moreover, these complexes exhibit two-dimensional second-order nonlinear optical (NLO) character. Thus, the studied complexes have a possibility to be excellent second-order NLO materials. Based on the two-level model, the variation of first hyperpolarizabilities of the studied complexes can be explained by the combined effect of the difference between the ground state and excited state dipole moment, the oscillator strength, and the cube of the transition energy.The first hyperpolarizability values of the v shaped complexes are larger than that of the linear shape complex, which indicates that larger intramolecular charge transfer for the v shaped complexes might occur under the external electric field. And the changes in first hyperpolarizability values can be explained by the combined effect of the difference between the ground state and excited state dipole moment, the oscillator strength, and the cube of the transition energy.
Co-reporter:Yuan-Mei Sang, Li-Kai Yan, Jian-Ping Wang, Na-Na Ma, Zhong-Min Su
Journal of Molecular Graphics and Modelling 2013 Volume 44() pp:26-32
Publication Date(Web):July 2013
DOI:10.1016/j.jmgm.2013.05.001
•The UV/ECD spectra of chiral POM enantiomer were studied using TDDFT method.•UV/ECD spectra of enantiomer R in gas phase and different solvents were calculated.•UV/ECD spectra of chiral POM in gas phase, polar, non-polar solvent are different.•Our work provides the first-hand structure–property information about chiral POMs.The ultraviolet–visible and electronic circular dichroism (UV–vis/ECD) spectra of diphosphonate-functionalized asymmetric cantilever-type chiral polyoxomolybdate (POM) enantiomer R-{Mo2O5[(Mo2O6)NH3CH2CH2CH2C(O)(PO3)2]2}6− (R) were systematically investigated using time-dependent density functional theory (TDDFT) method. From the view of molecular structure and relative energy, we inferred that there is likely a structural conversion from enantiomers R to S-{Mo2O5[(Mo2O6)NH3CH2CH2CH2C(O)(PO3)2]2}6− (S) via the intermediate configuration (IN). The ECD spectra of the enantiomer R were produced over the range of 3.0–6.3 eV. The UV–vis and ECD spectra of enantiomer R in the gas phase and different solvents were calculated. The results reveal that the UV–vis and ECD spectra of the chiral POM in gas phase, polar solvent, or non-polar solvent are different. The calculated electron density difference maps (EDDMs) display that the POM cluster is a chiroptical chromophore in studied compound.The electronic structures, chiroptical properties and the solvent effect on the CD spectra of diphosphonate-functionalized asymmetric polyoxomolybdates were systematically investigated using time-dependent density functional theory (TDDFT) method.
Co-reporter:Ting Zhang, Nana Ma, Likai Yan, Shizheng Wen, Tengying Ma, Zhongmin Su
Journal of Molecular Graphics and Modelling 2013 Volume 46() pp:59-64
Publication Date(Web):November 2013
DOI:10.1016/j.jmgm.2013.09.011
•The NLO properties of Keggin-type POMs organic–inorganic hybrids are investigated.•The metalloporphyrin is helpful in enhancing the optical nonlinearity.•The solvent effect affects the NLO response of the complex.The second-order nonlinear optical (NLO) properties of hybrid complexes via coordination interaction between porphyrin and Keggin-type polyoxometalates (POMs) α-[MSiW11O39]3− (M = NbV or VV) are investigated by time-dependent density functional theory (TDDFT). The calculated results show that this kind of organic–inorganic hybrid complexes possesses remarkably large molecular second-order NLO polarizability, especially for the ZnP3P-CC-4-Py-[VSiW11O39]3− (complex 4), which has a computed β0 value of 261,410 a.u. and might be an excellent second-order NLO material. The effects of substituted metal atom (M), metalloporphyrin, and π-conjugation on NLO response are analyzed, the substituted metal atom (M) with a large electronegativity, the metalloporphyrin, and the lengthening of π-conjugation are helpful in enhancing the optical nonlinearity of these systems, which reveal the general rules to design the complexes with large optical nonlinearities. Furthermore, the solvent effect largely affects the first-order hyperpolarizability of the complex, it implies that the second-order polarizabilities increased with the increase of the solvent in polarity.The second-order nonlinear optical (NLO) properties of hybrid complexes via coordination interaction between porphyrin and Keggin-type polyoxometalates (POMs) α-[MSiW11O39]3− (M = NbV or VV) are investigated by time-dependent density functional theory (TDDFT).
Co-reporter:Jing Wang, Sha Cong, Shizheng Wen, Likai Yan, and Zhongmin Su
The Journal of Physical Chemistry C 2013 Volume 117(Issue 5) pp:2245-2251
Publication Date(Web):January 11, 2013
DOI:10.1021/jp3106452
The novel dyes of organoimido-substituted hexamolybdates for positive type dye-sensitized solar cells (p-type DSSCs) have been studied on the basis of time-dependent density functional theory (TDDFT) calculations. The electronic absorption spectra, light harvesting efficiency (LHE), charge separation efficiency (CSE), and holes injecting efficiency (HJE) of designed systems have been systematically investigated. The results reveal that the long π-conjugated bridge and auxochrome play crucial roles in red-shifting the absorption bands and reinforcing the intensity of the bands. Based on [(n-C4H9)4N]2[Mo6O18(N-1-C10H6-2-CH3)], the designed systems 6 and 4 are good candidates for p-type DSSC dyes due to the strong absorption in the visible region as well as high LHE, CSE, and HJE. The maximum absorption of the one-electron-reduced system obviously red-shifts to the visible region. Therefore, the highly efficient dyes of p-type DSSC can be prepared by reducing POM-based organic–inorganic hybrids which have both long π-conjugated bridge and auxochrome.
Co-reporter:Yang-Yang Hu, Hong-Liang Xu, and Zhong-Min Su
The Journal of Physical Chemistry C 2013 Volume 117(Issue 1) pp:725-728
Publication Date(Web):December 11, 2012
DOI:10.1021/jp309686h
Co-reporter:Shuang-Yang Yang;Liang Zhao;Yu-Ai Duan;Yun Geng
Theoretical Chemistry Accounts 2013 Volume 132( Issue 9) pp:
Publication Date(Web):2013 September
DOI:10.1007/s00214-013-1377-1
The quantum mechanical and quantum mechanical/molecular mechanical methods were employed to investigate the photoluminescence and carrier transport properties of two bow-shaped thiophene compounds, whose optimized geometries were proved to have large deviation between the single-molecule and the solid-phase models. The results show that the molecular packings have large influence on the geometrical structures for both systems, but barely affect their energy levels of the frontier molecular orbitals and the reorganization energies, thus indicating little effect on the charge transport properties. While the obvious blue shifts of absorption and emission spectra in solid phase compared with single molecule are considered to relate with the intermolecular interactions, the difference in center thiophene units between two bow-shaped compounds induces the decided differences in optoelectronic properties, especially in carrier transport abilities.
Co-reporter:Ji Zhang;Yu-He Kan;Hai-Bin Li;Yun Geng;Yong Wu
Journal of Molecular Modeling 2013 Volume 19( Issue 4) pp:1597-1604
Publication Date(Web):2013 April
DOI:10.1007/s00894-012-1719-2
We report a DFT, TDDFT and DFTB investigation of the performance of two donor-π-acceptor (D-π-A)-type organic dyes bearing different electron-withdrawing groups (EWG) for dye-sensitized solar cells (DSSCs) to evaluate which EWG is better for an acrylic acid acceptor, i.e., Cyano (–CN) or o-nitrophenyl (o-NO2–Ph). A series of theoretical criteria applied successfully in our previous work to explain the different performance of organic dyes related to open-circuit photovoltage (Voc) and short-circuit current density (Jsc) were used to evaluate the performance of the dyes with just different EWG. Our calculated results reveal that dye 2 with o-NO2–Ph has a larger vertical dipole moment, more electrons transferred from the dye to the semiconductor and a lower degree of charge recombination, which could lead to larger Voc; while the larger driving force and comparable light harvesting efficiency could lead to higher Jsc , highlighting the potential of o-NO2–Ph as an EWG in an acrylic acid acceptor.
Co-reporter:Yuan-Mei Sang, Li-Kai Yan, Na-Na Ma, Jian-Ping Wang, and Zhong-Min Su
The Journal of Physical Chemistry A 2013 Volume 117(Issue 12) pp:2492-2498
Publication Date(Web):March 1, 2013
DOI:10.1021/jp400506z
The electronic circular dichroism (ECD) and UV–visible absorption (UV–vis) spectra of Strandberg-type polyoxometalates (POMs) (R, R)-[(R*PO3)2M5O15]2- (R* = CH3CH(NH3), (M = Mo, W)) have been explored using the time-dependent density functional theory (TDDFT) method. It demonstrates that the absolute configurations of chiral systems can be determined by chiroptical spectroscopic methods combined with DFT calculations. The calculated ECD spectra of the Strandberg-type molybdate were produced over the range of 3.3–6.5 eV, which are generally in agreement with the experimental spectra. In addition, the ECD spectra of (R, R)-[(R*PO3)2W5O15]2- (R* = CH3CH(NH3)) were produced over the range of 4.5–8.5 eV. The Becke’s half-and-half hybrid exchange-correlation functional (BHandHLYP) with the HF exchange fraction to 55% hybrid functional was found to well predict the excitation energies of studied systems. The origins of the ECD bands of two systems are mainly ascribed to charge-transfer (CT) transitions from oxygen atoms to metal atoms in polyanion. The results suggest that the polyanion are chiroptical chromophores. The polyanion plays a role as an optically active chromophore and contribute to the absorptions of ECD spectra. The difference of the UV–vis/ECD spectra between two systems shows that the transition metal atom significantly influences on the chiroptical properties of the studied Strandberg-type POMs.
Co-reporter:Dr. Chun-Yi Sun; Xin-Long Wang;Chao Qin;Jun-Ling Jin;Peng Huang ;Kui-Zhan Shao
Chemistry - A European Journal 2013 Volume 19( Issue 11) pp:3639-3645
Publication Date(Web):
DOI:10.1002/chem.201203080
Abstract
Two anionic metal–organic frameworks (MOFs) with 1D mesoporous tubes (1) and chiral mesoporous cages (2) have been rationally constructed by means of a predesigned size-extended hexatopic ligand, namely, 5,5′,5′′-(1,3,5-triazine-2,4,6-triyl)tris- (azanediyl)triisophthalate (TATAT). Charge neutrality is achieved by protonated dimethylamine cations. Notably, the two MOFs can be used to separate large molecules based on ionic selectivity rather than the size-exclusion effect so far reported in the literature. Owing to the imino triazine backbone and carboxyl groups of the hexatopic ligand, which provide important host–guest interactions, rare solvatochromic phenomena of 1 and 2 are observed on incorporating acetone and ethanol guests. Furthermore, guest-dependent luminescence properties of compound 2 were investigated, and the results show that luminescence intensity is significantly enhanced in toluene and benzene, while quenching effects are observed in acetone and ethanol. Thus, compound 2 may be a potential material for luminescent probes.
Co-reporter:Yu-Zhong Xie, Guo-Gang Shan, Zi-Yan Zhou, Zhong-Min Su
Sensors and Actuators B: Chemical 2013 177() pp: 41-49
Publication Date(Web):
DOI:10.1016/j.snb.2012.10.046
Co-reporter:Dr. Shu-Ran Zhang;Dr. Dong-Ying Du; Ke Tan;Dr. Jun-Sheng Qin;Dr. Hui-Qing Dong; Shun-Li Li;Dr. Wen-Wen He; Ya-Qian Lan;Dr. Ping Shen; Zhong-Min Su
Chemistry - A European Journal 2013 Volume 19( Issue 34) pp:11279-11286
Publication Date(Web):
DOI:10.1002/chem.201301536
Abstract
A new family of heterometal–organic frameworks has been prepared by two synthesis strategies, in which IFMC-26 and IFMC-27 are constructed by self-assembly and IFMC-28 is obtained by stepwise synthesis based on the metalloligand (IFMC=Institute of Functional Material Chemistry). IFMC-26 is a (3,6)-connected net and IFMC-27 is a (4,8)-connected 3D framework. The metalloligands {Ni(H4L)}(NO3)2 are connected by binuclear lanthanide clusters giving rise to a 2D sheet structure in IFMC-28. Notably, IFMC-26-EuxTby and IFMC-28-EuxTby have been obtained by changing the molar ratios of raw materials. Owing to the porosity of IFMC-26, Tb3+@IFMC-26-Eu and Eu3+@IFMC-26-Tb are obtained by postencapsulating TbIII and EuIII ions into the pores, respectively. Tunable luminescence in metal–organic frameworks is achieved by the two kinds of doping methods. In particular, the quantum yields of heterometal–organic frameworks are apparently enhanced by postencapsulation of LnIII ions.
Co-reporter:Wei-Chao Chen;Dr. Hao-Long Li; Xin-Long Wang;Dr. Kui-Zhan Shao; Zhong-Min Su; En-Bo Wang
Chemistry - A European Journal 2013 Volume 19( Issue 33) pp:11007-11015
Publication Date(Web):
DOI:10.1002/chem.201300615
Abstract
A versatile one-pot strategy was employed to synthesize three cerium(III)-stabilized polyoxotungstates nanoclusters by combining cerium linkers and SeO32−/TeO32− heteroanion templates: K32Na16[{(XO3)W10O34}8{Ce8(H2O)20}(WO2)4- (W4O12)]⋅n H2O [X=Se, n=81 (1); X=Te, n=114 (2)] and K12Na22[{(SeO3)W10O34}8{Ce8(H2O)20}(WO2)4- {(W4O6)Ce4(H2O)14(SeO3)4(NO3)2}]⋅ 79 H2O (3), which are the first lanthanide-containing polyoxotungstates with selenium or tellurium heteroatoms. The three clusters were characterized by single-crystal X-ray structure analysis, IR spectroscopy, thermogravimetric/differential thermal analysis, UV/Vis spectroscopy, ESI-MS, and X-ray photoelectron spectroscopy. Their electrochemical, photoluminescence, and magnetic properties were investigated. Their behavior in solution was studied by transmission electron microscopy, which showed that their single polyoxoanions assemble into intact, uniform-sized, purely inorganic hollow spheres in dilute water/acetone solution.
Co-reporter:Dr. Shizheng Wen;Dr. Guochun Yang; Likai Yan;Haibin Li; Zhongmin Su
ChemPhysChem 2013 Volume 14( Issue 3) pp:610-617
Publication Date(Web):
DOI:10.1002/cphc.201200770
Abstract
We design a new type of molecular diode, based on the organoimido derivatives of hexamolybdates, by exploring the rectifying performances using density functional theory combined with the non-equilibrium Green’s function. Asymmetric current–voltage characteristics were obtained for the models with an unexpected large rectification ratio. The rectifying behavior can be understood by the asymmetrical shift of the transmission peak observed under different polarities. It is interesting to find that the preferred electron-transport direction in our studied system is different from that of the organic D-bridge-A system. The results show that the studied organic–inorganic hybrid systems have an intrinsically robust rectifying ratio, which should be taken into consideration in the design of the molecular diodes.
Co-reporter:Dr. Peng Huang; Chao Qin;Xin-Long Wang;Chun-Yi Sun;Yan-Qing Jiao;Yan Xing;Kui-Zhan Shao
ChemPlusChem 2013 Volume 78( Issue 8) pp:775-779
Publication Date(Web):
DOI:10.1002/cplu.201300175
Co-reporter:Teng-Ying Ma, Na-Na Ma, Li-Kai Yan, Wei Guan, Zhong-Min Su
Journal of Molecular Graphics and Modelling 2013 40() pp: 110-115
Publication Date(Web):March 2013
DOI:10.1016/j.jmgm.2013.01.002
Co-reporter:Rong-Lin Zhong, Shi-Ling Sun, Hong-Liang Xu, Yong-Qing Qiu, and Zhong-Min Su
The Journal of Physical Chemistry C 2013 Volume 117(Issue 19) pp:10039-10044
Publication Date(Web):April 22, 2013
DOI:10.1021/jp402561h
An increasing number of scientists have focused on carbon–boron-nitride heteronanotubes because of their particularly adjustable properties, as shown in many fields. In this work, four isoelectronic models (BN-n, n = 1–4) were systematically investigated to explore the crucial factor for enhancing the static first hyperpolarizibility by doping the BN segment into the carbon nanotube (CNT) with differently connecting patterns. Theoretical results show that the N-connecting pattern might increase the contribution of the BN segment to the crucial transition states, which obviously increases the occupied orbital energy while the unoccupied orbital energy is slightly influenced. Correspondingly, the transition energy of BN-1 is smaller than that of BN-2. As a result, the static first hyperpolarizability of BN-1 is 1.05 × 104 au, which is remarkably larger than the 4.37 × 102 au of BN-2. The results indicate that, compared to the B-connecting pattern, the N-connecting pattern of the BN segment linking to the conjugated CNT segment is a more efficient way to enhance the first hyperpolarizability of heteronanotubes. It is our expectation that the new knowledge about the carbon–boron-nitride heteronanotubes could provide valuable information for scientists to develop the potential nonlinear optical nanomaterials by introducing BN segments into suitable positions of CNTs.
Co-reporter:Jie Wu, Yu-He Kan, Yong Wu, and Zhong-Min Su
The Journal of Physical Chemistry C 2013 Volume 117(Issue 16) pp:8420-8428
Publication Date(Web):April 1, 2013
DOI:10.1021/jp4008174
We theoretically designed a series of ambipolar host materials (1–8) which incorporate phosphine oxide and carbazole groups to the two ends of diphenyl (DP)-like bridges by para- and meta-connections, respectively. Density functional theory calculations were performed to investigate the influence of altering the DP-like bridges of these molecules on electronic structures and properties, and further to predict their performances as host materials in organic light-emitting diodes. The investigated results show the highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) of 1–8, distributed at the phenylcarbazole and the DP-like bridge, are responsible for hole and electron injection properties, respectively. The difference in the energies of HOMOs or LUMOs for 1–8 may be derived from different degrees of conjugation effect and electrostatic induction with altering the DP-like bridges of 1–8. The singlet states (S1), arising from the HOMO → LUMO transition, have intramolecular charge transfer character, which determines the small and different values of S1 energies. On the other hand, altering the DP-like bridges brings a great effect on triplet exciton distributions, and consequently different triplet energies. The different singlet/triplet energies for 1–8 make hosts 1–8 suitable for four reference guests with green/deep-blue light when scientists consider the matching of host and guest in singlet/triplet energies for efficient energy transfer.
Co-reporter:Peng Huang ; Chao Qin ; Zhong-Min Su ; Yan Xing ; Xin-Long Wang ; Kui-Zhan Shao ; Ya-Qian Lan ;En-Bo Wang
Journal of the American Chemical Society 2012 Volume 134(Issue 34) pp:14004-14010
Publication Date(Web):August 20, 2012
DOI:10.1021/ja303723u
Three novel polyoxoniobates, KNa2[Nb24O72H21]·38H2O (1), K2Na2[Nb32O96H28]·80H2O (2), and K12[Nb24O72H21]4·107H2O (3) with molecular triangle, molecular square, and cuboctahedral molecular cage geometries, respectively, have been successfully synthesized by conventional aqueous methods. All the compounds are built from [Nb7O22]9– fundamental building units. Compound 1 is the first isolated {Nb24O72} cluster, featuring three heptaniobate clusters linked in a ring by three additional NbO6 octahedra, while compound 2 is the largest isopolyoxoniobate cluster reported to date, consisting of four heptaniobate clusters linked by four additional NbO6 octahedra. Compound 3 is the largest solid aggregation of polyoxoniobates, assembled by four {KNb24O72} clusters joined by four K ions. To our knowledge, it is the first time these polyoxoniobate clusters have been crystallized with only alkali-metal counterions, thereby giving them the possibility of being redissolved in water. ESI-MS spectra indicate that compounds 1 and 2 remain structural integrity when the pure, crystalline polyanion salts are dissolved in water, while compound 3 is partially assembled into Nb24 fragments. The UV–vis diffuse reflectance spectra of these powder samples indicate that the corresponding well-defined optical absorption associated with Eg can be assessed at 3.35, 3.17, and 3.34 eV, respectively, revealing the presence of an optical band gap and the nature of semiconductivity with a wide band gap. UV-light photocatalytic H2 evolution activities were observed for these compounds with CoIII(dmgH)2pyCl as a cocatalyst and TEA as a sacrificial electron donor.
Co-reporter:Jun-Sheng Qin, Dong-Ying Du, Wen-Liang Li, Jing-Ping Zhang, Shun-Li Li, Zhong-Min Su, Xin-Long Wang, Qiang Xu, Kui-Zhan Shao and Ya-Qian Lan
Chemical Science 2012 vol. 3(Issue 6) pp:2114-2118
Publication Date(Web):07 Mar 2012
DOI:10.1039/C2SC00017B
A novel zeolite-like metal–organic framework (ZMOF) with sodalite topology, [Zn(HL)]·DMA (IFMC-1, L = 4,5-di(1H-tetrazol-5-yl)-2H-1,2,3-triazole and IFMC = Institute of Functional Material Chemistry), was solvothermally synthesized based on an N-rich aromatic ligand without a NH2 group. It exhibits high CO2 uptake and selective CO2/N2 adsorption capacity. For the first time, we investigated the influence of a large number of uncoordinated nitrogen atoms from aromatic rings for CO2 adsorption in ZMOFs. This result reveals that the high percentage of open N-donor sites leads to the high uptake capacity for CO2, even in the absence of any NH2 groups and open metal sites. In addition, it also exhibits efficient drug delivery capacity.
Co-reporter:Dong-Ying Du, Jun-Sheng Qin, Ting-Ting Wang, Shun-Li Li, Zhong-Min Su, Kui-Zhan Shao, Ya-Qian Lan, Xin-Long Wang and En-Bo Wang
Chemical Science 2012 vol. 3(Issue 3) pp:705-710
Publication Date(Web):19 Dec 2011
DOI:10.1039/C2SC00586G
Here, we synthesize a novel polyoxometalate-based crystalline tubular inorganic–organic compound, Mn[Zn(im)]2{[Na(H2O)]2[Mn(H2O)2][Zn(im)2][P4Mo6O31H6]2}·8H2O (IFMC-100) (im and IFMC correspond to imidazole and Institute of Functional Material Chemistry, respectively). Au-anchored tubular microreactor, Au@IFMC-100, has been prepared by simple immersion of IFMC-100 in an ethanol solution of HAuCl4 without any extra reducing agents, photochemical and electrochemical auxiliaries. Furthermore, IFMC-100 and Au@IFMC-100 have been employed as catalysts for the reduction of K3Fe(CN)6 and 4-nitrophenol with NaBH4 in aqueous solution, respectively. The results indicate the as-prepared Au@IFMC-100 microtubes exhibit enhanced catalytic performance in redox catalysis.
Co-reporter:Guang-Sheng Yang, Mei-Na Li, Shun-Li Li, Ya-Qian Lan, Wen-Wen He, Xin-Long Wang, Jun-Sheng Qin and Zhong-Min Su
Journal of Materials Chemistry A 2012 vol. 22(Issue 34) pp:17947-17953
Publication Date(Web):13 Jun 2012
DOI:10.1039/C2JM32990E
Three metal–organic frameworks (MOFs) comprising micropore, mesocage and nanotube structures have been prepared based on the same ligands and binuclear zinc secondary building units. We have successfully achieved flexible modulation of pore size from micropore to mesopore in a binary system by adjusting the reactant ratio. With the merit of a nanoscale channel, IFMC-8 can be applied as the host material for encapsulating Alq3 chromophores to exhibit tunable luminescence. The fluorescence emission of composite material Alq3@IFMC-8 has changed from green to blue. More importantly, the inclusion of IFMC-8 effectively prolonged the excited-state lifetime of Alq3 in ethanol. To the best of our knowledge, it is the first time that a MOF was studied as the host material for modulating the fluorescence properties of Alq3 chromophores.
Co-reporter:Dong-Ying Du, Jun-Sheng Qin, Chun-Gang Wang, Xian-Chun Liu, Shun-Li Li, Zhong-Min Su, Xin-Long Wang, Ya-Qian Lan and En-Bo Wang
Journal of Materials Chemistry A 2012 vol. 22(Issue 39) pp:21040-21044
Publication Date(Web):17 Aug 2012
DOI:10.1039/C2JM33959E
A 3D eight-connected redox-active polyoxometalate (POM)-based crystalline material, IFMC-101, has been synthesized based on reduced {P4Mo6O31H6}-based tetrameric clusters. IFMC-101 was used as a reducing agent and stabilizer to prepare Au and Pt nanoparticles (NPs) by auto-redox reactions without extra reduction assistance. For the first time, we have demonstrated a new strategy to prepare noble metal NP-loaded POM-based crystalline catalysts. These crystalline catalysts were employed toward the reduction of 4-nitrophenol by NaBH4 and the efficiency of the Au NP-loaded material, Au@IFMC-101, was nearly 9 times higher than that of the POM-based crystalline material, IFMC-101. This result reveals that Au@IFMC-101 exhibits enhanced activity owing to the synergistic catalysis of noble metal NPs and crystalline POM components.
Co-reporter:Xueqing Zhang, Hong Ren, Tingting Wang, Lingyu Zhang, Lu Li, Chungang Wang and Zhongmin Su
Journal of Materials Chemistry A 2012 vol. 22(Issue 26) pp:13380-13385
Publication Date(Web):11 May 2012
DOI:10.1039/C2JM31141K
Yolk–shell structured superparamagnetic iron oxide (SPIO) catalysts with mesoporous SnO2 shells with different thicknesses and tunable void spaces have been fabricated by a rapid and simple templating method. The resulting hybrid SPIO@SnO2 yolk–shell nanocapsules (NCs) exhibit high efficiency photocatalytic degradation of rhodamine B and could be reused up to five times by magnetic separation, showing their potential as candidates for practical applications in environmental protection. Moreover, the possible mechanism for the formation of the SPIO@SnO2 yolk–shell NCs was also discussed.
Co-reporter:Guo-Gang Shan, Hai-Bin Li, Dong-Xia Zhu, Zhong-Min Su and Yi Liao
Journal of Materials Chemistry A 2012 vol. 22(Issue 25) pp:12736-12744
Publication Date(Web):13 Apr 2012
DOI:10.1039/C2JM30480E
To make Ir(III)-based complexes potentially multifunctional materials, two new cationic Ir(III) complexes with a 2-(5-phenyl-2-phenyl-2H-1,2,4-triazol-3-yl)pyridine (Phtz) ancillary ligand were designed and synthesized. By introducing the pendant phenyl ring into the ancillary ligand, the two complexes possess desired intramolecular π–π stacking between the pendant phenyl ring of the Phtz ligand and one of the phenyl rings of the cyclometalated ligand, which renders the complexes more stable. Density functional theory calculation indicates that the intramolecular π–π interactions in both complexes can reduce the degradation reaction in metal-centered (3MC) states to some extent, which further implies their stability. With these results in combination with their reversible oxidation and reduction processes as well as excellent photophysical properties, the stable light-emitting cells (LECs) would be expected. Furthermore, the two synthesized complexes exhibit reversible piezochromism. Their emission color can be smartly switched by grinding and heating, which is visible to the naked eye. In light of our experimental results, the present piezochromic behavior is due to interconversion between crystalline and amorphous states.
Co-reporter:Xiao-Dan Tang, Yi Liao, Hong-Ze Gao, Yun Geng and Zhong-Min Su
Journal of Materials Chemistry A 2012 vol. 22(Issue 14) pp:6907-6918
Publication Date(Web):28 Feb 2012
DOI:10.1039/C2JM14871D
The bridging effect on the charge transport properties of cyclooctatetrathiophene and its derivatives (systems 1–4) was investigated at the level of density functional theory (DFT). Insights into their geometries, frontier molecular orbitals, reorganization energies, transfer integrals and band structures were provided in detail. Increasing charge mobilities for both holes and electrons were predicted in cyclooctatetrathiophene derivatives as the number of bridging sulfur atoms increased. The improved charge transport from system 1 to 4 can be interpreted from two contributions: (i) decreased reorganization energy with improved molecular planarity under the consideration of intermolecular interactions; and (ii) enhanced transfer integral derived from the π-stacking arrangement for 2 and 3, and multidimensional S⋯S interactions are found to contribute to charge transport in system 4 besides π⋯π interactions. The charge transport properties were also analyzed with a band-like model and the results were in agreement with those gained from the hopping model in the fact that the paths with large transfer integrals are all along the directions with large dispersions in the valence band or conduction band.
Co-reporter:Shunrui Luo, Fang Chai, Lingyu Zhang, Chungang Wang, Lu Li, Xianchun Liu and Zhongmin Su
Journal of Materials Chemistry A 2012 vol. 22(Issue 11) pp:4832-4836
Publication Date(Web):30 Jan 2012
DOI:10.1039/C2JM16476K
In this work, we have developed a facile and rapid synthesis of urchin-shaped Fe3O4@Bi2S3 core-shell hierarchical structures through a sonochemical method. The as-prepared Fe3O4@Bi2S3 hierarchical core-shell structures show excellent photocatalytic efficiency for the degradation of rhodamine B (RhB) and retained this photocatalytic activity after being recycled five times with the help of an external magnetic field. The possible mechanism for the formation of the urchin-shaped Fe3O4@Bi2S3 core-shell structures is also proposed. These novel urchin-shaped Fe3O4@Bi2S3 core-shell structures may have potential applications in water treatment, sensors, and energy storage.
Co-reporter:Rong-Lin Zhong, Hong-Liang Xu, Shabbir Muhammad, Ji Zhang and Zhong-Min Su
Journal of Materials Chemistry A 2012 vol. 22(Issue 5) pp:2196-2202
Publication Date(Web):15 Dec 2011
DOI:10.1039/C1JM14358A
Excess electron compounds have been proposed to be novel candidates of high-performance nonlinear optical (NLO) materials because of their large static first hyperpolarizabilities (β0). To enhance the stability of an unstable excess electron compound (LiCN⋯Li) with an extremely large β0 value (310196 a.u.), we designed a boron nitride nanotube (BNNT) as a protective shield molecule to encapsulate it (in theory). The stability of LiCN⋯Li was enhanced: the vertical ionization potentials (VIP) of LiCN⋯Li increased after encapsulating. Therefore, by comparison with LiCN⋯Li, the encapsulated complexes are more difficult to oxidize. Significantly, the BNNT encapsulated LiCN⋯Li complex exhibits a considerable β0 value (10645 a.u.), which is significantly (almost 380 times) larger than 28 a.u. of BNNT. Our further investigations into the intrinsic hyperpolarizabilites (βint) of these compounds show that there are clearly dependencies of the NLO response on the transition energy. Furthermore, it is easy to encapsulate LiCN⋯Li from the B-rich edge rather than N-rich edge of BNNT due to the lower energy barrier, which makes our calculations more useful to experimentalists who may try to synthesize these compounds. Knowledge of the encapsulation process of LiCN⋯Li within BNNT provides a new strategy for the design and synthesis of stable high-performance NLO materials.
Co-reporter:Ji Zhang, Hai-Bin Li, Shi-Ling Sun, Yun Geng, Yong Wu and Zhong-Min Su
Journal of Materials Chemistry A 2012 vol. 22(Issue 2) pp:568-576
Publication Date(Web):03 Nov 2011
DOI:10.1039/C1JM13028E
To rationalize the marked difference in the energy conversion efficiency of dye sensitized solar cells (DSSCs) based on organic dyes 1 and 2 different only in their π spacer, density functional theory (DFT) and time-dependent DFT calculations of the geometries, electronic structures and absorption spectra of the organic dyes before and after binding to titanium oxide were carried out. These enable us to determine factors such as dipole moments associated with the open-circuit photovoltage (Voc), and to quantify parameters such as the light harvesting efficiency, the electron injection efficiency associated with the short-circuit photocurrent density (Jsc). The results reveal that compared to 2 with a thiazole spacer, 1 with a thiophene spacer could cause a red shift of the absorption spectrum, increase the oscillator strength and improve the driving force for electron injection, thus leading to the larger Jsc, in good agreement with experimental data. As for Voc, our results stress that apart from the generally emphasized vertical dipole moment of the dyes pointing outward from the semiconductor surface, the number of photoinjected electrons from the dye to the semiconductor is also crucial to obtain high performance dyes with high Voc. After justifying the reliability of the quantum-chemical methods, we designed another four dyes with different π spacers to screen more efficient organic dyes. Fortunately, taking 1 as reference, we find that dye 4 with a thienothiophene spacer displays an enhanced Jsc and Voc, indicating that it will be a more efficient diarylamine-fluorene-based organic dye used in DSSCs, which will play a theoretical guiding role in the design and synthesis of new organic dyes.
Co-reporter:Yun Geng, Hai-Bin Li, Shui-Xing Wu and Zhong-Min Su
Journal of Materials Chemistry A 2012 vol. 22(Issue 39) pp:20840-20851
Publication Date(Web):25 Jun 2012
DOI:10.1039/C2JM33369D
Perylene diimide (PDI) and its derivatives hold great promise, since they are undeniably considered as an important family of organic n-type semiconductors with both high carrier mobilities and air stabilities comparable to p-type ones, although they traditionally stand out as a class of high-performance dyes and pigments. In this feature article, we summarize the influences of substituents on different positions (imide, ortho, bay) of PDI on their electronic and morphological (packing) properties, which are in close connection with the ability for carrier transport. Then representative molecular packing motifs for PDIs are also classified, with an emphasis on the intricate interplay of intermolecular interactions, packing motifs and electron transport properties of perylene imide related carrier transport materials from a theoretical point of view, towards paving the way for boosting and improving the electron transport mobilities and air stabilities of PDIs-based materials.
Co-reporter:Dong-Ying Du, Jun-Sheng Qin, Cheng-Xin Sun, Xin-Long Wang, Shu-Ran Zhang, Ping Shen, Shun-Li Li, Zhong-Min Su and Ya-Qian Lan
Journal of Materials Chemistry A 2012 vol. 22(Issue 37) pp:19673-19678
Publication Date(Web):31 Jul 2012
DOI:10.1039/C2JM34661C
Five heterometallic luminescent crystalline materials with the metalloligand, [Zn(HL)EuxTby(H2O)2][ZnBr4]·H2O (x = 1, y = 0, IFMC-21; x = 0.75, y = 0.25, IFMC-22; x = 0.5, y = 0.5, IFMC-23; x = 0.25, y = 0.75, IFMC-24; x = 0, y = 1, IFMC-25; H4L = 4,4′,4′′,4′′′-(2,2′,2′′,2′′′-(ethane-1,2-diylbis(azanetriyl))tetrakis(methylene)-tetrakis-(1H-benzo[d]imidazole-2,1-diyl))tetrakis(methylene)-tetrabenzoic acid; IFMC = Institute of Functional Material Chemistry), were prepared by the combination of hydrothermal and ionothermal methods for the first time. IFMC-21–25 can be obtained by introducing the desired Eu(III) and Tb(III) in the initial experiments. In these crystalline materials, the metalloligand Zn(HL) was connected by bi-lanthanide cores leading to a 2D sheet-structure and [ZnBr4]2− ions were distributed in the interspaces of the sheet. The luminescent properties of IFMC-21 to 25 were investigated and the results reveal that they exhibit characteristic Eu(III) and Tb(III) ion emissions, and the intensities of red and green arising from Eu(III) and Tb(III) emissions are shifted correspondingly by tuning the ratios of Eu(III):Tb(III).
Co-reporter:Kun Zhou, Chao Qin, Hai-Bin Li, Li-Kai Yan, Xin-Long Wang, Guo-Gang Shan, Zhong-Min Su, Chuang Xu and Xiu-Li Wang
Chemical Communications 2012 vol. 48(Issue 47) pp:5844-5846
Publication Date(Web):23 Apr 2012
DOI:10.1039/C2CC32321D
Bridging between silver clusters and polyoxoanion clusters, the first 1D assembly, [Ag34(StBu)26(W6O21)(CF3COO)](CF3COO)·Et3N·20CH3OH (1), based on POM-templated silver-thiolate nanoclusters featuring a [Ag34(StBu)26(CF3COO)]7+ shell and a [W6O21]6− core is reported. This novel core–shell nanocluster possesses nanoscopic morphology, displays intense deep-blue emission in solution under ambient conditions and also shows special electrochemical properties.
Co-reporter:Wen-Wen He, Shun-Li Li, Guang-Sheng Yang, Ya-Qian Lan, Zhong-Min Su and Qiang Fu
Chemical Communications 2012 vol. 48(Issue 80) pp:10001-10003
Publication Date(Web):13 Aug 2012
DOI:10.1039/C2CC34196D
A novel non-interpenetrating metal–organic framework IFMC-15 was successfully constructed based on octahedral cage-like building units and its outstanding performance in reversible adsorption of iodine was investigated.
Co-reporter:Peng Huang, Chao Qin, Xin-Long Wang, Chun-Yi Sun, Guang-Sheng Yang, Kui-Zhan Shao, Yan-Qing Jiao, Kun Zhou and Zhong-Min Su
Chemical Communications 2012 vol. 48(Issue 1) pp:103-105
Publication Date(Web):04 Nov 2011
DOI:10.1039/C1CC15684E
An unprecedented organic–inorganic hybrid {[Cu6L6(H2O)3][Nb10V4O40(OH)2]}2·13H2O (1) (L = 1,10-phenanthroline) containing the unreported {Nb10V4O40(OH)2}12− building blocks has been successfully synthesized and its photoluminescent properties, IR spectra, thermogravimetric analyses and single-crystal X-ray diffraction were investigated.
Co-reporter:Guo-Gang Shan, Hai-Bin Li, Hong-Tao Cao, Dong-Xia Zhu, Peng Li, Zhong-Min Su and Yi Liao
Chemical Communications 2012 vol. 48(Issue 14) pp:2000-2002
Publication Date(Web):11 Jan 2012
DOI:10.1039/C2CC15855H
We demonstrate that two new cationic Ir(III) complexes exhibit an interesting piezochromism, and their emission color can be smartly switched by grinding and heating. This is the first example that the Ir(III) complexes display piezochromic phosphorescence.
Co-reporter:Lei Chen, Ke Tan, Ya-Qian Lan, Shun-Li Li, Kui-Zhan Shao and Zhong-Min Su
Chemical Communications 2012 vol. 48(Issue 47) pp:5919-5921
Publication Date(Web):18 Apr 2012
DOI:10.1039/C2CC31257C
Two isostructural 2D → 2D parallel → 3D inclined interpenetrating polycatenane-like metal–organic frameworks were successfully constructed based on length-adjusted tricarboxylate ligands. With the merit of being microporous, IFMC-10 can serve as host for encapsulating lanthanide cations and I2 to exhibit luminescent sensing and rapid adsorption of iodine.
Co-reporter:Weili Zheng ; Shuixing Wu ; Shanshan Zhao ; Yun Geng ; Junling Jin ; Zhongmin Su ;Qiang Fu
Inorganic Chemistry 2012 Volume 51(Issue 7) pp:3972-3980
Publication Date(Web):March 13, 2012
DOI:10.1021/ic2011953
A compound having the capability of releasing NO upon exposure to visible or near-infrared (vis or NIR) light could be a potential candidate for photodynamic therapy (PDT), which is significant for humans. Here, we investigated a series of Mn(II) complexes (a–d) based on density functional theory (DFT) to illuminate the mechanism of their behavior of releasing NO. Their structural, spectroscopic, and photodissociable properties were calculated by quantum theoretical methods to give a detailed and warranted explanation of the performance of releasing NO. The results indicate that, for a–d, releasing NO was attributed to the electron transfer from dyz/dxz(Mn) orbitals to π*(NO) orbitals at the second excited triplet state (T2). Importantly, we confirmed the finding in the experiment that d could release NO upon exposure to the NIR region and, thus, may be a best candidate for PDT in a–d. Therefore, to take d for example, the analyses of the potential energy curves (PECs) of difference states and electron density difference between the T2 and the ground state (S0) were performed to further provide evidence of ligand dissociation and release of NO at the T2 state. Finally, we hope that our discussion can provide assistance to understand the behavior of the release of NO and design novel photodissociable transition metal nitrosyls for PDT applications.
Co-reporter:Xiang-Rong Hao, Xin-Long Wang, Kui-Zhan Shao, Guang-Sheng Yang, Zhong-Min Su and Gang Yuan
CrystEngComm 2012 vol. 14(Issue 17) pp:5596-5603
Publication Date(Web):14 May 2012
DOI:10.1039/C2CE25343G
Seven novel porous metal–organic frameworks (MOFs), Zn2(BTC)(NO3)(DMA)3 (1), Zn11(BTC)6(NO3)4(DEE)9 (2), Zn11(BTC)6(NO3)4(DEP)8 (3), Zn(BTC)·DMA·C2H8N (4), Zn3(BTC)3·3(C2H8N)·4(DMA) (5), Zn9(BTC)6(OH)2·2(C2H8N)·15(DEE) (6), and Zn9(BTC)5(OH)3(C2O4)·2(C4H12N)·5(DEE) (7) have been solvothermally synthesized using zinc nitrate, 1,3,5-benzenetricarboxylate acid (H3BTC) and differently sized solvents (DMF, DMA, DEE, DEP, DPE, DPP), showing that the solvent size can not only dramatically influence the pore size, but also allow access to new structures and topologies previously unrealized in MOFs. With increasing solvent size, the pore size of MOFs correspondingly changes from 9 Å to 23 Å along with different structures: three frameworks have cages larger than 16 Å, one has a 23 Å chiral cage, five are anionic frameworks, three have topologies heretofore unreported in MOFs and all structures are noninterpenetrating. The ion-exchange experiment estimated by HNMR analyses shows that dimethylamine ions in 5 can be exchanged by NH4+ ions.
Co-reporter:Gang Yuan, Kui-Zhan Shao, Dong-Ying Du, Xin-Long Wang, Zhong-Min Su and Jian-Fang Ma
CrystEngComm 2012 vol. 14(Issue 5) pp:1865-1873
Publication Date(Web):11 Jan 2012
DOI:10.1039/C1CE06178J
Seven mixed-ligand coordination polymers, namely [Cd2(HPIDC)2(bipy)]n (1), {[Cd3(HPIDC)2(btc)(H2O)4]·H2O}n (2), {[Cd2(HPIDC)2(bdc)1/2(H2O)Cl]·H2O}n (3), {[Cd2(PIDC)(Ac)(H2O)]·H2O}n (4), [Zn2(HPIDC)(ox)(H2O)2]n (5), {[Eu(HPIDC)(ox)1/2H2O]·2H2O}n (6), and {[Tb(HPIDC)(ox)1/2H2O]·2H2O}n (7) (H3PIDC = 2-(pyridin-4-yl)-1H-imidazole-4,5-dicarboxylic acid, bipy = 2,2′-bipyridine, H4btc = 1,2,4,5-benzenetetracarboxylic acid, H2bdc = 1,4-benzenedicarboxylic acid, H2ox = aoxalic acid) have been synthesized under hydrothermal conditions. Their structures have been determined by single crystal X-ray diffraction analyses and further characterized by elemental analyses, IR spectra, and thermogravimetric analyses (TGA). Complexes 1 and 2 display a two-dimensional (2D) layer formed by the inter-linking helices and ligands connecting di-metal units, respectively. Both 3 and 4 assume a 3D covalent framework built up from mixed ligands linking two types of rod-shaped SBUs with trinodal (3,4)-connected and (3,4,5)-connected nets. Complexes 5–7 feature a 3D network constructed from HPIDC2− and ox2− anions bridging metal centers exhibiting the CdSO4 and α-Po nets, respectively. Moreover, the photoluminescent properties of 1–7 have been studied in the solid state at room temperature.
Co-reporter:Shizheng Wen, Wei Guan, Jianping Wang, Zhongling Lang, Likai Yan and Zhongmin Su
Dalton Transactions 2012 vol. 41(Issue 15) pp:4602-4607
Publication Date(Web):01 Feb 2012
DOI:10.1039/C2DT12465C
The structural and electronic properties of [PW12O40]3− (PW12) anion deposited on a graphene layer are investigated by using periodic density functional theory. The equilibrium geometries of graphene–PW12 (G–PW12) are examined based on six different configurations. The adsorption energy and charge transfer between PW12 and graphene are calculated and analyzed. We found that the interaction between PW12 and graphene are noncovalent. The formation of G–PW12 complex is theoretically predicted to be feasible from an energetic perspective with electron transfer from the PW12 to graphene.
Co-reporter:Wei Guan, Zhijian Wu and Zhongmin Su
Dalton Transactions 2012 vol. 41(Issue 9) pp:2798-2803
Publication Date(Web):20 Jan 2012
DOI:10.1039/C2DT12068B
DFT investigations have been carried out on encapsulation of Lindqvist-type W6O192− anion inside hydrogenated (n,n) armchair single-walled carbon nanotubes (h-CNTs) with n = 8, 9, 10 to understand the confinement effect of the CNTs on the rotation of W6O192−. The energy-decomposition analysis (EDA) of interaction between W6O192− and CNTs shows that with the increase of confinement effect from n = 8, 9, to 10, the destabilizing ΔEPauli plays a more important role in the relative orientation of W6O192− inside CNTs. For W6O192−@(9,9) h-CNT, the most stable orientation appears at the y/z angle 45°/36°. The confinement effect reduces significantly the energy gap of W6O192−@(n,n) h-CNT (n = 8, 9, 10) compared with free W6O192−. Electron transfer from the W6O192− to CNT is observed.
Co-reporter:Hai-Ning Wang, Xing Meng, Xin-Long Wang, Guang-Sheng Yang and Zhong-Min Su
Dalton Transactions 2012 vol. 41(Issue 8) pp:2231-2233
Publication Date(Web):22 Dec 2011
DOI:10.1039/C2DT11872F
Three allomorphs with the same stoichiometry, [Mg3(H2O)4(5-aip)2(5-Haip)2]·4DMA, were solvothermally synthesized in the presence of different additives and represented the first 8-connected nanotubular networks in Mg-based metal–organic frameworks. The adsorption and delivery of drugs were also determined.
Co-reporter:Hai-Ning Wang, Xing Meng, Chao Qin, Xin-Long Wang, Guang-Sheng Yang and Zhong-Min Su
Dalton Transactions 2012 vol. 41(Issue 3) pp:1047-1053
Publication Date(Web):22 Nov 2011
DOI:10.1039/C1DT11304F
Four new compounds, [Cd(5-aip)(bpy)]·1.5DMA (1), [Cu(5-aip)(bpy)]·1.3DMA (2), [Co(5-aip)(bpy)]·1.6DMA (3), and [Cd(5-aip)(bpy)0.5(H2O)]·1.3DMA (4), based on 5-aminoisophthalic acid and 4,4′-bipyridine, have been synthesized by the solvothermal method and structurally determined using single crystal X-ray diffraction. Compounds 1–3 are structurally similar and show non-interpenetrating three-dimensional (3D) pillar-layer frameworks, while compound 4 displays a two-dimensional (2D) (3,4)-connected parallel non-interpenetrating architecture. In all these compounds, 1D rectangular channels are observed and the ligand 5-aminoisophthalic acid exhibits three kinds of coordination modes. Furthermore, 1 displays a single-crystal-to-single-crystal transformation when immersed in a methanol solution. More significantly, 1 can absorb and deliver I2 molecules by means of its channels, and could induce a reversible luminescent transformation from quenching to the initial state. The luminescent properties of 1 and 4 have also been studied.
Co-reporter:Guo-Gang Shan, Ling-Yu Zhang, Hai-Bin Li, Shuang Wang, Dong-Xia Zhu, Peng Li, Chun-Gang Wang, Zhong-Min Su and Yi Liao
Dalton Transactions 2012 vol. 41(Issue 2) pp:523-530
Publication Date(Web):01 Nov 2011
DOI:10.1039/C1DT11215E
We report the synthesis and characterization of two cationic iridium(III) complexes with dendritic carbazole ligands as ancillary ligands, namely, [Ir(ppy)2L3]PF6 (1) and [Ir(ppy)2L4]PF6 (2), where L3 and L4 represent 3,8-bis(3,6-di-tert-butyl-9H-carbazol-9-yl)-1,10-phenanthroline and 3,8-bis(3′,6′-di-tert-butyl-6-(3,6-di-tert-butyl-9H-carbazol-9-yl)-3,9′-bi(9H-carbazol)-9-yl)-1,10-phenanthroline, respectively. Their photophysical properties have been investigated and compared. The results have shown that complex 2 is aggregation-induced phosphorescent emission (AIPE) active and exhibits the highest photoluminescent quantum yield (PLQY) of 16.2% in neat film among the reported cationic Ir(III) complexes with AIPE activity. In addition, it also enjoys redox reversibility, good film-forming ability, excellent thermal stability as well as off/on luminescence switching properties, revealing its potential application as a candidate for light-emitting electrochemical cells and organic vapor sensing. To explore applications in biology, 2 was used to image cells.
Co-reporter:Zhong-Ling Lang, Wei Guan, Li-Kai Yan, Shi-Zheng Wen, Zhong-Min Su and Li-Zhu Hao
Dalton Transactions 2012 vol. 41(Issue 37) pp:11361-11368
Publication Date(Web):18 Jul 2012
DOI:10.1039/C2DT31166F
The formation mechanism is always a fundamental and confused issue for polyoxometalate chemistry. Two formation mechanisms (M1 and M2) of the Lindqvist anion [W6O19]2− have been adopted to investigate it's self-assembly reaction pathways at a density functional theory (DFT) level. The potential energy surfaces reveal that both the mechanisms are thermodynamically favorable and overall barrierless at room temperature, but M2 is slightly dominant to M1. The formation of the pentanuclear species [W5O16]2− and [W5O15(OH)]− are recognized as the rate-determining steps in the whole assembly polymerization processes. These two steps involve the highest energy barriers with 30.48 kcal mol−1 and 28.90 kcal mol−1, respectively, for M1 and M2. [W4O13]2− and [W4O12(OH)]− are proved to be the most stable building blocks. In addition, DFT results reveal that the formation of [W3O10]2− experiences a lower barrier along the chain channel.
Co-reporter:Guo-Gang Shan, Hai-Bin Li, Jun-Sheng Qin, Dong-Xia Zhu, Yi Liao and Zhong-Min Su
Dalton Transactions 2012 vol. 41(Issue 32) pp:9590-9593
Publication Date(Web):01 Jun 2012
DOI:10.1039/C2DT31013A
A new cationic Ir(III) complex based on a dendritic ancillary ligand has been designed and synthesized, which simultaneously exhibits piezochromic luminescent (PCL) behavior and aggregation-induced emission (AIE) property for the first time.
Co-reporter:Shanshan Zhao, Fei Yu, Guochun Yang, Hongyu Zhang, Zhongmin Su and Yue Wang
Dalton Transactions 2012 vol. 41(Issue 24) pp:7272-7277
Publication Date(Web):20 Apr 2012
DOI:10.1039/C2DT00009A
To deeply understand the charge-transporting nature of Pt(CNtBu)2(CN)2 nanowires induced by intermolecular Pt⋯Pt interactions, calculations based on first-principle band structure and Marcus theory have been performed. The calculated bandwidths of the valence band, conducting band, and the effective masses of hole and electron are almost equal. This suggests that this complex has ambipolar transport characteristics, in agreement with experimental results. Density of states analysis revealed that the hole transport resulted mainly from the Pt⋯Pt interactions, while the electron transport was derived mainly from the CN groups. The character of the frontier molecular orbitals, reorganization energies and transfer integrals in different directions also supports the calculated first-principle band structure. Moreover, an investigation into the intermolecular interaction energy of neighbors revealed that there is a remarkable relationship between the intermolecular interaction energy and the transfer integral.
Co-reporter:Chun-Yi Sun, Chao Qin, Xin-Long Wang, Guang-Sheng Yang, Kui-Zhan Shao, Ya-Qian Lan, Zhong-Min Su, Peng Huang, Chun-Gang Wang and En-Bo Wang
Dalton Transactions 2012 vol. 41(Issue 23) pp:6906-6909
Publication Date(Web):24 Apr 2012
DOI:10.1039/C2DT30357D
Zeolitic Imidazolate Framework-8 (ZIF-8), for the first time for ZIFs, exhibits a remarkable capacity for the anticancer drug 5-fluorouracil (5-FU), around 660 mg of 5-FU/g of ZIF-8, and presents a pH-triggered controlled drug release property. These prove ZIF-8 to be a valuable candidate for delivery of anticancer agents and reveal its potential applications in the treatment of cancer.
Co-reporter:Peng Huang, Chao Qin, Xin-Long Wang, Chun-Yi Sun, Yan Xing, Hai-Ning Wang, Kui-Zhan Shao and Zhong-Min Su
Dalton Transactions 2012 vol. 41(Issue 20) pp:6075-6077
Publication Date(Web):12 Apr 2012
DOI:10.1039/C2DT30265A
A new organic–inorganic hybrid, [Cu(en)2]3{[Cu(en)2][H6SiNb18O54]}·22H2O (1, en = ethylenediamine) containing the crescent-shaped polyoxoanion [H6SiNb18O54]8− and copper–organic cations has been successfully synthesized, and elemental analyses, IR spectra, thermogravimetric analyses and single-crystal X-ray diffraction were investigated.
Co-reporter:Dong-Lai Wang, Hong-Liang Xu, Zhong-Min Su and Guang Xin
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 43) pp:15099-15105
Publication Date(Web):17 Sep 2012
DOI:10.1039/C2CP42669B
Very recently, two novel Sc3NC-based cluster fullerenes Sc3NC@C80 (Wang et. al. J. Am. Chem. Soc. 2010, 132, 16362) and Sc3NC@C78 (Wu et. al. J. Phys. Chem. C 2011, 115, 23755) were prepared and characterized, respectively. Inspired by these findings, the possibility of encapsulating Sc3NC cluster in the C84 fullerene is performed using density functional theory (DFT). Firstly, the isolated pentagon rule (IPR) D2d (23) C84 fullerene is employed to encase the Sc3NC cluster: four possible endohedral metallofullerene isomers a–d are designed. The large binding energies (ranging from 163.7 to 210.0 kcal mol−1) indicate that the planar quinary cluster Sc3NC can be stably encapsulated in the C84 (isomer 23) cage. Further, we consider the incorporation of Sc3NC into the non-IPR Cs (51365) C84 cage leading to isomer e and show the high stability of isomer e, which has a larger binding energy, larger HOMO-LUMO gap, higher adiabatic (vertical) ionization potential, and lower adiabatic (vertical) electron affinity than the former four Sc3NC@C84 (isomer 23). Significantly, the predicted binding energy (294.2 kcal mol−1) of isomer e is even larger than that (289.2 and 277.7 kcal mol−1, respectively) of the synthesized Sc3NC@C80 and Sc3NC@C78, suggesting a considerable possibility for experimental realization. The 13C NMR chemical shifts and Raman spectra of this a new endofullerene have been explored to assist future experimental characterization.
Co-reporter:Nana Ma, Likai Yan, Wei Guan, Yongqing Qiu and Zhongmin Su
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 16) pp:5605-5612
Publication Date(Web):27 Feb 2012
DOI:10.1039/C2CP00054G
We report a theoretical study based on density functional theory (DFT) on the geometric and electronic structure, linear optical and second-order nonlinear optical properties of a series of new inorganic–organic hybrid hexamolybdate–organoimido–(car)boranes. By the incorporation of borane/carborane at the end of the phenyl ring of the organoimido segment, the studied systems show excellent nonlinear optical (NLO) response than the organoimido-substituted hexamolybdate. The computed static first hyperpolarizability βvec value of [Mo6O18(NC8H8)(B12H11)]4− (II) is largest, −167.2 × 10−30 esu, and a higher βvec value of [Mo6O18(NC8H8)(C2B10H11)]2− (III-2p) is 58.6 × 10−30 esu. Moreover, the time-dependent (TD)DFT calculation illustrates that the maximum absorption, which is helpful for the large NLO responses, is mainly assigned to the charge transfer (CT) from (car)borane and organoimido segment to the hexamolybdate cluster. The density of density (DOS) calculations further illustrate the excitation from valence orbitals of boron atoms to that of Mo and O atoms in hexamolybdate can be responsible for larger NLO responses. The linear and nonlinear optical properties of species III both vary with the position of the vertex on the carborane. Furthermore, the order of the βvec values is consistent with the bathochromic shift of the maximum absorption for our studied systems, and the studied systems show a wider transparency range extending into the entire visible and infrared (IR) region.
Co-reporter:Min Zhang, ZhongMin Su and GuanHua Chen
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 14) pp:4695-4702
Publication Date(Web):02 Feb 2012
DOI:10.1039/C2CP23164F
The electron excitations of Single-Walled Silicon Nanotubes (SWSiNTs), with sp2 and sp3 hybridization, were studied using the localized-density-matrix (LDM) method with INDO/S parameters. Strong anisotropic characteristics of the dynamic polarizabilities were found for all the nanotubes. The transitional intensity along the tubular axis is much larger than that perpendicular to the axis for all the nanotubes. The optical gaps of sp3-hybridized infinitely-long pentagonal SWSiNTs are near 3.0 eV and 4.7 eV owing to σ–σ* transitions along the direction of the tubular axis. The optical gaps of sp2-hybridized infinitely-long armchair SWSiNTs along the tube axis direction are about 0.7 eV and 2.4 eV for Si(3,3) SWSiNTs and 0.7 eV and 2.7 eV for Si(4,4) SWSiNTs. The former peak at 0.7 eV originated from π–π* electron transitions and the latter peak at 2.4 eV or 2.7 eV originated from σ–σ* electron transitions. Meanwhile, the intensities of π–π* electron transitions are stronger than those of σ–σ* electron transitions in SWSiNTs. The low sp2 transition energy derived from the weak overlap of unpaired pz orbitals of silicon atoms. Moreover, the electronic excitations of zigzag SWSiNTs are similar to those of armchair structures. This indicates that sp2-hybridized silicon nanotubes possess much greater potential for application in optical fields.
Co-reporter:Lian-Jie Li, Chao Qin, Xin-Long Wang, Kui-Zhan Shao, Zhong-Min Su and Peng-Jun Liu
CrystEngComm 2012 vol. 14(Issue 12) pp:4205-4209
Publication Date(Web):01 May 2012
DOI:10.1039/C2CE06451K
Reactions of 2,2′-(naphthalene-1,5-diylbis(oxy))diacetic acid (H2ndd) and two types of metal salts with different auxiliary ligands: 4,4′-bipyridine (4,4′-bipy), 1,4-di(1H-imidazol-1-yl)benzene (DIMB) and 1,4-bis[(1H-imidazol-1-yl)methyl]benzene (BIMB), yield two self-catenated networks and a classic pcu network. The structures of compounds 1–3 are elucidated by single crystal X-ray diffraction. Topological analysis reveals that the compound 1 is a new (4,8)-connected self-catenated network with point symbol of (32·42·52)(32·49·512·65)2 based on the trinuclear zinc clusters as an eight-connected node. 2 is an eight-connected self-catenated network with ilc topology and 3 is a 6-connected pcu network. Photoluminescent properties and thermogravimetric analyses for 1–3 were also investigated in detail.
Co-reporter:Jian-Ping Wang, Guo-Chun Yang, Li-Kai Yan, Wei Guan, Shi-Zheng Wen and Zhong-Min Su
Dalton Transactions 2012 vol. 41(Issue 33) pp:10097-10104
Publication Date(Web):28 May 2012
DOI:10.1039/C2DT30449J
We theoretically investigate a novel switching phenomenon based on the divacant Keggin-type polyoxotungstate bearing chiral organophosphonate [{NH2CH(CH3)PO}2(γ-SiW10O36)]4−, that is the synchronous chiroptical and nonlinear optical (NLO) switch triggered by redox. The ECD calculations on the Boltzmann weighted conformations of the three oxidation states of this chiral polyoxometalate (POM) clearly present a chiroptical switching process. The electronic transition and the bond-length alternation studies show that the chirality transfer from chiral carbon atom to POM cage increases as the polyanion is reduced. Simultaneously, the static first hyperpolarizability of studied chiral POM quadrupled from the oxidized state to the 1e-reduced state, and is further doubled to the 2e-reduced state, which is mainly due to the increasing electronic-dipole-allowed d–d charge transfer transitions in the POM cage. This work firstly reproduces the ECD spectrum of chiral POM with high accuracy and proves the possibility for confirming the molecular conformations of flexible chiral POMs in solution by the aid of ECD calculations. Most importantly, a sensitive diplex switch based on a chiral POM is predicted in theory, which may aid the design of novel POM-based switches.
Co-reporter:Lian-Jie Li, Chao Qin, Xin-Long Wang, Shuang Wang, Liang Zhao, Guang-Sheng Yang, Hai-Ning Wang, Gang Yuan, Kui-Zhan Shao and Zhong-Min Su
CrystEngComm 2012 vol. 14(Issue 1) pp:124-130
Publication Date(Web):14 Oct 2011
DOI:10.1039/C1CE05810J
Reactions of 1,4-bis(pyridin-4-ylmethoxy)benzene (L) and zinc acetate salts with different multi-caboxylate acids: 1,4-benzenedicarboxylic (p-H2BDC), isophthalic acid (m-H2BDC) and benzene-1,3,5-tricarboxylic acid (H3BTC) yield four entangled structures and one 3D supramolecular framework depending on pH variation. The structures of compounds 1–5 were elucidated by single crystal X-ray diffraction. Topology analysis revealed that the entangled structures of compounds 1–4 covered a range of poly-threading, self-penetrating and interpenetrating coordination. Interestingly, the poly-threaded arrays with cyclohexane-like windows in chair conformation in compound 2 was firstly observed. What's more, compound 4 exhibited a rare eight-connected self-penetrating network based on trinuclear zinc clusters as nodes with 42464 topology. Photoluminescent properties and thermogravimetric analyses for 1–5, were also investigated in detail.
Co-reporter:Guochun Yang, Yanling Si and Zhongmin Su
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 42) pp:8418-8425
Publication Date(Web):10 Sep 2012
DOI:10.1039/C2OB26374B
We have investigated the chiroptical, linear, and second-order nonlinear optical (NLO) properties of seven binaphthol derivatives and elucidated structure–property relationships from the micromechanism for the first time. The excitation energies, oscillator strengths, and rotational strengths of the 150 lowest energy electron excitations for the most stable conformers have been calculated at TDB3LYP/cc-pVDZ level of theory. The experimental UV–vis absorption energies were reproduced well by our calculations. The simulated circular dichroism (CD) spectra and calculated optical rotation (OR) values are in reasonable agreement with experimental ones. These results demonstrate that TDDFT calculations can not only describe the electron transition property but also can be used to assign the absolute configurations (ACs) of binaphthol derivatives with high confidence. Whereas OR values are more sensitive to the molecular structures than CD spectra. The electron transition property and chiroptical origin have been assigned and analyzed. These derivatives possess remarkably large molecular first hyperpolarizabilities, especially compound 7 which has a value of 241.65 × 10−30 esu. This value is about 60 times as large as that of highly π-delocalized phenyliminomethyl ferrocene complex. Moreover, compound 6 exhibits pronounced different second-order NLO response values from neutral state to the two cationic states (62+2+2+ and 64+4+4+), which indicates that this compound could act as a potential NLO switch material. The cooperativity of intramolecular charge transfer of the studied compounds was also discussed.
Co-reporter:Ji Zhang, Yu-He Kan, Hai-Bin Li, Yun Geng, Yong Wu, Zhong-Min Su
Dyes and Pigments 2012 Volume 95(Issue 2) pp:313-321
Publication Date(Web):November 2012
DOI:10.1016/j.dyepig.2012.05.020
Three organic donor-π-acceptor dyes 1-3 used for dye sensitized solar cells (DSSCs) with difference only in π spacer sequence were investigated via density functional theory (DFT) and time-dependent DFT calculations to shed light on how the π conjugation order influence the performance of the dyes. Key parameters in close connection with the open-circuit photovoltage (Voc) and the short-circuit current density (Jsc), including (i) light harvesting efficiency (LHE); (ii) injection driving force (ΔGinject.); (iii) reorganization energy (λreorg); (iv) number of photoinjected electrons (nc,inj); (v) the extent of charge recombination, and (vi) vertical dipole moment (μnormal), were discussed. The theoretical results reveal that compared with dyes 1 and 2, dye 3 has the largest Voc due to its largest μnormal and the slowest charge recombination. This conclusion is in good accordance with the experimental results and the theoretical criteria we used would be useful to design and fast screen other organic dyes.Graphical abstractConjugation order effects on the performance of the D-π-A dyes used in DSSCs: A DFT and TDDFT analysisHighlights► Conjugation order effects on DSSCs' efficiency were studied by DFT calculations. ► A set of key parameters affecting the efficiency of the cell have been discussed. ► Charge recombination is discussed through dye–I2 interaction. ► Dye 3 has the largest Voc due to the largest μnormal and slowest charge recombination.
Co-reporter:Yu-Ai Duan, Yun Geng, Hai-Bin Li, Xiao-Dan Tang, Jun-Ling Jin, Zhong-Min Su
Organic Electronics 2012 Volume 13(Issue 7) pp:1213-1222
Publication Date(Web):July 2012
DOI:10.1016/j.orgel.2012.03.026
When the oligothiophene is substituted by dicyanovinyl (DCV) or tricyanovinyl (TCV) group, how does its transport property change? Here, we will mainly focus on exploring the influence on charge transport properties of introducing a strong electron-withdrawing DCV/TCV group to the thiophene units within Marcus–Levich–Jortner formalism at the level of density functional theory. The results show that the introduction of cyanovinyl-substituents improves the molecular π-stacking, decreases the frontier molecular orbital energy levels and reorganization energies, and increases the transfer integrals and mobilities, comparing with their parent thiophene molecules. It is interesting to find the phenomenon that enriching intermolecular interactions can be favorable for controlling the transport channel and thus get high mobility, which would be shown by the angular resolution anisotropic mobilities analysis. Besides, the simulated packing motifs of dimers for 3a and 3b without crystal structures reported indicate that their packing may form the slip π–π stacking, and that 3b may be a good ambipolar material. In a word, compared with corresponding thiophene analogues and tetracyanoquinodimethane, these compounds may become the candidates for the n-type or ambipolar organic semiconductor materials.Graphical abstractHighlights► Cyanovinyl can improve and tune the type and size of mobility of oligothiophene. ► Enriching intermolecular interaction is conducive to obtain higher mobility. ► Distinct and anisotropic mobility is due to different intermolecular interaction.
Co-reporter:Muhammad Ramzan Saeed Ashraf Janjua;Muhammad Amin;Muhammad Ali;Beenish Bashir;Muhammad Usman Khan;Muhammad Awais Iqbal;Wei Guan;Likai Yan
European Journal of Inorganic Chemistry 2012 Volume 2012( Issue 4) pp:705-711
Publication Date(Web):
DOI:10.1002/ejic.201101092
Abstract
DFT calculations were carried out in order to study the second-order nonlinear optical (NLO) response of a series of proposed 2D polyoxometalate-based terpyridine-substituted compounds. These compounds can be formulated as [Mo6O17{N4C25H16(X)2}{N4C25H16(X)2}]2– (X = H, F, Cl, Br, I, CF3, or CN), which has a wedge Λ-shaped acceptor––π-conjugated bridge––donor––π-conjugated bridge––acceptor (A-π-D-π-A) configuration. The calculations showed that these compounds possess significantly large molecular second-order polarizabilities that range from approximately 1000 × 10–30 to 4300 × 10–30 esu. The combination of trifluoromethyl (CF3) and cyanide (CN) groups at the end of the terpyridine ligand strengthens the bridge conjugation, which is useful for the enhancement of the NLO response. In addition, the greatest contributions to the βvec values are dervied from the charge transfer (CT) from the Mo≡N bond and the organoimido ligand to the terpyridine-substituted segments. This report demonstrates that various combinations of the acceptor(s) remarkably affect the second-order NLO response. The electronic transitions to the crucial excited states indicated that the y polarized transition contributed to the off-diagonal second-order polarizabiliy tensor (βzyy) and that the z polarized transition accounted for the diagonal second-order polarizabiliy tensor (βzzz). Thus, itsteered towards in-plane nonlinear anisotropy (u = βzyy/βzzz) along with a good 2D second-order NLO response. These compounds can be used as good 2D second-order NLO materials from the point of view of their large β values.
Co-reporter:Yan Liu;Guochun Yang;Shiling Sun ;Zhongmin Su
Chinese Journal of Chemistry 2012 Volume 30( Issue 10) pp:2349-2355
Publication Date(Web):
DOI:10.1002/cjoc.201200514
Abstract
The structures and second-order nonlinear optical (NLO) properties of a series of chlorobenzyl-o-carboranes derivatives (1–12) containing different push-pull groups have been studied by density functional theory (DFT) calculation. Our theoretical calculations show that the static first hyperpolarizability (βtot) values gradually increase with increasing the π-conjugation length and the strength of electron donor group. Especially, compound 12 exhibits the largest βtot (62.404×10−30 esu) by introducing tetrathiafulvalene (TTF), which is about 76 times larger than that of compound 1 containing aryl. This means that the appropriate structural modification can substantially increase the first hyperpolarizabilities of the studied compounds. For the sake of understanding the origin of these large NLO responses, the frontier molecular orbitals (FMOs), electron density difference maps (EDDMs), orbital energy and electronic transition energy of the studied compounds are analyzed. According to the two-state model, the lower transition energy plays an important role in increasing the first hyperpolarizability values. This study may evoke possible ways to design preferable NLO materials.
Co-reporter:Gang Yuan, Kui-Zhan Shao, Lei Chen, Xin-Xin Liu, Zhong-Min Su, Jian-Fang Ma
Journal of Solid State Chemistry 2012 Volume 196() pp:87-92
Publication Date(Web):December 2012
DOI:10.1016/j.jssc.2012.06.016
Three new polymers, [Cd(L)2(H2O)2]n (1), [Cd3(L)2(μ3-OH)2(μ2-Cl)2(H2O)2]n (2), {[Cd2(L)2(nic)2(H2O)2]·H2O}n (3) (HL=5-(4-((1H-1,2,4-triazol-1-yl)methyl)phenyl)-1H-tetrazole, Hnic=nicotinic acid) have been prepared and structurally characterized. Compounds 1 and 2 display 2D monomolecular layers built by the inter-linking single helical chains and L− ligands connecting chain-like [Cd(μ3-OH)(μ2-Cl)]n secondary building units, respectively. Compound 3 is constructed from the mixed ligands and possesses a (3,4)-connected framework with (4·82)(4·82·103) topology. Moreover, the fluorescent properties of HL ligand and compounds 1–3 are also been investigated.Graphical abstractThree new coordination polymers based on the semi-rigid multidentate N-donor ligand have been successfully synthesized by hydrothermal reaction. Complexes 1 and 2 exhibit the 2D layers formed by inter-linking single helices and L− anions bridging 1D chain-like SBUs, respectively. Complex 3 is buit by L− and assistant nic− ligands connecting metal centers and possesses a (3,4)-connected framework with (4×82)(4×82×103) topology. Moreover, these complexes display fluorescent properties indicating that they may have potential applications as optical materialsHighlights► Three Cd-compounds were prepared from semi-rigid HL ligand with different N-containing groups. ► They exhibit diverse structures from 2D monomolecular layer to 3D covalent framework. ► The HL ligands displayed various coordination modes under different reaction conditions. ► These compounds exhibit good luminescent properties.
Co-reporter:Lian-Jie Li, Xin-Long Wang, Liang Zhao, Kui-Zhan Shao, Hai-Ning Wang, Gang Yuan, Zhong-Min Su
Inorganic Chemistry Communications 2012 Volume 15() pp:288-291
Publication Date(Web):January 2012
DOI:10.1016/j.inoche.2011.11.004
Two metal–organic frameworks, namely, [Zn(L)(phen)(H2O)]n (1) and [Cd(L)(phen)]n (2), where H2L = 2,2′-(naphthalene-1,5-diylbis(oxy))diacetic acid and phen = 1,10-phenanthroline, have been synthesized under hydrothermal conditions and characterized by single crystal X-ray diffraction analyses, powder X-ray diffraction and thermogravimetric analysis. Compound 1 can be described as a 3D supramolecular architecture formed via the strong π–π interactions. While, compound 2, obtained by changing the metal ions, exhibits a 3D metal–organic frameworks of PtS topology with the square window (the dimensions is 11.454 × 25.847 Å). The different structures between 1 and 2 indicate that metal ions have significant effects on the final complexes. Moreover, compounds 1 and 2 show good luminescent properties in the solid state at room temperature.Compound 1 shows a 1D chain structure. Compound 2 exhibits the 3D metal-organic frameworks of PtS topology. The different structures between 1 and 2 indicate that metal ions have significant effects on the final complexes.Highlights► Compound 1 shows a 3D supramolecular architecture formed via the strong π–π interactions. ► Compound 2 exhibits a 3D network of PtS topology. ► The different structures of 1 and 2 indicate that metal ions have significant effects on the final complexes.
Co-reporter:Lian-Jie Li, Xin-Long Wang, Kui-Zhan Shao, Zhong-Min Su, Qiang Fu
Inorganica Chimica Acta 2012 Volume 392() pp:77-83
Publication Date(Web):30 September 2012
DOI:10.1016/j.ica.2012.05.037
Three coordination polymers, namely, [Zn2(ndd)2(2,2′-bpy)2·2H2O]n (1) [Cd(ndd)(2,2′-bpy)]n (2), and [Cd(ndd)(1,4-pyb)]n (3), where H2ndd = 2,2′-(naphthalene-1,5-diylbis(oxy))diacetic acid, 1,4-pyb = 1,4-bis(pyridin-4-ylmethoxy)benzene and 2,2′-bpy = 2,2′-bipyridine, have been synthesized under hydrothermal conditions and characterized by single-crystal X-ray diffraction analyses, powder X-ray diffraction and thermogravimetric analysis. Compound 1 can be considered as a 1D chain, which is further linked through the strongly multiple π–π interactions into a 3D supramolecular structure. By changing the metal ions, compound 2 is the 2D square structure (10.96 × 15.20 Å) with the 44·62 topology. By changing the auxiliary ligand, compound 3 presents a new (4,5)-connected 3D network with the point symbol of (42·65·83)2·(66). Furthermore, compounds 1–3 show good fluorescence properties in the solid state at room temperature.Graphical abstractCompound 1 can be considered as 1D chain structure, which is further linked through the strongly multiple π–π interactions into 3D supramolecular structure. By changing the metal ions, compound 2 is the 2D square-shaped with the 44·62 topology. By changing the auxiliary ligand, compound 3 presents a new (4,5)-connected 3D network with the point symbol of (42·65·83)2·(66).Highlights► Compound 1 can be considered as 1D chain structure. ► Compound 2 is the 2D network with the 44·62 topology. ► Compound 3 presents a new (4,5)-connected 3D network.
Co-reporter:Xin-Xin Liu, Xin-Long Wang, Guang-Sheng Yang, Gang Yuan, Lian-Jie Li, Kui-Zhan Shao, Zhong-Min Su, Hai-Ming Xie
Inorganic Chemistry Communications 2012 Volume 23() pp:21-24
Publication Date(Web):September 2012
DOI:10.1016/j.inoche.2012.05.035
A new two-dimensional (2D) inorganic–organic hybrid compound H2{[Ni6(μ3-OH)3(H2O)4(en)(CH3COO)(IN)(B-PW9O34)]}·2H2O (en = ethylenediamine; HIN = isonicotinic acid) (1) has been hydrothermally synthesized and characterized by IR spectrum, electrochemical measurements, diffuse reflectance UV–vis spectrum and single-crystal X-ray diffraction. Compound 1 contains a nanometer-scale secondary building unit (SBU) [Ni6(μ3-OH)3(B-α-PW9O34)]2 ({Ni6PW9}2), in which two {Ni6PW9} units are linked together through Ni–O–W linkages. The adjacent SBUs are connected by IN and forming a 2D layer network. 1 exhibits semiconducting properties with the band gap Eg at 1.98 eV.Compound 1 contains a nanometer-scale secondary building unit (SBU) {Ni6PW9}2, in which two {Ni6PW9} units are linked together through Ni–O–W linkages. The adjacent SBUs are connected by IN and forming a 2D layer network.Highlights► We synthesized an inorganic–organic hybrid compound (1), which contains a nanometer-scale SBU {Ni6PW9}2. ► In 1, the adjacent SBUs are connected by IN (HIN = isonicotinic acid) and forming a 2D layer network. ► Compound 1 exhibits semiconducting properties with the band gap Eg at 1.98 eV.
Co-reporter:Lian-Jie Li, Xin-Long Wang, Kui-Zhan Shao, Zhong-Min Su
Inorganic Chemistry Communications 2012 Volume 26() pp:42-45
Publication Date(Web):December 2012
DOI:10.1016/j.inoche.2012.09.005
One novel metal–organic framework, namely, [Cd2(ndd)2(tib)2·H2O]n (1), [H2ndd = 2,2′-(naphthalene-1,5-diylbis(oxy))diacetic acid, tib = 1,3,5-tri(1H-imidazol-1-yl)benzene] has been prepared under hydrothermal condition and characterized by elemental analyses, infrared spectroscopy, powder X-ray diffraction and single-crystal X-ray diffraction analyses. Compound 1 presents a 3D 3-fold interpenetrated (3,5)-connected network with the point symbol of (42·65·83)(42·6) topology. Furthermore, the thermal stabilities and luminescent properties of compound 1 were studied.Compound 1 presents a 3D 3-fold interpenetrated (3,5)-connected network with the point symbol of (42·65·83)(42·6) topology. Furthermore, the thermal stabilities and photoluminescent properties of compound 1 were studied.Highlights► Compound 1 presents a 3D 3-fold interpenetrated metal–organic framework. ► Compound 1 shows (3,5)-connected network with the point symbol (42·65·83)(42·6) topology. ► Compound 1 shows good fluorescence properties in the solid state at room temperature.
Co-reporter:Guo-Dong Feng, Luan Jiang, Zong-Xiao Li, Qiang Chen, Xin-Long Wang, Kui-Zhan Shao, Chun-Yi Sun, Lian-Jie Li, Ji-Hong Yu, Zhong-Min Su
Inorganic Chemistry Communications 2012 Volume 24() pp:247-253
Publication Date(Web):October 2012
DOI:10.1016/j.inoche.2012.05.029
Three coordination polymers, i.e. [Co(HL)(HBDC)]n (1), [Co(HL)(BDC)0.5]n (2) and {[Zn2(H2L)(BTC)2](H4L)(H2O)2}n (3) (L = 2, 2′-(ethanediyl)bis(1H-benzimidazole), H2BDC = 1, 4-benzenedicarboxylic acid, H3BTC = 1, 3, 5-benzene tricarboxylic acid), have been synthesized under hydrothermal conditions based on bis(2-benzimidazole) and two multi-dentate carboxylate ligands, with different metal ions such as ZnII and CoII. The structures of these coordination polymers have been determined by X-ray single‐crystal diffraction. The diverse coordination modes of the H2L ligand and the varying geometries of multi-dentate carboxylate ligands play an important role in the construction of coordination polymers. Various assembly patterns have been observed, including one-dimensional (1D) chain (1), two-dimensional (2D) 3-connected network structure with honeycomb topology (2), and three-dimensional (3D) 3,4-connected porous structure with (63)(65,8) topology (3). Thermal analyses of the three polymers have been performed and discussed. Furthermore, strong room temperature photoluminescence of compound 3 was observed.Three new metal–organic frameworks based on the bis(2-benzimidazole), [Co(HL)(HBDC)]n (1), [Co(HL)(BDC)0.5]n (2) and {[Zn2(H2L)(BTC)2](H4L)(H2O)2}n (3), have been successfully synthesized under hydrothermal conditions and characterized by X-ray single-crystal diffractions. In addition, strong photoluminescence at room temperature of compound 3 was observed at room temperature.Highlights► Compound 1 shows a 1D chain architecture with L ligand hanging outside the chain structure. ► Compound 2 exhibits a 2D extra-large 32-membered ring network with honeycomb topology. ► Compound 3 exhibits a 3D 3, 4-connected porous structure with (63)(65,8) topology.
Co-reporter:Hong-Liang Xu;Rong-Lin Zhong;Li-Kai Yan
Journal of Physical Organic Chemistry 2012 Volume 25( Issue 2) pp:176-180
Publication Date(Web):
DOI:10.1002/poc.1914
Six N-substituted [n]cyclacene (n = 5, 6, 7,…,10) molecules were designed to study the relationship between the structure and first hyperpolarizability. Their static first hyperpolarizabilities (β0) were obtained by MP2/6-31 + g(d) level. Two interesting relationships between the β0 value and the structure have been found: (1) The β0 value increases with the increase of the number n when n is odd: 3155 ([5]cyclacene) < 48,905 ([7]cyclacene) < < 393,444 ([9]cyclacene), and when n is even: 357,620 ([6]cyclacene) < 618,608 ([8]cyclacene) < 3,513,644 a.u. ([10]cyclacene). (2) The β0 values (in the range of 357,620 ~ 3,513,644 a.u.) of the N-substituted [n]cyclacene (when n is odd) are much larger (in the range of 3155~393,444 a.u.) than that of the N-substituted [n]cyclacene (when n is even). Furthermore, their frequency-dependent β (−2ω; ω, ω) and β (−ω; ω, 0) (ω = 0.005, 0.01, and 0.0239 a.u.) were also estimated by Møller–Plesset perturbation/6-31 + g(d) level. Among the frequency-dependent β (ω), [10]cyclacene has the largest β (−ω; ω, 0) and β (−2ω; ω, ω) to be 1.2 × 108 (ω = 0.01) and 2.9 × 107 a.u. (ω = 0.005 a.u.), which are much larger than the static β0 = 3.5 × 106 a.u. by 34 and 8 times. Our present work may offer a new idea in the design of high-performance tubiform nonlinear optical materials. Copyright © 2011 John Wiley & Sons, Ltd.
Co-reporter:Zhong-Ling Lang, Wei Guan, Zhi-Jian Wu, Li-Kai Yan, Zhong-Min Su
Computational and Theoretical Chemistry 2012 Volume 999() pp:66-73
Publication Date(Web):1 November 2012
DOI:10.1016/j.comptc.2012.08.015
The formation mechanism is a fundamental scientific issue for polyoxometalate chemistry. The structural characterization and the thermodynamic behavior of various possible intermediates in the formation of α-Keggin-type [PW12O40]3− anion in aqueous solution were analyzed by density functional method. Based on two proposed mechanisms (order of reaction with heteroatom), thermodynamic analysis indicates that [HPO4]2−, [WO3(OH)]−, [W2O7]2−, [W3O10]2−, [W4O13]2−, [W5O16]2−, and [PW2O9]− will be mainly involved in the polymerization processes of [PW12O40]3− anion. The starting reaction from isodimer has been determined through transition state search for the initial steps. Heteroatom has been introduced in the heterotrimer [PW2O9]− at the second step which experiences almost the same barrier with isotrimer [W3O10]2−. From geometric topology and thermodynamics, the defect structures are more suitable for building blocks.Graphical abstractPresent DFT investigations provide the main polymerized channels for [PW12O40]3− formation. Thermodynamic analysis indicates that [HPO4]2−, [WO3(OH)]−, [W2O7]2−, [W3O10]2−, [W4O13]2−, [W5O16]2−, and [PW2O9]− will be mainly involved in the polymerization processes.Highlights► We study theoretically formation mechanism of α-Keggin-type [PW12O40]3− anion. ► We obtain various building units in the polymerization processes of PW12 anion. ► The starting reaction has been determined through transition state search. ► Heteroatom (P) has been introduced in the heterotrimer [PW2O9]− at the second step. ► Competition between hetero- and iso-polytungstate polymerizations is essential.
Co-reporter:Lili Chen, Binbin Wang, Liming Zhang, Dongxia Zhu, Peng Li, Zhongmin Su, Bin Li
Solid-State Electronics 2012 Volume 69() pp:67-71
Publication Date(Web):March 2012
DOI:10.1016/j.sse.2011.12.005
The photoluminescence (PL) and electroluminescence (EL) properties of a trimetallic dendritic europium (III) complex containing three metal cores as branching centers, tris(dibenzoylmethanato) (1,3,5-tris[2-(2′-pyridyl)benzimidazoly]methyl-benzene)-europium (III) (Eu3(DBM)9(TMMB)), have been investigated and reported. Red and white emissions have been acquired in the four EL devices of device A–D. Characteristic red emission peaking at 613 nm with four shoulder bands due to the 5D0 → 7Fj (j = 0–4) transitions of Eu3(DBM)9(TMMB) is observed in devices A–C. Device D with a simple non-doping device structure showed a white emission band. Also, the optical and the electrochemical properties of Eu3(DBM)9(TMMB) have been characterized to possess a better understanding on the intramolecular energy transfer process. It is concluded that dendritic structure with three DBM ligands as “absorption antennas” facilitates the energy absorption and transportation of the complex.Graphical abstractHighlights► A trimetallic dendritic europium (III) complex has been synthesized. ► Characteristic red emission peaking at 613 nm is observed in devices. ► The intramolecular energy transfer process has taken place. ► DBM as “absorption antennas” facilitates the energy absorption and transportation.
Co-reporter:Jian-Ping Wang, Li-Kai Yan, Guo-Chun Yang, Wei Guan, Zhong-Min Su
Journal of Molecular Graphics and Modelling 2012 Volume 35() pp:49-56
Publication Date(Web):May 2012
DOI:10.1016/j.jmgm.2011.12.011
The chiroptical properties of bisarylimidos bearing o-alkoxy chain-substituted polyoxomolybdates [Mo6O17(2,2′-NC6H4OCnH2nOC6H4N)]2− [n = 4(2), 6(3±), 8(4)] were investigated using the time-dependent density functional method. The results showed that the studied chiral polyoxometalates (POMs) manifested similar absorption sites but displayed different shapes and magnitudes in their electronic circular dichroism (ECD) spectra. The ECD spectra of the studied chiral POMs originated from charge-transfer (CT) transitions from arylimido fragments to the POM cages and from oxygen atoms to the molybdenum atoms in the POM cages. The o-alkoxy chain served as a scaffold for generating chirality rather than contributing to the ECD spectrum of the studied POMs. The induced chiralities of the POM cages were defined by the CT transitions, which were completely localized on the POM cages. Furthermore, the long-range corrected CAM-B3LYP hybrid functional and a basis set that is larger than Lanl2DZ should be used for ECD calculations of chiral POMs. Our work establishes the use of computational studies to investigate the chiroptical properties of chiral POMs and provides theoretical interpretations.Graphical abstractHighlights► The geometry, electronic, and chiroptical properties of polyoxometalatocyclophanes are studied. ► The rigidity of POM in solution leads to the intramolecular chirality. ► The ECD spectra of POMs vary in signs and magnitudes, while show similar absorption sites. ► The o-alkoxy chain is scaffold for generating chirality, rather than contributes to ECD absorption. ► The induced chirality of POM cage is confirmed by electron transition assignment.
Co-reporter:Jian-Ping Wang, Li-Kai Yan, Wei Guan, Shi-Zheng Wen, Zhong-Min Su
Journal of Molecular Graphics and Modelling 2012 Volume 32() pp:1-8
Publication Date(Web):February 2012
DOI:10.1016/j.jmgm.2011.09.005
In this paper, density functional theory is used to investigate the linear optical and nonlinear optical (NLO) properties of a series of Λ-type chiral compounds composed of two Lindqvist-type polyoxometalates (POMs) linked by 1,1′-binaphthyl derivatives through arylimido. It shows that compound 1 which has two POMs on 6-6′-sites of 1,1′-binaphthyl possesses large static first hyperpolarizability and the strongest two-dimensional NLO response among studied compounds. The organic substituents on 2-2′-sites of 1,1′-binaphthyl twofold control the NLO responses of studied compounds. They act as electron acceptors or donors therefore suppress or enhance the NLO responses of studied compounds, and they restrain the torsion angles between two naphthyl rings at certain degrees which are inversely proportional to the NLO responses. Compound 6 with remarkable NLO response is obtained as ferrocene substitutes on 2-2′-sites of 1,1′-binaphthyl. Additionally, the electronic circular dichroism (ECD) spectra of studied compounds are simulated with CAM-B3LYP and B3LYP hybrid functionals. The results agree well with the experimental ECD spectra. The charge-transfer transitions from organic fragment to POM are responsible for the ECD differences between molecular hybrids and their precursors. It is confirmed that these Λ-type chiral compounds are potentially high-dimensional NLO materials and the structure–property relationship of these compounds is presented.Graphical abstractHighlights► The structure–property relationship of 1,1′-binaphthyl-hexamolybdate clusters is disclosed by linear and nonlinear optical (NLO) studies. ► These compounds are confirmed to be high-dimensional NLO materials. ► The substituents on 2,2′-sites of 1,1′-binaphthyl two-fold control the second-order NLO responses of these compounds. ► Among them, the ferrocene substituent gives the largest NLO response.
Co-reporter:Heng-Qing Wu;Shi-Ling Sun;Rong-Lin Zhong;Hong-Liang Xu
Journal of Molecular Modeling 2012 Volume 18( Issue 11) pp:4901-4907
Publication Date(Web):2012 November
DOI:10.1007/s00894-012-1478-0
In the present work, Li@porphyrins and their derivatives were designed in order to explore the effect of dehydrogenation/hydrogenation on linear and nonlinear optical properties. Their stable structures were obtained by the M06-2X method. Moreover, the M06-2X method showed that dehydrogenation/hydrogenation has greatly influences polarizabilities (α0 values) and hyperpolarizabilities (βtot and γtot values): α0 values ranged from 331 to 389 au, βtot values from 0 to 2465 au, and γtot values from −21.2 × 104 to 21.4 × 104 au. This new knowledge of the effect of dehydrogenation/hydrogenation on nonlinear optical properties may prove beneficial to the design and development of high-performance porphyrin materials.
Co-reporter:Jinlian Li;Jia Fu;Jianping Wang;Donghua Hu;Zhongmin Su
Medicinal Chemistry Research 2012 Volume 21( Issue 12) pp:4010-4016
Publication Date(Web):2012 December
DOI:10.1007/s00044-011-9927-3
Human DNA G-quadruplex has been an attractive drug target for cancer therapeutic intervention. Especially the G-quadruplex groove has been paid growing attention to because of its selectivity. In this work, molecular dynamics (MD) simulations of the complexes of parallel G-quadruplexes ([d(TGGGGT)]4 and [d(GGGGGG)]4) with distamycin A (Dist-A) dimmers were performed. The characteristic of drug binding in G-quadruplex grooves was investigated. The simulations reveal the propensity of Dist-A dimmer toward the end of the groove. The electronic properties of three successive G-tetrads are estimated by density functional theory (DFT) method at the B3LYP/6-31G (d, p) level. The results show that the N3 atoms in two terminal G-tetrads possess more negative charges than it in the middle G-tetrad, which is one reason for driving cation ligand toward the end of groove. This study provides the factors of affecting the binding of drugs in the G-quadruplex groove and is of significance for drug design based on the structure of G-quadruplex groove.
Co-reporter:Fang Zhang 张芳;Jin Huang;Hao Zhang
Journal of Wuhan University of Technology-Mater. Sci. Ed. 2012 Volume 27( Issue 4) pp:608-614
Publication Date(Web):2012 August
DOI:10.1007/s11595-012-0514-3
Polyurethanes/multi-walled carbon nanotube (PU/CNT) composites were prepared with a help of ultrasonically dispersing CNT in the traditional procedure of synthesizing polyurethane. In this case, the various loading levels, sizes and surface-modified groups were considered to regulate the mechanical performances of the PU/CNT nanocomposites. Moreover, the structure and mechanical properties of all the PU/CNT nanocomposites were investigated by attenuated total reflection-Fourier transform infrared spectroscopy, dynamic mechanical analysis, scanning electron microscope, transmission electron microscope, and tensile testing. The experimental results showed that a moderate loading-level of 0.1wt% and a diameter of 10–15 nm for CNT could produce the maximum tensile strength and elongation while it was worth noting that the surface carboxylation of CNT could further enhance the tensile strength and elongation of the PU/CNT nanocomposites.
Co-reporter:Wei Tao, Yu-He Kan, Shui-Xing Wu, Hai-Bin Li, Li-Kai Yan, Shi-Ling Sun, Zhong-Min Su
Journal of Molecular Graphics and Modelling 2012 Volume 33() pp:26-34
Publication Date(Web):March 2012
DOI:10.1016/j.jmgm.2011.10.008
The vertical excitation energies of tetrathiafulvalene (TTF)-annulated zinc porphyrazine (ZnPzTTF) were investigated using time-dependent density functional theory (TDDFT) calculations and compared to the experimental UV–vis spectra. To examine the effects of the aza substitutions and TTF groups on the molecular properties, zinc complexes of porphyrin (ZnP), porphyrazine (ZnPz) and tetraTTF-annulated porphyrin (ZnPTTF) were also selected for comparison. It was shown that numerous electronic transitions with TTF-to-porphyrin or porphyrazine charge transfer character exist and the Q band of ZnPzTTF is dominated by TTF-to-porphyrazine charge transfer transition mixed with porphyrazine core unit itself except for classic porphyrazine π → π* transitions. The Q band of ZnPzTTF mixes with other configurations, which breaks down the Gouterman's classic four-orbital model for the spectral interpretation. The data suggest that TDDFT/SAOP performs best for Q and B bands of ZnPzTTF with the maximum error in excitation energy being 0.17 eV. The CAM-B3LYP, ωB97XD and M06-2X calculations qualitatively predict that the low-lying electronic transitions of ZnPzTTF with TTF-to-porphyrazine charge transfer character located below the Q band. The broad and intense red-shifted Q band suggests that ZnPzTTF can be a candidate for dye-sensitized solar cells.Graphical abstractHighlights► The Q band of ZnPzTTF is dominated by TTF-to-porphyrazine charge transfer mixed with porphyrazine core unit itself except for classic porphyrazine π → π* transitions. ► ZnPzTTF has a broad and red-shifted Q band with strong intensity, and so has potential as a sensitizer for dye-sensitized solar cells. ► TDDFT/SAOP performs best for the Q and B bands of ZnPzTTF in terms of agreement with experimental values. ► The low-lying CT states of ZnPzTTF occur below the Q band.
Co-reporter:Shizheng Wen, Wei Guan, Zhongmin Su, Likai Yan, Stefano Sanvito
Journal of Molecular Graphics and Modelling 2012 Volume 38() pp:220-225
Publication Date(Web):September 2012
DOI:10.1016/j.jmgm.2012.05.005
The transport properties of Lindqvist type-polyoxometalates H2M6O19 (M = Mo, W) sandwiched between carbon nanotube electrodes are investigated by using density functional theory combined with the non-equilibrium Green's function method. It is found that the precise position of the protonation has little effect on the transport properties of H2Mo6O19, as it is established by investigating two different geometries. Furthermore we have discovered that H2Mo6O19 and H2W6O19 display similar conduction profiles with the main conduction mechanism being quantum tunneling. With a large energy gap and robust structural stability these molecules appear to be good candidate for high bias applications.Graphical abstractThe transport properties of Lindqvist type-polyoxometalates H2M6O19 (M = Mo, W) sandwiched between carbon nanotube electrodes are investigated using density functional theory combined with the non-equilibrium Green's function method. The H2Mo6O19 and H2W6O19 display similar conduction profiles with quantum tunneling as main conduction mechanism.Highlights► The transport properties of Lindqvist type-polyoxometalates based molecular junctions are simulated for the first time. ► We conclude that the main conduction mechanism of the molecules is quantum tunneling with similar conduction profiles. ► It finds that the precise position of the protonation has little effect on the transport properties.
Co-reporter:Sha Cong;LiKai Yan;Ping Song;Wei Guan;ZhongMin Su
Science Bulletin 2012 Volume 57( Issue 9) pp:976-982
Publication Date(Web):2012 March
DOI:10.1007/s11434-011-4971-4
The electronic properties and stabilities of five [Nb2W4O18OCH3]3− isomers have been investigated using a density functional theory method. The results show that the isomer with the methoxy group occupying a bridging position between two tungsten atoms (two tungsten atoms in the plane that contains two niobium atoms) in the [Nb2W4O18OCH3]3− framework is the most stable isomer in acetonitrile. The stability of the one-electron-reduced isomers changes little. The most stable one-electron-reduced isomer has the methoxy group occupying a bridging position between niobium atoms in the [Nb2W4O18OCH3]4− framework. The M-Ob (M = Nb, W; b denotes bridging) bond lengths in anions in which the metal atoms are connected by a methoxy group are longer than those in [Nb2W4O19]4−. The highest occupied molecular orbitals (HOMO) in [Nb2W4O19]4− mainly delocalize over the bridging oxygen atoms of two niobium atoms and two tungsten atoms located in the equatorial plane, and the bridging oxygen atoms on the axial surface. The lowest unoccupied molecular orbitals (LUMO) of [Nb2W4O19]4− are mainly concentrated on the tungsten atoms and antibonding oxygen atoms. Methoxy substitution modifies the electronic properties of the [Nb2W4O18OCH3]3− isomers. The HOMOs in the five isomers formally delocalize over the bridging oxygen atoms, which are distant from the surface containing the methoxy group and four metal atoms. The LUMOs delocalize over the d-shells of the four metal atoms that are close to the methoxy group, and the p-orbitals of oxygen. One-electron reduction occurred at the tungsten atoms, not the niobium atoms.
Co-reporter:Xing Meng, Hai-Ning Wang, Xin-Long Wang, Guang-Sheng Yang, Shuang Wang, Kui-Zhan Shao, Zhong-Min Su
Inorganica Chimica Acta 2012 390() pp: 135-142
Publication Date(Web):
DOI:10.1016/j.ica.2012.04.011
Co-reporter:Yan-Qing Jiao, Chao Qin, Chun-Yi Sun, Kui-Zhan Shao, Peng-Jun Liu, Peng Huang, Kun Zhou, Zhong-Min Su
Inorganic Chemistry Communications 2012 20() pp: 273-276
Publication Date(Web):
DOI:10.1016/j.inoche.2012.03.025
Co-reporter:Hong-Liang Xu, Cui-Cui Zhang, Shi-Ling Sun, and Zhong-Min Su
Organometallics 2012 Volume 31(Issue 12) pp:4409-4414
Publication Date(Web):June 11, 2012
DOI:10.1021/om2012858
Assembly with large nonlinear optical response of the novel sandwich-like supermolecules CpLi-C60 (Cp = cyclopentadienide) has been investigated for two pathways. First, the half-sandwich Li salt CpLi is formed, and then the sandwich-like supermolecules CpLi-C60(trans) and CpLi-C60(cis) are obtained by affixing the C60 to CpLi. The results indicate that the βtot values of CpLi-C60(trans) (14 093 au) and CpLi-C60(cis) (15 560 au) increase sharply to about 22 times and 25 times greater than that of CpLi (624 au), respectively. This shows that the whole supermolecule is lighted by C60. Second, a Li atom is added to C60 to form the half-sandwich-like Li-C60, and then we attach the Cp to Li-C60 to form CpLi-C60(trans) and CpLi-C60(cis). The results indicate that the βtot values of CpLi-C60(trans) and CpLi-C60(cis) increase sharply to about 10 times and 11 times greater than that of Li-C60 (1387 au), respectively. Then, the two supermolecule Li salts are expected to be molecular switches by rotation of Cp. Notably, the interaction energies (Eint) of CpLi-C60(trans) and CpLi-C60(cis) are about −101 and −100 kcal/mol, which implies that they are more stable than Li-C60 (−38 kcal/mol). The present work proposes a new strategy for designing high-performance nonlinear optical materials and provides a new way to assemble sandwich supermolecule salts.
Co-reporter:Guo-Gang Shan, Hai-Bin Li, Zhong-Cheng Mu, Dong-Xia Zhu, Zhong-Min Su, Yi Liao
Journal of Organometallic Chemistry 2012 702() pp: 27-35
Publication Date(Web):
DOI:10.1016/j.jorganchem.2011.12.020
Co-reporter:Yuan-Mei Sang, Li-Kai Yan, Jian-Ping Wang, and Zhong-Min Su
The Journal of Physical Chemistry A 2012 Volume 116(Issue 16) pp:4152-4158
Publication Date(Web):March 23, 2012
DOI:10.1021/jp211262b
The UV/CD spectra of tin-bearing acetonyl-substituted Wells–Dawson polyoxotungstates α1- and α2-[P2W17O61{SnCH2CH2C(═O)}]6– were systematically investigated using the time-dependent density functional theory (TDDFT) method. The electronic circular dichroism (ECD) spectra were produced over the range of 3.3–5.8 eV. The calculated ECD spectra of the α1-R isomer were generally in agreement with the experimental spectra. The CAM-B3LYP hybrid functional was found to predict the excitation energies of tin-containing polyoxotungstates well. The fact that the UV/ECD spectra of α1-isomers are different from those of α2-isomers demonstrates the effect of the tin substitution site on the chiroptical properties of the studied isomers. The origins of the ECD bands are mainly ascribed to charge-transfer (CT) transitions from oxygen atoms to W atoms, from organic fragments to W atoms, or from the combination of two CT transitions. The results suggest that the organic fragment and polyoxometalate (POM) cage are chiroptical chromophores.
Co-reporter:Dr. Zhi-Ming Zhang;Dr. Shuang Yao;Dr. Yang-Guang Li;Xin-Bao Han; Zhong-Min Su;Zhi-Shu Wang; En-Bo Wang
Chemistry - A European Journal 2012 Volume 18( Issue 30) pp:9184-9188
Publication Date(Web):
DOI:10.1002/chem.201200555
Co-reporter:Rong-Lin Zhong;Dr. Hong-Liang Xu;Dr. Shi-Ling Sun; Yong-Qing Qiu ; Zhong-Min Su
Chemistry - A European Journal 2012 Volume 18( Issue 36) pp:11350-11355
Publication Date(Web):
DOI:10.1002/chem.201201570
Abstract
The unusual properties of species with excess electrons have attracted a lot of interest in recent years due to their wide applications in many promising fields. In this work, we find that the excess electron could be effectively bound by the B atoms of boron nitride nanotube (BNNT), which is inverted pyramidally distributed from B-rich edge to N-rich edge. Further, Li@B-BNNT and Li@N-BNNT are designed by doping the Li atom to the two edges of BNNT, respectively. Because of the interaction between the Li atom and BNNT, the 2s valence electron of Li becomes a loosely bound excess electron. Interestingly, the distribution of the excess electron in Li@N-BNNT is more diffuse and pyramidal from B-rich edge to N-rich edge, which is fascinating compared with Li@B-BNNT. Correspondingly, the transition energy of Li@N-BNNT is 0.99 eV, which is obviously smaller than 2.65 eV of Li@B-BNNT. As a result, the first hyperpolarizability (3.40×104 a.u.) of Li@N-BNNT is dramatically larger (25 times) than 1.35×103 a.u. of Li@B-BNNT. Significantly, we find that the pyramidal distribution of the excess electron is the key factor to determine the first hyperpolarizability, which reveals useful information for scientists to develop new electro-optic applications of BNNTs.
Co-reporter:Lu Li;Dr. Tingting Wang;Lingyu Zhang;Dr. Zhongmin Su;Dr. Chungang Wang;Dr. Rongshun Wang
Chemistry - A European Journal 2012 Volume 18( Issue 36) pp:11417-11422
Publication Date(Web):
DOI:10.1002/chem.201200791
Abstract
A method is reported for the first time for the selected-control, large-scale synthesis of monodispersed Fe3O4@C core–shell spheres, chains, and rings with tunable magnetic properties based on structural evolution from eccentric Fe2O3@poly(acrylic acid) core–shell nanoparticles. The Fe3O4@C core–shell spheres, chains, and rings were investigated as anode materials for lithium-ion batteries. Furthermore, a possible formation mechanism of Fe3O4@C core–shell chains and rings has also been proposed.
Co-reporter:Haiyan Liu;Dr. Tingting Wang;Lingyu Zhang;Lu Li;Dr. Y. Andrew Wang;Dr. Chungang Wang;Dr. Zhongmin Su
Chemistry - A European Journal 2012 Volume 18( Issue 12) pp:3745-3752
Publication Date(Web):
DOI:10.1002/chem.201103066
Abstract
The selected-control preparation of uniform core–shell and yolk–shell architectures, which combine the multiple functions of a superparamagnetic iron oxide (SPIO) core and europium-doped yttrium oxide (Y2O3:Eu) shell in a single material with tunable fluorescence and magnetic properties, has been successfully achieved by controlling the heat-treatment conditions. Furthermore, the shell thickness and interior cavity of SPIO@Y2O3:Eu core–shell and yolk–shell nanostructures can be precisely tuned. Importantly, as-prepared SPIO@Y2O3:Eu yolk–shell nanocapsules (NCs) modified with amino groups as cancer-cell fluorescence imaging agents are also demonstrated. To the best of our knowledge, this is the first report on the selected-control fabrication of uniform SPIO@Y2O3:Eu core–shell nanoparticles and yolk–shell NCs. The combined magnetic manipulation and optical monitoring of magnetic–fluorescent SPIO@Y2O3:Eu yolk–shell NCs will open up many exciting opportunities in dual imaging for targeted delivery and thermal therapy.
Co-reporter:Lingyu Zhang;Dr. Tingting Wang;Lei Yang;Cong Liu;Dr. Chungang Wang;Haiyan Liu;Dr. Y. Andrew Wang;Dr. Zhongmin Su
Chemistry - A European Journal 2012 Volume 18( Issue 39) pp:12512-12521
Publication Date(Web):
DOI:10.1002/chem.201200030
Abstract
Hollow mesoporous SiO2 (mSiO2) nanostructures with movable nanoparticles (NPs) as cores, so-called yolk-shell nanocapsules (NCs), have attracted great research interest. However, a highly efficient, simple and general way to produce yolk-mSiO2 shell NCs with tunable functional cores and shell compositions is still a great challenge. A facile, general and reproducible strategy has been developed for fabricating discrete, monodisperse and highly uniform yolk-shell NCs under mild conditions, composed of mSiO2 shells and diverse functional NP cores with different compositions and shapes. These NPs can be Fe3O4 NPs, gold nanorods (GNRs), and rare-earth upconversion NRs, endowing the yolk-mSiO2 shell NCs with magnetic, plasmonic, and upconversion fluorescent properties. In addition, multifunctional yolk-shell NCs with tunable interior hollow spaces and mSiO2 shell thickness can be precisely controlled. More importantly, fluorescent-magnetic-biotargeting multifunctional polyethyleneimine (PEI)-modified fluorescent Fe3O4@mSiO2 yolk-shell nanobioprobes as an example for simultaneous targeted fluorescence imaging and magnetically guided drug delivery to liver cancer cells is also demonstrated. This synthetic approach can be easily extended to the fabrication of multifunctional yolk@mSiO2 shell nanostructures that encapsulate various functional movable NP cores, which construct a potential platform for the simultaneous targeted delivery of drug/gene/DNA/siRNA and bio-imaging.
Co-reporter:Rong-Lin Zhong;Dr. Hong-Liang Xu;Dr. Shi-Ling Sun; Yong-Qing Qiu ; Zhong-Min Su
Chemistry - A European Journal 2012 Volume 18( Issue 36) pp:
Publication Date(Web):
DOI:10.1002/chem.201290154
Co-reporter:Jun-Ling Jin;Hai-Bin Li;Dr. Yun Geng;Yong Wu;Yu-Ai Duan ; Dr. Zhong-Min Su
ChemPhysChem 2012 Volume 13( Issue 16) pp:3714-3722
Publication Date(Web):
DOI:10.1002/cphc.201200384
Abstract
The geometric and electronic structures and photophysical properties of anilido-pyridine boron difluoride dyes 1–4, a series of scarce 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY) derivatives with large Stokes shift, are investigated by employing density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations to shed light on the origin of their large Stokes shifts. To this end, a suitable functional is first determined based on functional tests and a recently proposed index—the charge-transfer distance. It is found that PBE0 provides satisfactory overall results. An in-depth insight into Huang–Rhys (HR) factors, Wiberg bond indices, and transition density matrices is provided to scrutinize the geometric distortions and the character of excited states pertaining to absorption and emission. The results show that the pronounced geometric distortion due to the rotation of unlocked phenyl groups and intramolecular charge transfer are responsible for the large Stokes shift of 1 and 2, while 3 shows a relatively blue-shifted emission wavelength due to its mild geometric distortion upon photoemission, although it has a comparable energy gap to 1. Finally, compound 4, which is designed to realize the rare red emission in BODIPY derivatives, shows desirable and expected properties, such as high Stokes shift (4847 cm−1), red emission at 660 nm, and reasonable fluorescence efficiency. These properties give it great potential as an ideal emitter in organic light-emitting diodes. The theoretical results could complement and assist in the development of BODIPY-based dyes with both large Stokes shift and high quantum efficiency.
Co-reporter:Rong-Lin Zhong;Dr. Hong-Liang Xu; Zhong-Min Su; Zhi-Ru Li;Dr. Shi-Ling Sun; Yong-Qing Qiu
ChemPhysChem 2012 Volume 13( Issue 9) pp:
Publication Date(Web):
DOI:10.1002/cphc.201290042
Co-reporter:Rong-Lin Zhong;Dr. Hong-Liang Xu; Zhong-Min Su; Zhi-Ru Li;Dr. Shi-Ling Sun; Yong-Qing Qiu
ChemPhysChem 2012 Volume 13( Issue 9) pp:2349-2353
Publication Date(Web):
DOI:10.1002/cphc.201200213
Abstract
Much effort has been devoted to investigating the unusual properties of the π electrons in Möbius cyclacenes, which are localized in a special region. However, the localized π electrons are a disadvantage for applications in optoelectronics, because intramolecular charge transfer is limited. This raises the question of how the intramolecular charge transfer of a Möbius cyclacene with clearly localized π electrons can be enhanced. To this end, [8]Möbius cyclacene ([8]MC) is used as a conjugated bridge in a donor–π-conjugated bridge–acceptor (D–π–A) system, and NH2-6-[8]MC-10-NO2 exhibits a fascinating spiral charge-transfer transition character that results in a significant difference in dipole moments Δμ between the ground state and the crucial excited state. The Δμ value of 6.832 D for NH2-6-[8]MC-10-NO2 is clearly larger than that of 0.209 D for [8]MC. Correspondingly, the first hyperpolarizability of NH2-6-[8]MC-10-NO2 of 12 467 a.u. is dramatically larger than that of 261 a.u. for [8]MC. Thus, constructing a D–π–A framework is an effective strategy to induce greater spiral intramolecular charge transfer in MC although the π electrons are localized in a special region. This new insight into the properties of π electrons in Möbius cyclacenes may provide valuable information for their applications in optoelectronics.
Co-reporter:Guo-Gang Shan, Hai-Bin Li, Hong-Tao Cao, Dong-Xia Zhu, Zhong-Min Su, Yi Liao
Journal of Organometallic Chemistry 2012 713() pp: 20-26
Publication Date(Web):
DOI:10.1016/j.jorganchem.2012.04.018
Co-reporter:Dr. Dong-Lai Wang ;Dr. Hong-Liang Xu; Zhong-Min Su;Dr. Shabbir Muhammad;Dr. Dong-Yan Hou
ChemPhysChem 2012 Volume 13( Issue 5) pp:1232-1239
Publication Date(Web):
DOI:10.1002/cphc.201100774
Abstract
Drying-tube-shaped single-walled carbon nanotubes (SWCNTs) with multiple carbon ad-dimer (CD) defects are obtained from armchair (n,n,m) SWCNTs (n=4, 5, 6, 7, 8; m=7, 13). According to the isolated-pentagon rule (IPR) the drying-tube-shaped SWCNTs are unstable non-IPR species, and their hydrogenated, fluorinated, and chlorinated derivatives are investigated. Interestingly, chemisorptions of hydrogen, fluorine, and chlorine atoms on the drying tube-shaped SWCNTs are exothermic processes. Compared to the reaction energies for binding of H, F, and Cl atoms to perfect and Stone–Wales-defective armchair (5,5) nanotubes, binding of F with the multiply CD defective SWCNTs is stronger than with perfect and Stone–Wales-defective nanotubes. The reaction energy for per F2 addition is between 85 and 88 kcal mol−1 more negative than that per H2 addition. Electronic structure analysis of their energy gaps shows that the CD defects have a tendency to decrease the energy gap from 1.98–2.52 to 0.80–1.17 eV. After hydrogenation, fluorination, and chlorination, the energy gaps of the drying-tube-shaped SWCNTs with multiple CD defects are substantially increased to 1.65–3.85 eV. Furthermore, analyses of thermodynamic stability and nucleus-independent chemical shifts (NICS) are performed to analyze the stability of these molecules.
Co-reporter:Chun-Yi Sun;Chao Qin;Chun-Gang Wang;Shuang Wang;Xin-Long Wang;Guang-Sheng Yang;Kui-Zhan Shao;Ya-Qian Lan ;En-Bo Wang
Advanced Materials 2011 Volume 23( Issue 47) pp:5629-5632
Publication Date(Web):
DOI:10.1002/adma.201102538
Co-reporter:Yun Geng, Shui-Xing Wu, Hai-Bin Li, Xiao-Dan Tang, Yong Wu, Zhong-Min Su and Yi Liao
Journal of Materials Chemistry A 2011 vol. 21(Issue 39) pp:15558-15566
Publication Date(Web):26 Aug 2011
DOI:10.1039/C1JM12483H
Three naphthalene tetracarboxylic diimide derivatives 1–3 with high electron mobilities and long-term ambient stabilities were investigated employing Marcus–Levich–Jortner formalism at the density functional theory (DFT) level. The complicated relationships among molecular packings, intermolecular interactions, and transport properties for these compounds were focused on and analyzed through investigating the sensitivities of transfer integrals to intermolecular relative orientations, the optimizations of the major transport pathways and the calculations of intermolecular interaction energies by using dispersion-corrected DFT. The results show that the transfer integrals are sensitive to the subtle changes of relative orientations of molecules, especially for core-chlorinated compounds, and there is an interplay between intermolecular interaction and molecular packing. It is found that the transfer integrals associated with the molecular packing motifs of these systems determine their electron mobilities. Interestingly, further discussions on band structures, the anisotropies and temperature dependences of mobilities, and the comparisons of mobilities before and after optimization indicate that the intermolecular packing motifs in the film state may be different from those in the crystalline state for 2. Finally, we hope that our conjecture would facilitate the future design and preparation of high-performance charge-transport materials.
Co-reporter:Tingting Wang, Fang Chai, Qin Fu, Lingyu Zhang, Haiyan Liu, Lu Li, Yi Liao, Zhongmin Su, Chungang Wang, Beiye Duan and Dongxue Ren
Journal of Materials Chemistry A 2011 vol. 21(Issue 14) pp:5299-5306
Publication Date(Web):18 Feb 2011
DOI:10.1039/C0JM04115G
In this article, we report the fabrication of monodisperse hollow mesoporous silica (HMS) nanocages with uniform size possessing a hollow cubic core and mesoporous shell with penetrating pore channels based on a template-coating-etching process. It is worthwhile noting the obtained HMS nanocages with cubic void space and highly porous shell endow the structures with much higher storage capacity and sustained release of anticancer drugs. More importantly, the therapeutic efficacy of doxorubicin-loaded HMS nanocages was evaluated in vitro and in vivo for liver cancer therapy. The results show that the doxorubicin-loaded HMS nanocages have good cell uptake and can induce efficient cell deathinvitro. Taken together, this study demonstrates that the biocompatible HMS nanocages can effectively deliver drugs to the tumors and suppress tumor growth compared to free doxorubicinin vivo. Additionally, the synthetic strategy has also extended to fabrication of the uniform and monodisperse HMS nanocapsules with spherical shape.
Co-reporter:Yun Geng, Jianping Wang, Shuixing Wu, Haibin Li, Fei Yu, Guochun Yang, Hongze Gao and Zhongmin Su
Journal of Materials Chemistry A 2011 vol. 21(Issue 1) pp:134-143
Publication Date(Web):19 Oct 2010
DOI:10.1039/C0JM02119A
Seven perylene bisimide derivatives with different molecular packings and intermolecular interactions were investigated in detail within Marcus-Levich-Jortner formalism at the level of density functional theory (DFT). In theory, we further proved the report that different halogen substitutions in the core position of perylene bisimide lead to different molecular packings in their single crystals and thus obviously different electron transport properties. Here, insight into the geometries, the character of the frontier molecular orbitals, the decompositions of reorganization energies and transfer integrals in different directions was provided to shed light on the relationship between structures and properties. The molecular dynamics (MD) simulations and band structures calculations were also employed to give a multiscale understanding of their transport properties. The results show that there are small discrepancies of the intramolecular electron reorganization energies among these compounds and the transfer integrals determine their electron transport properties. Compounds 1a, 3a and 3b, with typical “brick” packing, π-stacked face-to-face packing and “herringbone” packing, respectively, have larger electron mobilities among these systems and possess different transport dimensionalities. Moreover, we also find there is close relationship between the intermolecular interaction energy and the transfer integral.
Co-reporter:Hai-Ning Wang, Xing Meng, Guang-Sheng Yang, Xin-Long Wang, Kui-Zhan Shao, Zhong-Min Su and Chun-Gang Wang
Chemical Communications 2011 vol. 47(Issue 25) pp:7128-7130
Publication Date(Web):26 May 2011
DOI:10.1039/C1CC11932J
A 12-connected network with fcu topology was firstly reported focusing on using predesigned metal–organic polyhedron (MOP) as the precursor, and its adsorption and delivery of the drug 5-fluorouracil (5-FU) was also determined.
Co-reporter:Dong-Ying Du, Jun-Sheng Qin, Yang-Guang Li, Shun-Li Li, Ya-Qian Lan, Xin-Long Wang, Kui-Zhan Shao, Zhong-Min Su and En-Bo Wang
Chemical Communications 2011 vol. 47(Issue 10) pp:2832-2834
Publication Date(Web):24 Jan 2011
DOI:10.1039/C0CC04343E
The first 3D uninodal eight-connected {P4Mo6O31H6}-based pure inorganic framework linked by transition metal ions has been synthesized and its electrochemical behavior and diffuse reflectance UV-Vis spectrum were investigated.
Co-reporter:Tingting Wang, Lingyu Zhang, Zhongmin Su, Chungang Wang, Yi Liao, and Qin Fu
ACS Applied Materials & Interfaces 2011 Volume 3(Issue 7) pp:2479
Publication Date(Web):May 24, 2011
DOI:10.1021/am200364e
Highly uniform and multifunctional hollow mesoporous silica nanocages that combined excellent properties (good biocompatibility, fluorescence imaging, drug delivery, and dual-mode cancer therapy) in one single system were synthesized. Dye molecules labeled in the nanocages could be used as traceable detectors in fluorescence imaging. A chemotherapeutic drug, doxorubicin (DOX), has been loaded into the nanocages with a high storage capacity due to the large cubic cavities and could be released through the penetrating mesoporous channels in a sustained fashion. Hematoporphyrin molecules were also covalently doped in the nanocages and allowed for photodynamic therapy. More importantly, a cooperative, synergistic therapy combining chemotherapy and photodynamic therapy exhibited high therapeutic efficacy for cancer therapy in vitro.Keywords: cancer therapy; drug delivery; fluorescence imaging; mesoporous silica nanocage; photodynamic therapy; sustained release;
Co-reporter:Fu-Qiang Zhang ; Wei Guan ; Li-Kan Yan ; Yin-Tang Zhang ; Mao-Tian Xu ; Ebenezer Hayfron-Benjamin
Inorganic Chemistry 2011 Volume 50(Issue 11) pp:4967-4977
Publication Date(Web):April 28, 2011
DOI:10.1021/ic200203s
Density functional theory calculations have been carried out to investigate α-, β-, γ-, α*-, β*-, and γ*-[(PO4)2W18O54]6– Wells–Dawson isomers, which exhibited stability in the order of α > β > γ > γ* > β* > α*, reproduced the experimental observations (α > β > γ), and confirmed the hypothesis of Contant and Thouvenot (γ* > β* > α*). Energy decomposition analysis reveals that both the spatial arrangement of the host W18O54 cage (eclipsed or staggered) and its structural distortion induced by the encapsulated guest anions are two dominant factors in control of the stability order, while the influences of host–guest interaction and distortion of the guest anions are very small. A building block decomposition approach is designed and provides an effective means to clarify the detailed relationship between the local distortion and energy. By using this method, it is found that the eclipsed belt, and in particular the staggered belt, significantly distort the two caps inside the Wells–Dawson structure. Notably, there is a direct relationship between the overall stability and distortion in the belts, which is proven to be partly originating from the dominance in the quantity of the belt building blocks over that of the caps (12:6). Besides, half-unit {XW9} decomposition confirms that [(XO4)2W18O54]n− (X = Si, Ge, Al, and Ga) are thermodynamically instable because of the notable electrostatic repulsion between two {XW9} units induced by the highly charged guest anions.
Co-reporter:Shun-Li Li, Ke Tan, Ya-Qian Lan, Jun-Sheng Qin, Mei-Na Li, Dong-Ying Du and Zhong-Min Su
CrystEngComm 2011 vol. 13(Issue 15) pp:4945-4949
Publication Date(Web):09 Jun 2011
DOI:10.1039/C1CE05277B
A novel metal–organic compound, namely [Zn6(bib)6L6]·7H2O (1) where H2L = 4-(4-carboxybenzyloxy)benzoic acid, and bib = 4,4′-bis(imidazol-1-ylmethyl)benzene, has been synthesized under hydrothermal conditions. Its structure was determined by single-crystal X-ray diffraction analysis and further characterized by elemental analysis and thermogravimetric (TG) analysis. In the structure of 1, two bib ligands cuddle two metal ions to form a ring, and are linked by deprotonated L2− to form a 6-fold interpenetrated 3D framework. From the topological analysis, each ZnII ion is considered as a three-connected node, and a pair of bib ligands and L2− anions are linkers. The framework of compound 1 can be classified as a (62·10)(6·102)(102·12) topology, which is a novel 3-connected [3 + 3] 6-fold interpenetrated net showing both polyrotaxane and polycatenane characters. In addition, the luminescent properties of the compound are discussed.
Co-reporter:Guang-Sheng Yang, Hong-Ying Zang, Ya-Qian Lan, Xin-Long Wang, Chun-Jie Jiang, Zhong-Min Su and Lian-De Zhu
CrystEngComm 2011 vol. 13(Issue 5) pp:1461-1466
Publication Date(Web):18 Nov 2010
DOI:10.1039/C0CE00259C
Here, we report two novel POM-based MOFs: a nano-caged hybrid compound [Ni2(BIMB)2(MoVI4MoV2O19)] (1), whose cages are based on [Mo6O19]4− units and a [Mo6O19]2−-templated framework [Zn4(BIMB)4(PO4)2(Mo6O19)]·2H2O (2) (BIMB = 1,4-bis(1-imidazolyl)benzene). According to topological analysis, the first is a 3D (4,6)-connected self-penetrating framework with polycatenanes motif. In contrast, the second metal–organic framework has no interpenetration for the templated [Mo6O19]2− occupying the cavity. The UV-vis absorption spectrum of 1 was investigated to prove the existence of MoV atoms and the typical peak at 577 nm can be attributed to MoV → MoVI charge transfer. Furthermore, the diffuse reflectivity spectrum shows that its band gap can be assessed as 3.06 eV, which can be regarded as a wide gap semiconductor. Additionally, cyclic voltammogram of compound 1 shows two redox peaks in the range of 0–800 mV in H2SO4 aqueous solution.
Co-reporter:Kui-Zhan Shao, Ya-Hui Zhao, Ya-Qian Lan, Xin-Long Wang, Zhong-Min Su and Rong-Shun Wang
CrystEngComm 2011 vol. 13(Issue 3) pp:889-896
Publication Date(Web):18 Oct 2010
DOI:10.1039/C0CE00042F
An investigation into the molecular tectonics of metal–organic frameworks (MOFs) is reported on the basis of N-donor ligand modulated polynuclear zinc clusters and different aromatic polycarboxylic ligands. A series of three-dimensional (3D) coordination frameworks, [Zn2(BDC)1.5(L)(OH)]·H2O (1), [Zn2(BOABA)(L)(OH)]·2H2O (2), [Zn2(BOABA)(L)(OH)]·4H2O (3), [Zn3(BTC)2(OH)]·0.25H2O·[N(C4H9)4] (4) and [Zn2(BTEC)0.5(L)(OH)2] (5), were synthesized by self-assembly of zinc ions with a new N-donor ligand 4,5-diazafluoren-9-oxime (L) and the aromatic polycarboxylic ligands 1,4-benzenedicarboxylic acid (H2BDC), 3,5-bis-oxyacetate-benzoic acid (H3BOABA), 1,3,5-benzenetricarboxylic acid (H3BTC), and 1,2,4,5-benzenetetracarboxylic acid (H4BTEC). Compound 1 exhibits a twofold interpenetrated α-polonium-type network based on tetranuclear Zn clusters as six-connected vertices and BDC ligands as linkers. Compound 2 also consists of tetranuclear units, it shows a (3,6)-connected rutile network, where tetranuclear zinc clusters act as six-connected nodes and BOABA ligands act as three-connected nodes. Compound 3 is an isomer of compound 2, due to the different configuration of tetranuclear zinc, it displays a novel (3,6)-connected network with a complex (4·62)2(42·69·84) topology. In 4, the connection between the trinuclear zinc clusters and the BTC ligands results in an infinite 3D (3,6)-connected network with point symbol (4·62)(63)(4·611·83). Compound 5 constitutes a lvt net, which is built from tetranuclear clusters and BTEC as four-connected nodes, respectively. The results indicate that various polynuclear zinc clusters are modulated by L ligands combining with Zn atoms via chelation or monodentate coordination, or acting as a structure-directing agent. Meanwhile, aromatic multicarboxylic acids also play important roles in the construction of the compounds with various structures.
Co-reporter:Xing Meng, Chao Qin, Xin-Long Wang, Zhong-Min Su, Bo Li and Qi-Hua Yang
Dalton Transactions 2011 vol. 40(Issue 39) pp:9964-9966
Publication Date(Web):09 Sep 2011
DOI:10.1039/C1DT11227A
Immobilization of the chiral salen-metal complex [MnIII(salen)(H2O)2ClO4] on the Keggin-type polyoxometalate (POM) skeletons leads to the isolation of POM derivatives functionalized with chiral salen-metal complexes, which represent the first examples of introducing chiral salen-metal complexes into the POM systems.
Co-reporter:Yong Wu, Shui-Xing Wu, Hai-Bin Li, Yun Geng and Zhong-Min Su
Dalton Transactions 2011 vol. 40(Issue 17) pp:4480-4488
Publication Date(Web):15 Mar 2011
DOI:10.1039/C0DT01299H
The electronic structures and photophysical properties of eight Pt-complexes with different N-heterocyclic carbene ligands and potential to serve as light emitting diode materials were investigated by density functional theory and time-dependent density functional theory, employing the BP86 functional for geometry optimisations, SAOP potential for excited state calculations and all-electron TZ2P basis set throughout. Non-radiative and radiative decay rate constants were determined for each system through analyses of the geometric relaxations, d-orbital splitting and spin–orbit couplings at the optimised S0 and T1 geometries. Three Pt-systems bound to two N-heterocyclic carbenes were shown to be nonemissive, while a fourth was shown to be emissive from the T1 excited state. Similar T1-initated emission was observed for three other Pt-systems investigated, each bound to four N-heterocyclic carbenes, while a fourth similarly tetra-ligated system showed T2-initation of emission. The results highlight the coupling of ligand-identity to photophysical properties and more importantly, the potential for rational optimisation and tuning of emission wavelengths and phosphorescent efficiencies. Encouragingly, two of the tetra-N-heterocyclic carbene ligated systems show strong potential to serve as highly-efficient blue and green light emitting materials, respectively.
Co-reporter:Hong-Ying Zang, Ya-Qian Lan, Shun-Li Li, Guang-Sheng Yang, Kui-Zhan Shao, Xin-Long Wang, Li-Kai Yan and Zhong-Min Su
Dalton Transactions 2011 vol. 40(Issue 13) pp:3176-3182
Publication Date(Web):21 Feb 2011
DOI:10.1039/C0DT00656D
With the bottom-up design principle, we use metal-ions to bridge the predesigned tectons (1 [(H2L1)2(Mo8O26)]·4H2O and 3 [(H2L2)(L2)0.5(Mo8O26)0.5]·H2O) so that two higher dimensional γ-octamolybdate based inorganic–organic hybrid compounds 2 [CuI2(L1)3(Mo8O26)0.5] and 4 [Ni(L2)2(HL2)2(Mo8O26)]·4H2O are successfully obtained.
Co-reporter:Chun-Guang Liu, Wei Guan, Li-Kai Yan and Zhong-Min Su
Dalton Transactions 2011 vol. 40(Issue 12) pp:2967-2974
Publication Date(Web):15 Feb 2011
DOI:10.1039/C0DT01085E
High-valent transition-metal-substituted Keggin-type polyoxometalates (POMs) are active and robust oxidation catalyst. The important oxidized intermediates of these POM complexes are very difficult to be characterized by using the experimental method, and thus no detail information is available on such species. In the present paper, density functional theory (DFT) calculations have been carried out to characterize the electronic structures of a series of mono-ruthenium-substituted Keggin-type POMs. We find that the aquaruthenium(II/III/IV) species possess dxy2dxz2dyz2, dxy2dxz2dyz1, and dxy2dxz1dyz1 electronic configuration, respectively, and hydroxyl/oxoruthenium(IV/V/VI) species possess dxy2dxz1π*yz1, dxy2π*xz1π*yz1, dxy1π*xz1π*yz1, and dxy1π*xz1π*yz0 electronic configuration, respectively. Mulliken spin population shows that spin density is localized on the ruthenium center in aquaruthenium(II/III/IV) POM complexes, and the RuOa unit in hydroxyl/oxoruthenium(IV/V/VI) POM complexes. The Oa atom has substantial radical character in oxoruthenium(IV/V) species, and the radical character of the Oa atom are significantly weakened in the oxoruthenium(VI) species. The relevant energy of the important Ru–Oa π*-antibonding unoccupied orbitals with high RuOa compositions of oxoruthenium(IV/V/VI) POM complexes decrease in the order: oxoruthenium(IV) > oxoruthenium(V) > oxoruthenium(VI). The pH-independent multiple reduction energies for Ru(III/II), Ru(V/IV), and Ru(VI/V) couples are calculated, which is in agreement with the experimental data.
Co-reporter:Guo-Gang Shan, Dong-Xia Zhu, Hai-Bin Li, Peng Li, Zhong-Min Su and Yi Liao
Dalton Transactions 2011 vol. 40(Issue 12) pp:2947-2953
Publication Date(Web):15 Feb 2011
DOI:10.1039/C0DT01559H
Three cationic iridium complexes containing 4,7-bis(3,6-di-tert-butyl-9H-carbazol-9-yl)-1,10-phenanthroline (L1) and 4,7-bis(3′,6′-di-tert-butyl-6-(3,6-di-tert-butyl-9H-carbazol-9-yl)-3,9′-bi(9H-carbazol)-9-yl)-1,10-phenanthroline (L2) as the ancillary ligands, namely, [Ir(ppy)2(L1)]PF6 (1), [Ir(ppy)2(L2)]PF6 (2) and [Ir(oxd)2(L2)]PF6 (3) (ppy is 2-phenylpyridine, oxd is 2,5-diphenyl-1,3,4-oxadiazole), have been designed and prepared. With more intramolecular rotational units on the ancillary ligand (L2), 2 and 3 possess a unique aggregation-induced phosphorescent emission (AIPE) property. This phenomenon was unprecedentedly observed in the cationic iridium(III) complexes. In order to investigate the underlying mechanism of this AIPE behavior, their photophysical, temperature-dependent aggregation properties as well as theoretical calculations, were performed. The results suggest that restricted intramolecular rotation is responsible for the AIPE of cationic complexes. Moreover, photoluminescent quantum yields in the neat film, thermal stabilities and off/on luminescence switching of 2 were investigated, revealing its potential application as a candidate for LECs and organic vapor sensing.
Co-reporter:Mei-Na Li, Dong-Ying Du, Guang-Sheng Yang, Shun-Li Li, Ya-Qian Lan, Kui-Zhan Shao, Jun-Sheng Qin, and Zhong-Min Su
Crystal Growth & Design 2011 Volume 11(Issue 6) pp:2510-2514
Publication Date(Web):April 27, 2011
DOI:10.1021/cg2002223
A new 3D chiral metal–organic framework [Co2(BTT)(OH)(H2O)5]·5CH3OH·6DMA (1) has been synthesized using an achiral tritopic bridging ligand, 1,3,5-tris(2H-tetrazol-5-yl)benzene (H3BTT). 1 possesses a (10,3)-a net containing large helical channels of 17.43 Å and exhibits a high solvent-accessible volume (calculated 79%). The N2 adsorption and magnetic properties for 1 have been examined.
Co-reporter:Jun-Sheng Qin, Dong-Ying Du, Shun-Li Li, Ya-Qian Lan, Kui-Zhan Shao and Zhong-Min Su
CrystEngComm 2011 vol. 13(Issue 3) pp:779-786
Publication Date(Web):01 Oct 2010
DOI:10.1039/C0CE00260G
Three organic–inorganic hybrid compounds have been designed and synthesized based on different vanadate chains, Zn(II) ions and flexible ligands at different pH values under hydrothermal conditions, namely, [Zn2(bbi)2(V3O9)(OH)]·H2O (1), [Zn2(bbi)2(V4O12)] (2), and [Zn(bbi)(V2O6)] (3), where bbi is 1,1′-(1,4-butanediyl)bis(imidazole). In 1, helical chains [(V3O9)3−]∞ were connected by [Zn2(OH)]3+ dimers giving rise to a 2D chiral layer which was further bridged via bbi ligands to construct a 3D POM-based chiral framework. There are two opposite helical chains [(V4O12)4−]∞ in 2. The [(V4O12)4−]∞ chains with the same chirality were connected each other by Zn2 cations to generate a 2D chiral layer, respectively. The adjacent chiral layers were linked by Zn1 ions to generate a 3D non-centrosymmetric framework with large channel encased by bbi. Similarly, the [(V2O6)2−]∞ chains with the same chirality in 3 were connected by zinc atoms to form a 2D chiral layer which was further linked through bbi to generate a 3D centrosymmetric network. Compounds 2 and 3 are supramolecular isomers. With adjusting the pH value of reaction mixture, we have achieved three new hybrids crystallized from chiral, non-centrosymmetric to centrosymmetric space group.
Co-reporter:Hai-jun Pang, Hui-yuan Ma, Jun Peng, Chun-jing Zhang, Peng-peng Zhang and Zhong-min Su
CrystEngComm 2011 vol. 13(Issue 23) pp:7079-7085
Publication Date(Web):06 Oct 2011
DOI:10.1039/C1CE05648D
Three new compounds, [Cu(bimb)]2(HPW12O40)·3H2O (1), (H2bimb)2SiW12O40 (2) and (H2bimb)3CoW12O40 (3) (bimb = 1,4-bis(imidazol-1-ylmethyl)biphenyl), have been synthesized under the same hydrothermal conditions. Structural characterizations by single-crystal X-ray diffraction show that compound 1 possesses a porous metal–organic pseudo-rotaxane framework in which [PW12O40]3−clusters are encapsulated, whereas compounds 2 and 3 are common supramolecular compounds. The distinct structure of compound 1 suggests that the porous framework may selectively encapsulate the polyoxometalate clusters, although the three polyoxometalate clusters have subtle structural differences. Catalytic photodegradation of organic dye RhB by compounds 1–3 has been studied.
Co-reporter:Ting-Ting Wang, Fang Chai, Chun-Gang Wang, Lu Li, Hai-Yan Liu, Ling-Yu Zhang, Zhong-Min Su, Yi Liao
Journal of Colloid and Interface Science 2011 Volume 358(Issue 1) pp:109-115
Publication Date(Web):1 June 2011
DOI:10.1016/j.jcis.2011.02.023
Multifunctional uniform and versatile hollow and rattle-type nanocapsules composed of spindle-shaped Au nanoparticles as cores and fluorescent mesoporous silica shells with tunable optical and fluorescent properties have been developed by controlled etching Au nanorods (AuNRs) coated with mesoporous SiO2 (AuNR@mSiO2) via a small amount of aqua regia (volume ratio HCl/HNO3 = 3/1) as an etching agent in a facile way. The etching process can be tracked by UV–Vis absorption and fluorescence spectroscopy and the size of cavities in the hollow/rattle-type particles can be tuned by controlling the reaction time. The dye molecules incorporated in mSiO2 walls enabled the nanocapsules to be utilized as a fluorescent imaging agent in cancer cell imaging. Furthermore, such hollow/rattle-structured nanocapsules have the merit of enhanced drug loading capacity acting as carriers for the loading and delivery of an anticancer drug, doxorubicin hydrochloride (DOX), with higher storage for cancer therapy. Herein, the combined functionalities of simultaneous cell imaging and drug delivery of the synthesized nanocapsules have been demonstrated, which provide a very promising candidate for application in optical imaging and drug delivery for cancer cells.Graphical abstractMultifunctional uniform and versatile hollow and rattle-type nanocapsules composed of spindle-shaped Au nanoparticles as cores and fluorescent mesoporous silica shells with tunable optical and fluorescent properties have been developed by controlled etching Au nanorods coated with mesoporous SiO2 in a facile way. The combined functionalities of simultaneous cell imaging and drug delivery of the synthesized nanocapsules have also been demonstrated..Research highlights► Multifunctional uniform and versatile hollow and rattle-type mesoporous silica nanocapsules were fabricated in a facile way. ► The obtained multifunctional rattle-type mesoporous silica nanocapsules have tunable optical and fluorescent properties. ► The fluorescent rattle-type nanocapsules with Au nanoparticles were used for drug delivery and fluorescence imaging for cancer cells.
Co-reporter:Chun-Guang Liu;Wei Guan;Li-Kai Yan
European Journal of Inorganic Chemistry 2011 Volume 2011( Issue 4) pp:489-494
Publication Date(Web):
DOI:10.1002/ejic.201000907
Abstract
Density functional theory (DFT) calculations were performed for three ruthenium/nitrous oxide(N2O) adducts, RuIICl2(η1-N2O)(P–N)(PPh3) {P–N = [o-(dimethylamino)phenyl]diphenylphosphane} (1), RuII(η1-N2O)(TMP)(THF) (TMP = dianion of tetramesitylporphyrin, THF = tetrahydrofuran) (2), and [PW11O39{RuII/(η1-N2O/H2O)}]n– (3). The bonding interactions between the N2O molecule and the metal–ruthenium fragment were determined from the energy-decomposition analysis (EDA) and their electronic structures. The results show that the bonding in the ruthenium(II)/N2O adducts 1–3 can be interpreted in terms of weak donor–acceptor interactions between the N2O molecule and the ruthenium(II) center. The geometrical optimization of the polyoxometalate (POM) adduct 3 indicates a bent Ru–NNO linkage, which effectively enhances donor–acceptor interactions with the quasi π-symmetry orbital. The mono-ruthenium(II) substituted Keggin-type POM adduct 3 is a potential reagent for the activation of the N2O molecule because of the strong Ru–NNO bond and the significant RuNN–O π*-antibonding orbital character relative to adducts 1 and 2, according to our DFT calculations.
Co-reporter:Gang Yuan, Kui-Zhan Shao, Dong-Ying Du, Xin-Long Wang, Zhong-Min Su
Solid State Sciences 2011 Volume 13(Issue 5) pp:1083-1091
Publication Date(Web):May 2011
DOI:10.1016/j.solidstatesciences.2011.01.014
Six new compounds, namely, {[Cd3(Himpy)3(tda)2]·3H2O}n (1), {[Zn3(bipy)2(tda)2(H2O)2]·4H2O}n (2), {[Cd3(bipy)3(tda)2]·4H2O}n (3), {[Cd3(tda)2(H2O)3Cl]·H2O}n (4), {[Zn2(tz)(tda)(H2O)2]·H2O}n (5) and {[Cd7(pz)(tda)4(OAc)(H2O)7]·3H2O}n (6) [H3tda = 1H-1,2,3-triazole-4,5-dicarboxylic acid, Himpy = 2-(1H-imidazol-2-yl)pyridine, bipy = 2,2′-bipyridine, Htz = 1H-1,2,4-triazole, H2pz = piperazine] have been prepared under hydrothermal condition and characterized by elemental analyses, infrared spectroscopy, powder X-ray diffraction and single-crystal X-ray diffraction analyses. Compound 1 is a 1D column-like structure and displays a 3D supramolecular network via the π···π stacking interaction. The compounds 2 and 3 exhibit similar 2D layer-like structure, which further extend to 3D supermolecular structure by the π···π stacking interaction. All of compounds 4–6 display 3D framework with diverse topology constructed from the tda3− ligands in different coordination modes and secondary ligands (or bridging atom) connecting metal ions. Furthermore, the thermal stabilities and photoluminescent properties of compounds 1–6 were studied.
Co-reporter:Xing Meng, Hai-Ning Wang, Guang-Sheng Yang, Shuang Wang, Xin-Long Wang, Kui-Zhan Shao, Zhong-Min Su
Inorganic Chemistry Communications 2011 Volume 14(Issue 9) pp:1418-1421
Publication Date(Web):September 2011
DOI:10.1016/j.inoche.2011.05.036
Two new inorganic–organic hybrid supramolecular compounds based on Keggin-type polyoxoanions, namely [Mn(salen)(CH3OH)2]3[PMo12O40] (1) and [Mn(salen)(CH3OH)2]3[PW12O40] (2) (H2salen = N,N′-ethylenebis(salicylideneimine)) have been synthesized and characterized by single-crystal X-ray diffraction analysis, elemental analysis, IR spectroscopy, and TG analysis. Two compounds reported herein represent the first examples of integrating metal–Schiff-base segments with Keggin polyanions characterized via single-crystal X-ray diffraction. Such hybrid materials display photocatalytic and electrocatalytic bifunctional catalytic activities.Two new inorganic–organic hybrid supramolecular compounds, namely [Mn(salen)(CH3OH)2]3[PMo12O40] (1) and [Mn(salen)(CH3OH)2]3[PW12O40] (2) (H2salen = N,N′-ethylenebis(salicylideneimine)) have been successfully synthesized. They represent the first examples of integrating metal–Schiff-base segments with Keggin heteropolyanions characterized via single-crystal X-ray diffraction which display photocatalytic and electrocatalytic bifunctional catalytic activities.Highlights► Two new inorganic–organic hybrid supramolecular compounds have been synthesized. ► They are successfully characterized via single-crystal X-ray diffraction. ► These compounds represent the first examples of integrating metal–Schiff-base segments with Keggin heteropolyanions. ► Both display photocatalytic and electrocatalytic bifunctional catalytic activities.
Co-reporter:Luan Jiang, Zong-Xiao Li, Yan Wang, Guo-Dong Feng, Wei-Xing Zhao, Kui-Zhan Shao, Chun-Yi Sun, Lian-Jie Li, Zhong-Min Su
Inorganic Chemistry Communications 2011 Volume 14(Issue 7) pp:1077-1081
Publication Date(Web):July 2011
DOI:10.1016/j.inoche.2011.03.032
Two new metal-organic frameworks, [Cd2(H2C3PIm)(BDC)4(H2O)3]n (1) and [Zn3(H2C3PIm)3 (BTC)2(H2O)2]n (2) (H2C3PIm = 2,2′-(1,3-propanediyl)bis(1 H-benzimidazole), H2BDC = 1,4-benzenedicarboxylic acid, H3BTC = 1,3,5-benzene tricarboxylic acid), have been synthesized hydrothermally and their structures are characterized by single crystal X-ray analysis. Compound 1 presents a 1D columnar structure, and compound 2 exhibits a 2D sheet with novel extra-large rings of 4.82 topology. Both complexes exhibit strong photoluminescence at room temperature.Two novel metal-organic frameworks based on the bis(2-benzimidazole), [Cd2(H2C3PIm)(BDC)4(H2O)3]n (1), [Zn3(H2C3PIm)3 (BTC)2(H2O)2]n (2), have been successfully prepared under hydrothermal conditions and characterized. Their fluorescence spectra have been studied, both complexes exhibit strong photoluminescence at room temperature.Research Highlights► Two new interesting Cd(II)-containing and Zn(II)-containing polymeric complexes , [Cd2(H2C3PIm)(BDC)4(H2O)3]n (1) and [Zn3(H2C3PIm)3(BTC)2(H2O)2]n (2) have been prepared. ► 1 presents a 1D columnar structure though three types of coordination modes with Cd ion, while compound 2 exhibits 2D grid structure with novel 32-membered and 64-membere rings of 4.82 topology. ► Both two complexes exhibit strong photoluminescence at room temperature.
Co-reporter:Junsheng Qin, Dongying Du, Lei Chen, Xiuyun Sun, Yaqian Lan, Zhongmin Su
Journal of Solid State Chemistry 2011 Volume 184(Issue 2) pp:373-378
Publication Date(Web):February 2011
DOI:10.1016/j.jssc.2010.11.012
Reactions of the tripodal bridging ligand 5-(4-carboxy–phenoxy)-isophthalic acid (abbreviated as H3cpia) with lanthanide salts lead to the formation of a family of different coordination polymers, that is, [Ln(cpia)(H2O)2]n·nH2O (Ln=Ce (1), Pr (2), Nd (3), Sm (4), Eu (5), Gd (6), Dy (7), Er (8), Tm (9) and Y (10)) in the presence of formic acid or diethylamine, which are characterized by elemental analysis, IR spectrum, thermogravimetric analysis (TGA), XRPD spectrum and single-crystal X-ray diffraction. Compounds 1–10 are isostructural and exhibit three-dimensional microporous frameworks. Furthermore, the photoluminescent properties of 4, 5 and 7 have been studied in detail.Graphical abstractReactions of the tripodal bridging ligand (H3cpia) with lanthanide ions lead to the formation of a series of coordination polymers in the presence of formic acid or diethylamine.Research Highlights► Ten new lanthanides-based coordination polymers (1–10) have been synthesized. ► 1–10 exhibit 3D (4,8)-connected fluorite topology networks with 1D channel parallel to the b-axis. ► Compounds 4, 5 and 7 exhibit characteristic luminescence of Sm3+, Eu3+ and Dy3+ ions, respectively.
Co-reporter:Shuang Wang, Gang Yuan, Chunyi Sun, Guangsheng Yang, Kuizhan Shao, Xinlong Wang, Yaqian Lan, Zhongmin Su
Inorganic Chemistry Communications 2011 Volume 14(Issue 2) pp:347-350
Publication Date(Web):February 2011
DOI:10.1016/j.inoche.2010.11.023
One 2D coordination polymer, [Ag2(Lp)1.5(NO3)](NO3) (1) (Lp = p − [CH(pz)2]2C6H4,), has been successfully synthesized under solvothermal conditions by the reaction of arene-linked p-bis[bis(1-pyrazolyl)methyl]benzene ligand with AgNO3. Compound 1 presents the first two-dimensional (2D) framework of trinuclear Ag cluster and Lp ligand, and exhibits a three-dimensional (3D) supramolecular structure supported by the weak interactions between neighboring sheets. It was characterized by IR, PXRD, single-crystal X-ray diffraction and thermogravimetric analysis. The fluorescence property of compound 1 has also been studied.One novel 2D supramolecular compound based on the arene-linked bis(pyrazolyl)methane ligand and trinuclear Ag cluster has been successfully synthesized under the solvothermal conditions. The fluorescence property of this complex has also been studied.Highlights► In this paper, we reported and characterized one 2D supramolecular polymer utilizing arene-linked bis(pyrazolyl)methane ligand and silver cations. ► This complex based on trinuclear Ag cluster provided a new coordination mode of arene-linked bis(pyrazolyl)methane ligand and exhibited the first example of 2D structure in the system of arene-linked bis(pyrazolyl)methane ligand. ► The framework further extended into a three-dimensional (3D) supramolecular structure by weak interaction.
Co-reporter:Fei Yu, Xiaodan Tang, Guochun Yang, Yuai Duan, Zhongmin Su
Chemical Physics Letters 2011 Volume 506(4–6) pp:255-259
Publication Date(Web):20 April 2011
DOI:10.1016/j.cplett.2011.03.030
Abstract
A novel C60 derivative-1,4-bis(pentafluorobenzyl)[60]-fullerene (C60(CH2C6F5)2) has been recently synthesized and can be utilized for high-performance organic photovoltaic devices. Its charge transport properties have been systemically investigated by band model and hopping model, respectively. Both models demonstrate that both electron and hole are favor of transporting, and C60(CH2C6F5)2 has the potential to be used as ambipolar transport material. The density of states, frontier molecular orbitals, and transfer integrals in main pathways show that it is the fullerene–fullerene face-to-face interaction, not C6F5–fullerene interaction determines the charge transport properties.
Co-reporter:Chun-Yi Sun, Shuang Wang, Guang-Sheng Yang, Xin-Long Wang, Kui-Zhan Shao, Zhong-Min Su
Inorganic Chemistry Communications 2011 Volume 14(Issue 6) pp:893-896
Publication Date(Web):June 2011
DOI:10.1016/j.inoche.2011.03.021
A non-interpenetrated 3D porous metal–organic framework [(CH3)2NH2]2[Cd(TATAT)2/3]·4DMF (1) (TATAT = 5,5′,5″-(1,3,5-triazine-2,4,6-triyl)tris(azanediyl)triisophthalate) has been successfully synthesized under solvothermal conditions and characterized by elemental analysis, TGA, and single-crystal X-ray diffraction analysis. Compound 1 exhibits α-Al2O3 (corundum) topology and represents the first metal–organic analog presently known with a natural material topology based on 6-connected organic ligand and tetrahedral metal ion. In addition, the thermal stabilities of the complex and the acid-treated product have been investigated and the results indicate that acid treatment is an efficient way for this compound to improve its thermal stability.A non-interpenetrated 3D porous metal–organic framework (1) has been successfully synthesized. 1 exhibits α-Al2O3 (corundum) topology and represents the first metal–organic analog presently known with a natural material topology which is based on octahedral organic ligand and tetrahedral single metal ion.Research highlights► A non-interpenetrated 3D porous metal–organic frameworks (1) was synthesized. ► 1 is based on octahedral organic ligand and tetrahedral single metal ion. ► 1 exhibits peculiar α-Al2O3 (corundum) topology. ► Thermal stability of 1 and acid treated product have been determined. ► Acid treatment is a efficient way for 1 having a higher thermal stability.
Co-reporter:Fei Yu, Guochun Yang, Zhongmin Su
Synthetic Metals 2011 Volume 161(11–12) pp:1073-1078
Publication Date(Web):June 2011
DOI:10.1016/j.synthmet.2011.03.018
To get insights of the effect of multiple intermolecular interactions on the charge transport ability of polymorphs, we systematically investigated nine crystals in two kinds of polymorphs within the framework of band model, in which the type and strength of the weak interactions are different. The results show that: (i) the dispersions are relative to the π-stacking area. The bigger area the is, the larger dispersion the is. Thus, it can enhance the charge transport ability. (ii) Hydrogen bonding interactions have a great influence on the dispersion of valence band (VB) rather than conduction band (CB). (iii) When π-stacking and hydrogen bonding interactions coexist in the polymorphs, they enhance the dispersions of CB and restrain that of VB. (iv) When the type of weak interactions increases, the dispersions will be enlarged. Understanding the effect of multiple weak interactions on the polymorphs is a theoretical guide to design novel organic semiconductors with efficient charge transport ability.Graphical abstractHighlights► The effect of multiple intermolecular interactions on charge transfer was studied. ► The band structure shows that dispersion is proportional to the π-stacking area. ► The hydrogen bonding interactions have a great influence on the dispersion of VB. ► The dispersion of CB is enhanced and VB is restrained when the interactions coexist. ► When the type of weak interactions increases, the dispersions will be enlarged.
Co-reporter:Liang Zhao, Min Zhang, Lili Shi, Shiling Sun, Shuixing Wu, Chunguang Liu, Zhongmin Su
Synthetic Metals 2011 Volume 161(21–22) pp:2185-2191
Publication Date(Web):November–December 2011
DOI:10.1016/j.synthmet.2011.07.004
Systematic density functional investigations on the structural and electronic properties of C55O5 and C54O6, two open-cage oxo-fullerene derivatives, have been performed. Those formed from topmost pentagon and hexagon of C60 cleaved with oxygen atoms, have a large circular opening 4.92 Å and 5.99 Å, respectively. The size of the opening is large enough to allow guest atoms or small molecules penetrating, thus C55O5 and C54O6 may act as promising candidates for fuel storage. The calculations on their nonlinear optical (NLO) properties with ZINDO/SCI–SOS method show that the investigated compounds possess remarkably larger static first hyperpolarizability (β) values amounting to −239.48 and −339.83 × 10−30 esu for C55O5 and C54O6, respectively, which are comparable to the reported exohedral C60 derivatives. It opens a new route to explore new type of NLO materials based on C60 derivatives.Highlights► Open-cage oxo-fullerene derivatives are more stable than fullerene. ► The formation of C55O5 may be easier than that of C54O6. ► They show remarkable static first hyperpolarizability. ► New route to explore novel NLO materials based on fullerene derivatives.
Co-reporter:Jinlian Li, Xiaoqiang Jin, LiHong Hu, Jianping Wang, Zhongmin Su
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 23) pp:6969-6972
Publication Date(Web):1 December 2011
DOI:10.1016/j.bmcl.2011.09.125
DNA G-quadruplex is an attractive drug target for anticancer therapy. Most G-quadruplex ligands have little selectivity, due to π-stacking interaction with common G-tetrads surface. Thanks to the varieties of G-quadruplex grooves, the groove-binding ligand is expected to create high selectivity. Therefore, developing novel molecular geometries that target G-quadruplex groove has been paid growing attention. In this work, steroid FG, a special nonplanar and nonaromatic small molecule, interacting with different conformations of G-quadruplexes has been studied by molecular docking and molecular dynamics simulations. The results showed the selectivity of the hydrophobic group of steroid FG for the wide groove of antiparallel G-quadruplex. The methyl groups on the tetracyclic ring of steroid represent the specific binding ability for the small hydrophobic cavity formed by reversed stacking of G-tetrads in antiparallel G-quadruplex groove. This work provides new insight for developing new classes of G-quadruplex groove-binding ligands.Steroid FG binding in the wide groove of antiparallel G-quadruplex.
Co-reporter:Hong-Ying Zang, Dong-Ying Du, Shun-Li Li, Ya-Qian Lan, Guang-Sheng Yang, Li-Kai Yan, Kui-Zhan Shao, Zhong-Min Su
Journal of Solid State Chemistry 2011 Volume 184(Issue 5) pp:1141-1147
Publication Date(Web):May 2011
DOI:10.1016/j.jssc.2011.02.026
With BIMB ligand, we have successfully obtained and characterized three novel entangled inorganic–organic hybrid compounds by choosing different metal ions, that is, [Ni(BIMB)2(γ-Mo8O26)0.5]·3H2O (1), [Zn(BIMB)2(γ-Mo8O26)0.5] (2), and [Cu3(BIMB)4(H2O)(δ-Mo8O26)Cl2]·3H2O (3) (where BIMB=1,4-bis(1-imidazolyl)benzene). Compound 1 is a 2-fold interpenetration 4-connected 3D framework with the short Schläfli symbol of (4×64×8)2(42×62×82), in which octamolybdate anion shows γ-isomer; 2 exhibits a (5,6)-connected 3D self-penetrating topological motif with the short Schläfli symbol of (4×57×62)2(42×511×72), and 3 shows a (4,6)-connected self-penetrating 3D framework with the short Schläfli symbol of (42·52·6·7) (44·5·69·8) (54·62) whose octamolybdate has δ-isomer. In addition, the optical band gaps of these three compounds have been measured, which are 2.98 eV for 1, 3.42 eV for 2, and 2.88 eV for 3. Moreover, 2 has photoluminescent property, which can be attributed to ligand-to-metal charge-transfer (LMCT) band.Graphical abstractWith BIMB ligand, we have obtained and characterized three novel POM-based entangled hybrid compounds. Compound 1 is a 2-fold interpenetration 3D framework; 2 and 3 exhibit self-penetrating 3D polycatenated frameworks.Highlights► Three novel octamolybdate-based entanglement frameworks successfully obtained by choosing the rigid ligand 1,4-bis(1-imidazolyl)benzene and different metal ions. Compound 1 is a 2-fold interpenetration 4-connected 3D framework; 2 exhibits a (5,6)-connected 3D self-penetrating topological net, and 3 shows a (4,6)-connected self-penetrating 3D framework. ► In compounds 1 and 2, the octamolybdate anions show the γ-isomer, while in 3 the octamolybdate anion is a δ-isomer. ► Optical band gap of these three compounds have been investigated and the results show that they are all wide gap semiconductors, which are 2.98 eV for 1, 3.42 eV for 2, and 2.88 eV for 3.
Co-reporter:Chunsheng Qin, Yanling Si, Guochun Yang, Zhongmin Su
Computational and Theoretical Chemistry 2011 Volume 966(1–3) pp:14-19
Publication Date(Web):June 2011
DOI:10.1016/j.comptc.2011.01.031
A theoretical study of polynuclear lithium compounds has shown that these species display large calculated nonlinear optical (NLO) responses. These compounds are based on aromatic subunits connected through polyhedral inorganic core (Li7O6 or Li8O6). These compounds show the calculated first hyperpolarizabilities (β) ranging from 262.55 to 16336.35 × 10−33 esu. The results show that subtle structural modification can substantially enhance the first hyperpolarizability. A basis for understanding the origin of these large NLO responses is proposed based on consideration of the molecular orbitals and electronic transition features of the compounds and the two-state model. Charge transfer from central core to the peripheral phenyl groups plays a key role in the nonlinear optical response. Moreover, the effects of different functionals and basis sets on first hyperpolarizability were systemically investigated.
Co-reporter:Dong-Lai Wang, Hong-Liang Xu, Yang-Yang Hu, Zhong-Min Su
Computational and Theoretical Chemistry 2011 Volume 966(1–3) pp:1-8
Publication Date(Web):June 2011
DOI:10.1016/j.comptc.2011.01.019
Density functional theory (DFT) calculations on the finite clusters of perfect and defective armchair (5, 5) and zigzag (9, 0) single-walled carbon nanotubes (SWCNTs) terminated by hydrogen atoms and fullerene hemispheres were carried out at the B3LYP/6-31G* level of theory. The most negative potentials and their positions, reactivity index in SWCNTs with no defects, with a carbon ad-dimer (CD) defect and with a Stone–Wales (SW) defect were compared. The results indicate that the CD defective sites in both the open-ended and cap-ended SWCNTs are chemically more reactive than the SW defective sites and the perfect sites. In contrast, the SW defect in the cap-ended armchair (5, 5), zigzag (9, 0) as well as the open-ended armchair (5, 5) SWCNTs has slight effect on the molecular electrostatic potential (MEP) compared with the MEP of pristine SWCNTs; the SW defective sites become more reactive than the perfect sites for open-ended zigzag (9, 0) SWCNT.
Co-reporter:Dong-Lai Wang, Hong-Liang Xu, Zhong-Min Su, Dong-Yan Hou
Computational and Theoretical Chemistry 2011 Volume 978(1–3) pp:166-171
Publication Date(Web):30 December 2011
DOI:10.1016/j.comptc.2011.10.005
The 89 classical C44 isomers, its anions and derivatives such as C44Xn (X = H, F, and Cl; n = 14 and 16) and M@C44 (M = He, Li, and Ca) have been systematically investigated by ab initio and density function methods. Although the D2:75 species has the least number of adjacent pentagons, the MP2 geometry optimizations suggest that the D3h:72 structure is favored by ∼11 kcal/mol over the D2:75 isomer. Systematic investigations of the electronic properties show that neutral D3h :72 is highly aromatic. In contrast, the two lowest energy isomers of C442-, D2:75 and D2:89, are highly aromatic. Compared with C50X10 (D5h), the large energy gaps and the aromatic character of C44X14 (D3h:72) indicate that they possess high stabilities. Furthermore, the predicted reaction energies of C44X14 (D3h:72) derivatives are highly exothermic, suggesting a considerable possibility for experimental realization. Investigations also show that the D2:75 endohedral structure is most stable for Ca@C44.Graphical abstractTo obtain the most stable C44q- (q = 0, 1, and 2) isomers, all 89 neutral, singly and doubly charged C44 fullerenes have been orderly optimized using B3LYP/3-21G, HF/6-31G*, B3LYP/6-31G*, B3LYP/6-31+G*, MP2/6-31G*, and MP2/TZVP methods. Exohedral derivatives C44Xn (X = H, F, and Cl; n = 14 and 16) and endohedral fullerenes M@C44 (M = He, Li, and Ca) based on the first four most stable parent cages have also been studied at B3LYP/6-31G* and MP2/6-31G*.Highlights► The MP2/6-31G* and MP2/TZVP calculations show that D3h:72 C44 is the most stable structure. ► D2:75 and D2:89 C442− are predicted to be the two most stable isomers, and they are highly aromatic. ► The calculated AEAs of the D2:75 and D2:89 isomers are in good agreement with experimental data. ► Exohedral derivatives C44X14 formed from the initial D3h:72 C44 are molecules with high stability. ► D2:75 is suggested as the most probable isomer for the experimentally observed Ca@C44.
Co-reporter:Fei Yu;Guochun Yang;Shuixing Wu;Yun Geng;Zhongmin Su
Theoretical Chemistry Accounts 2011 Volume 129( Issue 1) pp:45-51
Publication Date(Web):2011 May
DOI:10.1007/s00214-010-0883-7
Due to the different molecular stacking conformations, two kinds of intermolecular interactions, arene–arene π-stacking interaction and Cu–Cu interaction coexist in the polymorphs of [C6F5Cu]2(4,4′-bipy) crystals, 3-α and 3-β. However, the relative magnitude of the two kinds of intermolecular interactions in 3-α and 3-β is different. With the help of first-principle band structure calculations, the relationship between the charge transport abilities and the intermolecular interactions in the two polymorphs was investigated for the first time. The analysis of band structures and Г point wave functions of the band-edge state in the valence band of crystal 3-α shows that the Cu–Cu interaction so-called cuprophilic interaction determines the hole transport ability, although this interaction is weaker than that in crystal 2 of C6F5Cu(py) discussed in our previous work, which is a promising hole transport material. For polymorph crystal 3-β, the wave functions of LUMO are mainly localized on the bipyridine (bpy) groups, which are result from the arene–arene π-stacking interaction between the bpy groups. Such a π–π stacking interaction dominates the electron transport ability in the conduction band of 3-β and makes the electron main carrier for transporting. The results are also supported by the analysis of effective masses and density of states (DOS). Thus, the charge transport properties are dominated by different intermolecular interactions due to the different molecule stacking in the two polymorphs.
Co-reporter:Yun Geng;Haibin Li;Shuixing Wu;Yuai Duan;Zhongmin Su
Theoretical Chemistry Accounts 2011 Volume 129( Issue 2) pp:247-255
Publication Date(Web):2011 May
DOI:10.1007/s00214-011-0928-6
Fused thiophenes have been an important class of materials due to their intriguing organic optoelectronic application. Here, comparative theoretical investigation on the fluorescence and charge transport properties of dithienothiophene compounds (1 and 2) and their dioxide derivatives (3 and 4) was carried out to shed light on the role of the thienyl-S,S-dioxide unit. The lower HOMO, LUMO energy levels, and red-shift spectra (absorption and emission) of 3 and 4 compared with 1 and 2 were attributed to the electron-withdrawing nature of the thienyl-S,S-dioxide unit. The phenomenon that fluorescence quantum yield of 4 was significantly increased through thienyl-S,S-dioxidation, compared with those of 1 and 2, was analyzed by the evaluations of the radiative decay rates and the radiationless decay rates in theory at the single molecule level and the simulation of absorption spectrum of dimer of 1. For 1, a much higher hole mobility (0.12 cm2/V.s) calculated by carrier hopping model than the experimental value ~10−4 cm2/V.s was also further elucidated by molecular dynamics simulation. Furthermore, a preliminary investigation of the transport property of 3 was performed by combining the molecular dynamics simulation with dispersion-corrected B3LYP functional to provide insight into the effect of thienyl-S,S-dioxidation on the charge transport.
Co-reporter:Guochun Yang;Yanling Si;Yun Geng;Fei Yu;Qingxiu Wu
Theoretical Chemistry Accounts 2011 Volume 128( Issue 2) pp:257-264
Publication Date(Web):2011 January
DOI:10.1007/s00214-010-0841-4
The charge transport and photophysical properties of N-heteroquinones, which can function as n-type organic semiconductors in organic field-effect transistors (OFETs) with high electron mobility, were systematically investigated using hopping model, band theory, and time-dependent density functional theory (TDDFT). The calculated absorption spectra and electron mobility are in good agreement with experimental results. To the studied compounds, subtle structural modifications can greatly reduce the reorganization energy. There are two main kinds of intermolecular interaction forces of the studied compounds in the crystal, which result from intermolecular π–π and hydrogen bonds interactions, respectively. The results of hopping model show that the electron transport properties are mainly determined by pathways containing intermolecular π–π interactions, and hole transport properties are mainly determined by pathways containing intermolecular hydrogen bonds from the standpoint of transfer integral. Moreover, electronic transfer integral value increases with the enhancement of intermolecular overlap corresponding to the overlap extent of π–π packing. Hole transfer integral value decreases with decreasing the number of hydrogen bonds. This means that charge transport properties can be efficiently tuned by controlling the relative positions of the molecules and the number of hydrogen bonds. The analysis of band structure also supports the conclusion of hopping model.
Co-reporter:XiaoNa Sun;YongQing Qiu;ShiLing Sun;ChunGuang Liu;YanQing Du
Science China Chemistry 2011 Volume 54( Issue 7) pp:1086-1093
Publication Date(Web):2011 July
DOI:10.1007/s11426-011-4293-z
The polarizabilities and hyperpolarizabilities of the tetrahydropyrrole diradical in different electronic states have been investigated using ab initio and density functional theory (DFT) methods combined with the finite field (FF) approach. The polarizability average value αs is a maximum for the singlet state, while that for the closed-shell is a minimum. The trend in second hyperpolarizability average value γ is in good agreement with that for αs. The γ values of the singlet and triplet states are, respectively, about 3 and 2 times larger than that of the closed-shell. The order of the first hyperpolarizability total effective value βtot is βtot (closed-shell) > βtot (singlet) > βtot (triplet). The αs, βtot, and γ values of different electronic states obtained using the B3LYP and MP4SDQ methods are close to those obtained using the reliable CCSD method. The nonlinear optical (NLO) properties of two systems isoelectronic with the tetrahydropyrrole diradical — cyclopentane and tetrahydrofuran diradicals — show that the polarizabilities and hyperpolarizabilities of these systems are all smaller than those of the tetrahydropyrrole diradical in the three electronic states.
Co-reporter:Hong-Liang Xu;Shi-Ling Sun;Shabbir Muhammad
Theoretical Chemistry Accounts 2011 Volume 128( Issue 2) pp:241-248
Publication Date(Web):2011 January
DOI:10.1007/s00214-010-0837-0
Significant alkali-metal-doped effects on the structure and the first hyperpolarizability (β0) of effective multi-nitrogen complexant tris[(2-imidazolyl)methyl]amine (TIMA) are investigated. Three imidazoles of TIMA like three blades of propeller connect with methyls by the C–C single bonds. Because of the three C–C single-bond cooperative rotations, the TIMA behaves with great flexibility, and it is a high-performance multi-nitrogen complexant for the alkali metal doping. Thus, the new complexes Am-TIMA (Am = Li, Na, and K) with electride characteristic have diffuse excess electron than the reported electride-type system due to the strong interaction between the complexant TIMA and alkali metal. For the first hyperpolarizability, three engaging electrides Am-TIMA with the diffuse excess electrons exhibit considerably large β0 values using the MP2 (full) method and the β0 values of new electrides are greatly larger (3,464–29,705 times) than that (338 au) of TIMA. Surprisingly, the K-TIMA sets a new record β0 value to be 1.00 × 107 au which far exceeds than that (3,694–76,978 au) of the reported electride-type system Li@calix[4]pyrrole (J Am Chem Soc 127:10977–10981, 2005) and Lin−H−(CF2−CH2)3−H (n = 1, 2) (J Am Chem Soc 129:2967–2970, 2007) and 31,123 au of the organometallic system (J Am Chem Soc 121:4047–4053, 1999) Ru(trans-4,4′-diethylaminostyryl-2,2′-bipyridine)32+, as well as 1.23 × 106 au of the large donor-CNT systems (Nano Lett 8:2814–2818, 2008). Clearly, the alkali-metal-doped effect on the first hyperpolarizability is very dramatic for the high-performance multi-nitrogen complexant TIMA. Considering simple possibility from molecule to material, the β0 values of optimized Li-TIMA-dimer and Li-TIMA-tetramer are investigated by BHandHLYP method. Interestingly, results show that the order of β0 value is Li-TIMA-monomer < Li-TIMA-dimer < Li-TIMA-tetramer. So the new three-propeller-blade-shaped electrides can be considered as candidates for high-performance nonlinear optical materials.
Co-reporter:Dr. Rong-Lin Zhong;Dr. Ji Zhang;Dr. Shabbir Muhammad;Dr. Yang-Yang Hu;Dr. Hong-Liang Xu; Zhong-Min Su
Chemistry - A European Journal 2011 Volume 17( Issue 42) pp:11773-11779
Publication Date(Web):
DOI:10.1002/chem.201101430
Abstract
On the basis of the famous staggered biphenalenyl diradical π dimer 1, the eclipsed biphenalenyl (1 a), with no centrosymmetry, was obtained by rotating a layer of 1 by 60° around its central axis. Furthermore, the central carbon atoms of 1 and 1 a were substituted by boron and nitrogen atoms to form 2 and 2 a with a novel 2e–12c bond. We found that the novel 2e–12c bond is formed by the electron pair of the occupied orbital of the phenalenyl monomer substituted by the nitrogen atom and the unoccupied orbital of the phenalenyl monomer substituted by the boron atom. As a result of the novel 2e–12c bond, 2 and 2 a exhibit a fascinating interlayer charge-transfer transition character, which results in a significant difference in the dipole moments (Δμ) between the ground state and the crucial excited state. The values of Δμ for 2 and 2 a are 6.4315 and 6.9253 Debye, clearly larger than the values of 0 and 0.0015 Debye for 1 and 1 a. Significantly, the boron/nitrogen substitution effect can greatly enhance the first hyperpolarizabilities (β0) of 2 and 2 a with a novel 2e–12c bond compared with 1 and 1 a with a traditional 2e–12c bond: 0 and 19 a.u. for 1 and 1 a are much lower than 3516 and 12272 a.u. for 2 and 2 a. Furthermore, the interaction energies (Eint)of 2 and 2 a are larger than those of 1 and 1 a, which could be considered as a signature of reliability for the newly designed dimers. Our present work will be beneficial for further theoretical and experimental studies on the properties of molecules with the novel 2e–12c bond.
Co-reporter:Dr. Yan Shen; Jun Peng;Dr. Haijun Pang;Dr. Pengpeng Zhang;Dan Chen;Changyun Chen;Huanqiu Zhang;Cuili Meng ; Zhongmin Su
Chemistry - A European Journal 2011 Volume 17( Issue 13) pp:3657-3662
Publication Date(Web):
DOI:10.1002/chem.201001837
Abstract
In this paper, the preparation of ascorbic acid (AA)-doped polyoxometalate (SiW12-AA) microtubes is described. The SiW12-AA microtubes convert to heteropoly blue microtubes upon exposure to ammonia gas, which is an ammonia-triggered solid–solid redox reaction between AA molecules and polyoxometalates, and can possibly be applied to a chemical sensor for detecting ammonia and volatile organic amines. Furthermore, the SiW12-AA microtubes have been applied to the in situ synthesis of Ag nanoparticles (NPs) through the redox reaction between the AA component and Ag+ ions occurring on the surfaces of the SiW12-AA microtubes to give silver NPs immobilized on polyoxometalate microtubes (Ag@SiW12).
Co-reporter:Dr. Yinghui Wang ; Bin Li;Liming Zhang;Qinghui Zuo ;Peng Li ;Dr. Jun Zhang; Zhongmin Su
ChemPhysChem 2011 Volume 12( Issue 2) pp:349-355
Publication Date(Web):
DOI:10.1002/cphc.201000884
Abstract
An optical oxygen sensor based on an EuIII complex/polystyrene (PS) composite nanofibrous membrane is prepared by electrospinning. The emission intensity of [Eu(TTA)3(phencarz)] (TTA=2-thenoyltrifluoroacetonate, phencarz=2-(N-ethylcarbazolyl-4)imidazo[4,5-f]1,10-phenanthroline) decreases with increasing oxygen concentration, and thus the [Eu(TTA)3 (phencarz)]/PS composite nanofibrous membranes can be used as an optical oxygen-sensing material based on emission quenching caused by oxygen. Elemental analysis, UV/Vis absorption spectra, scanning electron microscopy (SEM), fluorescence microscopy, luminescence-intensity quenching Stern–Volmer plots, and excited-state decay analysis are used to characterize the obtained oxygen-sensing materials. A high sensitivity (IN2/IO2) of 3.38 and short response and recovery times (t=5.0, t=8.0 s) are obtained. These results are the best values reported for oxygen sensors based on EuIII complexes. The high surface area-to-volume ratio and porous structure of the electrospun nanofibrous membranes are taken to be responsible for the outstanding performance.
Co-reporter:Chun-Guang Liu ; Zhong-Min Su ; Xiao-Hui Guan ;Shabbir Muhammad
The Journal of Physical Chemistry C 2011 Volume 115(Issue 48) pp:23946-23954
Publication Date(Web):October 28, 2011
DOI:10.1021/jp2049958
The switching of second-order nonlinear optical (NLO) properties for a tetrathiafulvalene (TTF) derivative across the six stable states has been studied by using the density functional theory (DFT) calculations. The redox-active TTF unit and a photoisomerized chromophore 1,2-dithienylperfluorocyclopentene (DTE) have been implemented to switch the second-order NLO responses. Our DFT calculations with three functionals demonstrate that introduction of the DTE moiety into the π-conjugated bridge can significantly enhance the second-order NLO response relevant to the donor/acceptor end in this work. Our DFT calculations illustrate that photoisomerization bring forth a large change in the geometry of the series of compounds. The closed-ring form possesses a good π-conjugation relative to the open-ring form and thus a large second-order NLO response. The electronic structure analysis shows that the TTF unit will perform as an oxidation center in the one- and two-electron-oxidation processes. The one- and two-electron-oxidized species have better planar structures of TTF unit than its neutral compound, which ultimately leads to the low excited energy and enhances the static first hyperpolarizability. Our present DFT calculations using three functionals show that the TTF derivative 4 can switch the second-order NLO properties across six stable states, which is a rare example in previously reported second-order NLO switches.
Co-reporter:Yang-Yang Hu ; Shi-Ling Sun ; Rong-Lin Zhong ; Hong-Liang Xu
The Journal of Physical Chemistry C 2011 Volume 115(Issue 38) pp:18545-18551
Publication Date(Web):August 24, 2011
DOI:10.1021/jp2069336
Trumpet-shaped carbon nanocone (CNC) is used as a π-conjugated bridge to design high-performance nonlinear optical (NLO) material. Owing to the attractive trumpet-shaped structure, O2N–CNC–NH2 exhibits nonlinear optical behavior that is both surprisingly and qualitatively distinct from conventional π-conjugated organic species. It has been shown that the electron-acceptor nitryl (−NO2) at the apex of the trumpet and the electron-donor amino (−NH2) at the bottom edge of the trumpet to form O2N–CNC–NH2 is beneficial to improve the NLO response of CNC. Significantly, the first hyperpolarizabilities (βtot) of O2N–CNC–NH2 show an arc-shaped change as −NH2 substituting different H atoms along the arc-shaped bottom edge of CNC. Interestingly, the βtot values of H–CNC–NH2 also show an arc-shaped change. By comparison of the βtot values of O2N–CNC–NH2 and H–CNC–NH2, it has been found that no matter which bottom H of H–CNC–H is substituted by NH2, the βtot value of H–CNC–NH2 will be increased about 1.6 times after substituting the top H with −NO2 into the corresponding molecule. This study may stimulate the search for new types of NLO materials based on CNC for application.
Co-reporter:Hong-Liang Xu ; Rong-Lin Zhong ; Shi-Ling Sun
The Journal of Physical Chemistry C 2011 Volume 115(Issue 33) pp:16340-16346
Publication Date(Web):July 15, 2011
DOI:10.1021/jp204740d
Very recently, tubiform multilithium salts have been investigated as new candidates for high-performance nonlinear optical (NLO) materials because of their large static first hyperpolarizability (β0) ( J. Phys. Chem. C 2009, 113, 4984−4986). The interesting question of how to further enhance the β0 value of the multilithium salt, by widening or lengthening the tubiform cyclacene, is studied in this work. On one hand, the effect of widening on the β0 value of tubiform multilithium salts is investigated using Lin-[n]cyclacene (n = 5–8). We found that the β0 value (15497 au) of Li8-[8]cyclacene is 3 times larger than the 5028 au value of the Li5-[5]cyclacene, with the total number of carbon atoms increasing from 20 to 32. On the other hand, lengthening Li5-[5]cyclacene to form Li5–CNT (5,0) results in the dramatically enhanced β0 value of 146642 au, which is 29 times larger than the value for the Li5-[5]cyclacene, even though the total number of carbon atoms is increased only from 20 to 30. Further, the second harmonic generation (SHG) β(−2ω;ω,ω) and the electro-optical Pockels effect (EOPE) β(−ω;ω,0) increase with an increase in frequency (ω) from 0.0000 to 0.0200 au. Because cyclacenes have been proposed to be super-short single-walled carbon nanotubes, it is our expectation that this work could provide more useful information for the development of nonlinear optical nanomaterials.
Co-reporter:Chun-Guang Liu ; Xiao-Hui Guan
The Journal of Physical Chemistry C 2011 Volume 115(Issue 13) pp:6024-6032
Publication Date(Web):March 14, 2011
DOI:10.1021/jp111797n
The redox-switchable 2D second-order nonlinear optical (NLO) property of a series of tetrathiafulvalene (TTF) derivatives has been studied based on the density functional theory (DFT) calculations. The redox-active TTF unit has been considered as a manipulative center for switching the 2D second-order NLO properties. Our DFT calculations show that introduction of the TTF unit cannot effectively enhance the second-order NLO properties relative to the reference system 1 because the nonplane embowed arrangement of the TTF unit reduces the electron donor capacity. The electronic structure analysis shows that the TTF unit acts as the oxidized center in one- and two-electron-oxidized processes for 5. A significant transformation on the structure of the TTF unit, the TTF unit changes from the embowed structure to a planar structure, has been found in the series of oxidized processes according to DFT-optimized calculations. This leads to the low excited energy and different charge transfer features of the oxidized species relative to its reduced parents, and thus enhances the static first hyperpolarizabilities. The β value of one- and two-electron-oxidized species is at least ∼15 and ∼8.6 times as large as that of its reduced parents according to our DFT calculations. Simultaneously, the oxidized process increases the contributions from the y-polarized transition, and thus improves the 2D second-order NLO property.
Co-reporter:Guochun Yang, Yanling Si, and Zhongmin Su
The Journal of Physical Chemistry A 2011 Volume 115(Issue 46) pp:13356-13363
Publication Date(Web):October 14, 2011
DOI:10.1021/jp204860x
Time-dependent density functional theory (TDDFT) calculations have been used to investigate UV/CD spectra and nonlinear optical (NLO) property of the C60-fullerene bisadduct (R,R,f,sA)-[CD(+)280] for the first time. The electron transition natures of the four main measured bands are analyzed, and their results are used to designate the excited states involved in an electron-transfer process of the studied compound. On a comparative scale, the predicted excitation energies and oscillator strengths are in reasonable agreement with the observed values, demonstrating the efficiency of TDDFT in predicting the localized and charge transfer transitions. The good agreement between the experimental and the simulated CD spectra shows that TDDFT calculations can be used to assign the absolute configurations (ACs) of chiral fullerene C60 derivatives with high confidence. The observed large dissymmetry ratio g (g = Δε/ε) at about 700 nm results from the orbital characters of the local fullerene excited state, which leads to large transition magnetic dipole moment and small transition electronic dipole moment. The different functionals and solvent effects on UV/CD spectra were also considered. The studied compound has a possibility to be an excellent second-order NLO material from the standpoint of transparency and large second-order polarizability value.
Co-reporter:Shabbir Muhammad, Hongliang Xu, and Zhongmin Su
The Journal of Physical Chemistry A 2011 Volume 115(Issue 5) pp:923-931
Publication Date(Web):January 12, 2011
DOI:10.1021/jp110401f
We present the design of fluoro derivatives of B10H14 and Li@B10H14 baskets. A synergistic effect of conical push and inward pull (reported independently in previous lithium nonlinear optical (NLO) complexes) has been explored in these derivatives to achieve a robustly large NLO response and a higher vertical ionization potential. Li@1,3,6,9-F4B10H10, Li@6,9-F2B10H12, and Li@2,4,6,9-F4B10H10 exhibit first hyperpolarizability (β0) values as large as 181 624, 133 199, and 32 314 au; their vertical ionization potentials are 6.45, 6.30, and 6.78 eV, respectively. These values are significantly higher than those previously reported in Li-doped fluorocarbon chains at the same MP2/6-31+G* level of theory (Xu, H. L.; Li, Z. R.; Wu, D.; Wang, B. Q.; Li, Y.; Gu, F. L.; Aoki, Y. J. Am. Chem. Soc. 2007, 129, 2967). They also exceed those from our earlier designed Li@B10H14 basket (Muhammad, S.; Xu, H. L.; Liao, Y.; Kan, Y. H.; Su, Z. M. J. Am. Chem. Soc. 2009, 131, 2967). In addition, new quantum chemical calculations of enthalpies of reaction (ΔrH°) at 298 K for B10H14 and its lithium/fluoro derivatives highlight the changes in their thermodynamical aspects. The calculated enthalpies of lithiation reactions are −10.04, −11.29, and −13.18 kcal/mol for B10H14, 6,9-F2B10H12, and 2,4-F2B10H12, respectively, demonstrating a higher probability of fluoro decaboranes for reaction with lithium. The obtained results not only explain the effect of position and number dependence of substituted fluoro atom(s) in B10H14 and Li@B10H14 but also elucidate a synergistic behavior to polarize a lithium excess electron for high NLO responses and vertical ionization potentials.
Co-reporter:Cui-Cui Zhang, Hong-Liang Xu, Yang-Yang Hu, Shi-Ling Sun, and Zhong-Min Su
The Journal of Physical Chemistry A 2011 Volume 115(Issue 10) pp:2035-2040
Publication Date(Web):February 21, 2011
DOI:10.1021/jp110412n
On the basis of the n-acenes (n = 1, 2, 3 and 4), the α-Li@n-acenes and β-Li@n-acenes salts were selected to investigate how increasing the number n of conjugated benzenoid rings affects the linear and nonlinear optical responses. The α-Li@n-acenes and β-Li@n-acenes salts are obtained by a lithium atom substituting the α-H and β-H, respectively. In the present work, both ab initio (HF and MP2) and DFT (B3LYP, BhandHLYP, M05-2X, and CAM-B3LYP) methods are adopted to calculate the polarizability (α0) and first hyperpolarizability (βtot) of the α-Li@n-acenes and β-Li@n-acenes salts. MP2 results show that the α0 values of both classes of lithium salts increase with increasing number n of conjugated benzenoid rings. Interestingly, we found that the βtot values of α-Li@n-acenes and β-Li@n-acenes salts take on opposite trends: the βtot values of α-Li@n-acenes are decreasing slowly (2187 for α-Li@benzene > 1978 for α-Li@naphthalene > 1898 for α-Li@anthrecene > 1830 au for α-Li@tetracene) and inceasing remarkably (2738 for β-Li@naphthalene < 3186 for β-Li@anthrecene < 3314 au for β-Li@tetracene) for β-Li@n-acenes. Furthermore, we found that the βtot values (2738−3314 au) of the β-Li@n-acenes are larger than those of the α-Li@n-acenes (1830−2187 au). On the other hand, comparing the results of different methods, the βtot values obtained by the M05-2X and CAM-B3LYP methods reproduce the polarizability and first hyperpolarizability of the α-Li@n-acenes and β-Li@n-acenes salts well, which test and verify the results of the MP2 method. Our present work may be beneficial to development of high-performance organic NLO optical materials.
Co-reporter:Dong-Ying Du, Jun-Sheng Qin, Gang Yuan, Ya-Qian Lan, Xin-Long Wang, Kui-Zhan Shao, Zhong-Min Su
Solid State Sciences 2011 13(5) pp: 1115-1121
Publication Date(Web):
DOI:10.1016/j.solidstatesciences.2011.01.007
Co-reporter:Hongze Gao, Hongyu Zhang, Houyu Zhang, Yun Gen, and Zhong-Min Su
The Journal of Physical Chemistry A 2011 Volume 115(Issue 33) pp:9259-9264
Publication Date(Web):August 3, 2011
DOI:10.1021/jp202976m
The charge carrier transporting ability in the polymorphism of tris(8-hydroxyquinolinato)aluminum(III) (Alq3) has been studied using density functional theory (DFT) and Marcus charge transport theory. α- and β-Alq3 composed of mer-Alq3 molecules have stronger electron-transporting property (n-type materials) compared with their hole-transporting ability. In contrast, γ- and δ-Alq3 formed by fac-Alq3 molecules possess stronger hole-transporting character than their electron-transporting ability. The detailed theoretical calculations indicate the reason lies in the differences of HOMO and LUMO distribution states of the two kinds of isomers, and the different molecular packing modes of charge-transporting pathways for different phases.
Co-reporter:Xianchun Liu, Yan Xing, Xinlong Wang, Hongbin Xu, Xizheng Liu, Kuizhan Shao and Zhongmin Su
Chemical Communications 2010 vol. 46(Issue 15) pp:2614-2616
Publication Date(Web):24 Feb 2010
DOI:10.1039/B923028A
A new chiral open-framework cobalt phosphite [C6N2H14][Co(HPO3)2] (1) with helical channels has been synthesized under solvothermal conditions in the presence of achiral organic amine. The circular dichroism (CD) spectrum indicates that the bulk crystals have optical activity. Magnetic measurements reveal that 1 is a weak ferromagnet.
Co-reporter:Hai-jun Pang, Jun Peng, Chun-jing Zhang, Yang-guang Li, Peng-peng Zhang, Hui-yuan Ma and Zhong-min Su
Chemical Communications 2010 vol. 46(Issue 28) pp:5097-5099
Publication Date(Web):15 Jun 2010
DOI:10.1039/C003048A
A novel compound, [Cu(bimb)]2(HPW12O40)·3H2O (bimb = 1,4-bis(imidazol-1-ylmethyl)biphenyl) with a polyoxometalate-encapsulated 3D metal–organic pseudo-rotaxane framework that can be described as a 2-fold dianet topology showing well-defined 1D nano-scale tunnels, has been synthesized hydrothermally, and its de-/rehydration behavior has been investigated.
Co-reporter:Fang Chai, Chungang Wang, Tingting Wang, Lu Li and Zhongmin Su
ACS Applied Materials & Interfaces 2010 Volume 2(Issue 5) pp:1466
Publication Date(Web):April 29, 2010
DOI:10.1021/am100107k
A facile, cost-effective and sensitive colorimetric detection method for Pb2+ has been developed by using glutathione functionalized gold nanoparticles (GSH-GNPs). The sensitivity and selectivity of detection were investigated in detail. The GSH-GNPs could be induced to aggregate immediately in the presence of Pb2+, especially after the addition of 1 M NaCl aqueous solution. The Pb2+ could be detected by colorimetric response of GNPs that could be monitored by a UV−vis spectrophotometer or even naked eyes, and the detection limit could reach 100 nM. The GSH-GNPs bound by Pb2+ showed excellent selectivity compared to other metal ions (Hg2+, Mg2+, Zn2+, Ni2+, Cu2+, Co2+, Ca2+, Mn2+, Fe2+, Cd2+, Ba2+, and Cr3+), which led to prominent color change. This provided a simple and effective colorimetric sensor (no enzyme or DNA) for on-site and real-time detection of Pb2+. Most importantly, this probe was also applied to determine the Pb2+ in the lake samples with low interference and high sensitivity.Keywords: colorimetry; detection; glutathione; gold nanoparticle; lead(II) ion
Co-reporter:Fu-Qiang Zhang ; Wei Guan ; Yin-Tang Zhang ; Mao-Tian Xu ; Jian Li
Inorganic Chemistry 2010 Volume 49(Issue 12) pp:5472-5481
Publication Date(Web):May 25, 2010
DOI:10.1021/ic100343t
Density functional theory calculations have been carried out to investigate the α, β, γ, δ, and ε isomers of [(MnO4)Me12Sb12O24]6− (Me = CH3) anions, which are simplified Baker−Figgis models of Keggin-type antimonate complexes in experiments. It is found that the stability order of the five isomers (α < β < γ < δ < ε) perfectly reverses to the well-known trend of the classical Keggin [PW12O40]3− anions (α > β > γ > δ > ε), despite their significant similarities in frameworks. On the basis of the building block decomposition method, the stabilizing effect of the edge-sharing [Sb2(μ-O)2Me2] fragment inside γ, δ, and ε structures is confirmed and found to originate from its two energy-favorable components rather than itself as an indivisible unit. Similar behavior is also held by the destabilizing [W2(μ-O)2O2] fragment in [PW12O40]3−; however, the well-accepted electrostatic repulsion between the short WVI−WVI contacts cannot be taken as direct evidence. Notably, in the assembly of the [(MnO4)Me12Sb12O24]6− structure, all of the octahedral building units incline to compress axially and elongate horizontally, and this is exactly opposite to the deformation pattern observed in the building blocks of Keggin tungstates, which tend to elongate axially and compress horizontally, thus giving rise to the inverted stability order. Furthermore, energy decomposition analysis reveals that the intrinsic property of the anion comes from the spatial arrangements of the metal−oxygen cage and does not change significantly with the type and charge of the encapsulated anion.
Co-reporter:Hong-Ying Zang, Ke Tan, Wei Guan, Shun-Li Li, Guang-Sheng Yang, Kui-Zhan Shao, Li-Kai Yan and Zhong-Min Su
CrystEngComm 2010 vol. 12(Issue 11) pp:3684-3690
Publication Date(Web):07 Jul 2010
DOI:10.1039/C001593H
Three novel compounds co-existing isomers or forms of polymolybdate, namely, {[Ni(γ-Mo8O26)(H2L1)2(H2O)2](γ-Mo8O26)(H2L1)}·2H2O (1), {[Co(γ-Mo8O26)(H2L1)2(H2O)2](β-Mo8O26)[Co(H2O)6]}·6H2O (2) and [CuI6L24(β-Mo8O26)(Mo6O19)] (3), where L1 = 1,1′-bis(pyridin-3-ylmethyl)-2,2′-biimidazole, L2 = 3-((1H-1,2,4-triazol-1-yl)methyl)pyridine, have been successfully synthesized. Crystal structure analysis reveals that 1 is a 1D + 1D supramolecular structure containing both γ-[Mo8O26N2] and γ-[Mo8O28] units; 2 has γ-[Mo8O26]4− as well as β-[Mo8O26]4− anions and 3 is a (3,4,6)-connected 2D network containing both β-[Mo8O26]4− and [Mo6O19]2− anions. According to quantum chemical calculations, the transformation from β-[Mo8O26]4− to [Mo6O19]2− is spontaneous in aqueous solution. Furthermore, we have investigated the optical band gap of compounds 1 and 3, with 3 showing a lower band gap (1.69 eV). Additionally, the photoluminescent property of 3 is also studied, which can be assigned to metal-to-ligand charge transfer (MLCT).
Co-reporter:Shuang Wang, Hongying Zang, Chunyi Sun, Guangjuan Xu, Xinlong Wang, Kuizhan Shao, Yaqian Lan and Zhongmin Su
CrystEngComm 2010 vol. 12(Issue 11) pp:3458-3462
Publication Date(Web):06 Jul 2010
DOI:10.1039/C0CE00137F
Two genuine supramolecular isomers (Lp = p-[CH(pz)2]2C6H4), have been successfully obtained under similar solvothermal conditions by the reaction of arene-linked p-bis[bis(1-pyrazolyl)methyl]benzene ligand with AgNO3. Both of the compounds are made of unusual meso-helical chains and present a three-dimensional (3D) supramolecular structure. Compounds 1 and 2 were characterized by elemental analyses, IR spectra, single-crystal X-ray diffraction and thermogravimetric (TG) analyses. The fluorescence property of them has also been studied.
Co-reporter:Jing-Dong Feng, Kui-Zhan Shao, Shu-Wei Tang, Rong-Shun Wang and Zhong-Min Su
CrystEngComm 2010 vol. 12(Issue 5) pp:1401-1403
Publication Date(Web):09 Apr 2010
DOI:10.1039/B923583C
The first ionothermal case of open-framework metal phosphite, Zn3(HPO3)4·2C6H11N2, denoted NIS-3, was prepared using 1-ethyl-3-methyl imidazolium bromide as solvent and template.
Co-reporter:Gang Yuan, Kui-Zhan Shao, Xin-Long Wang, Ya-Qian Lan, Dong-Ying Du and Zhong-Min Su
CrystEngComm 2010 vol. 12(Issue 4) pp:1147-1152
Publication Date(Web):02 Dec 2009
DOI:10.1039/B917779E
Seven novel coordination polymers, [Ln(tda)(H2O)]n·2nH2O [Ln = La(1), Ce(2), Pr(3), Nd(4), Eu(5), Sm(6) and Gd(7)] (H3tda = 1H-1,2,3-triazole-4,5-dicarboxylic acid), have been synthesized under hydrothermal conditions and characterized by elemental analysis, IR, thermogravimetric analysis (TGA), and single-crystal X-ray diffraction. Compounds 1–7 are isostructural and exhibit novel 3D chiral framework based on the linkage of chiral columns constructed from the interweaving two different kinds of helical chains. Interesting channels exist like labyrinths in the frameworks formed by interconnecting 16Ln-based cage-like building units. Furthermore, the photoluminescence property of 6 and the magnetic property of 7 have been studied.
Co-reporter:Hong-Ying Zang, Ya-Qian Lan, Guang-Sheng Yang, Xin-Long Wang, Kui-Zhan Shao, Guang-Juan Xu and Zhong-Min Su
CrystEngComm 2010 vol. 12(Issue 2) pp:434-445
Publication Date(Web):15 Sep 2009
DOI:10.1039/B911881K
Six new transition-metal complexes modified octamolybdate compounds have been successfully synthesized with two kinds of V-shaped flexible organic ligands under hydrothermal conditions, namely, [CuII(HL1)2(H2O)2(Mo8O26)] (1), [CuII(L1)2(Mo8O26)0.5] (2), [CuII(HL2)2(Mo8O26)]·2H2O (3), [CuII2(L2)4(Mo8O26)]·3H2O (4), [CuI3(L2)2(Mo8O26)0.5Cl] (5), [CuI4(L2)4(Mo8O26)] (6) where L1 = 3-((1H-imidazol-1-yl)methyl)pyridine, L2 = 3-((1H-1,2,4-triazol-1-yl)methyl)pyridine. Their crystal structures have been determined by X-ray single-crystal diffraction, elemental analyses, IR spectra, and thermogravimetric analyses (TGA). With L1 ligand, two compounds were obtained: compound 1 is a 1D β-octamolybdate based chain and compound 2 is a 2D 4-connected network most strikingly, whose octamolybdate anion shows a θ-isomer. With L2 ligand, compounds 3, 4, 5 and 6 were obtained, respectively. In compound 3, 1D β-octamolybdate-based metal–organic chains are further extended by hydrogen bonds to build a 3D supramolecular structure, while in compound 4, every two adjacent double-helical metal–organic chains are bridged by β-octamolybdate anions to generate a 2D 3-connected network. When CuSO4·5H2O was substituted by CuCl, two CuI compounds were attained. Interestingly, in compound 5 the octamolybdate anion threads through the metal–organic loop presenting a fascinating 2D polythreading topology, and in compound 6 the octamolybdate anions pillar the railroad-like metal–organic strands to generate a 2D 3-connected network. The structural differences among compounds 1–6 indicate the importance of the different coordination mode of organic ligands and various polyoxoanions, and of the pH values of the systems for the framework formation. Furthermore, the semiconducting properties of compounds 1–5 were investigated, and photoluminescence as well as photoconductivity for compound 5 were also studied. Compound 5 has a comparatively lower band gap and is photoconductive in the UV-vis range, so it can be potentially applied as a semiconductor of photoelectronic detectors.
Co-reporter:Shabbir Muhammad, Hongliang Xu, Muhammad Ramzan Saeed Ashraf Janjua, Zhongmin Su and Muhammad Nadeem
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 18) pp:4791-4799
Publication Date(Web):24 Mar 2010
DOI:10.1039/B924241D
A novel sequence for reversible second-order nonlinear optical (NLO) molecular switching with protonation/deprotonation has been achieved and tuned as well. The NLO switching with first hyperpolarizabilities (β0) as low as 14 × 10−30 esu (Off-phase) and as large as 1189 × 10−30 esu (On-phase) have been computed by using density functional theory (DFT). Remarkably large differences between the β0 values of benzimidazole containing chromophores and their deprotonated anions have shown their significant potential for a new type of NLO molecular switching, as (1-(4-methoxyphenyl)-2-(2-thienyl)pyrrolyl)-1,3-benzimidazole anion (1−) has a β0 value computed to be 61 × 10−30 esu, which is 4 times larger than its neutral molecule 1. This β0 value has been tuned up to 2028 × 10−30 esu by effective substitutions in the derivatives of 1− (1a−, 1b−, 1c−, and 1d−). Interestingly, the substituted compounds have illustrated robustly large off–on NLO switching with a difference in β0 values of 7, 63, 85 and 75 times larger than their neutral counterparts, respectively. TD-DFT calculations along with natural bond orbital (NBO), frontier molecular orbitals (FMOs) and molecular electrostatic potential (MEP) analyses show that the abstraction of an imido proton brings about a change in push–pull configurations resulting in a red shift for both absorption and emission spectra which subsequently leads to a high performance second-order NLO molecular switching. A similar trend of NLO switching in F− compounds of these chromophores has also been observed with significantly large β0 values having analogous electro-optical properties like deprotonated anions. Furthermore, gas-phase acidity (GPA) calculations for the neutral molecule 1 and its derivatives (1a, 1b, 1c, and 1d) have also revealed that these are rationally potent nitrogen acids and can easily be dissociated to produce stable deprotonated anions.
Co-reporter:Jing-Dong Feng, Shu-Wei Tang, Kui-Zhan Shao, Rong-Shun Wang, Chan Yao, Hai-Ming Xie and Zhong-Min Su
CrystEngComm 2010 vol. 12(Issue 11) pp:3448-3451
Publication Date(Web):25 Jun 2010
DOI:10.1039/C002882G
A novel non-centrosymmetric zinc phosphate, Zn(HPO4)(H2PO4)·C6H11N2 (denoted NIS-4), was ionothermally synthesized. The framework exhibits fascinating helical channels and remarkably low framework density (FD). Meanwhile, an ionic liquid cation plays a significant structure-directing role during the course of crystallization.
Co-reporter:Ai-xiang Tian, Jun Ying, Jun Peng, Jing-quan Sha, Zhong-min Su, Hai-jun Pang, Peng-peng Zhang, Yuan Chen, Min Zhu and Yan Shen
Crystal Growth & Design 2010 Volume 10(Issue 3) pp:1104-1110
Publication Date(Web):February 3, 2010
DOI:10.1021/cg900806k
Three inorganic−organic hybrids based on polyoxometalate (POM), [Cu6(bbtz)6(HPM12O40)]·2H2O (M = Mo, 1; W, 2) and [Cu6(trz)2(bbtz)2(SiW12O40)] (3) (trz = 1-H-1,2,4-triazole, bbtz = 1,4-bis(1,2,4-triazol-1-ylmethyl)benzene), were synthesized under hydrothermal conditions, and structurally characterized. Through the use of the flexible ligand bbtz, isostructural compounds 1 and 2 with interpenetrating structures were obtained. In compound 1, ladder-like chains exist, in which the PMo12 anions act as “middle rails”. These chains are linked by wave-like [Cu(bbtz)]nn+ lines to construct a three-dimensional (3D) framework. Two such frameworks penetrate each other to construct a 2-fold interpenetrating structure. By introducing the rigid ligand trz, compound 3 with an un-interpenetrating structure is obtained. In compound 3, two-dimensional (2D) (63)2 metal organic framework (MOF) layers exist, which are linked by Keggin anions to construct a 3D (44·62)(63)2 framework. The differences between these compounds should be ascribed to the introduction of the rigid molecule trz that plays a role in restraining the formation of interpenetrating structures.
Co-reporter:Shun-Li Li, Ke Tan, Ya-Qian Lan, Jun-Sheng Qin, Mei-Na Li, Dong-Ying Du, Hong-Ying Zang and Zhong-Min Su
Crystal Growth & Design 2010 Volume 10(Issue 4) pp:1699-1705
Publication Date(Web):February 25, 2010
DOI:10.1021/cg9012763
Six binary metal−organic compounds, namely, [Cd(L)2] (1), α-[Cd(L)2(H2O)] (2), β-[Cd(L)2(H2O)] (3a and 3b), γ-[Cd(L)2(H2O)] (4), and [Cd(L)2(H2O)2] (5), where HL = 4-(pyridin-3-ylmethoxy)benzoic acid, have been synthesized under hydrothermal conditions. Their structures were determined by single-crystal X-ray diffraction analyses and further characterized by elemental analyses, IR spectra, and thermogravimetric (TG) analyses. The structure of 1 is a two-dimensional (2D) → 2D 3-fold parallel interpenetrating network with (4,4) grid. Compounds 2, 3, and 4 are supramolecular isomers formed by Cd(II) cations, L− ligand, and water molecule, showing a 2D → 2D 2-fold parallel interpenetrating (4,4) network for 2, single (4,4) network for 3a and 3b, and (66) topological net for 4 through using different reaction materials. Compounds 3a and 3b show chiral structures by spontaneous resolution using an achiral ligand. In compound 5, Cd(II) cations are linked by L− ligands to form an infinite chain. Comparing these structures, the number of coordination water molecules, the coordination fashion of organic ligands, and reaction conditions play fundamental roles in the formation of the final products. In addition, the luminescent properties of these compounds are discussed.
Co-reporter:Li-Li Shi, Yun Geng, Hong-Ze Gao, Zhong-Min Su and Zhi-Jian Wu
Dalton Transactions 2010 vol. 39(Issue 33) pp:7733-7740
Publication Date(Web):23 Jul 2010
DOI:10.1039/C0DT00146E
The complexes AlQ3 and Ir(ppy)3 (Q = 8-hydroxyquinolate; ppy = 2-phenylpyridyl) are typical green emitting fluorescence and phosphorescence materials, respectively. Here we hybridize Ir(ppy)3 with AlQ3 to investigate the optoelectronic properties of the Ir(III)-centred derivatives including (ppy)2IrQ, (ppy)IrQ2 and IrQ3 by using density functional methods. Our calculations show that the derivative Ir(III) complexes are red emitting phosphorescence materials. The characters of the lowest triplet excited states for these Ir(III) complexes are mainly dominated by the 8-hydroxyquinolate ligand. IrQ3 and (ppy)2IrQ possess good electron transfer performance, while (ppy)IrQ2 might have hole transport properties.
Co-reporter:Chan Yao, Li-Kai Yan, Wei Guan, Chun-Guang Liu, Ping Song and Zhong-Min Su
Dalton Transactions 2010 vol. 39(Issue 33) pp:7645-7649
Publication Date(Web):16 Apr 2010
DOI:10.1039/C002547J
The second-order nonlinear optical (NLO) properties of porphyrin–metal–polyoxometalate (por–metal–POM) sandwich structures [(por)M(PW11O39)]5− (por = TPP, TPyP, TPPF20, M = Hf; por = TPP, M = Zr) and [(TPP)Hf(XW11O36)]6− (X = Si, Ge) are investigated by time-dependent density functional theory (TDDFT). The character of charge-transfer transition indicates that the porphyrin ligand acts as an electron acceptor and the lacunary Keggin-type POM acts as an electron donor. Our results show that this kind of organic–inorganic hybrid compound possesses remarkably large molecular second-order NLO polarizability, ∼100 × 10−30 esu, and might be an excellent second-order NLO material. Furthermore, the NLO response can be tuned by the element substituents. The computed β0 values increase with the auxiliary electron-accepting group on the porphyrin ring (TPPF20 > TPyP > TPP) and a heavy central heteroatom (Ge > Si > P). The present investigation provides important insight into the NLO properties of this class of por–metal–POM sandwich compound.
Co-reporter:Ping Song, Wei Guan, Likai Yan, Chunguang Liu, Chan Yao and Zhongmin Su
Dalton Transactions 2010 vol. 39(Issue 15) pp:3706-3713
Publication Date(Web):08 Mar 2010
DOI:10.1039/B922876D
The polarizability and redox properties for the tetranuclear vanadium–oxide–carboxylates have been investigated using density functional theory (DFT). These so-called inorganic crown ethers possess large polarizabilities, which are equivalent to fullerene and generate large polarization on the guest anions into the host. The dipole-induced dipole interaction between the host and the guest anions induces charge transfer from the guest to the host, which is enhanced with the increasing polarization. Moreover, the redox potentials are sensitive to different guests inside the host bowl, and they shift negatively as compared to the isolated host bowl. In contrast, the modification of the methyl group by CH2tBu on the rim of the bowl has evoked higher polarizability (over 560 a.u.) with larger polarization on the guest anions and more negative redox potentials. The weak interaction energies in accord with the organic crown ether incorporating alkali metal ions indicate that the guest anions can move freely inside and outside the host bowl, so this kind of inorganic crown ether may exhibit potential guest-switchable redox properties based on reversible complexation–decomplexation and will be expected to find applications in ion recognition and selectivity studies based on the sensitivity to different guests.
Co-reporter:Shuang Yao, Zhiming Zhang, Yangguang Li, Ying Lu, Enbo Wang and Zhongmin Su
Crystal Growth & Design 2010 Volume 10(Issue 1) pp:135
Publication Date(Web):December 1, 2009
DOI:10.1021/cg900745z
Reactions of hexavacant polyoxotungstate [H2P2W12O48]12− ({P2W12}) with transition-metal and lanthanide-metal cations assisted with organic molecules (tartrate or dimethylammonium chloride) lead to the isolation of two polyoxotungstates: K3Na8[K3⊂{GdMn(H2O)10}{HMnGd2(Tart)O2(H2O)15}{P6W42O151(H2O)7}]·44H2O (1) and K3Na10[K3⊂{GdCo(H2O)11}2{P6W41O148(H2O)7}]·43H2O (2). Single-crystal X-ray diffraction analyses reveal that both 1 and 2 contain a crown-type polyoxoanion shell [{WO(H2O)}3(P2W12O48)3]30− ({P6W39}), which consists of three {P2W12} units connected by three {WO(H2O)} linkers. Compound 1 has a two-dimensional porous framework constructed from 3d-4f cluster-containing {P6W39} aggregates and mixed 3d and 4f linkers. Compound 2 exhibits a one-dimensional chain-like structure composed of di-Co-containing {P6W39} aggregates and {Gd(H2O)7}3+ bridging units. Magnetic studies of compounds 1 and 2 indicate that antiferromagnetic interactions exist in these two compounds.
Co-reporter:Yongli Shi, Yuqin Fu, Changli Lü, Li Hui, Zhongmin Su
Dyes and Pigments 2010 Volume 85(1–2) pp:66-72
Publication Date(Web):April 2010
DOI:10.1016/j.dyepig.2009.10.005
A series of transparent, highly fluorescent, organic–inorganic nanocomposite films were prepared by incorporating mercaptoethanol-capped ZnS nanoparticles into a copolymer of trialkoxysilane-capped poly(MMA-co-Hq-CH2-HEMA) carrying an 8-hydroxyquinoline (Hq) unit, followed by ligand exchange and sol–gel processing. Electron microscopy revelaed that the ZnS nanoparticles were uniformly dispersed in the organic–inorganic hybrid matrix regardless of the content and matrix composition. The hybrid nanocomposites had good optical transparency in the visible region. The nanocomposites that contained the ZnS nanoparticles were stable and displayed high fluorescence emission at 500 nm, which differed from that of hybrid materials obtained by simply blending the zinc ions or bis(8-hydroxyquinoline) zinc compound with the copolymer matrix. Thermogravimetric analysis indicated that the nanocomposite materials had high thermal stability.
Co-reporter:Muhammad Ramzan Saeed Ashraf Janjua;Wei Guan;Likai Yan;Abdul Karim;Jamshed Akbar
European Journal of Inorganic Chemistry 2010 Volume 2010( Issue 22) pp:3466-3472
Publication Date(Web):
DOI:10.1002/ejic.201000428
Abstract
A dramatic increase in the second-order nonlinear optical (NLO) response of terpyridine-substituted hexamolybdates has been observed from 886.55 × 10–30 esu (system 1) to 4622.92 × 10–30 esu (system 7). The dipole polarizabilities and second-order nonlinear optical (NLO) properties of terpyridine derivatives of hexamolybdates have been investigated by using time-dependent density functional theory (TDDFT). The quantum mechanical design suggests that [Mo6O18(N4C25H14(CF3)2 (CN)2)]2– (system 7) is the best choice among all studied systems to improve nonlinearity. The electron-withdrawing ability of electron-acceptor groups (F, Cl, Br, I, CF3, and CN) at the end of the terpyridine ligand directs the charge transfer (CT) from the POM cluster to the terpyridine segment along the z axis, which leads to an efficient second-order NLO molecular design. These small changes in molecular composition by substitution may have disproportionately huge effects on the NLO properties, which can be attributed to the so-called “butterfly effect”.
Co-reporter:Yan-Hong Xu, Ya-Qian Lan, Kui-Zhan Shao, Zhong-Min Su, Yi Liao
Journal of Solid State Chemistry 2010 Volume 183(Issue 4) pp:849-857
Publication Date(Web):April 2010
DOI:10.1016/j.jssc.2010.01.027
Five novel ZnII-(pyridyl)imidazole derivative coordination polymers, [Zn(L)2] (1), [Zn2(μ3-OH)L(m-BDC)] (2), [Zn2(μ3-OH)L(p-BDC)]·H2O (3), [Zn2L(BTC)(H2O)]·2.5H2O (4) and [Zn3.5(μ3-OH)L2(BTEC)(H2O)]·H2O (5) (L=4-((2-(pyridine-2-yl)-1H-imidazol-1-yl)methyl)benzoic acid, p-H2BDC=1,4-benzenedicarboxylic acid, m-H2BDC=1,3-benzenedicarboxylic acid, H3BTC=1,3,5-benzenetricarboxylic acid, H4BTEC=1,2,4,5-benzenetetracarboxylic acid), were successfully synthesized under hydrothermal conditions through varying auxiliary aromatic-acid ligands and structurally characterized by X-ray crystallography. Compound 1 exhibits a 1D chain linked via double L bridges. Compound 2 features a well-known pcu topology with bent dicarboxylate ligand (m-H2BDC) as an auxiliary ligand, while 3 displays a bcu network with linear dicarboxylate ligand (p-H2BDC) as an auxiliary ligand. The structure of compound 4 is a novel 3D (3,5)-connected network with (4·62)(4·64·82·10·122) topology. It is interesting that compound 5 shows an intricate (3,4,8)-connected framework with (4·62)(42·63·8)(42·64)(42·618·7·86·10) topology. In addition, their infrared spectra (IR), X-ray powder diffraction (XPRD) and photoluminescent properties were also investigated in detail.Five novel ZnII-organic architectures have been hydrothermally synthesized through varying auxiliary aromatic-acid ligands and characterized by X-ray diffraction, the photoluminescence properties of compounds 1–5 were studied.
Co-reporter:Mei-Na Li, Guang-Sheng Yang, Shun-Li Li, Hong-Ying Zang, Guang-Juan Xu, Kui-Zhan Shao, Zhong-Min Su
Inorganic Chemistry Communications 2010 Volume 13(Issue 10) pp:1203-1206
Publication Date(Web):October 2010
DOI:10.1016/j.inoche.2010.06.050
Two new coordination polymers, namely [Zn3(BTT)(μ3-OH)3] (1), and [Mn3(BTT)(μ3-OH)3] (2), where H3BTT = 1,3,5-Tris(2H-tetrazol-5-yl)benzene, have been synthesized under hydrothermal conditions and characterized by single crystal X-ray diffraction. The structure of compound 1 can be described as tetranuclear zinc cluster linked with zinc (Zn2II) atoms to form a 2D layer, and the adjacent parallel layers bridged by the rigid H3BTT ligand to show 3D network. Taking Mn ions in replace of Zn ions, a new compound 2 is successfully obtained which is isostructural with 1. In addition, photoluminescent property for 1, magnetic properties for 2 and thermogravimetric analyses for 1 and 2 are investigated in detail.Two novel isostructural compounds, namely [Zn3(BTT)(μ3-OH)3] (1), [Mn3(BTT)(μ3-OH)3] (2) by combining polytetrazolate rigid ligand. The μ3-OH group linked MII (Zn and Mn) atoms to form interesting 2D layers structure and the BTT bridged adjacent layers to give a 3D network with tetranuclear metal–oxygen clusters and 16-membered metallocycles.
Co-reporter:Dongying Du, Yaqian Lan, Xinlong Wang, Kuizhan Shao, Haining Wang, Zhongmin Su
Solid State Sciences 2010 Volume 12(Issue 1) pp:128-133
Publication Date(Web):January 2010
DOI:10.1016/j.solidstatesciences.2009.10.017
The reactions between trivacant Keggin polyanions and transition metal ions in the presence of organoamine resulted in the isolation of two new polyoxotungstates, that is, [Hen]2[Cu(en)2(H2O)]2{[Cu(en)2]2[X2W23CuO79]}·4H2O (X = Ge, 1; X = Si, 2; and en = 1,2-ethylenediamine). Single-crystal X-ray diffraction analyses reveal that the polyanions in 1 and 2 are constructed by saturated Keggin anions condensed through sharing common oxygen atoms into Keggin dimers, which are further fused by copper(II) bridging fragments into one-dimensional (1D) ladder-like chains and then extended to a 3D supramolecular framework through hydrogen bond interactions. The IR spectrum and X-ray powder diffraction have been studied in detail for compounds 1 and 2. The UV spectrum, X-ray photoelectron spectroscopy (XPS), thermogravimetric analysis (TGA), the cyclic voltammetry and electrocatalytic activity toward the reduction of nitrite have also been studied for compound 1.
Co-reporter:Hongze Gao, Rigen Mo, Houyu Zhang, Yue Wang, Zhong-Min Su
Synthetic Metals 2010 Volume 160(9–10) pp:1015-1021
Publication Date(Web):May 2010
DOI:10.1016/j.synthmet.2010.02.019
The tris(1-phenylpyrazolato,N,C2′)iridium(III) Ir(ppz)3, (fac-Ir(ppz)3, 1; mer-Ir(ppz)3, 2) and iridium(III)bis(1-phenylpyrazolato,N,C2′) (2,2,6,6-tetramethyl-3,5-heptane-dionato-O,O) ppz2Ir(dpm) (C-cis,N-trans-ppz2Ir(dpm), 3; C-cis,N-cis-ppz2Ir(dpm), 4) have been investigated theoretically to explore their electronic structures, spectroscopic and electron blocking properties. A detailed comparison of the electronic structure characteristics of the two isomers has been addressed for pointing out differences in absorption and emission properties. The geometries and electronic structures are investigated at B3LYP and CIS levels for ground and excited states, respectively. At the TD-DFT and PCM levels, 1–4 give rise to absorptions at 329, 346, 355 and 347 nm, respectively, and phosphorescent emissions at 377, 461 and 405 nm for 1–3, respectively. The transitions of 1–2 are attributed to [d(Ir) + π(phenyl)] → [π*(pyrazolyl)] charge transition, whereas 3–4 are related to [d(Ir) + π(phenyl)] → [π*(pyrazolyl) + π*(dpm)]. The reorganization energies computed for hole (λhole) except 2 are smaller than that of N,N′-diphenyl-N,N′-bis(1,1′-biphenyl)-4,4′-diamine which is a typical hole transport material. Fac-Ir(ppz)3 is the most efficient electron blocking material among the four complexes.
Co-reporter:Guang-Juan Xu, Ya-Hui Zhao, Kui-Zhan Shao, Ya-Qian Lan, Xin-Long Wang, Zhong-Min Su, Li-Kai Yan
Journal of Molecular Structure 2010 Volume 983(1–3) pp:93-98
Publication Date(Web):1 November 2010
DOI:10.1016/j.molstruc.2010.08.037
To investigate the influence of spatial extended direction on the resulting structure, three new compounds have been synthesized by the reactions of ZnII or CdII salts and 3,5-bis(pyridin-4-ylmethoxy)benzoic acid (HL1) or 3,4-bis(pyridin-4-ylmethoxy)benzoic acid (HL2) with two rationally selected multicarboxylate ligands. Without a secondary ligand, an 1D ribbon structure [Zn(L1)2]·H2O (1) furnished, the hydrogen bonds extended the structure to a 2D layer. Employing 4,4′-biphenyldicarboxylic acid (H2bpdc), a 2D layer structure [Zn(L1)(bpdc)0.5] (2) resulted. The use of 1,3,5-benzenetricarboxylic acid (H3btc) led to a 3D compound [Cd2(L2)(btc)(H2O)] (3). It can be observed from the architectures of 1–3 that secondary ligands had great effects on the spatial connective fashions, resulting in the formation of various dimensional compounds.
Co-reporter:Hong-Ying Zang, Ke Tan, Ya-Qian Lan, Guang-Sheng Yang, Kui-Zhan Shao, Zhong-Min Su
Inorganic Chemistry Communications 2010 Volume 13(Issue 12) pp:1473-1475
Publication Date(Web):December 2010
DOI:10.1016/j.inoche.2010.08.020
Co-reporter:Lianjiang Su, Hongying Zang, Hongsheng Liu, Limin Wang, Weihong Li, Zhongmin Su
Inorganic Chemistry Communications 2010 Volume 13(Issue 8) pp:981-984
Publication Date(Web):August 2010
DOI:10.1016/j.inoche.2010.05.012
Two novel inorganic-organic hybrid compounds (Ni3(Hfcz)4(V3O9)2∙ 2H2O) (1) and Co5(H2O)4(fcz)2(V4O12)2(2) have been successfully constructed, based on [Ni2(V3O9)2]2− “bow tie” clusters and {Co2V2} pure inorganic cages. Structural analysis reveals that the clusters and cages are expanded to a 2D network and 3D framework via NiII and CoII, respectively. In addition, we investigated the optical band gaps of the two compounds: 2.92 eV and 2.23 eV.
Co-reporter:Guang-Juan Xu, Ya-Hui Zhao, Kui-Zhan Shao, Ya-Qian Lan, Peng Li, Zhong-Min Su
Inorganic Chemistry Communications 2010 Volume 13(Issue 8) pp:932-934
Publication Date(Web):August 2010
DOI:10.1016/j.inoche.2010.04.029
A new 2D 5-connected coordination polymer [Zn3(HL2)(1,4-bdc)3] (1) (HL2 = 3,4-bis(pyridin-4-ylmethoxy)benzoic acid, 1,4-H2bdc = 1,4-benzenedicarboxylate) has been hydrothermally constructed using HL2, 1,4-bdc ligands as linear linkers and Zn3-clusters as 5-connected nodes. In addition, powder X-ray diffraction, photoluminescent property and thermogravimetric analysis for 1 are investigated in detail.A new 2D 5-connected coordination polymer [Zn3(HL2)(1,4-bdc)3] (1) (HL2 = 3,4-bis(pyridin-4-ylmethoxy)benzoic acid, 1,4-H2bdc = 1,4-benzenedicarboxylate) has been hydrothermally constructed using HL2, 1,4-bdc as linear linkers and Zn3-clusters as 5-connected nodes. In addition, powder X-ray diffraction, photoluminescent property and thermogravimetric analysis for 1 are investigated in detail.
Co-reporter:Hong-Ying Zang, Ya-Qian Lan, Zhong-Min Su, Guang-Sheng Yang, Guang-Juan Xu, Dong-Ying Du, Lei Chen, Li-Kai Yan
Inorganica Chimica Acta 2010 Volume 363(Issue 1) pp:118-126
Publication Date(Web):4 January 2010
DOI:10.1016/j.ica.2009.09.036
Four octamolybdate-based compounds, that is, CuII2(L1)4(Mo8O26) (1), CuII2(HL2)4(Mo8O26)2 (2), [CuIIL2(H2O)(Mo8O26)0.5]·2H2O (3) and [CuIIL2(H2O)(Mo8O26)0.5]·2H2O (4) (L1 = 2-(2-pyridyl)imidazole, L2 = 2-(1-(pyridine-3-ylmethyl)-1H-imidazol-2-yl)pyridine), have been hydrothermally synthesized via changing the reaction conditions and structurally characterized by single-crystal X-ray diffraction. With L1 ligand, we obtained compound 1, which is a 0D molecule and extends to a 3D supramolecular structure via hydrogen-bonding interactions. By using L2 instead of L1 ligand, compound 2 comes into being which is as well a discrete molecule and further extended to a 3D supramolecular structure by hydrogen bonds. Intriguingly, compounds 3 and 4 are supramolecular isomers: the former is a 2D 4-connected network and the latter is a 3D (3,4)-connected framework. The measurements of diffuse reflectance for compounds 1–4 indicate that they are potential wide gap semiconductors.Four new octamolybdate-based compounds have been attained via changing several influencing factors and characterized by single-crystal X-ray diffraction. Compounds 1 and 2 are discrete molecules and further extended to 3D supramolecular structures by hydrogen bonds. Intriguingly, compounds 3 and 4 are supramolecular isomers: the former is a 2D 4-connected network and the latter is a 3D (3,4)-connected framework.
Co-reporter:Dong-Ying Du, Jun-Sheng Qin, Shun-Li Li, Xin-Long Wang, Guang-Sheng Yang, Yang-Guang Li, Kui-Zhan Shao, Zhong-Min Su
Inorganica Chimica Acta 2010 Volume 363(Issue 14) pp:3823-3831
Publication Date(Web):25 November 2010
DOI:10.1016/j.ica.2010.07.028
Co-reporter:Hai-Ning Wang, Jun-Sheng Qin, Dong-Ying Du, Guang-Juan Xu, Xin-Long Wang, Kui-Zhan Shao, Gang Yuan, Lian-Jie Li, Zhong-Min Su
Inorganic Chemistry Communications 2010 Volume 13(Issue 11) pp:1227-1230
Publication Date(Web):November 2010
DOI:10.1016/j.inoche.2010.06.011
A new Zn(II) coordination polymer, [Zn3(pzdc)4]·en (1) (H2pzdc = pyrazine-2,3-dicarboxylic acid, en = ethylenediamine), has been prepared under hydrothermal condition. X-ray diffraction analysis shows that 1 has a 3D structure with micropores (calcd. 4.074 × 6.444 Å2) along the [1 0 0] direction filled with en guest molecules. Notably, the pzdc groups connect Zn(II) cations to form a macrometallocycle as the subunit, which is further connected to generate the final framework. Additionally, the luminescent property of compound 1 has been investigated in detail.A new Zn(II) coordination polymer (1), has been prepared. Compound 1 has a 3D structure with micropores along the [1 0 0] direction filled with en guest molecules. Notably, the pzdc groups connect Zn(II) cations to form a macrometallocycle as the subunit further connected to generate the final framework.
Co-reporter:JianPing Wang;DongHua Hu;ZhongMin Su
Science Bulletin 2010 Volume 55( Issue 23) pp:2497-2504
Publication Date(Web):2010 August
DOI:10.1007/s11434-010-3271-8
Polyoxometalate (POM) has promising antiviral activities. It shows broad-spectrum inhibiting ability, high efficiency, and low toxicity. Experimental assays show that titanium containing polyoxotungstates have anti-influenza-virus activity. In this paper, the binding mechanisms of five isomers of di-Ti-substituted polyoxotungstate, [α-1,2-PTi2W10O40]7− (α-1,2), [α-1,6-PTi2W10O40]7− (α-1,6), [α-1,5-PTi2W10O40]7− (α-1,5), [α-1,4-PTi2W10O40]7− (α-1,4) and [α-1,11-PTi2W10O40]7− (α-1,11), to five subtypes of influenza virus A neuraminidase (FluV-A NA) were investigated in the context of aqueous solution by using molecular docking and molecular dynamics studies. The results show that the isomer α-1,2 is superior to other isomers as a potential inhibitor to neuraminidase. The positively charged arginine residues around the active site of NA could be induced by negatively charged POM to adapt themselves and could form salt bridge interactions and hydrogen bond interactions with POM. The binding free energies of POM/NA complexes range from −5.36 to −8.31 kcal mol−1. The electrostatic interactions are found to be the driving force during the binding process of POM to NA. The conformational analysis shows that POM tends to bind primarily with N1 and N8 at the edge of the active pocket, which causes the conformational change of the pincers structure comprising residue 347 and loop 150. Whereas, the active pockets of N2, N9 and N4 are found to be more spacious, which allows POM to enter into the active pockets directly and anchor there firmly. This study shows that negatively charged ligand as POM could induce the reorganization of the active site of NA and highlights POM as a promising inhibitor to NA despite the ever increasing mutants of NA.
Co-reporter:Ping Song, Wei Guan, Chunguang Liu, Likai Yan, Chan Yao, Zhongmin Su
Journal of Molecular Structure: THEOCHEM 2010 Volume 947(1–3) pp:9-15
Publication Date(Web):15 May 2010
DOI:10.1016/j.theochem.2010.01.026
Mono- and multi-organoimido substituted derivatives for Lindqvist-type molybdate were carried out to search the preferred contribution factor on the redox property and the second-order nonlinear optical response. It was indicated that organic conjugated groups were much more sensitive to the redox property and nonlinear optical response than non-conjugated ones. The increment of NAr ligands, i.e. the greater net charge donation between organic segment and inorganic polyanion cluster, drove the first reduction potential dramatically negative. In contrast, the lengthening of conjugated chain resulted in remarkable improvement on second-order nonlinear optical response than in reduction potentials. In addition, MoN in mono-organoimido substituted derivatives has localized the electrons for HOMO on π-orbital of the first phenyl ring adjacent to N1 together with small part of π-pd orbital between Mo1 and N1, and the electron transition tends to transfer to organic conjugated groups. The two distinct paths may offer favorable direction for experiment.
Co-reporter:Ziyan Zhou, Guogang Shan, Yi Liao, Wei Xing, Shuangyang Yang, Zhongmin Su
Journal of Molecular Structure: THEOCHEM 2010 Volume 945(1–3) pp:110-115
Publication Date(Web):15 April 2010
DOI:10.1016/j.theochem.2010.01.018
The absorption and emission spectra of salicylanilide (SAN) in methanol show strong blue-shift compared with those in cyclohexane. To make clear this phenomenon, the ground (S0) and excited state (S1) proton transfer reactions in cyclohexane and in methanol solutions were investigated, respectively. In cyclohexane, the intramolecular mechanism was carried out for the isolated SAN. Whereas in methanol solution, the role of the methanol molecule in proton transfer reaction was supposed that it directly participated the reaction path, called methanol-assisted mechanism. The computational results show that barrier height for the ground state proton transfer is too high to happen, but is energetically favored in S1 for both isolated SAN and methanol-assisted system. In addition, it indicates that the hydrogen-bonding between SAN and methanol molecule can dramatically lower the barrier by methanol-assisted mechanism in S1. Herein, we also have applied a combination of the continuum dielectric and the explicit solvent model to study the spectra of SAN in methanol, which exhibit that the interaction between SAN and one methanol molecule yields a hydrogen-bonding complex responsible for the blue-shifted wavelength of absorption and emission spectra in methanol solvent.
Co-reporter:Muhammad Ramzan Saeed Ashraf Janjua, Wei Guan, Likai Yan, Zhong-Min Su, Muhammad Ali, Iftikhar Hussain Bukhari
Journal of Molecular Graphics and Modelling 2010 Volume 28(Issue 8) pp:735-745
Publication Date(Web):June 2010
DOI:10.1016/j.jmgm.2010.01.011
We have explored an innovative, versatile, and novel molecular hybrid containing polyoxometalate (POM) cluster linked with terpyridine ligand via π-bridged donor–acceptor (D–A) configuration. The dipole polarizabilities, density of states, and second-order nonlinear optical (NLO) properties of terpyridine-substituted hexamolybdates have been investigated by using time-dependent density functional response theory (TDDFT). This class of organic–inorganic hybrid compounds possesses a robustly large molecular second-order NLO response, especially [Mo6O18(N4C25H16I2)]2− (system 5) and [Mo6O17(N4C25H16(CN)2)(N4C25H16(CN)2)]2− (system 10) with the static second-order polarizability (βvec) computed to be 1209.25 × 10−30 esu and 1622.67 × 10−30 esu respectively. Thus, these systems have the possibility to be excellent second-order nonlinear optical materials. Analysis of the major contributions to the βvec value suggests that the charge transfer (CT) from POM-cluster to terpyridine ligand (D–A) along the z-axis plays the key role in the NLO response, POM-cluster (hexamolybdates) acts as a donor (D) whereas terpyridine ligand acts as an acceptor (A) in all the studied systems. The computed βvec values increase by the incorporation of electron acceptors (halogen = F, Cl, Br and I) at the terminus of terpyridine ligand. Furthermore, substitution of trifluoromethoxy (–OCF3), trifluoromethyl (–CF3), and cyanide (–CN) at the end of terpyridine ligand respectively enhances the optical nonlinearity. Orbital analysis shows that the degree of CT between POM and terpyridine segment was increased in 2D and organometallic/POM hybrid systems. The present investigation provides important and thought provoking insight into the robustly large NLO properties of terpyridine-substituted hexamolybdates.
Co-reporter:Yang-Yang Hu ; Shi-Ling Sun ; Shabbir Muhammad ; Hong-Liang Xu
The Journal of Physical Chemistry C 2010 Volume 114(Issue 46) pp:19792-19798
Publication Date(Web):November 3, 2010
DOI:10.1021/jp105045j
How do the number and location of lithium atoms affect the first hyperpolarizability (βtot) of graphene? In this paper, based on pentacene, a series of graphene (multi)lithium salts Lin@pentacene (n = 1, 2, 3, 4, 5, and 6) have been designed to answer this question. βtot obviously increases stepwise with an increase in the number of lithium atoms: 1369−1843 for Li@pentacence < 3510−4081 for Li2@pentacence < 6933−7934 for Li3@pentacence < 11 188−12 145 for Li4@pentacence < 14 904 au for Li5@pentacence, which are much larger than pentacence. This pattern suggests that the lithium salt effect on the first hyperpolarizability is very large. Unexpectedly, when an additional lithium atom is doped into the graphene multilithium salt Li5@pentacence, which leads to Li6@pentacence, the βtot value dramatically increases to a value of 4 501 764 au with a remarkable increase of 302-fold in contrast to Li5@pentacence. On the other hand, when the number of lithium atoms is equal, the location of lithium atoms also affects the βtot value: the closer the lithium atoms are clustered, the larger the βtot value: for Li3@pentacence, 6933 au of system 10 < 7401 au of system 9 < 7934 au of system 8. Furthermore, their transition energies (ΔE) are also obtained. The results show that ΔE decreases stepwise with an increase in the number of the lithium atoms, and ΔE of Li6@pentacence sharply decreases to 0.299 eV, which may explain the huge βtot value. This study may stimulate the search for new types of graphene NLO materials based on alkali metals for NLO application.
Co-reporter:Jie Wu;Shuixing Wu;Yun Geng;Guochun Yang
Theoretical Chemistry Accounts 2010 Volume 127( Issue 4) pp:419-427
Publication Date(Web):2010 November
DOI:10.1007/s00214-010-0730-x
Phosphole-based systems due to the unique electronic and optical properties have recently been paid much attention as optoelectronic materials. In this work, the relationship among the electronic structure, charge injection, and transport was investigated for five derivatives of dithieno[3,2-b:2′,3′-d]phosphole (systems 1–5). The structures of systems 1–5 in the ground (S0) and the lowest singlet excited (S1) states were optimized at the HF/6-31G* and CIS/6-31G* levels of theory, respectively. Based on these structures, electronic spectra were calculated by time-dependent density functional theory. The simulated emission peaks of five phosphole derivatives locating at the blue–green region (448–516 nm), are in good agreement with the experimental data. Compared with tris-(8-quinolinolate) aluminum (III) (Alq3), normally used as an excellent electron transporter, systems 1–5 show a significant improvement in electron affinity (EA) due to σ*–π* hyperconjugation, which can effectively promote ability of electron injection. The small differences between λh and λe for systems 1–5 (0.06–0.14 eV) facilitate charge transfer balance, which suggests systems 1–5 can act as potential ambipolar materials. Owing to good rigidity, low-lying LUMO levels, delocalized frontier molecular orbitals, and the small reorganization energies, the five derivatives of dithieno[3,2-b:2′,3′-d]phosphole are expected to be high-efficiency blue materials in single-layer OLEDs.
Co-reporter:Chan Yao;Li-Kai Yan;Wei Guan;Chun-Guang Liu;Ping Song
Journal of Cluster Science 2010 Volume 21( Issue 2) pp:69-80
Publication Date(Web):2010 June
DOI:10.1007/s10876-010-0300-3
The second-order nonlinear optical (NLO) properties of Keggin-type organosilicone derivatives [PW11O39(RSi)2O]3− are investigated by time-dependent density functional theory (TDDFT). Our results indicated that the length of conjugated chain R has a crucial effect on the charge transfer (CT). The direction of CT alters with the end-cap-substituted electron donating or accepting moieties (NH2 or NO2), until the chain length reach a certain length, as the CT originates from the heteropolyanion to organosilicone moiety along the chain, further chain lengthening leaves this behavior invariant. The derivatives with long chain substitution and end-cap-substituted electron acceptor (NO2) display excellent second-order NLO responses. The system 18 with the relevant long conjugated polyphenylethynyl chain and end-cap-substituted electron acceptor (NO2) has the largest β0 value, 4538.1 × 10−30 esu. The present investigation provides important insight into the characteristic of CT and NLO properties of Keggin-type organosilicone derivatives.
Co-reporter:Fang Chai;Tingting Wang;Lu Li;Haiyan Liu;Lingyu Zhang
Nanoscale Research Letters 2010 Volume 5( Issue 11) pp:
Publication Date(Web):2010 November
DOI:10.1007/s11671-010-9730-y
A simple, cost-effective yet rapid and sensitive sensor for on-site and real-time Hg2+ detection based on bovine serum albumin functionalized fluorescent gold nanoparticles as novel and environmentally friendly fluorescent probes was developed. Using this probe, aqueous Hg2+ can be detected at 0.1 nM in a facile way based on fluorescence quenching. This probe was also applied to determine the Hg2+ in the lake samples, and the results demonstrate low interference and high sensitivity.
Co-reporter:Ping Song, Li-Kai Yan, Wei Guan, Chun-Guang Liu, Chan Yao, Zhong-Min Su
Journal of Molecular Graphics and Modelling 2010 Volume 29(Issue 1) pp:13-20
Publication Date(Web):24 August 2010
DOI:10.1016/j.jmgm.2010.04.006
The Λ-shaped phenanthroline-hexamolybdate compounds that are based on the reversible Mo-centered redox process were investigated. The attachment of hexamolybdate terminals to phenanthroline by a π-conjugated phenylamine bridge generated the organic ligand-centered or Ni-centered HOMO and the transition metal Mo-centered LUMO. The population in HOMO and LUMO predicted the reversible MoVI/V redox process and the ligand-to-metal charge transfer (LMCT) to a polyanion acceptor, which consequently evoked a significant second-order nonlinear optical (NLO) response. Moreover, the electron transition of these compounds exhibited a large βzyy tensor along the y-axis, which confirms a promising two-dimensional (2D) character with sizable anisotropy values. Interestingly, the addition of electrons into the high-valence Mo atom in the hexamolybdate acceptor evoked dramatic enhancements in the NLO response for the reduction states in contrast to the response of the corresponding oxidation states. The reduction states in system I exhibited second-order NLO responses about 200 times larger than the oxidation states. In addition, the attachment of the Ni atom in compound IIared enhanced the NLO response to nearly 1019 times greater than the response of the corresponding oxidation state compound IIa. The Ni atom as the electron donor plays an important role in the major electron transition for the reduction states in system II. Therefore, the NLO response of such compounds can be reversibly switched through the transition metal MoVI/V redox that is effectively coupled with the LMCT transition. Thus, the NLO activity can be controlled by a one-electron redox process, and the redox-active phenanthroline-hexamolybdate compounds are promising candidates for 2D redox-switching NLO materials in novel optoelectronic applications.
Co-reporter:Bo Liu;Shuang Wei;Yan Xing Dr.;Dan Liu Dr.;Zhan Shi ;Xianchun Liu;Xiujuan Sun;Suying Hou;Zhongmin Su
Chemistry - A European Journal 2010 Volume 16( Issue 22) pp:6625-6631
Publication Date(Web):
DOI:10.1002/chem.200903384
Abstract
The preparation of exquisite hierarchical worm-like Co1−xS (x=0.75) microtubes by a one-pot complex–surfactant-assisted hydrothermal method is successfully achieved for the first time. The hierarchical structures of the microtube wall are assembled from numerous interleaving hexagonal nanoplates. X-ray diffraction, X-ray photoelectron spectroscopy, scanning electron microscopy, transmission electron microscopy, and high-resolution transmission electron microscopy were used to characterize the samples. The experimental results indicate that the “soft template” surfactant cetyltrimethylammonium bromide and the chelating ethylenediamine both play important roles for the formation of hierarchical Co1−xS microtubes. A possible formation mechanism for the growth processes is proposed. Additionally, the electrochemical and magnetic properties of Co1−xS microtubes were systematically studied.
Co-reporter:Gang Yuan;KuiZhan Shao;DongYing Du;XinLong Wang;ZhongMin Su
Science China Chemistry 2010 Volume 53( Issue 10) pp:2177-2182
Publication Date(Web):2010 October
DOI:10.1007/s11426-010-4127-4
Two novel three-dimensional (3-D) coordination polymers, [Pb(HTDA)]n (1) and [Co5(TDA)2(H2TDA)2(H2O)8]n (2) [H3TDA = 1H-1,2,3-triazole-4,5-dicarboxylic acid], have been prepared by hydrothermal reactions and characterized by elemental analysis, infrared spectroscopy and single-crystal X-ray diffraction. Compound 1 is constructed from rod-shaped secondary building units (SBUs) and exhibits a 3-D network with (410·65)(410·63·82) topology. Compound 2 is built up from ligands bridging three different cobalt ions and exhibits a 3-D network with (4·82)3(4·82·103) topology. In addition, the thermal stabilities of the two compounds, the photoluminescence properties of compound 1 and the magnetic properties of compound 2 have been studied.
Co-reporter:Yaying Du, Yuqin Fu, Xiaohui Guo, Hui Li, Changli Lü, Zhongmin Su
Microporous and Mesoporous Materials 2010 130(1–3) pp: 122-129
Publication Date(Web):
DOI:10.1016/j.micromeso.2009.10.022
Co-reporter:Likai Yan, Mingshun Jin, Ping Song and Zhongmin Su
The Journal of Physical Chemistry B 2010 Volume 114(Issue 11) pp:3754-3758
Publication Date(Web):March 3, 2010
DOI:10.1021/jp909336z
The organoimido functionalization of polyoxometalates (POMs) has drawn tremendous attention due to particular merits in fabricating POM-based hybrid materials with finely tunable properties. The electronic properties, orbital and bonding characters of unprecedented bridging organoimido-substituted hexamolybdate are investigated using density functional theory methods. Among the organoimido-bridged hexamolybdates, [Mo6O16(2,6-Me2-NC6H3)2(μ2-2,6-Me2-NC6H3)]2− (3-Ar-1), which features two terminal and one bridging organoimido ligand, is more favorable. The calculations confirm that the three-center (3c) π bond originates from the coplanarity of bridging nitrogen atom with two Mo atoms and the hybridization of bridging nitrogen. The 3c bond stabilizes the organoimido-bridged anion 3-Ar-1. Compared with cis-bifunctionalized organoimido derivative [Mo6O17(2,6-Me2-NC6H3)2]2− (2-Ar), the bonding interaction between terminal organoimido ligand and hexamolybdate cluster in 3-Ar-1 is strengthened by the bridging organoimido. The results are in good agreement with the analysis of the Wiberg bond index of the Mo−N bond. The organoimido segment modifies the occupied molecular orbitals of organoimido hexamolybdates. The unoccupied molecular orbitals in 3-Ar-1 are largely nonbonding Op and Mod orbitals in character, which resemble those of 2-Ar.
Co-reporter:Lili Shi, Bo Hong, Wei Guan, Zhijian Wu and Zhongmin Su
The Journal of Physical Chemistry A 2010 Volume 114(Issue 24) pp:6559-6564
Publication Date(Web):May 26, 2010
DOI:10.1021/jp1010617
The electronic structures and optoelectronic properties of several blue-emitting phosphors (dfppy)2Ir(pyN2), (dfppy)Ir(pyN2)2, and (fpmb)2Ir(pyN3) [dfppyH: 2-(2,4-difluorophenyl)pyridine; pyN2H: 5-(2-pyridyl)-3-trifluoromethylpyrazole; Hfpmb: 1-(4-fluorophenyl)-2,3-dihydro-3-methyl-1H-benzo[d]imidazole; and pyN3H: 2-(5-(trifluoromethyl)-2H-1,2,4-triazol-3-yl)pyridine] are investigated with density functional theory. The injection abilities of holes and electrons are estimated by evaluating the ionization potentials and electron affinities. It is found that the properties of the ligands have great influence on the photophysical properties, such as energy gap, absorption spectra, emission spectra, etc. The assumed complex (dfppy)2Ir(pyN2) is found to be a good candidate for blue-emitting material. We suggest that the luminescent properties of this class of materials can be tuned by modifications of the corresponding ligands.
Co-reporter:Yinghui Wang, Bin Li, Yanhong Liu, Liming Zhang, Qinghui Zuo, Linfang Shi and Zhongmin Su
Chemical Communications 2009 (Issue 39) pp:5868-5870
Publication Date(Web):12 Aug 2009
DOI:10.1039/B910305H
The first optical oxygen sensor based on Cu(I) complex–polystyrene composite nanofibrous membranes, showing high sensitivity (I0/I100 = 15.56), good linear Stern–Volmer characteristics (R2 = 0.9966) and short response/recovery time (t↓ (s) = 7 and t↑ (s) = 14), has been prepared; these results represent the best values reported for oxygen sensors based on Cu(I) complexes.
Co-reporter:Xin-Long Wang, Chao Qin, Ya-Qian Lan, Kui-Zhan Shao, Zhong-Min Su and En-Bo Wang
Chemical Communications 2009 (Issue 4) pp:410-412
Publication Date(Web):14 Nov 2008
DOI:10.1039/B815629H
The first uninodal 10-connected metal–organic framework, based on pentanuclear cadmium cluster building blocks, exhibits an unprecedented γ-Pu topology, which adds a new member to the series of metal–organic analogues which have a natural materials topology.
Co-reporter:Chun-Guang Liu, Zhong-Min Su, Wei Guan and Li-Kai Yan
Inorganic Chemistry 2009 Volume 48(Issue 2) pp:541-548
Publication Date(Web):December 12, 2008
DOI:10.1021/ic8012443
High-valent MVI≡N (M = Ru, Os) species are important reagents in nitrogen transfer reactions; the unique withdrawing properties of polyoxometalate (POMs) ligands would possibly modify the reactivity of the MVI≡N functional group. In the present paper, density functional theory (DFT) and natural bond orbital (NBO) analysis have been employed to calculate electronic structures, MVI−N bonding, and redox properties of high-valent metal nitrido derivatives of Keggin-type POMs, [PW11O39 {MVIN}]4− (M = Ru, Os, Re). Our calculations show that [PW11O39{RuN}]4− possesses stronger antibonding interaction between metal and nitrogen atoms compared with anions [PW11O39{OsN}]4− and [PW11O39{ReN}]4−. A large increase in the Ru−N bond length of anion [PW11O39{RuN}]4− in the excited states has been found; the effective order and composition of the molecular orbital in anion [PW11O39{RuN}]4− is a key factor in determination of the increase of the Ru−N bond length in the excited states. The substitution effects of central tetrahedron heteroatoms (XO4, X = Al, Si, P, As) in anions [XW11O39{RuN}]4− affect the relative energy of the LUMO; the relevant orbital energy increases in the order Al(III) < Si(IV) < P(V) ≈ As(V). The RuN unit is the reduced center. NBO analysis of the extent of the bonding interaction between the ruthenium and the nitrogen centers in [PW11O39{RuVIN}]4− shows that the Ru−N bond possesses a covalent feature and displays triple-, double-, and single-bond character when moving along the change of spin state (11 → 31 → 51).
Co-reporter:Chun-Guang Liu ; Wei Guan ; Ping Song ; Zhong-Min Su ; Chan Yao ;En-Bo Wang
Inorganic Chemistry 2009 Volume 48(Issue 17) pp:8115-8119
Publication Date(Web):July 29, 2009
DOI:10.1021/ic900829z
The donor-conjugated bridge−acceptor (D-A) model, as a simple molecular scheme, has been successfully used in the development of second-order organic compound, organometallic compound, and metal complex nonlinear optical (NLO) materials. However, for the totally inorganic molecules, the use of this model is still prohibitive. In the present paper, time-dependent density functional theory (TDDFT) was used to investigate the second-order NLO properties of vanadium- and molybdenum-trisubstituted Keggin and Wells−Dawson polyoxometalates (POMs). The results show that these POM clusters possess D-A structures. The oxygen atoms in the cap region and metal (vanadium and molybdenum) atoms in another cap region in these POM clusters can be viewed as the electron donor and acceptor, respectively. The vanadium ion derivatives possess larger second-order NLO responses and dipole moment than molybdenum ions derivatives; thus, the three vanadium atoms in the cap region act as a strong acceptor related to the three molybdenum atoms in cap region in our D-A scheme. The vanadomolybdate with Wells−Dawson structure displays the good second-order NLO response because of the relevant long conjugated bridge and strong acceptor. This D-A model may be an effective approach for optimizing the first hyperpolarizabilities of inorganic POM clusters.
Co-reporter:Chun-Guang Liu, Wei Guan, Ping Song, Li-Kai Yan and Zhong-Min Su
Inorganic Chemistry 2009 Volume 48(Issue 14) pp:6548-6554
Publication Date(Web):June 12, 2009
DOI:10.1021/ic9004906
The redox-active tetrathiafulvalene (TTF) is a good electron donor, and porphyrin is highly delocalized in cyclic π-conjugated systems. The direct combination of the two interesting building units into the same molecule provides an intriguing molecular system for designing nonlinear optical (NLO) molecular materials. In the present paper, the second-order NLO properties of a series of monoTTF-porphyrins and metalloporphyrins have been calculated by density functional theory (DFT) combined with the finite field (FF) method. Our calculations show that these compounds possess considerably large static first hyperpolarizabilities, ∼400 × 10−30 esu. Since the TTF unit is able to exist in three different stable redox states (TTF, TTF•+, and TTF2+), the redox switching of the NLO response of the zincII derivative of monoTTF-metalloporphyrin has been studied, and a substantial enhancement in static first hyperpolarizability has been obtained in its oxidized species according to our DFT-FF calculations. The β values of one- and two-electron-oxidized species are 3.6 and 8.7 times as large as that of the neutral compound, especially for two-electron-oxidized species, with a value of 3384 × 10−30 esu. This value is about 3 times that for a push−pull metalloporphyrin, which has an exceptionally large hyperpolarizability among reported organic NLO chromophores. Meanwhile, to give a more intuitive description of band assignments of the electron spectrum and trends in NLO behavior of these compounds, the time-dependent (TD)DFT method has been adopted to calculate the electron spectrum. The TDDFT calculations well-reproduce the soret band and Q-type bands of the monoTTF-porphyrin, and these absorption bands can be assigned to the π → π* transition of the porphyrin core. On the other hand, the oxidized process significantly affects the geometrical structures of the TTF unit and porphyrin ring, and the two-electron-oxidized species has a planar TTF unit and a high conjugative porphyrin ring. This effect reduces the excited energy, changes the CT feature, and thus enhances its static first hyperpolarizability.
Co-reporter:Kui-Zhan Shao, Ya-Hui Zhao, Xin-Long Wang, Ya-Qian Lan, De-Jun Wang, Zhong-Min Su and Rong-Shun Wang
Inorganic Chemistry 2009 Volume 48(Issue 1) pp:10-12
Publication Date(Web):December 5, 2008
DOI:10.1021/ic801439q
A polynuclear zinc compound, [Zn7(BTA)7(OABDC)(μ3-OH)2(μ2-OH)2·H2O] (1), has been prepared by using benzotriazole (HBTA) and 5-oxyacetatoisophthalic acid (H3OABDC) as ligands under hydrothermal conditions. For compound 1, an unprecedented metallophthalocyanine-like “Zn2(μ3-OH)2⊂[Zn4BTA4]” subunit is constructed from η3-BTA ligands and Zn atoms and further linked via μ2-OH, outer four-connected Zn atoms, and 5-oxyacetateisophthalic acid to form a novel three-dimensional framework.
Co-reporter:Ai-xiang Tian, Jun Ying, Jun Peng, Jing-quan Sha, Hai-jun Pang, Peng-peng Zhang, Yuan Chen, Min Zhu and Zhong-min Su
Inorganic Chemistry 2009 Volume 48(Issue 1) pp:100-110
Publication Date(Web):December 1, 2008
DOI:10.1021/ic801338b
Four inorganic−organic hybrid compounds, [CuI4(bte)4(SiW12O40)] (1), [CuII2(bte)4(SiW12O40)]·4H2O (2) [bte = 1,2-bis(1,2,4-triazol-1-yl)ethane], [CuI4(btb)2(SiW12O40)]·2H2O (3), and [CuII2(btb)4(SiW12O40)]·2H2O (4) [btb = 1,4-bis(1,2,4-triazol-1-yl)butane], were hydrothermally synthesized through use of the same Keggin polyoxometalate as the template and tuning the molar ratio of the bis(triazole) ligand to the CuII ion. The ratio of the bis(triazole) ligand to CuII has a crucial influence on the structures of this series. Single-crystal X-ray diffraction analyses indicate that compound 1 is constructed by tetranuclear ring-connecting chains and polymerized [Cu(bte)]+ chains, between which SiW12 anions are inserted to form a three-dimensional (3D) structure. Compound 2 shows a (44·62) two-dimensional grid sheet. The discrete SiW12 anions are sandwiched by the sheets, just like “hamburgers”. Compound 3 presents channel-like [Cu2(btb)]2+ polymerized chains, which are further connected by SiW12 anions to construct a 3D framework. Compound 4 exhibits a (66) 3D Cu-btb framework with hexagonal channels, into which the tetradentate SiW12 anions are incorporated. The thermal stabilities of the compounds are discussed.
Co-reporter:Ting Gao, Li-Li Shi, Hai-Bin Li, Shan-Shan Zhao, Hui Li, Shi-Ling Sun, Zhong-Min Su and Ying-Hua Lu
Physical Chemistry Chemical Physics 2009 vol. 11(Issue 25) pp:5124-5129
Publication Date(Web):23 Mar 2009
DOI:10.1039/B812492B
The combination of genetic algorithm and back-propagation neural network correction approaches (GABP) has successfully improved the calculation accuracy of absorption energies. In this paper, the absorption energies of 160 organic molecules are corrected to test this method. Firstly, the GABP1 is introduced to determine the quantitative relationship between the experimental results and calculations obtained by using quantum chemical methods. After GABP1 correction, the root-mean-square (RMS) deviations of the calculated absorption energies reduce from 0.32, 0.95 and 0.46 eV to 0.14, 0.19 and 0.18 eV for B3LYP/6-31G(d), B3LYP/STO-3G and ZINDO methods, respectively. The corrected results of B3LYP/6-31G(d)-GABP1 are in good agreement with experimental results. Then, the GABP2 is introduced to determine the quantitative relationship between the results of B3LYP/6-31G(d)-GABP1 method and calculations of the low accuracy methods (B3LYP/STO-3G and ZINDO). After GABP2 correction, the RMS deviations of the calculated absorption energies reduce to 0.20 and 0.19 eV for B3LYP/STO-3G and ZINDO methods, respectively. The results show that the RMS deviations after GABP1 and GABP2 correction are similar for B3LYP/STO-3G and ZINDO methods. Thus, the B3LYP/6-31G(d)-GABP1 is a better method to predict absorption energies and can be used as the approximation of experimental results where the experimental results are unknown or uncertain by experimental method. This method may be used for predicting absorption energies of larger organic molecules that are unavailable by experimental methods and by high-accuracy theoretical methods with larger basis sets. The performance of this method was demonstrated by application to the absorption energy of the aldehyde carbazole precursor.
Co-reporter:Suyue Li, Ming Li, Jingui Qin, Mingliang Tong, Xiaoming Chen, Tao Liu, Yang Fu, Shuixing Wu and Zhongmin Su
CrystEngComm 2009 vol. 11(Issue 4) pp:589-596
Publication Date(Web):05 Dec 2008
DOI:10.1039/B811833G
Three new nonlinear optical (NLO) chromophores (1–3) with the same dialkylamino group as an electron donor and the same 2-dicyanomethylene-3-cyano-4-methyl-2, 5-dihydrofuran (TCF) as an electron acceptor have been synthesized and characterized. The only difference in these three molecules lies in the length and nature of conjugation bridges. X-Ray single-crystal structural analyses show that 1 displays the best coplanarity between the two building blocks (conjugative bridge and TCF acceptor moieties) among three chromophores. The crystal packing reveals various antiparallel dimer structures which are stabilized by intermolecular π–π stacking, C–H⋯N and C–H⋯O interactions. Density functional theory (DFT) calculations demonstrate a linear relation between the first-order hyperpolarizability (β) and the bond length alternation (BLA) for all investigated chromophores.
Co-reporter:Xiang-Rong Hao, Xin-Long Wang, Zhong-Min Su, Kui-Zhan Shao, Ya-Hui Zhao, Ya-Qian Lan and Yao-Mei Fu
Dalton Transactions 2009 (Issue 40) pp:8562-8566
Publication Date(Web):01 Sep 2009
DOI:10.1039/B906728K
Two unprecedented 3D porous anionic metal–organic frameworks, [Me2NH2]2[Cd2(bpdc)3]·4dma 1 and [Me2NH2]2[Cd2(NH2bdc)3]·4dma 2 (dma = N,N′-dimethylacetamide, bpdc = 4,4′-biphenyldicarboxylate, NH2bdc = 2-amino-1,4-benzenedicarboxylate) have been solvothermally synthesized with a dimethylammonium cations template. Both 1 and 2 are constructed from low-symmetry SBUs. 1 has a chiral framework with helical nanotube-like channels, and 2 has a MOF-5-like motif. The fluorescence, N2 adsorption and ion-exchange properties for 1 have been examined.
Co-reporter:Chun-Guang Liu, Wei Guan, Li-Kai Yan, Ping Song and Zhong-Min Su
Dalton Transactions 2009 (Issue 31) pp:6208-6213
Publication Date(Web):24 Jun 2009
DOI:10.1039/B902837D
In the present paper, the electronic structures and bonding features between the ruthenium atom and nitrido ligand of known nitrido ruthenium (VI) porphyrin and hexavalent ruthenium nitrido derivative of Keggin typical polyoxometalate (POM) have been investigated by using density functional theory (DFT) calculations and natural bond orbital (NBO) analysis. The results show that the Keggin POM complex and porphyrin complex have the RuN triple bond, the NBO calculations yield a nitrido ligand charge close to zero in both complexes because of the charge transfer from the rich-electron nitrido ligand to the ruthenium center. The molecular orbitals predict that the ruthenium porphyrin complex would generate oxidized species with no change in the oxidation state of the metal center; however, the ruthenium center of the POM complex would be oxidized in a one-electron-oxidation process because of the different HOMO character, and moreover, the spin unrestricted calculations confirm this prediction made by molecular orbital analysis. On the other hand, both complexes have the same reduced center, the RuN unit, and the one-electron-reduction process weakens the bonding interaction between the ruthenium atom and nitrido ligand for both complexes because the antibonding LUMO is occupied. Compared with the porphyrin complex, the POM complex possesses a higher RuN composition and lower energy π* antibonding orbital according to gas-phase and solvent calculations.
Co-reporter:Ya-Qian Lan, Shun-Li Li, Kui-Zhan Shao, Xin-Long Wang, Xiang-Rong Hao and Zhong-Min Su
Dalton Transactions 2009 (Issue 6) pp:940-947
Publication Date(Web):16 Dec 2008
DOI:10.1039/B810287B
Two POM-pillared 3D porous compounds, [CuICuII(CuIIfcz)2(H2O)5(PMoVI10MoV2O40)]·6H2O (1) and [CuI2(CuIIfcz)2(H2O)2(PMoVI8VV3VIV3O42)]·6H2O (2) (Hfcz = fluconazole, (1-(2,4-difluorophenyl)-1,1-bis[(1H-1,2,4-triazol-1-yl)methyl]benzyl alcohol) have been constructed based on different polyanions, (Cufcz)22+ macrocations and copper cations by the hydrothermal method. The (Cufcz)22+ macrocations link Cu cations to generate a 2D wavelike cationic sheet. Then the POM anions act as pillars to the cationic sheet to form different POM-pillared 3D frameworks. In compound 1, the polyanion exhibits a rare coordination mode and acts as a penta-dentate ligand, which acts as to pillars to the cationic sheet to form an unprecedented 3D (3,4,5,6)-connected open framework with (3·6·7)(32·6·73)(33·4·62·73·8)(34·42·62·76·8)(32·62·76·84·10) topology. In compound 2, polyanions covalently link cationic sheets to extend to an unusual 3D (3,4,6)-connected framework with the (52·6)(52·62·7·9)(54·64·74·93) topology. To the best of our knowledge, it is the first time that POM-pillared 3D metal–organic frameworks have been realized by combining POMs with deliberately designed macrocations and transition-metal ions, using a rational approach for synthesis of POM-based open metal–organic frameworks. In addition, the electrochemical behaviors of compounds 1 and 2 have been investigated.
Co-reporter:Ya-Qian Lan, Shun-Li Li, Yao-Mei Fu, Dong-Ying Du, Hong-Ying Zang, Kui-Zhan Shao, Zhong-Min Su and Qiang Fu
Crystal Growth & Design 2009 Volume 9(Issue 3) pp:1353
Publication Date(Web):January 27, 2009
DOI:10.1021/cg8005234
A series of mixed-ligand coordination complexes, namely, [Zn(L1)2(p-BDC)] (1), [Cd(L1)2(p-BDC)] (2), [Zn(L2)(p-BDC)] (3), [Cd(L2)(p-BDC)] (4), and [Zn2(L3)(p-BDC)1.5(OH)]·H2O (5) (L1 = 2-chloro-5-((2-(pyridin-2-yl)-1H-imidazol-1-yl)methyl)pyridine, L2 = 2-(1-(pyridin-3-ylmethyl)-1H-imidazol-2-yl)pyridine, L3 = 2-(1-(pyridin-4-ylmethyl)-1H-imidazol-2-yl)pyridine, and p-BDC = 1,4-benzenedicarboxylate), have been synthesized under hydrothermal conditions. Their structures have been determined by single crystal X-ray diffraction analyses and further characterized by elemental analyses, IR spectra, and thermogravimetric (TG) analyses. Compounds 1 and 2 are isostructural and show infinite chains which extend into two-dimensional (2D) supramolecular sheets by π···π interactions. For 3, L2 ligands link ZnII ions to form dimeric units [Zn2(L2)2] which are connected by p-BDC anions to form 63 layers which interpenetrate each other in a vertical manner to generate a rare 2D → three-dimensional (3D) polycatenated net. In 4, similar dimeric units [Cd2(L2)2] formed by L2 ligands and Cd cations which are linked by p-BDC ligands to give a unique 3D 3-fold interpenetrated (10,3)-b network structure. In 5, two kinds of p-BDC ligands coordinate to Zn cations to form a 2-fold interpenetrating network with α-Po topological structure, in which each L3 ligand coordinates to a Zn ion acting as a terminal ligand. By careful inspection of the structures of 1−5, we believe that the coordination geometries of metal centers and different substituent groups of N-donor ligands adopting various coordination modes are crucial factors for the formation of the different structures. The photoluminescent properties of L1−L3 and 1−5 have been studied in the solid state at room temperature.
Co-reporter:Jia Zhuang;Likai Yan;Chunguang Liu ;Zhongmin Su
European Journal of Inorganic Chemistry 2009 Volume 2009( Issue 17) pp:2529-2535
Publication Date(Web):
DOI:10.1002/ejic.200900011
Abstract
The bonding characteristics, first hyperpolarizabilities, and origin of the nonlinear optical (NLO) properties of aryloxido and salicylaldehydo derivatives of [XW5O18]3– (X = Zr or Ti) have been investigated using density functional theory (DFT). The flexible bonding behavior of the linkage oxygen (OL) atom attracted our attention initially, and NBO analysis revealed that there aret two different kinds of π-interactions involving the OL atom in these systems. In two aryloxido derivatives (systems 1 and 2), OL–metal π-interactions are largely OL–M p-d in character, whereas in the salicylaldehydo derivatives (system 3) we observed another kind of π-interaction, namely OL–C p-p π-bonding. The differences in molecular composition in our studied systems are very slight. However, the β0 values differ significantly. Thus, system 3 has the largest β0 value(559.27 × 10–30 esu), which is 170 times larger than that of system 1. This variation can be traced to the different electronic transitions and charge-transfer characteristics. The heteropolyanion cluster and organic ligand are both electron-rich groups. A small change in molecular composition or architecture can therefore significantly modify the configuration and overlapping of molecular orbitals, thus causing a “butterfly effect” upon the electronic transitions and charge-transfer characteristics.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2009)
Co-reporter:Muhammad Ramzan Saeed Ashraf Janjua;Wei Guan;Chun Guang Liu;Shabbir Muhammad;Likai Yan ;Zhongmin Su
European Journal of Inorganic Chemistry 2009 Volume 2009( Issue 34) pp:5181-5188
Publication Date(Web):
DOI:10.1002/ejic.200900713
Abstract
The second-order polarizabilities, transition moments, and density of states of aryldiazenido hexamolybdates derivatives were investigated by density functional theory (DFT). System 2 [Mo6O18(N2C6H5)]3– has a considerably large second-order polarizability, 14.50 × 10–30 esu, and it is larger than that of system 1 [Mo6O18(N2C6H4NO2)]3– due to the absence of nitro group in the aryldiazenido ligand. The aryldiazenido ligand acts as an electron donor and the polyanion acts as an electron acceptor. The substitution of an amino(-NH2) group in the ortho/para positions on the aryldiazenido segment leads to a substantially higher nonlinear optical (NLO) response. The introduction of an electron donor(-NH2) in the ortho, meta, para, and ortho/para positions on the aryldiazenido ligand significantly enhances the second-order polarizabilities of aryldiazenido hexamolybdates in comparison to the electron acceptor (-NO2) as in system 1, because the electron-donating ability was reasonably enhanced when the electron donor is attached to the aryldiazenido ligand. Furthermore, orbital analysis shows that incorporation of another phenyl (aromatic) ring in the aryldiazenido ligand leads to a maximum NLO response by reverting the direction and degree of charge transfer (CT), which might result from the C=C π-conjugated bridge. System 8 [Mo6O18 (N2C14H11)]3– possesses a strikingly large and conspicuous static second-order polarizability (βvec) computed to be 210.21 × 10–30 esu. The NLO response can be tuned by subtle changes in the aryldiazenido segment; the present investigation provides important insight into the NLO properties of (aryldiazenido) hexamolybdate derivatives.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2009)
Co-reporter:Hai-jun Pang;Chun-jing Zhang;Jun Peng;Yong-hui Wang;Jing-quan Sha;Ai-xiang Tian;Peng-peng Zhang;Yuan Chen;Min Zhu
European Journal of Inorganic Chemistry 2009 Volume 2009( Issue 34) pp:5175-5180
Publication Date(Web):
DOI:10.1002/ejic.200900559
Abstract
Two isostructural compounds, {[Cu2(DF)2H2O]2SiW12O40}·2H2O (1) and {[Cu2(DF)2H2O]2GeMo12O40}·2H2O (2) (DF = 4,5-diazafluoren-9-one), were synthesized and characterized by routine methods and single-crystal X-ray diffraction. In the compounds, each Keggin cluster links five copper dimers [Cu2(DF)2H2O]2+ in an unusual asymmetrical pentacoordination mode to form 3D networks with left- and right-handed helical chains. Notably, by comparing 1 and 2 with other helical compounds with Keggin clusters as connectors, we found that the pitches of those compounds are mainly dominated by the coordination modes of the Keggin clusters, suggesting that the Keggin clusters are pitch-tunable synthons. Furthermore, the luminescent and electrochemical properties of the title compound were studied. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2009)
Co-reporter:Dan Gu, Guochun Yang, Yi He, Bin Qi, Guang Wang, Zhongmin Su
Synthetic Metals 2009 Volume 159(23–24) pp:2497-2501
Publication Date(Web):December 2009
DOI:10.1016/j.synthmet.2009.08.040
A triphenylamine-containing pH chemosensor, N,N′-di[3-(diphenylamine)benzyl]-piperazine, was designed and synthesized with Ullmann reaction. The compound crystallize in the monoclinic space group P21/n: a = 9.8004(12) Å, b = 13.3321(17) Å, c = 12.9575(16) Å, β = 103.510(2)°, V = 1646.2(4) Å3, Z = 2 and ρ = 1.212 Mg/m3. The synthesized compound excited by UV light produce intensive emission and a quantum yield of 0.56 relative to quinine sulfate was achieved. The fluorescence intensity of synthesized compound in water/DMF (4:1, v/v) under excitation of 307 nm decreased with the decrease of pH, experimental and computational studies further confirmed that the protonation of N-triphenylamine leads to the fluorescence quenching. The results clearly indicate the potential of synthesized compound as highly efficient “on–off” switcher for protons.
Co-reporter:Guang-Juan Xu, Ya-Hui Zhao, Kui-Zhan Shao, Lei Chen, Hong-Ying Zang, Zhong-Min Su, Dong-Ying Du
Inorganic Chemistry Communications 2009 Volume 12(Issue 10) pp:1024-1026
Publication Date(Web):October 2009
DOI:10.1016/j.inoche.2009.08.012
Co-reporter:Gang Sun;Min Zhang;Guochun Yang;Liang Zhao;Qiang Fu;Zhongmin Su
Chinese Journal of Chemistry 2009 Volume 27( Issue 10) pp:1891-1896
Publication Date(Web):
DOI:10.1002/cjoc.200990317
Abstract
An unsystematic molecule PPV-Alq3 [3-(4-((E)-2-(8-hydroxy-3-(4-styrylstyryl)quinolin-Alq2-6-yl)vinyl)- styryl)-6-(4-styrylstyryl)quinolin-8-olate-Alq2; q=8-quinolinolate], which combines poly(p-phenylenevinylene) with tris(8-quinolinolate)aluminum, has been studied using a localized-density-matrix method. The absorption spectra and electronic transition properties were analyzed and compared with both intermediate neglect of the differential overlap method and the localized-density-matrix method. Great efforts have been made on investigating conjugated system on the absorption properties as these can be particularly important for many applications. Two different absorptions of the special molecule, tris(8-quinolinolate)aluminum grafted on poly(p-phenylenevinylene) units, were further discussed with density matrices. For the molecule, the first absorption peak is at 413 nm near the purple light. Two 8-hydroxyquinolines have very slight electronic transition properties. Another absorption peak is at 237 nm. The second characteristic peak of molecule is completely different from that of the first one, which comes from contribution of 8-hyroxyquinolines in the two different side chains. Our studies show that electronic transition properties of poly(p-phenylenevinylene) can be effectively tuned by grafting tris(8-quinolinolate)-aluminum on poly(p-phenylenevinylene) from the standpoint of transition energies, frontier molecular orbitals and density matrices.
Co-reporter:Yan-Hong Xu, Ya-Qian Lan, Xin-Long Wang, Hong-Ying Zang, Kui-Zhan Shao, Yi Liao, Zhong-Min Su
Solid State Sciences 2009 Volume 11(Issue 3) pp:635-642
Publication Date(Web):March 2009
DOI:10.1016/j.solidstatesciences.2008.10.008
Five novel coordination polymers, [Zn(imbz)2]n (1), {[Zn(imbz)2]·H2O}n (2), [Zn(imbz)(μ2-OH)]n (3), [Zn3(imbt)2(p-bdc)3]n (4), [Zn4(μ3-OH)2(imbt)2(p-bdc)3]n (5), (imbt = 4′-(imidazol-1-ylmethyl)benzonitrile, imbz− = 4′-(imidazol-1-ylmethyl)benzoate and p-bdc = terephthalic acid) have been hydrothermally prepared through systematically changing the pH values of reaction mixture, and structurally characterized by elemental analysis, IR spectroscopy and single-crystal X-ray crystallography. Compounds 1 and 2 exhibit similar 2D (4,4) grid structures, whereas compound 2 contains a right-handed helix along b-axis. Compound 3 has a distorted diamond framework which was constructed via imbz− ligands and μ2-OH groups linking metal atoms. Compound 4 shows a 2D 6-connected network with trinuclear zinc clusters as secondary building units (SBUs), whereas 5 shows a distorted α-Po with tetranuclear zinc clusters as SBUs, in which p-bdc ligands act as bridges. Moreover, compounds 1–5 all exhibit strong blue photoluminescence in the solid state at room temperature.
Co-reporter:Chunsheng Qin, Guochun Yang, Liang Zhao, Shiling Sun, Yongqing Qiu, Zhongmin Su, Yulan Zhu
Synthetic Metals 2009 Volume 159(21–22) pp:2406-2409
Publication Date(Web):November 2009
DOI:10.1016/j.synthmet.2009.07.015
The equilibrium geometries of gold (III) alkyl complexes are optimized by DFT/B3LYP method. On the basis of the optimized structures, the electronic structures and second-order nonlinear optical properties are calculated by using time-dependent density-functional theory (TDDFT) combined with the sum-over-states (SOS) method. The results show that these complexes possess remarkably larger molecular second-order polarizabilities compared with the typical organometallic and organic complexes. The analysis suggests that charge transfer from the z-axis directions plays a key role in the second-order nonlinear optical response. Moreover, different ancillary ligands can substantially adjust the second-order nonlinear optical response. Thus, it can be concluded that these complexes will be hopeful candidates for the second-order nonlinear optical materials from the standpoint of high transparency, relatively large β values and small dispersion behaviors.
Co-reporter:Hongze Gao, Houyu Zhang, Rigen Mo, Shiling Sun, Zhong-Min Su, Yue Wang
Synthetic Metals 2009 Volume 159(17–18) pp:1767-1771
Publication Date(Web):September 2009
DOI:10.1016/j.synthmet.2009.05.023
Co-reporter:Yong-Ming Lu, Ya-Qian Lan, Yan-Hong Xu, Zhong-Min Su, Shun-Li Li, Hong-Ying Zang, Guang-Juan Xu
Journal of Solid State Chemistry 2009 Volume 182(Issue 11) pp:3105-3112
Publication Date(Web):November 2009
DOI:10.1016/j.jssc.2009.08.018
To investigate the relationship between topological types and molecular building blocks (MBBs), we have designed and synthesized a series of three-dimensional (3D) interpenetrating metal-organic frameworks based on different polygons or polyhedra under hydrothermal conditions, namely [Cd(bpib)0.5(L1)] (1), [Cd(bpib)0.5(L2)]·H2O (2), [Cd(bpib)0.5(L3)] (3) and [Cd(bib)0.5(L1)] (4), where bpib=1,4-bis(2-(pyridin-2-yl)-1H-imidazol-1-yl)butane, bib=1,4-bis(1H-imidazol-1-yl)butane, H2L1=4-(4-carboxybenzyloxy)benzoic acid, H2L2=4,4′-(ethane-1,2-diylbis(oxy))dibenzoic acid and H2L3=4,4′-(1,4-phenylenebis(methylene))bis(oxy)dibenzoic acid, respectively. Their structures have been determined by single crystal X-ray diffraction analyses and further characterized by elemental analyses, IR spectra, and thermogravimetric (TG) analyses. Compounds 1–3 display α-Po topological nets with different degrees of interpenetration based on the similar octahedral [Cd2(–COO)4] building blocks. Compound 4 is a six-fold interpenetrating diamondoid net based on tetrahedral MBBs. By careful inspection of these structures, we find that various carboxylic ligands and N-donor ligands with different coordination modes and conformations, and metal centers with different geometries are important for the formation of the different MBBs. It is believed that different topological types lie on different MBBs with various polygons or polyhedra. Such as four- and six-connected topologies are formed by tetrahedral and octahedral building blocks. In addition, with the increase of carboxylic ligands’ length, the degrees of interpenetration have been changed in the α-Po topological nets. And the luminescent properties of these compounds have been investigated in detail.A series of three-dimensional interpenetrating metal-organic frameworks based on different polygons or polyhedra has been synthesized. The crystal structures and topological analysis of these compounds, along with a systematic investigation of the relationship between topological types and molecular building blocks, will be discussed.
Co-reporter:Guang-Juan Xu, Ya-Hui Zhao, Kui-Zhan Shao, Gang Yuan, Zhong-Min Su, Li-Kai Yan
Inorganic Chemistry Communications 2009 Volume 12(Issue 10) pp:969-971
Publication Date(Web):October 2009
DOI:10.1016/j.inoche.2009.07.023
A novel 3D polymer [Zn(L1)(1,4-BDC)0.5] (1) (HL1 = 3,5-bis(pyridin-4-ylmethoxy)benzoic acid, 1,4-BDC = 1,4-benzenedicarboxylate anion) has been isolated under hydrothermal condition and characterized by single crystal X-ray diffraction. Compound 1 displays a 3D 4-fold interpenetrating (3,4)-connected net with (4 · 82)(4 · 85) topology. In addition, powder X-ray diffraction, photoluminescent property and thermogravimetric analysis for 1 are investigated in detail.A novel 3D polymer [Zn(L1)(1,4-BDC)0.5] (1) (HL1 = 3,5-bis(pyridin-4-ylmethoxy)benzoic acid, 1,4-BDC = 1,4-benzenedicarboxylate anion) has been isolated under hydrothermal condition and characterized by single crystal X-ray diffraction. Compound 1 displays a 3D 4-fold interpenetrating (3,4)-connected net with (4 · 82)(4 · 85) topology. In addition, powder X-ray diffraction, photoluminescent property and thermogravimetric analysis for 1 are investigated in detail.
Co-reporter:Yan-Hong Xu, Ya-Qian Lan, Ya-Hui Zhao, Dong-Ying Du, Guang-Juan Xu, Kui-Zhan Shao, Zhong-Min Su, Yi Liao
Inorganic Chemistry Communications 2009 Volume 12(Issue 2) pp:169-172
Publication Date(Web):February 2009
DOI:10.1016/j.inoche.2008.11.030
Two Zn(II)-2,2′-biimidazole derivative supramolecular isomers 1 and 2 formulated as [Zn(L)] · 2H2O (H2L = 4,4′-(1H,1′H-2,2′-biimidazole-1,1′-diylbis(methylene))dibenzoic acid) have been synthesized, and structurally characterized by elemental analysis, IR spectroscopy, single-crystal X-ray crystallography. Complex 1 exhibits a 2D four-connected network with (43.63) topology when the zinc center and the L2− ligand act as four-connected nodes (or a distorted (4,4) grid if the [Zn2(imidazole)4] units are considered as four-connected nodes). Complex 2 shows a novel four-connected twofold interpenetrated net with (42.62.82) topology when the zinc center and the L2− ligand act as four-connected nodes (or a twofold interpenetrated α-Po topology when [Zn2(imidazole)4] units are identified as six-connected nodes). Moreover, complexes 1 and 2 both exhibit strong luminescent properties at room temperature.Two novel supramolecular isomers with their formulas as [Zn(L)] · 2H2O, have been prepared by hydrothermal reaction. Complex 1 shows a 2D grid and complex 2 exhibits an interesting twofold interpenetrated 3D network structure. Moreover, two complexes both exhibit good room-temperature phosphorescence emissions.
Co-reporter:Hong-Ying Zang, Ya-Qian Lan, Guang-Sheng Yang, Zhong-Min Su, Xin-Long Wang, Kui-Zhan Shao, Li-Kai Yan
Journal of Molecular Structure 2009 Volume 935(1–3) pp:69-74
Publication Date(Web):29 October 2009
DOI:10.1016/j.molstruc.2009.06.045
Two new octamolybdate-based inorganic–organic hybrid compounds NiII(HL)2(H2O)2(β-Mo8O26) (1) and CuI4L4(β-Mo8O26) (2) (L = 3-((1H-1,2,4-triazol-1-yl)methyl)pyridine) have been hydrothermally synthesized and structurally characterized. Compound 1 is built up of octamolybdate anions [β-Mo8O26]4− covalently linked by [Ni(HL)2(H2O)2]4+ cations into 1D infinite chains and was further connected by hydrogen bonds into a 3D supramolecular structure. Compound 2 is constructed from [β-Mo8O26]4− building blocks and [Cu2L2]2n4n+ strands to form a 3-connected 2D network. Furthermore, the diffuse reflectance spectra reveal that these two inorganic–organic hybrid compounds are potential wide gap semiconductor materials.
Co-reporter:Shabbir Muhammad, Muhammad Ramzan Saeed Ashraf Janjua and Zhongmin Su
The Journal of Physical Chemistry C 2009 Volume 113(Issue 28) pp:12551-12557
Publication Date(Web):June 22, 2009
DOI:10.1021/jp903075s
In the present investigation, a new sequence has been presented for the reversible switching and modulation of nonlinear optical properties (NLO). The static first hyperpolarizabilities (β) of four dibenzoborole derivatives have been computed by DFT. The β values of these dibenzoborole derivatives have shown a significant increase with the attachment of F− and/or one electron reduction. For example, 5-fluoro-5-(2,4,6-triisopropylphenyl)-2,8-dimethoxy-3,7-bithienyl-5H-dibenzo[d,b] borole ion (3·F−) and 5-fluoro-5-phenyl-3,7-bis-dinitrothienyl-5H-dibenzo[d,b]bor- ole ion (4·F−) both showed β values as large as 64 × 10−30 and 272 × 10−30 esu, that is, about 12 times and 4 times larger than their counterparts 5-(2,4,6-triisopropylphenyl)-2,8-dimethoxy-3,7-bithienyl-5H-dibenzo[d,b]borole (3) and 5-phenyl-3,7-bis-dinitrothienyl-5H-dibenzo[d,b]borole (4) (without F−), respectively. Similarly, systems 3Red and 4Red (one electron reduced) also showed 47 times and 15 times larger β values than their neutral forms, respectively. Interestingly, this NLO switching is two-dimensional in characteristic in which large off-diagonal hyperpolarizability tensors can be related to the charge-transfer transitions that are polarized perpendicular to the molecular dipolar axis. Density of states (DOS) and frontier molecular orbital (FMO) analysis show that the binding of F− at a boron atom and/or one electron reduction process turn off the pπ→π* conjugation of vacant p-orbital of boron atom in LUMOs, resulting in a higher extent of perpendicular charge transfer (CT) and lager β values. The present investigation reveals a new idea and different means for multifunctional use of the present dibenzoborole class, especially (already synthesized) 3·F− as two-dimensional NLO molecular switch with Cartesian nonlinear anisotropy as large as η = 10.48.
Co-reporter:Hong-Liang Xu, Zhi-Ru Li, Zhong-Min Su, Shabbir Muhammad, Feng Long Gu and Kikuo Harigaya
The Journal of Physical Chemistry C 2009 Volume 113(Issue 34) pp:15380-15383
Publication Date(Web):August 5, 2009
DOI:10.1021/jp901358f
Four knot-isomers of Möbius cyclacene are composed of 15 nitrogen-substituted benzo rings. They are non-Möbius cyclacenes without a knot (0), Möbius cyclacenes with a knot (1), non-Möbius cyclacenes with two knots (2), and Möbius cyclacenes with three knots (3). Their structures and nonlinear optical properties are systematically studied. The order of first hyperpolarizability (β0) is 4693 (0) < 10 484 (2) < 25 419 (3) < 60 846 au (1). The β0 values of the knot-isomers with knot(s) are larger than that of the knot-isomer without a knot. It shows that the β0 value can be dramatically increased (13 times) by twisting the knot(s) to the cyclacene. Two noticeable relationships between the number of knots and the first hyperpolarizability have been observed: (i) the β0 values of one surface Möbius cyclacene (1 and 3) with an odd number of knots are larger than that of two-surface non-Möbius cyclacenes (0 and 2) with an even number of knots. (ii) For the one-surface Möbius cyclacenes, the β0 value for 1 with one knot is larger than that for 3 with three knots. On the other hand, the largest component of β0 is alternated for the four knot-isomers. The largest components are βz for the 0 and βy for the 1 and 2. The largest component turns back to the βz for the 3.
Co-reporter:Chun-Guang Liu, Wei Guan, Li-Kai Yan, Zhong-Min Su, Ping Song and En-Bo Wang
The Journal of Physical Chemistry C 2009 Volume 113(Issue 45) pp:19672-19676
Publication Date(Web):October 14, 2009
DOI:10.1021/jp906979z
To date, the most widely used second-order nonlinear optical (NLO) materials are the totally inorganic crystals. However, the small photoelectric coefficients of inorganic NLO materials are the bottleneck in practical applications. The donor−conjugated bridge−acceptor (D−A) model, which is successfully used in the development of organic second-order NLO materials, is still prohibitive in totally inorganic molecules. In the present paper, time-dependent density functional (TDDFT) has been employed to investigate the second-order NLO properties of a series of transition-metal-trisubstituted polyoxometalates (POMs)−diphosphate clusters. We find that these totally inorganic POM clusters possess D−A structure, and the large static first hyperpolarizability can be effectively designed based on this D−A model. The results show that the substituted transition metal centers can be viewed as electron acceptor, and the POM cluster serves as both electron donor and conjugated bridge. The three vanadium atoms derivative of 30-molybdobipyrophosphate POM cluster displays large static first hyperpolarizability by ∼700 × 10−30 esu, and it is ∼70 times as large as that of typical organic NLO molecule p-nitroaniline (PNA) according to LB94/TZP calculations. Thus, this POM cluster seems to be promising totally inorganic materials for application in nonlinear optics.
Co-reporter:Shabbir Muhammad;Chunguang Liu;Liang Zhao;Shuixing Wu
Theoretical Chemistry Accounts 2009 Volume 122( Issue 1-2) pp:77-86
Publication Date(Web):2009 January
DOI:10.1007/s00214-008-0486-8
The interaction between chemosensor, N-(2-methyl-1,3-dioxo-indan-5-yl)-benzamide (1) and different halide ions (F−, Cl− and Br−) has been investigated using density functional theory (DFT). A clear insight of the sensor anion binding process has been presented. Our calculations revealed that the observed colorimetric and fluorescent signals are induced due to the ground state deprotonation of the sensor molecule caused by F− which has two times higher binding affinity than other halide ions (Cl− and Br−). Derivatives of system 1 have been made to find a better sensor with higher binding affinity and longer wavelength of absorption. All the derivatives are better sensors than the parent 1 except 4-methyl-N-(2-methyl-1,3-dioxo-indan-5-yl)-benzamide (2). Among these derivatives, trimethyl-[4-(2-methyl-1,3-dioxo-indan-5-ylcarbamoyl)-phenyl]-ammonium (8) and (5-benzoylamino-1,3-dioxo-indan-2-yl)-trimethyl-ammonium (9) showed a change to higher binding energies of about 58 Kcal/mol and longer absorption wavelengths of 53 nm after deprotonation process than the parent system 1 which is highly demanded in selective chemical sensing. Systems 8, 9 and their deprotonated zwitterionic forms (8z and 9z) have also been studied for their nonlinear optical responses. Systems 8, 9 showed significantly good first hyperpolarizability (β) of 84 × 10−30 and 40 × 10−30 esu, respectively. These β values increase in zwitterionic states up to 216 × 10−30 and 109 × 10−30 esu, respectively after deprotonation with F−, representing a new signal of deprotonation.
Co-reporter:L. L. Shi;T. Li;S. S. Zhao;H. Li;Zhongmin Su
Theoretical Chemistry Accounts 2009 Volume 124( Issue 1-2) pp:29-36
Publication Date(Web):2009 September
DOI:10.1007/s00214-009-0573-5
In this work, we theoretically investigate the effect of phenyl group on the electronic and phosphorescent properties of cyclometalated platinum(II) complexes, thereby designing an efficient blue emitting material. Three platinum(II) complexes Pt(N^N^N)Cl (N^N^N = terpyridine), Pt(N^C^N)Cl (N^C^N = 1,3-di(2-pyridyl)-benzene) and Pt(N^N^C)Cl (N^N^C = 6-phenyl-2,2′-bipyridines) are chosen as the models. Their electronic and phosphorescent properties are investigated utilizing quantum theoretical calculations. The results reveal that the phenyl group significantly affects the molecular and electronic structures, charge distribution and phosphorescent properties. The coordination bond length trans to phenyl group is the longest among the same type of bonds owing to the trans influence of phenyl group. Moreover, the phenyl group largely restricts the geometry relaxation of cyclometalated ligand. The strong σ-donor ability of Pt–C bond makes more electrons center at Pt atom and the fragments trans to phenyl group. In comparison with Pt(N^N^N)Cl and Pt(N^N^C)Cl, the complex Pt(N^C^N)Cl has the smallest excited-state geometry relaxation and the biggest emission energy and spatial overlap between the transition orbitals in the emission process. As a result, Pt(N^C^N)Cl has the largest emission efficiency, which well agrees with the experimental observation. Based on these calculation results, a potentially efficient blue-emitting material is designed via replacing pyridine groups in Pt(N^C^N)Cl by 3-methylimidazolin-2-ylidene.
Co-reporter:W. Guan;C. G. Liu;P. Song;G. C. Yang;Z. M. Su
Theoretical Chemistry Accounts 2009 Volume 122( Issue 5-6) pp:265-273
Publication Date(Web):2009 April
DOI:10.1007/s00214-009-0505-4
To analyze the effect of redox state changes on the second-order nonlinear optical (NLO) responses of organoimido-functionalized Keggin-type heteropolyanions, the excitation properties and static second-order polarizabilities of fully oxidized state, the first and second reduced states were calculated by means of the time-dependent density functional theory (TDDFT) method combined with the sum-over-states (SOS) formalism. The incorporation of extra electrons causes significant enhancement in the second-order NLO activity. The reduced complexes show more than three times the efficiency of fully oxidized state. Moreover, the NLO activities for PW11ReVNPh system can also be modified by controlling the spin multiplicity. The high spin state (33) has twice larger βvec value than the low spin state (13). The characteristic of the charge-transfer transition corresponding to the dominant contributions to the βvec values indicates that metal-centered redox processes influence the intramolecular donor or acceptor character, which accordingly leads to the variations in the computed β values. Owing to the reversible and manipulable redox processes, these kinds of the POM-based hybrid complexes could comprise a promising family of three-state redox-switchable molecular device combining chromic, magnetic, and NLO output.
Co-reporter:Ping Song;LiKai Yan;Wei Guan;JingDong Feng;ChunGuang Liu
Science Bulletin 2009 Volume 54( Issue 2) pp:203-211
Publication Date(Web):2009 January
DOI:10.1007/s11434-008-0500-5
Keggin-type phenylimido-polyoxometalates α-[PM12O39NPh]3− (M = W and Mo) have been systematically investigated on the electronic structures, redox as well as nonlinear optical (NLO) properties by density functional theory (DFT). The strong M≡N bond confirmed by natural bond orbital (NBO) analysis comprises one s bond and two π bonds, the same as Mo≡N in [Mo6O18NPh]2−. Furthermore, phenylimido segment effectively modifies the electronic properties of α-[PM12O39NPh]3−. On one hand, when enlarging the inorganic cluster from {Mo6O18} to {PMo12O39}, the energy gap between HOMO and LUMO in α-[PMo12O39NPh]3− decreased, resulting in enormously anodic shift for the reduction potential, while the excitation energy is less and the total second-order polarizability β0 is up to 438-3×10−30 esu, which is nearly 10 times larger than that of [Mo6O18NPh]2−. On the other hand, when metal W in α-[PM12O39NPh]3− is substituted by Mo, the interaction between Mo and N is enhanced and the redox ability becomes stronger. The β0 value for α-[PMo12O39NPh]3− is more than 5 times higher than that of α-[PW12O39NPh]3−. It indicates that changing appropriate metal or enlarging the inorganic cluster will improve the redox properties and second-order nonlinear response. Moreover, the electron transition for three compounds mentioned above occurred mainly from organoimido segment (as the electron donor) to polyanion cluster (as the acceptor). As a result, α-[PMo12O39NPh]3− may be a promising candidate for oxidant and nonlinear optical material.
Co-reporter:Huaqiao Tan;Yangguang Li ;Weilin Chen;Ding Liu;Zhongmin Su ;Ying Lu Dr.;Enbo Wang
Chemistry - A European Journal 2009 Volume 15( Issue 41) pp:10940-10947
Publication Date(Web):
DOI:10.1002/chem.200901112
Abstract
Five compounds based on [MnMo9O32]6−: (Himi)6[MnMo9O32] (1) (imi=imidazole), Na2(Himi)4[MnMo9O32]⋅2 H2O (2), Na3(Himi)3[MnMo9O32] (3), D-NH4Mn2.5[MnMo9O32]⋅11 H2O (4 a), and L-NH4Mn2.5[MnMo9O32]⋅11 H2O (4 b) were prepared and characterized. X-ray crystallographic analysis revealed that compounds 1 and 2 with imidazole molecules as linkers are racemic compounds; compound 3 is a racemic solid solution of Na+ cations and the polyoxoanion [MnMo9O32]6−; and compounds 4 a and 4 b are enantiomers. In compound 4, the homochiral polyoxoanions [MnMo9O32]6− are connected by Mn2+ cations to form a unique (45⋅6)(47⋅68) topology net framework. By adjusting the linkers from imidazole molecules to Na+ and finally Mn2+ cations, the chiral polyoxoanions [MnMo9O32]6− were changed from a racemic compound to a conglomerate. This means that spontaneous resolution can be efficiently realized by connecting homochiral polyoxoanions into one-dimensional (1D), 2D, and 3D structures, with an emphasis on using appropriate linkers with substantial interaction strength, directionality, and enantioselectivity.
Co-reporter:Y. L. Si;C. G. Liu;E. B. Wang;Z. M. Su
Theoretical Chemistry Accounts 2009 Volume 122( Issue 3-4) pp:217-226
Publication Date(Web):2009 March
DOI:10.1007/s00214-008-0501-0
To probe the cooperativity of charge transfer between organoimido and hexamolybdate, and enhance the second-order nonlinear optical (NLO) response of organoimido derivatives of hexamolybdates, electronic structures and second-order NLO properties of a series of charge-transfer covalently bonded organoimido derived hexamolybdate complexes with donor-(π conjugated bridge)-acceptor-(π conjugated bridge)-donor or acceptor-(π conjugated bridge)-donor-(π conjugated bridge)-acceptor structures were studied by density functional theory. Studies show that different combinations of the donor, acceptor, heterocycle, –C≡C– and –N=N– moieties, and orientation of heterocycle remarkably affect the second-order NLO responses. The complexes containing electronic acceptor matched with the direction of charge transfer possess remarkable large molecular second-order polarizabilities. Electronic transitions to crucial excited states show that x-polarized transition, contributed to the off-diagonal second-order polarizabiliy tensor (βzxx), possesses lower excited energy compared with z-polarized transition which accounted for the diagonal second-order polarizabiliy tensor (βzzz) and thus led to the large in-plane nonlinear anisotropy (u = βzxx/βzzz) value, as well as good two-dimensional (2-D) second-order NLO properties. These complexes can be used as excellent 2-D second-order NLO materials from the standpoint of both large β and u values.
Co-reporter:Shi-Ling Sun, Chun-Sheng Qin, Yong-Qing Qiu, Guo-Chun Yang, Zhong-Min Su
Journal of Organometallic Chemistry 2009 694(9–10) pp: 1266-1272
Publication Date(Web):
DOI:10.1016/j.jorganchem.2008.11.053
Co-reporter:Muhammad Ramzan Saeed Ashraf Janjua, Chun-Guan Liu, Wei Guan, Jia Zhuang, Shabbir Muhammad, Li-Kai Yan and Zhong-Min Su
The Journal of Physical Chemistry A 2009 Volume 113(Issue 15) pp:3576-3587
Publication Date(Web):March 19, 2009
DOI:10.1021/jp808707q
The dipole polarizabilities, dipole moments, density of states, and second-order nonlinear optical (NLO) properties of organoimido derivatives of hexamolybdates have been investigated by using time-dependent density functional response theory. This class of organic−inorganic hybrid compounds possesses remarkably large and eye-catching molecular second-order NLO response, especially [Mo6O17(NC16H12NO2)(FeNC10H9)]2− (7) and [Mo6O17(NC16H12NO2)(NC6H2(NH2)3)]2− (6) with static second-order polarizability (βvec) computed to be 15766.27 × 10−30 esu and 6299.59 × 10−30 esu, respectively. Thus, these systems have the possibility to be excellent second-order nonlinear optical materials. Analysis of the major contributions to the βvec value suggests that the charge transfer (CT) from polyanion to organic segment (D−A) along the z-axis plays the key role in NLO response; the polyanion acts as a donor (D) whereas organoimido acts as an acceptor (A) in all the studied systems. The computed βvec values increase by incorporation of an electron acceptor (−NO2) at the end of the phenyl ring of the organoimido segment. Furthermore, substitution of amino (−NH2) or ferrocenyl (−FeC10H9) at the outer side of polyanion and an electron acceptor (−NO2) at the end of the phenyl ring in organoimido segment simultaneously is more important to enhance the optical nonlinearity. Orbital analysis shows that the degree of CT between the polyanion and organoimido segments was increased when ferrocenyl donor was introduced. The present investigation provides important insight into the remarkably large NLO properties of organoimido-substituted hexamolybdates.
Co-reporter:Xin-Long Wang ;Chao Qin ;Shui-Xing Wu Dr.;Kui-Zhan Shao Dr.;Ya-Qian Lan Dr.;Shuang Wang Dr.;Dong-Xia Zhu , ;En-Bo Wang
Angewandte Chemie International Edition 2009 Volume 48( Issue 29) pp:5291-5295
Publication Date(Web):
DOI:10.1002/anie.200902274
Co-reporter:Kother Osman Ashiry, Ya-Hui Zhao, Kui-Zhan Shao, Zhong-Min Su, Guang-Juan Xu
Polyhedron 2009 28(5) pp: 975-979
Publication Date(Web):
DOI:10.1016/j.poly.2008.12.056
Co-reporter:Xin-Long Wang ;Chao Qin ;Shui-Xing Wu Dr.;Kui-Zhan Shao Dr.;Ya-Qian Lan Dr.;Shuang Wang Dr.;Dong-Xia Zhu , ;En-Bo Wang
Angewandte Chemie 2009 Volume 121( Issue 29) pp:5395-5399
Publication Date(Web):
DOI:10.1002/ange.200902274
Co-reporter:Guang-Juan Xu, Ya-Hui Zhao, Kui-Zhan Shao, Guang-Sheng Yang, Ya-Qian Lan, Zhong-Min Su, Li-Kai Yan
Polyhedron 2009 28(14) pp: 3155-3161
Publication Date(Web):
DOI:10.1016/j.poly.2009.07.011
Co-reporter:Ya-Qian Lan, Shun-Li Li, Zhong-Min Su, Kui-Zhan Shao, Jian-Fang Ma, Xin-Long Wang and En-Bo Wang
Chemical Communications 2008 (Issue 1) pp:58-60
Publication Date(Web):14 Nov 2007
DOI:10.1039/B714413J
Two enantiomerically 3D chiral POM-based architectures have been constructed based on the achiral ligand bbi, [V10O26]4− polyoxoanion and mixed valence Cu(I/II) without a chiral auxiliary, and they represent the first examples of enantiomerically 3D POM-based compounds using achiral ligands.
Co-reporter:Wei Guan, Guochun Yang, Chunguang Liu, Ping Song, Liang Fang, Likai Yan and Zhongmin Su
Inorganic Chemistry 2008 Volume 47(Issue 12) pp:5245-5252
Publication Date(Web):May 21, 2008
DOI:10.1021/ic8001527
In this paper, the relationship between the reversible redox properties and the second-order nonlinear optical (NLO) responses for the title series of complexes has been systematically investigated by using the time-dependent density functional theory (TDDFT) method combined with the sum-over-states (SOS) formalism. The results reveal that the successive reduction processes of five PW11ReN redox states should be PW11ReVII (1) → PW11ReVI (2) → PW11ReV (3) → PW11ReV1e (4) → PW11ReV2e (5). Furthermore, their electrochemical properties have been reproduced successfully. It is noteworthy that the second-order NLO behaviors can be switched by reversible redox for the present studied complexes. Full oxidation constitutes a convenient way to switch off the second-order polarizability (system 1). The incorporation of extra electrons causes significant enhancement in the second-order NLO activity, especially for the third reduced state (system 4), whose static second-order polarizability (βvec) is about 144 times larger than that of fully oxidized 1. The characteristic of the charge-transfer transition corresponding to the dominant contributions to the βvec values indicates that metal-centered redox processes influence the intramolecular donor or acceptor character. Therefore, these kinds of complexes with the facile and reversible redox states could become excellent switchable NLO materials.
Co-reporter:Ya-Qian Lan ; Shun-Li Li ; Xin-Long Wang ; Kui-Zhan Shao ; Dong-Ying Du ; Hong-Ying Zang
Inorganic Chemistry 2008 Volume 47(Issue 18) pp:8179-8187
Publication Date(Web):August 13, 2008
DOI:10.1021/ic800702d
Six polyoxometalate (POM)-based hybrid materials have been designed and synthesized based on octamolybdate building blocks and copper-organic units at different pH values under hydrothermal conditions, namely, [H2bbi][CuII(bbi)2(β-Mo8O26)] (1), [CuII(bbi)2(H2O)(β-Mo8O26)0.5] (2), [CuII(bbi)2(α-Mo8O26)][CuI(bbi)]2 (3), [CuIICuI(bbi)3(α-Mo8O26)][CuI(bbi)] (4), [CuI(bbi)]2[CuI2(bbi)2(δ-Mo8O26)0.5][α-Mo8O26]0.5 (5), and [CuI(bbi)][CuI(bbi)(θ-Mo8O26)0.5] (6), where bbi is 1,1′-(1,4-butanediyl)bis(imidazole). Their crystal structures have been determined by X-ray diffraction. In compound 1, the bbi ligands with bis-monodentate coordination modes link CuII cations to generate a 2D copper-organic unit with (4, 4) net, which is pillared by the (β-Mo8O26)4− anions to form a 3D framework with α-Po topology. The similar copper-organic units are connected alternately by (β-Mo8O26)4− anions to generate a 3D 2-fold interpenetrating (4,6)-connected framework with (44·62)(44·610·8) topology in compound 2. Compounds 3 and 4 are supramolecular isomers with polythreaded topology. If CuI···O interactions are considered, the structure of 3 is a novel self-penetrating (3,4,6)-connected framework with (52·8)2(54·6·8)(44·610·10) topology, and the structure of 4 is a (4,6)-connected framework with (42·63·7)(5·64·8)(42·56·66·8)(42·56·64·7·82) topology. Different from compounds 3 and 4, compounds 5 and 6 are supramolecular isomers with polythreaded topology based on different octamolybdate isomers. By careful inspection of the structures of 1−6, it is believed that various copper-organic units, which are formed by bbi ligands combined with CuII/CuI cations, octamolybdates with different types and coordination modes, and the nonbonding interactions between polyanions and copper-organic units are important for the formation of the different structures. In addition, with step by step increasing of the amount of organic amine, we have achieved the transformation of CuII ions into CuI ones in different degrees in POMs-based metal-organic frameworks (MOFs) for the first time. The infrared spectra, X-ray powder diffraction, and thermogravimetric analyses have been investigated in detail for all compounds, and the luminescent properties have been also been investigated for compounds 3 and 4.
Co-reporter:Xinyu Zhao, Dadong Liang, Shuxia Liu, Chunyan Sun, Ruige Cao, Chaoying Gao, Yuanhang Ren and Zhongmin Su
Inorganic Chemistry 2008 Volume 47(Issue 16) pp:7133-7138
Publication Date(Web):July 12, 2008
DOI:10.1021/ic800131r
The Dawson anion P2W18O626− has been used as a noncoordinating polyoxoanion template for the construction of two metal−organic frameworks, namely, [M2(bpy)3(H2O)2(ox)][P2W18O62]2(H2-bpy)·nH2O (M = Co(II), n = 3 (1); M = Ni(II), n = 2 (2)) (bpy = 4,4′-bipyridine; ox = C2O42−). Single-crystal X-ray analysis reveals that both of the structures exhibit 3D host frameworks constructed from the oxalate-bridged binuclear superoctahedron secondary building units (SBUs) and bpy linkers and the voids of which are occupied by Dawson anions, guest bpy, and water molecules. Magnetic studies reveal that there are antiferromagnetic exchange interactions among the transition-metal centers in compounds 1 and 2. Furthermore, a compound 1-modified carbon paste electrode (1-CPE) displays good electrocatalytic activity toward the reduction of nitrite.
Co-reporter:Li-Li Shi ; Yi Liao ; Guo-Chun Yang ; Zhong-Min Su ;Shan-Shan Zhao
Inorganic Chemistry 2008 Volume 47(Issue 7) pp:2347-2355
Publication Date(Web):March 6, 2008
DOI:10.1021/ic7018154
We report a quantum-chemical study of the electronic and optical properties of several platinum(II) dimers, [Pt(pip2NCN)]2(L)2+ (pip2NCNH = 1,3-bis(piperidylmethyl)benzene, L represents the bridging ligands pyrazine, 4,4′-bipyridine, or trans-1,2-bis(4-pyridyl)ethylene). The theoretical calculations reveal that as the π-conjugated length of bridging ligand increases, the energies of HOMOs and LUMOs, bonding energy of Pt−Nbridge, and the largest absorption strength increase whereas the ionization potentials decrease. According to the inner reorganization energy and density of states, we presume the hole-transporting properties of these dimers is better than the electron-transporting, and their inner reorganization energies for hole transport are lower than that of 4,4′-bis(phenyl-m-tolylamino)biphenyl (TPD), a well-known hole-transporting material. These platinum(II) dimers, especially [Pt(pip2NCN)]2(bpe)2+, hold promise for use as a new kind of third-order nonlinear optical material, owing to their large third-order polarizabilty value and high transparency. Moreover, the optoelectronic properties of these complexes are easy to tailor by modifying the peripheral and central ligands. These theoretical results are beneficial to the design of new functional materials with excellent optoelectronic properties.
Co-reporter:Ya-Qian Lan ; Shun-Li Li ; Jun-Sheng Qin ; Dong-Ying Du ; Xin-Long Wang ; Zhong-Min Su ;Qiang Fu
Inorganic Chemistry 2008 Volume 47(Issue 22) pp:10600-10610
Publication Date(Web):October 23, 2008
DOI:10.1021/ic801275w
A series of mixed-ligand coordination complexes, namely, [Cd2(bimb)2(L1)2] (1), [Cd(bpimb)0.5(L2)(H2O)] (2), [Zn5(bpib)2(L3)4(OH)2(H2O)2] (3), [Zn(bpib)0.5(L4)] (4), and [Cd(bib)(L4)] (5), where bimb = 1,4-bis((1H-imidazol-1-yl)methyl)benzene, bpimb = 1,4-bis((2-(pyridin-2-yl)-1H-imidazol-1-yl)methyl)benzene, bpib = 1,4-bis(2-(pyridin-2-yl)-1H-imidazol-1-yl)butane, bib = 1,4-bis(1H-imidazol-1-yl)butane, H2L1 = 4-((4-(dihydroxymethyl)phenoxy)methyl)benzoic acid, H2L2 = 4,4′-methylenebis(oxy)dibenzoic acid, H2L3 = 3,3′-methylenebis(oxy)dibenzoic acid, and H2L4 = 4,4′-(2,2′-oxybis(ethane-2,1-diyl)bis(oxy))dibenzoic acid, have been synthesized under hydrothermal conditions. Their structures have been determined by single-crystal X-ray diffraction analyses and further characterized by elemental analyses, IR spectra, and thermogravimetric (TG) analyses. In 1, (L1)2− anions link the metal−neutral ligand subunits to generate a 2-fold parallel interpenetrating net with the 63 topology. In 2−4, neutral ligands connect the various metal−carboxylic ligand subunits to give a 2-fold parallel interpenetrating net with (4,4) topology in 2, a 2-fold parallel interpenetrating net with (3,6)-connected topology in 3, and a 3-fold parallel interpenetrating net with (4,4) topology in 4. Compounds 1−4 display both polyrotaxane and polycatenane characters. Compound 5 is a 5-fold parallel interpenetrating net with (4,4) topology. By careful inspection of these structures, we find that different topological structures showing both polyrotaxane and polycatenane characters have been achieved with increase of the carboxylic ligand length. It is believed that various carboxylic ligands and N-donor ligands with different coordination modes and conformations are important for the formation of the different structures. In addition, the luminescent properties of these compounds are discussed.
Co-reporter:Ya-Qian Lan, Shun-Li Li, Yao-Mei Fu, Yan-Hong Xu, Lu Li, Zhong-Min Su and Qiang Fu
Dalton Transactions 2008 (Issue 47) pp:6796-6807
Publication Date(Web):22 Oct 2008
DOI:10.1039/B809336A
A series of mixed-ligand coordination complexes, namely [Zn(L1)(oba)] (1), [Cd(L1)(oba)] (2), [Zn2(L2)(oba)2]·8H2O (3), [Cd2(L2)(oba)2]·2H2O (4), [Zn3(L3)(oba)3] (5), [Cd2(L3)(oba)2]·(L3) (6), [Cd(L4)(oba)]·H2O (7) and [Cd(L5)(oba)]·3H2O (8), where L1 = 2-(2-pyridyl)imidazole, L2 = 1,4-bis[2-(2-pyridyl)imidazol-1-yl]butane, L3 = 1,4-bis[2-(2-pyridyl)imidazol-1-ylmethyl]benzene, L4 = 1,3-bis[2-(2-pyridyl)imidazol-1-ylmethyl]benzene, L5 = 1,2-bis[2-(2-pyridyl)imidazol-1-ylmethyl]benzene and H2oba = 4,4′-oxydibenzoic acid, have been synthesized under hydrothermal conditions. Their structures have been determined by single crystal X-ray diffraction analyses and further characterized by elemental analyses, IR spectra, and thermogravimetric (TG) analyses. In compounds 1 and 2, oba2−, L1 ligand and ZnII or CdII ions assemble to form the parallel chains or parallel sheets which are linked by the weak hydrogen bonding and π⋯π stacking interactions to give the 2D supramolecular sheet or 3D supramolecular net, respectively. For 3, L2 ligands connect [Zn(oba)] chains to generate a unusual (10,3)-b topological structure which is the first example for eight-fold interpenetrating framework based on the (10,3)-b net. In 4, L2 ligands link [Cd(oba)] double-chains to give a 2D sheet which is assembled by π⋯π stacking interactions to obtain a 3D supramolecular net. In 5, L3 ligands link ZnII ions from α-Po net formed by ZnII ions and oba2− anions to show a novel 3D 8-connected self-penetrating framework with the unreported (416·611·8) topological structure. In 6, the double-chains constructed by CdII and oba2− anions are linked by one kind of L3 ligand to form a layer-like structure which is assembled by π⋯π stacking interactions to show a 3D supramolecular structure. In 7, oba2− anions coordinate to CdII cations to form chains which are connected by L4 to form a four-fold interpenetrating diamond network. In 8, the weak hydrogen bonding and π⋯π stacking interactions connect the [Cd(L5)(oba)] chains to give a 2D supramolecular sheet. By careful inspections of the structures of 1–8, we believe that the different flexible and angular neutral ligands, coordination geometries of metal centers and weak interactions (hydrogen bonds and π⋯π stacking interactions) are crucial factors for the formation of the different structures. The photoluminescent properties of 1–8 have been studied in the solid state at room temperature.
Co-reporter:Ya-Qian Lan, Shun-Li Li, Kui-Zhan Shao, Xin-Long Wang and Zhong-Min Su
Dalton Transactions 2008 (Issue 29) pp:3824-3835
Publication Date(Web):13 Jun 2008
DOI:10.1039/B802626B
Five POM-based hybrid materials have been designed and synthesized based on different metal ions under hydrothermal conditions, namely, [Zn(Hfcz)(H2O)3](H3fcz)(SiMo12O40)·3H2O (1), [Cd2(Hfcz)6(H2O)2](SiMo12O40)·H2O (2), [Co2(Hfcz)2(SiW12O40)](H3fcz)2(SiW12O40)·10H2O (3), [Ni2(Hfcz)4(H2O)2](SiW12O40)·5H2O (4) and [Ag4(Hfcz)2(SiMo12O40)] (5), where Hfcz is fluconazole [2-(2,4-difluorophenyl)-1,3-di(1H-1,2,4-triazol-1-yl)propan-2-ol]. Their crystal structures have been determined by X-ray diffraction, elemental analyses, IR spectra, and thermogravimetric analyses (TGA). There are 1D mono and double chain-like metal–organic units in compounds 1 and 2, respectively. Polyoxometalates and metal–organic units co-crystallize through hydrogen bonds. In compound 3, metal–organic sheets are pillared by one kind of polyanion through covalent connections to generate a sandwich double-sheet. The other kind of polyanion acts as a counter-ion and lies in two adjacent sandwich double-sheets through non-covalent interactions. Polyanions covalently link metal–organic sheets to extend to an unusual 3D 5-connected framework with the (44·66) topology in 4. In compound 5, polyanions link metal–organic chains to form a sheet through covalent connections. It is interesting that compound 5 shows an intricate (4,5,10)-connected framework with (44·62)4(48·62)2(414·619·812) topology based on two kinds of Ag cations as four-connected and five-connected nodes, and polyanions as ten-connected nodes, when Ag⋯O interactions are considered. It represents the highest connected network topology presently known for polyoxometalate systems. The structural differences among 1–5 indicate the importance of different metal–organic units, coordination modes of polyanions for framework formation, and the interactions between polyanions and metal–organic units. In addition, the luminescent properties of compounds 1, 2 and 5, and electrochemical behaviours of compounds 1–5 have been investigated.
Co-reporter:Kui-Zhan Shao, Ya-Hui Zhao, Yan Xing, Ya-Qian Lan, Xin-Long Wang, Zhong-Min Su and Rong-Shun Wang
Crystal Growth & Design 2008 Volume 8(Issue 8) pp:2986
Publication Date(Web):July 18, 2008
DOI:10.1021/cg800103b
A novel chiral coordination polymer with a bikitaite zeolite framework has been constructed based on asymmetrical tetrahedral building blocks, in which the original chiralities derive from the configurational effect of benzotriazole ligands and transfer to the whole network through four distinct single helical chains.
Co-reporter:Ya-Qian Lan, Shun-Li Li, Kui-Zhan Shao, Xin-Long Wang, Dong-Ying Du, Zhong-Min Su and De-Jun Wang
Crystal Growth & Design 2008 Volume 8(Issue 10) pp:3490-3492
Publication Date(Web):August 29, 2008
DOI:10.1021/cg800169n
A (3,12)-connected 3D metal-organic framework has been synthesized based on octanuclear zinc clusters as 12-connected nodes and flexible carboxylate ligands as 3-connected nodes, and it represents the highest connected binodal network topology presently known for metal-organic frameworks.
Co-reporter:Gang Yuan, Kui-Zhan Shao, Xin-Long Wang, Ya-Qian Lan, Ya-Hui Zhao, Zhong-Min Su
Inorganic Chemistry Communications 2008 Volume 11(Issue 10) pp:1246-1249
Publication Date(Web):October 2008
DOI:10.1016/j.inoche.2008.07.018
A novel 3D coordination polymer, [Zn5(hqc)4(μ3-OH)2]n (1), has been prepared by hydrothermal reaction and characterized by elemental analyses, TGA, and single-crystal X-ray diffraction analysis. Compound 1 is built by hqc2− ligands bridging pentanuclear Zn(II) SBUs, [Zn5(μ3-OH)2], and there is coexistence of left-, and right-handed double helical chains in the 3D network. It represents the first pentanuclear Zn(II) coordination polymers with double helices constructed from quinoline-based carboxylic acid. In addition, the fluorescent property of compound 1 was also determined, which exhibits blue fluorescence at 441 nm in the solid-state upon excitation at 330 nm.A novel 3D coordination polymer, [Zn5(hqc)4(μ3-OH)2]n (1), has been prepared by hydrothermal reaction. The hqc2− as bridging ligand links pentanuclear Zn(II) cluster to construct a three-dimensional framework. In this framework, there exists an intriguing double helical motif.
Co-reporter:Kother Osman Ashiry, Ya-Hui Zhao, Kui-Zhan Shao, Zhong-Min Su, Yao-Mei Fu, Xiang-Rong Hao
Inorganic Chemistry Communications 2008 Volume 11(Issue 10) pp:1181-1183
Publication Date(Web):October 2008
DOI:10.1016/j.inoche.2008.06.027
A new metal–organic frameworks (MOFs), [Cd3(pmb)2(bpdc)2] 2H2O (1) (pmb = 3,5-bis(4-pyridylmethylenoxyl)benzoate, bpdc = 2,2′-biphenyldicarboxylate), has been synthesized by hydrothermal method and structurally characterized through IR, TG and single-crystal X-ray analysis. The infinite wave-like 1D metal–oxygen chains made by Cd3N4O12 clusters in 1 bridged by the carboxylate of bpdc ligand, construct a 2D framework in which interesting meso-helical chains are constructed by metal centers and pmb ligands. Solid state luminescent spectrum of 1 also has been investigated at room temperature.The metal–organic framework (MOF), [Cd3(pmb)2(bpdc)2] · 2H2O (1), has been hydrothermally synthesized and structurally characterized. The infinite wave-like 1D metal–oxygen chains made by Cd3N4O12 clusters in 1 construct a 2D framework bridged by the carboxylate of bpdc ligand, in which interesting meso-helical chains are constructed by metal centers and pmb ligands along the c-axis.
Co-reporter:Liang Fang;Wei Guan;LiKai Yan;GuoChun Yang
Science China Chemistry 2008 Volume 51( Issue 12) pp:1174-1181
Publication Date(Web):2008 December
DOI:10.1007/s11426-008-0123-3
The magnetic exchange interactions between the dimanganese(II)-substituted complexes and the heteropolymolyanion, [MnII2(Xn+Mo9O33)2]2(n−10)− (X = PV(I), AsV(II) and SeVI(III)), are investigated by using density functional theory combined with broken-symmetry approach (DFT-BS) method. The calculated magnetic exchange coupling constant (J) of complex II is in reasonable agreement with the responding experimental value and the negative J values indicate that antiferromagnetic exchange interactions exist in these complexes. Furthermore, the influence of the central heteroatom on the exchange coupling within the dimanganese core unit is studied from standpoints of geometry, spin density and frontier orbitals. It demonstrates that the change of the heteroatom X via PV-AsV-SeVI elongates the distances of Mn1...Mn2 and shortens the distances of Ob...Ob, and reduces the effectiveness of the superexchange pathways, consequently, decreasing the magnitude of the antiferromagnetic coupling constant, J, of these species.
Co-reporter:L. Fang;G. C. Yang;Y. Q. Qiu;Z. M. Su
Theoretical Chemistry Accounts 2008 Volume 119( Issue 4) pp:329-333
Publication Date(Web):2008 March
DOI:10.1007/s00214-007-0388-1
We investigate the nonlinear third-order polarizabilities of novel sandwichlike clusters [Al4MAl4]n− (n = 0–2, M = Ti, V and Cr). The calculations have been performed by employing time-dependent density functional theory combined with sum-over-states method. The results show that these complexes possess remarkably large third-order static polarizability, and change of a metal centre has a great influence on the third-order nonlinear optical properties. The calculated third-order polarizability follows: [Al4CrAl4] > [Al4VAl4]− > [Al4TiAl4]2−. Analysis of the main contributions to the third-order polarizability suggests that charge transfer (\({\rm Al}_{4}^{2-} \rightarrow {\rm M}\)) along the z-axis direction plays a key role in the nonlinear optical response.
Co-reporter:Liang Zhao, Guochun Yang, Zhongmin Su, Likai Yan
Journal of Molecular Structure: THEOCHEM 2008 Volume 855(1–3) pp:69-76
Publication Date(Web):30 April 2008
DOI:10.1016/j.theochem.2008.01.006
A comparative study of one-photon absorption (OPA) and two-photon absorption (TPA) properties of three-branched oligofluorenes with boron center (CBTFn) and two-branched (CBDFn) as well as one-branched (CBSFn) counterparts is presented based on the correlated quantum chemical calculations. The roles of boron center in the multi-branched oligofluorenes on OPA and TPA properties are discussed first. Theoretical calculations results reveal that the maximal two-photon absorption cross sections (δmax) values of CBSFn, CBDFn and CBTFn increase obviously with the increase of conjugation length and branch number. Among them, CBTFn has the largest δmax value and maintains good transparency when n increases. The comparisons of δmax values with simplified exciton model show that the effective electronic coupling between boron center and peripheral fluorene is relatively strong. In addition, the cooperative coupling in CBTFn is larger than that in CBDFn. Therefore, CBTFn is a kind of promising TPA material for optical power limiting.
Co-reporter:Yan-Chun Liu, Yu-He Kan, Shui-Xing Wu, Guo-Chun Yang, Liang Zhao, Min Zhang, Wei Guan and Zhong-Min Su
The Journal of Physical Chemistry A 2008 Volume 112(Issue 35) pp:8086-8092
Publication Date(Web):August 12, 2008
DOI:10.1021/jp801305e
Geometry structures, electronic spectra, and third-order nonlinear optical (NLO) properties of Fe(η5-C55X5)2 (X = CH, N, B) have first been investigated by time-dependent density functional theory. We analyzed the intramolecular interactions between ferrocene and the C50 moiety. The calculated electronic absorption spectrum indicates that the short wavelength transitions are ascribed to the C50 moiety mixed charge transfer transition of ferrocene itself, while the low energy excitation transitions are ascribed to the unique charge transfer transition from ferrocene to C50 moiety in these systems. The third-order polarizability γ values based on sum of states (SOS) method show that this class of ferrocene/fullerene hybrid molecule possesses a remarkably large third-order NLO response, especially for Fe(η5-C55B5)2 with the static third-order polarizability (γav) computed to be −10410 × 10−36 esu and the intrinsic second hypepolarizability to be 0.250. Thus, these complexes have the potential to be used for excellent third-order nonlinear optical materials. Analysis of the major contributions to the γav value suggest that the charge transfer from ferrocene to C50 moiety along the z-axis (through Fe atom and the centers of two hybrid fullerenes) play the key role in the NLO response. Furthermore, boron substitution is an effective way of enhancing the optical nonlinearity compared to CH and N substitution, owing to smaller energy gap and better conjugation through the whole molecule.
Co-reporter:Hongze Gao, Chunsheng Qin, Houyu Zhang, Shuixing Wu, Zhong-Min Su and Yue Wang
The Journal of Physical Chemistry A 2008 Volume 112(Issue 38) pp:9097-9103
Publication Date(Web):August 27, 2008
DOI:10.1021/jp804308e
The structural, electronic, and carrier transport properties of bathocuproine (BCP), which is a typical hole/exciton-blocking material applied in organic light-emitting diodes (OLEDs), have been investigated based on density functional theory (DFT) and ab initio HF method. The detail characterizations of frontier electronic structure and lowest-energy optical transitions have been studied by means of time-dependent density functional theory (TD-DFT). Five BCP analogues, o-phenanthroline (1), 2,9-dimethyl-1,10-phenanthroline (2), 2,9-diphenyl-1,10-phenanthroline (3), 4,7-diphenyl-1,10-phenanthroline (4), and 2,9-bis(trifluoromethyl)-1,10-phenanthroline (5) have also been studied in order to select more suitable candidates of efficient hole-blocking materials. The calculated results showed that rigid planar structures, conjugate degrees, and substitute groups play crucial roles in the hole/exciton-blocking and electron-transport properties of these materials. The calculated geometries, ionization energies (IP), and energy gap between the singlet ground state and triplet excited state (ET1) were well in agreement with the experimental results. On the basis of the incoherent transport model, the calculated electron mobility of BCP is 1.79 × 10−2 cm2/(V s), which is comparable to experimental results of 1.1 × 10−3 cm2/(V s). The electron mobilities for compounds 1, 4, and 5 are 3.45 × 10−2, 2.90 × 10−2, and 1.40 × 10−2 cm2/(V s), respectively. The calculated results indicated that compounds 1, 4, and 5 may be more effective hole/exciton-blocking materials than BCP.
Co-reporter:Ya-Qian Lan Dr.;Shun-Li Li Dr.;Xin-Long Wang Dr.;Kui-Zhan Shao Dr.;Dong-Ying Du, ;En-Bo Wang
Chemistry - A European Journal 2008 Volume 14( Issue 32) pp:9999-10006
Publication Date(Web):
DOI:10.1002/chem.200800666
Abstract
Four enantiomerically pure 3D chiral POM-based compounds, [Ni2(bbi)2(H2O)4V4O12]⋅2 H2O (1 a and 1 b) and [Co(bbi)(H2O)V2O6] (2 a and 2 b) (bbi=1,1′-(1,4-butanediyl)bisimidazole) based on the achiral ligand, different vanadate chains, and different metal centers have been synthesized by hydrothermal methods. Single-crystal X-ray diffraction analyses revealed that 1 a and 1 b, and 2 a and 2 b, respectively, are enantiomers. In 1 a and 1 b two kinds of vanadate chains with different screw axes link Ni cations to generate 3D chiral inorganic skeletons, which are connected by the achiral bbi ligands to form complicated 3D 3,4-connected chiral self-penetrating frameworks with (72⋅8)(72⋅82⋅92)(73⋅82⋅10) topology. They represent the first examples of chiral self-penetrating frameworks known for polyoxometalate (POM) systems. Contrary to 1 a and 1 b, in 2 a and 2 b the vanadate chains link CoII cations to generate 3D chiral inorganic skeletons, which are assembled from two kinds of heterometallic helical units of opposite chirality along the c axes. The chiral inorganic skeletons are connected by bbi to form 3D 3,4-connected chiral POM-based frameworks with (62⋅8)2(62⋅82⋅102) topology. It is believed that the asymmetrical coordination modes of the metal cations in 1 a–2 b generate the initial chiral centers, and that the formation of the various helical units and the hydrogen bond interactions are responsible for preservation of the chirality and spontaneous resolution when the chirality is extended into the homochiral 3D-networks. This is the first known report of chiral POM-based compounds consisting of 3D chiral inorganic skeletons being obtained by spontaneous resolution upon crystallization in the absence of any chiral source, which may provide a rational strategy for synthesis of chiral POM-based compounds by using achiral ligands and POM helical units.
Co-reporter:Li-Kai Yan, Ming-Shun Jin, Jia Zhuang, Chun-Guang Liu, Zhong-Min Su and Chia-Chung Sun
The Journal of Physical Chemistry A 2008 Volume 112(Issue 40) pp:9919-9923
Publication Date(Web):September 4, 2008
DOI:10.1021/jp804342h
The static first hyperpolarizabilities and origin of nonlinear optical (NLO) properties of [(2-methylnaphthyl)imido]hexamolybdates derivatives have been investigated by density functional theory (DFT). The [(2-methylnaphthyl)imido]hexamolybdate has considerable large first hyperpolarizability, 6.780 × 10−30 esu, and it is larger than that of [(2,6-dimethylphenyl)arylimido]hexamolybdate due to the double aromatic rings in the naphthylimido ligand. The naphthylimido ligand acts as an electron-donor and the polyanion acts as an electron-acceptor. The substituent position on the naphthylimido is a key factor to determine the first hyperpolarizability of (naphthylimido)hexamolybdate derivatives. The derivative, which the iodine atom locates on the para nitrogen on the naphthylimido ligand, has the largest β0 value among the iodine-substituted derivatives. It suggests that the iodine atom is quasi linear with nitrogen and Mo, which is bonded to the nitrogen atom, could generate a large static electronic field and give the large contribution to NLO response. The introducing of electron-donors significantly enhances the first hyperpolarizabilities of (naphthylimido)hexamolybdates comparing with the electron-acceptors as the electron-donating ability is significantly enhanced when the electron-donor is attached to the naphthylimido segment. The present investigation provides important insight into NLO properties of (arylimido)molybdate derivatives.
Co-reporter:Xin-Long Wang, Chao Qin, En-Bo Wang and Zhong-Min Su
Chemical Communications 2007 (Issue 41) pp:4245-4247
Publication Date(Web):08 Aug 2007
DOI:10.1039/B709563E
Threading molecular square “beads” on a twofold interpenetrated diamondoid skeleton gives a new type of 3D metal–organic polyrotaxane framework with large channels, in which nanosized Keggin anions as guests are encapsulated for the first time.
Co-reporter:Ya-Qian Lan, Xin-Long Wang, Shun-Li Li, Zhong-Min Su, Kui-Zhan Shao and En-Bo Wang
Chemical Communications 2007 (Issue 46) pp:4863-4865
Publication Date(Web):19 Sep 2007
DOI:10.1039/B709835A
The first (6,8)-connected self-penetrating metal–organic framework has been constructed using an asymmetric neutral ligand based on dinuclear zinc clusters, as six-connected nodes, and trinuclear zinc clusters as eight-connected nodes, representing the highest-connected binodal network topology presently known for self-penetrating systems.
Co-reporter:Xiang-Rong Hao, Xin-Long Wang, Chao Qin, Zhong-Min Su, En-Bo Wang, Ya-Qian Lan and Kui-Zhan Shao
Chemical Communications 2007 (Issue 44) pp:4620-4622
Publication Date(Web):20 Sep 2007
DOI:10.1039/B711405B
A 3D chiral nanoporous coordination framework consisting of homochiral nanotubes assembled from octuple helices with a 19.4 Å by 22.4 Å aperture is formed by the parallel alignment of eight infinite helical chains.
Co-reporter:Donghua Hu, Chen Shao, Wei Guan, Zhongmin Su, Jiazhong Sun
Journal of Inorganic Biochemistry 2007 Volume 101(Issue 1) pp:89-94
Publication Date(Web):January 2007
DOI:10.1016/j.jinorgbio.2006.08.013
Ti-containing α-Keggin polyoxometalates (POMs) have been proved with properties of both anti-tumor and anti-HIV (human immunodeficiency virus). The potential anti-SARS (severe acute respiratory syndrome) activity of the POMs [α-PTi2W10O40]7− isomers was investigated in this paper by molecular modeling method. The SARS 3c like protease, namely the SARS 3CLpro is the key function protease for virus replication as well as transcription and thus can be taken as one of the key targets for anti-SARS drug design. Affinity/Insight II was used to explore possible binding locations for POMs/3CLpro interaction. Charges in the POMs were obtained from density-functional theory (DFT) method. The results show that POMs bind with 3CLpro in the active site region with high affinity; POMs are more prone to bind with 3CLpro than with some organic compounds; for the POMs/3CLprocomplex, the OTi2 in POMs is the vital element for electrostatic interaction, and the electrostatic binding energy is strong enough to keep the complex stable.
Co-reporter:Liang Zhao;Li Mu;Guo-Chun Yang;Chun-Sheng Qin;Chen Shao
Chinese Journal of Chemistry 2007 Volume 25(Issue 4) pp:
Publication Date(Web):5 APR 2007
DOI:10.1002/cjoc.200790088
Based on the equilibrium structures from quantum mechanics AM1 method, employing INDO/CI method and the sum-over-state (SOS) formula, the one-photon absorption (OPA) and two-photon absorption (TPA) properties as well as the second hyperpolarizabilities were discussed in detail for a kind of PPV derivative poly 2-(9-phenylan- thracen-10-yl)-1,4-phenylenevinylene (P1). The results indicate that the two-photon cross section (δ) increases with the increasing of the number of repeating segment (n), however only a slight increase corresponds to the increasing of molecular weight when the repeating unit number arrives at a certain number. From this point of view, the TPA cross section values of P1 were extrapolated through the linear fit of δ value vs. 1/n. The δ value of P1 is as high as 181 GM (1 GM10−50 cm4·s·photon−1). Concerning the influence of pendent group and extension of π-conjuga- tion on δ values, another four oligomers P2, PPV, P1-1, and P2-1 were investigated for comparison. The calculation results reveal that the position and property of pendent group have great influence on the OPA and TPA properties. A most crucial role for relatively larger δ value was played by the extension of π-conjugation.
Co-reporter:P. Song;W. Guan;C. Yao;Z. M. Su;Z. J. Wu;J. D. Feng
Theoretical Chemistry Accounts 2007 Volume 117( Issue 3) pp:407-415
Publication Date(Web):2007 March
DOI:10.1007/s00214-006-0168-3
Bond distances, dissociation energies, ionization potentials and electron affinities of 4d transition metal monoxides from YO to CdO and their positive and negative ions were studied by use of density functional methods B3LYP, BLYP, B3PW91, BPW91, B3P86, BP86, SVWN, MPW1PW91 and PBE1PBE. It was found that calculated properties are highly dependent on the functionals employed, especially for dissociation energy. For most neutral species, pure density functionals BLYP, BPW91 and BP86 have good performance in predicting dissociation energy than hybrid density functionals B3LYP, B3PW91 and B3P86. In addition, BLYP gives the largest bond distance compared with other density functional methods, while SVWN gives shortest bond distance, largest dissociation energy and electron affinity. For the ground state, the spin multiplicity of the charged species can be obtained by ± 1 of their corresponding neutral species.
Co-reporter:Z. J. Wu;W. Guan;J. Meng;Z. M. Su
Journal of Cluster Science 2007 Volume 18( Issue 2) pp:444-458
Publication Date(Web):2007 June
DOI:10.1007/s10876-007-0108-y
Bond distances, vibrational frequencies, electron affinities, ionization potentials, dissociation energies and dipole moments of the title molecules in neutral, positively and negatively charged ions were studied by use of density functional method. Ground electronic state was assigned for each molecule. The bonding patterns were analyzed and compared with both the available data and across the series. It was found that besides ionic component, covalent bonds are formed between the metal s, d and f orbitals and oxygen p orbitals. Contrary to the well known lanthanide contraction, the bond distance is not regular from LaO to LuO for both neutral and charged molecules. An obvious population at 5d orbital was observed through the lanthanide series. 4f electrons also participate the chemical bonding for CeO to NdO and TbO to TmO. For EuO, GdO, YbO and LuO, 4f electrons tend to be localized. The spin multiplicity is regular for neutral and charged molecules. The spin multiplicity of the charged molecules can be obtained by −1 (or +1 for TbO+, DyO+, YbO− and YbO+) compared with the corresponding neutral molecules.
Co-reporter:Y. L. Teng;Y. H. Kan;Z. M. Su;Y. Liao;S. Y. Yang
Theoretical Chemistry Accounts 2007 Volume 117( Issue 1) pp:1-5
Publication Date(Web):2007 January
DOI:10.1007/s00214-005-0025-9
The molecular structures of the ground state and the first singlet excited state for diphenylboron analogs of Alq3 [Ph2Bq where q is 8-hydroxyquinoline (QH)] and its three derivatives were optimized with the Density Functional Theory and ab initio “configuration interaction with single excitations” method, respectively. The frontier molecular orbital characteristics of Ph2Bq were analyzed systematically in order to study the electronic transition mechanism. Electronic and spectroscopic properties of complexes have been investigated with Time-Dependent Density Functional Theory, which indicates that the emissions of Ph2Bq and its derivatives originate from the electronic π → π* transitions within the QH ligands. That means that one might tune the emission wavelengths and improve charge transfer properties through the effect of substituent on the 8-hydroxyquinoline ligand. Similar calculations were carried out for isolated QH and its three derivatives for comparison. We found that the highest occupied molecular orbital and the lowest unoccupied molecular orbital of Ph2Bq are similar to those of QH and their spectroscopic properties change similarly when they are substituted by the same group, which suggests that one can search possibility of a red or blue emission from Ph2Bq derivatives by analyzing QH and its derivatives.
Co-reporter:C. Yao;W. Guan;P. Song;Z. M. Su;J. D. Feng;L. K. Yan
Theoretical Chemistry Accounts 2007 Volume 117( Issue 1) pp:115-122
Publication Date(Web):2007 January
DOI:10.1007/s00214-006-0147-8
Bond distances, vibrational frequencies, electron affinities, ionization potentials, and dissociation energies of the diatomic 5d transition metal (except La) monoxides and their positively and negatively charged ions were studied by use of density functional methods B3LYP, BLYP, B3PW91, BPW91, B3P86, BP86, MPW1PW91, PBE1PBE, and SVWN. Our calculation shows that for each individual species, the calculated properties are quite sensitive to the method used. Compared with hybrid density functional method B3PW91 (B3P86), pure density functional method BPW91 (BP86) gives longer bond distance (lower vibrational frequency) from HfO to PtO for neutral species, HfO+ to IrO+ for cationic species, and HfO− to AuO− for anionic species. While for B3LYP and BLYP, the trend was observed for cationic species from HfO+ to IrO+ and anionic species from HfO− to AuO− (except TaO−), but not for neutrals. Pure density function methods BLYP, BPW91, and BP86 give larger dissociation energy compared with hybrid density functional methods B3LYP, B3PW91, and B3P86. SVWN in most cases gives the smallest bond distance, while BLYP gives the largest value. MPW1PW91 and PBE1PBE show the same performance in predicting the spectroscopic constants. In addition, useful empirical criteria that one has obtained the ground states of a species and its ions are the spin multiplicities of a neutral and its single charged ions which differs by ±1.
Co-reporter:H.-M. Xie;L.-Y. Zhang;J.-R. Ying;R.-S. Wang;A. F. Jalbout;X.-M. Pan;G.-L. Yang;Z.-M. Su;H.-Y. Yu
Advanced Materials 2006 Volume 18(Issue 19) pp:2609-2613
Publication Date(Web):26 SEP 2006
DOI:10.1002/adma.200600578
A novel core/shell compound has been developed by coating a spherical LiFePO4 structure with a specific π-bond character planar polymer (polyacene, PAS). The electronic conductivity, low-temperature character, and tap density of the LiFePO4–PAS composite were significantly improved compared to LiFePO4, which may lead to use in lithium-ion battery applications.
Co-reporter:Wei Guan;Guo-Chun Yang;Li-Kai Yan
European Journal of Inorganic Chemistry 2006 Volume 2006(Issue 20) pp:
Publication Date(Web):11 AUG 2006
DOI:10.1002/ejic.200600450
Systematic DFT calculations have been carried out on the lacunary α-Keggin polyoxometalate derivatives [PW11O39]7–, [XW9O34]n– (X = AlIII, SiIV, GeIV, PV, AsV, and SbV), [XW9M2O39]n–, and [XW9M3O40]n– (X = PV and SiIV, M = MoVI, VV, NbV, and TaV) to investigate the geometric structure and element substitution effects on the molecular nonlinear optical response. Analysis of the computed static second-order polarizability (β0) predicts that the molecular nonlinear optical activity of lacunary Keggin polyoxometalate derivatives can be modified by replacing the central heteroatom and the addenda metal atom. Substitution of the central Al atom or the addenda V atom causes significant enhancement in the molecular nonlinearity. Moreover, the β0 values are substantially dependent on the defect structures. This class of inorganic complexes possesses remarkably large molecular optical nonlinearity, especially for the partial substitution complex [SiW9Nb2O39]10– (IIIc), which has a computed β0 value of 2071.0 a.u. Thus, lacunary Keggin polyoxometalates could become excellent candidates in the field of second-order NLO. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2006)
Co-reporter:Li-Kai Yan;Zhuo Dou;Wei Guan;Shao-Qing Shi
European Journal of Inorganic Chemistry 2006 Volume 2006(Issue 24) pp:
Publication Date(Web):19 OCT 2006
DOI:10.1002/ejic.200600720
DFT calculations were carried out to investigate the electronic and redox properties of nitrido-functionalized polyoxometalate species, [PW11O39(ReN)]n– (n = 3, 4, 5) and [PW11O39(OsN)]2–. The rhenium- and osmium-nitrido effectively modify the electronic properties. The LUMOs in fully oxidized [PW11O39(ReN)]3– and [PW11O39(OsN)]2– are mainly concentrated on the Re and Os centers. The high-valent transition metals Re and Os modify the components and energies of the LUMO. The LUMO energies of [PW11O39(ReN)]3– and [PW11O39(OsN)]2– are lower than that of related Keggin [PW12O40]3–. In addition, Re centers will prefer to accept the electrons in the first and second reduced process of fully oxidized [PW11O39(ReN)]3–. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2006)
Co-reporter:S.Y. Yang, Y.H. Kan, G.C. Yang, Z.M. Su, L. Zhao
Chemical Physics Letters 2006 Volume 429(1–3) pp:180-184
Publication Date(Web):29 September 2006
DOI:10.1016/j.cplett.2006.07.078
Modifications of the optical properties and charge mobility of spiro-bithiophene (SCPDT) were investigated by time-dependent density functional theory (TD-DFT). Geometry optimizations have been performed for the ground state and excited state of bithiophene (BT), 4H-cyclopenta[2,1-b:3,4-b′]dithiophene (CPDT) and SCPDT. Vertical excitations and emission transition energies have been calculated. The computed vertical and emission transition energies of BT and CPDT are in good agreement with the experimental data. Compared to CPDT and BT, SCPDT is expected to be an effective EL material by considering that smaller reorganization energy is presented and fluorescence quantum efficiency of non-spiro isolated fragment is preserved in the molecule with spiro-structure.In BT intersystem crossing between the S1 singlet state and a triplet (T2) state is expected to be very efficient and in SCPDT is much lower, that explained quantum efficiencies of SCPDT are much higher. Furthermore, in view of their same s–t split energy 0.12 eV, SCPDT and CPDT that is explained that quantum efficiencies corresponding to the isolated branches are preserved.
Co-reporter:Xinlong Wang, Chao Qin, Enbo Wang, Lin Xu, Zhongmin Su
Journal of Molecular Structure 2006 Volume 796(1–3) pp:172-178
Publication Date(Web):30 August 2006
DOI:10.1016/j.molstruc.2006.02.048
Two novel three-dimensional d10 metal coordination polymers [Zn2(pydc)2(DMF)3]n (1) and [Cd2(pydc)2(H2O)]n (2) (H2pydc=pyridine-3,4-dicarboxylic acid, DMF=N,N-dimethylformamide) have been synthesized under solvothermal and hydrothermal conditions, and characterized by elemental analysis, IR, TG analysis and single crystal X-ray diffraction. Compound 1 exhibits a complex three-dimensional framework containing one-dimensional elliptic channels occupied by coordinated DMF molecules. In complex 2, adjacent sheets constructed by carboxyl-bridged Cd–O–Cd chains are pillared by pydc ligands into a three-dimensional network. From a topological point of view, the two three-dimensional nets display (3,4)-connected (6.82)2(6.85) and 4-connected (4.85)(42.6.83)2(43.62.8) topologies, respectively, which are unprecedented within coordination frames. Furthermore, both compounds show intense fluorescent emissions upon photoexcitation at room temperature.
Co-reporter:Liang Zhao, Guochun Yang, Zhongmin Su, Chunsheng Qin, Shuangyang Yang
Synthetic Metals 2006 Volume 156(18–20) pp:1218-1224
Publication Date(Web):1 November 2006
DOI:10.1016/j.synthmet.2006.09.006
One- and two-photon absorption properties of a very intriguing perpendicular π-electron system 5,6,11,12-tetraphenylnaphthacene (rubrene) and its derivatives have been discussed from the theoretical investigation view. Based on the correct geometries, the INDO/SDCI method was adopted to determine the one-photon absorption (OPA) and two-photon absorption (TPA) properties. The elongation of π-conjugation and the perpendicular phenyl groups on two-photon absorption cross section (δmax) of rubrene are investigated in detail. The results show that the extension of π-conjugation plays an important role on the magnitude of δmax. In addition, the perpendicular phenyl groups favor to enhance the δmax of rubrene, which can be seen from the dominant charge transfer (CT) contributing to TPA obviously. This CT process is a symmetrical intramolecular CT process from the pendent phenyl groups to the main π-backbone. At the same time, the relationship between substituents attached to rubrene and two-photon absorption cross section is also systematically discussed.
Co-reporter:Jing-Dong Feng;Ya-Qian Lan;Yu-He Kan;Yi Liao;Li-Kai Yan;Yu-Lan Zhu
Chinese Journal of Chemistry 2006 Volume 24(Issue 1) pp:119-123
Publication Date(Web):17 JAN 2006
DOI:10.1002/cjoc.200690005
AM1 semi-empirical method was used to optimize the barbituric acid derivatives substituted with glucosyl B1–5(series B), and the thiobarbituric acid derivatives substituted with glucosyl T1–5 (seriesT). Based on the optimized structures, INDO/CI method was adopted to calculate the electronic spectra. Meanwhile, the second-order nonlinear optical (NLO) coefficients βμ were calculated with the sum-over-state (SOS) formula. The results show that when the number of glucosyl units was increased, |βμ| values of the barbituric and thiobarbituric acid derivatives were both enhanced, especially for thiobarbituric acid derivatives. It indicates that non-conjugated substituted group could also improve NLO properties of materials when the number of repeated units was increased. Additionally, the absorption bands appearing in UV area are consistent with the proper change of the number of glucosyl units, and consequently it can be concluded that the high transparencies of all systems were scarcely varied.
Co-reporter:Xin-Long Wang Dr.;Chao Qin Dr.;En-Bo Wang ;Yang-Guang Li Dr.;Lin Xu
Angewandte Chemie International Edition 2006 Volume 45(Issue 44) pp:
Publication Date(Web):6 NOV 2006
DOI:10.1002/anie.200603250
Bridging two traditional but distinct research areas—polyoxometalate chemistry and cuprous halide clusters—the (4,12)-connected 3D framework (NH4)[Cu24I10L12][PMoV2MoVI10O40]3 has been built by covalent linkage of nanoscale Keggin anions and [Cu24I10L12]14+ clusters (see picture; L is a multidentate N-heterocyclic ligand); it exhibits remarkable photoluminescent and electrochemical properties.
Co-reporter:Dong-Rong Xiao Dr.;En-Bo Wang ;Hai-Yan An Dr.;Yang-Guang Li Dr. ;Chun-Yan Sun
Chemistry - A European Journal 2006 Volume 12(Issue 25) pp:
Publication Date(Web):14 JUN 2006
DOI:10.1002/chem.200501308
Rational self-assembly of a long V-shaped 3,3′,4,4′-benzophenonetetracarboxylate (bptc) ligand and metal salts in the presence of linear bidentate ligand yield a series of novel pillared helical-layer complexes, namely, [Cu2(bptc)(bpy)2] (1), [M3(Hbptc)2(bpy)3(H2O)4]⋅2 H2O (M = Fe(2) and Ni(3)), [Co2(bptc)(bpy)(H2O)]⋅0.5 bpy (4), [Cd2(bptc)(bpy)(H2O)2]⋅H2O (5), [Mn2(bptc)(bpy)1.5(H2O)3] (6) and [M2(bptc)(bpy)0.5(H2O)5]⋅0.5 bpy (M = Mn(7), Mg(8) and Co(9), bpy=4,4′-bipyridine). Their structures were determined by single-crystal X-ray diffraction analyses and further characterized by elemental analyses, IR spectra, and thermogravimetric (TG) analyses. The structure of 1 consists of two types of chiral layers, one left-handed and the other right-handed, which are connected by bpy pillars to generate a novel 3D open framework featuring four distinct helical chains. Compounds 2 and 3 are isostructural and feature 3D structures formed from the interconnection of arm-shaped helical layers with bpy pillars. Compound 4 is a pillared helical double-layer complex containing four different types of helices, among which the nine-fold interwoven helices constructed from triple-stranded helical motifs are unprecedented. Compound 5 exhibits a novel 3D covalent framework which features nanosized tubular channels. These channels are built from helical layers pillared by bptc ligands. The structure of 6 is constructed from {Mn(bptc)(H2O)}n2n− layers, which consist of left- and right-handed helical chains, pillared by [Mn2(bpy)3(H2O)4]4+ complexes into a 3D framework. To the best of our knowledge, compounds 1–6 are the first examples of pillared helical-layer coordination polymers. Compounds 7–9 are isostructural and exhibit interesting 2D helical double-layer structures, which are constructed from {M(bptc)(H2O)2}n2n− ribbons cross-linked by [M2(bpy)(H2O)6]4+ complexes. Furthermore, the 3D supramolecular structures of 7–9 are similar to the 3D structure of 6, and the 2D structure of 7 can be transformed into the 3D structure of 6 at higher reaction temperature. By inspection of the structures of 1–9, it is believed that the V-shaped bptc ligand and V-shaped phthalic group of the bptc ligand are important for the formation of the helical structures. The magnetic behavior of compounds 1, 2, 4, 6, and 9 was studied and indicated the existence of antiferromagnetic interactions. Moreover, compound 5 shows intense photoluminescence at room temperature.
Co-reporter:Xin-Long Wang Dr.;Chao Qin Dr.;En-Bo Wang
Chemistry - A European Journal 2006 Volume 12(Issue 10) pp:
Publication Date(Web):7 FEB 2006
DOI:10.1002/chem.200501242
To investigate the relationship between network connectivity and metal nuclearity, we designed and synthesized a series of three-dimensional (3D) entangled coordination frameworks based on different metal cores, namely [Zn2(bdc)2(L)2]⋅2H2O (1), [Zn(bdc)(L)0.5] (2), [Zn(oba)(L)0.5] (3) and [Cd3(bdc)3(L)2(H2O)2] (4) by self-assembly of d10 metal salts with the flexible long-chain ligand 1,4-bis(1,2,4-triazol-1-yl)butane (L), and with the rigid and nonrigid aromatic dicarboxylate ligands 1,4-benzenedicarboxylate (bdc) and 4,4′-oxybis(benzoate) (oba). Compound 1 exhibits a threefold interpenetrated diamondoid array typically based on a tetrahedral second building unit (SBU) at a single Zn center. Compound 2 adopts a threefold interpenetrated α-polonium-type network that is built from bimetallic cores as six-connected vertices. The structure of 3 also consists of dinuclear units; it comprises a novel (3,4)-connected threefold interpenetrated net with complex (4⋅6⋅10)(4⋅62⋅103) topology when single zinc centers act as four-connected nodes (or the α-Po topology if dinuclear units are considered as six-connected nodes). Compound 4, derived from a crosslinked fivefold interpenetrated diamond-like substructure, is an unusual example of a self-penetrating coordination framework displaying an unprecedented eight-connected 42068 topology with trinuclear cadmium clusters as eight-connected nodes which, to our knowledge, not only defines a new topology for eight-connected coordination networks, but also represents the highest connected topology presently known for self-penetrating systems. Detailed structural comparison of these complexes indicates that the increase in metal nuclearity induces the progressive increase in the connectivities of the ultimate nets: that is, the metal nuclearity plays a significant role in tuning the connectivity of a specific network. The thermal and luminescent properties of these compounds are discussed.
Co-reporter:Hai-Yan An Dr.;En-Bo Wang ;Dong-Rong Xiao Dr.;Yang-Guang Li Dr. ;Lin Xu
Angewandte Chemie 2006 Volume 118(Issue 6) pp:
Publication Date(Web):30 DEC 2005
DOI:10.1002/ange.200503657
Ein Dreh nach rechts oder nach links: Beide Enantiomere eines neuen chiralen Materials wurden auf rationalem Weg synthetisiert, wobei enantiomerenreine Prolinliganden, Kupferionen und Keggin-Polyoxometallatanionen als Bausteine dienten. Die Strukturen der Verbindungen sind durch offene Gerüste und helicale Kanäle gekennzeichnet.
Co-reporter:Hai-Yan An, En-Bo Wang, Dong-Rong Xiao, Yang-Guang Li, Zhong-Min Su,Lin Xu
Angewandte Chemie International Edition 2006 45(6) pp:904-908
Publication Date(Web):
DOI:10.1002/anie.200503657
Co-reporter:Xin-Long Wang, Chao Qin, En-Bo Wang, Yang-Guang Li and Zhong-Min Su
Chemical Communications 2005 (Issue 43) pp:5450-5452
Publication Date(Web):05 Oct 2005
DOI:10.1039/B510089E
An unprecedented fivefold interpenetrated lvt network, containing the rare racemic motifs originated from nine interwoven helices, is reported, which represents the highest degree of interpenetration presently known for 3D nets containing only planar four-coordinate nodes.
Co-reporter:Min Zhang, Zhong-Min Su, Li-Kai Yan, Yong-Qing Qiu, Guan-Hua Chen, Rong-Shun Wang
Chemical Physics Letters 2005 Volume 408(1–3) pp:145-149
Publication Date(Web):7 June 2005
DOI:10.1016/j.cplett.2005.04.025
Abstract
Three different nanotube structures, armchair, zigzag and wurtzite, were studied using B3LYP/6-31G(d) for carbon, BN, AlN and GaN nanotubes, respectively. Our calculations found that AlN and GaN can implausibly form the usual tubular morphologies of carbon and BN nanotubes. The same conclusion was confirmed based on analyzing the different configurations of benzene, borazine, and the analogies of the hexagonal Al3N3H6 and Ga3N3H6 at the same level of calculations.
Co-reporter:Yi Liao, Zhong-Min Su, Yu-He Kan, Dong-Xia Zhu, Yue Wang, Jia-Cong Shen
Journal of Molecular Structure: THEOCHEM 2005 Volume 731(1–3) pp:123-126
Publication Date(Web):24 October 2005
DOI:10.1016/j.theochem.2005.04.003
Geometrical structures of boron hydroxyphenylpyridine (dppyBF) excimers have been optimized at the B3LYP/6-31G* levels of theory. Two stable excimer conformers, crossed and parallel-displaced configuration, were obtained with similar energies. Electronic spectrum properties of two conformers were studied by TD-DFT methods. It is shown that both excimer present red-shifted emission peaks relative to that of monomer (dppy)BF, which mainly originate from the lowest unoccupied molecular orbital (LUMO) of excited (dppy)BF* segment to the highest occupied molecular orbital (HOMO) of ground (dppy)BF segment in excimer.
Co-reporter:Xin-Long Wang Dr.;Chao Qin Dr.;En-Bo Wang ;Yang-Guang Li Dr. ;Lin Xu ;Lucia Carlucci Dr.
Angewandte Chemie 2005 Volume 117(Issue 36) pp:
Publication Date(Web):8 SEP 2005
DOI:10.1002/ange.200501373
Verschiedenartig vernetzt: Nickelsalze, 4,4′-Bipyridin und der V-förmige 4,4′-Oxybis(benzoat)-Ligand reagieren selbstorganisiert zu drei verschiedenen Koordinationsnetzwerken: Die parallele Verkettung von Nanoröhren ergibt eine Schichtstruktur (siehe Bild), eine vielfach 2D3D-verflochtene Anordnung verbindet fünf polymere Einheiten, und ein binodales 3D-Durchdringungsnetzwerk zeigt eine beispiellose (4.82)(4.64.84.10)-Topologie.
Co-reporter:Xin-Long Wang Dr.;Chao Qin Dr.;En-Bo Wang ;Yang-Guang Li Dr. ;Lin Xu ;Lucia Carlucci Dr.
Angewandte Chemie International Edition 2005 Volume 44(Issue 36) pp:
Publication Date(Web):8 SEP 2005
DOI:10.1002/anie.200501373
Caught in a net: Self-assembly of nickel salts with 4,4′-bipyridine and the long V-shaped 4,4′-oxybis(benzoate) ligand has led to three different coordination networks: a parallel polycatenation of nanotube motifs to give a layered framework (see picture), a 2D3D polythreaded array involving five polymeric units at a time, and a 3D self-penetrating binodal network with an unprecedented (4.82)(4.64.84.10) topology.
Co-reporter:Dong-Rong Xiao Dr.;En-Bo Wang ;Hai-Yan An Dr. ;Yang-Guang Li Dr.;Lei Gao;Chun-Yan Sun;Lin Xu
Chemistry - A European Journal 2005 Volume 11(Issue 22) pp:
Publication Date(Web):10 AUG 2005
DOI:10.1002/chem.200500548
Reactions of the antimicrobial fluoroquinolone ciprofloxacin (cfH) with metal salts in the presence of aromatic polycarboxylate ligands or under basic conditions produce fourteen new metal–cfH complexes, namely, [Ba2(cf)2(1,4-bdc)(H2O)2]⋅H2O (1), [Sr6(cf)6(1,4-bdc)3(H2O)6]⋅2 H2O (2), [M2(cfH)2(bptc)(H2O)2]⋅8 H2O (M = Mn(3) and Cd(4)), [M(cfH)(1,3-bdc)] (M = Mn(5), Co(6), and Zn(7)), [Zn2(cfH)4(1,4-bdc)](1,4-bdc)⋅13 H2O (8), [Ca(cfH)2(1,2-Hbdc)2]⋅2 H2O (9) and [M(cf)2]⋅2.5 H2O (M = Mn(10), Co(11), Zn(12), Cd(13), and Mg(14)) (1,4-bdc = 1,4-benzenedicarboxylate, bptc = 3,3′,4,4′-benzophenonetetracarboxylate, 1,3-bdc = 1,3-benzenedicarboxylate, 1,2-bdc = 1,2-benzenedicarboxylate). Their structures were determined by single-crystal X-ray diffraction analyses and further characterized by elemental analyses, IR spectra, and thermogravimetric analyses. The structures of 1 and 2 consist of unique two-dimensional arm-shaped layers. Compounds 3 and 4 are isostructural and feature one-dimensional structures formed from the interconnection of [M2(cfH)2(H2O)2] dimers with bptc ligands. Compounds 5–7 are isostructural and contain double-chain-like ribbons constructed from [M2(cfH)2(CO2)2] dimers and 1,3-bdc. Compound 8 consists of a pair of [Zn(cfH)2]2+ fragments bridged by a 1,4-bdc into a dinuclear dumbbell structure. Compound 9 is a neutral monomeric complex. To the best of our knowledge, compounds 1–9 are the first examples of metal-quinolone complexes that contain aromatic polycarboxylate ligands. Compounds 10–14 are isostructural and exhibit interesting two-dimensional rhombic grids featuring large cavities with dimensions of 13.6×13.6 Å. Up to now, polymeric extended metal–cfH complexes have never been reported.
Co-reporter:Hong-Ze Gao, Zhong-Min Su
Journal of Molecular Structure: THEOCHEM 2005 Volume 722(1–3) pp:161-168
Publication Date(Web):2 May 2005
DOI:10.1016/j.theochem.2004.12.040
By means of ab initio HF and DFT B3LYP methods, the structure of bis(2-methyl-8-quinolinolato)gallium(III) chlorine complex(GaMq2Cl) was optimized and the electronic transition mechanism was studied in the complex. The lowest singlet excited state (S1) of GaMq2Cl has been studied by the singles configuration interaction (CIS) method and time-dependent density functional theory (TD-DFT). The lowest singlet electronic transition (S0→S1) of GaMq2Cl is π–π* electronic transitions and primarily localized on the phenol and pyridyl ligands. The emission of GaMq2Cl is due to the electron transitions from the phenol donor to the pyridyl acceptor including C→C and O→N transference. Two possible electron transfer pathways are presented, one by carbon, oxygen and nitrogen atoms, and the other via metal cation Ga3+. The comparison between the CIS optimized excited-state structure and the Hartree-Fock ground-state structure indicates that the geometric shift is mainly confined to the one quinoline and these changes can be easily understood in terms of the nodal patterns of the highest occupied and lowest unoccupied molecular orbitals. TD-B3-LYP calculations predict an emission wavelength of 504.57 nm. This is comparable to GaMq2Cl 492 nm observed experimentally for photoluminescence. Lending theoretical corroboration to recent experimental observations and supposition, the nature of the electron transition mechanism was revealed.
Co-reporter:Gang Sun;Yong-Qing Qiu;Hai-Zhu Sun;Jing-Dong Feng;Yu-Lan Zhu
Chinese Journal of Chemistry 2004 Volume 22(Issue 5) pp:
Publication Date(Web):26 AUG 2010
DOI:10.1002/cjoc.20040220506
The structures of barbituric acid derivatives substituted with Schiff base were optimized using ab initio HF method at 6-31G basis set. Based on the optimized structures, the electronic spectra were obtained by INDO/CI method. The second-order nonlinear optical (NLO) coefficients βμ were calculated according to the sum-over-states (SOS) formula. In addition, the effect of conjugation on electronic specra and second-order NLO coefficients was investigated. The influence of exchange between C and N atoms as well as the substituted effect on the barbituric acid was discussed. It was indicated that the exchange between C and N atoms on Schiff base is important for enhancing the NLO coefficient of the whole molecule with donor and acceptor (D-A). Meanwhile significant changes in electron donation and acception were observed as substituents changes positions. Among the designed models, molecule lb has maximal βμ value of 124.65×10−-30 esu. About molecule lb, barbituric acid is considered as an accepted electronic group and the position of N atom on Schiff base is close to it.
Co-reporter:Xin-Long Wang Dr.;Chao Qin Dr.;En-Bo Wang ;Lin Xu ;Chang-Wen Hu
Angewandte Chemie 2004 Volume 116(Issue 38) pp:
Publication Date(Web):22 SEP 2004
DOI:10.1002/ange.200460758
Durchdringungsfreie Strukturen sind die Produkte der Reaktion von Cadmiumsalzen mit langen flexiblen Liganden. In einem System entsteht aus achiralen Komponenten ein außergewöhnliches Netz, in dem chemisch unabhängige homochirale Helices neunfach verzahnt, also physikalisch verwoben sind (siehe Bild). Eine vergleichbar verschlungene Architektur war bisher für durchdringungsfreie Strukturen nicht bekannt.
Co-reporter:Xin-Long Wang Dr.;Chao Qin Dr.;En-Bo Wang ;Lin Xu ;Chang-Wen Hu
Angewandte Chemie International Edition 2004 Volume 43(Issue 38) pp:
Publication Date(Web):22 SEP 2004
DOI:10.1002/anie.200460758
Noninterpenetrating structures are formed by self-assembly of cadmium salts and long flexible ligands. In one case exceptional ninefold interlocked homochiral helices are built from achiral components (see picture). The helices are chemically independent but physically interwoven. This represents the highest degree of entanglement presently known for a noninterpenetrating system.
Co-reporter:Hong-Ze Gao, Zhong-Min Su, Hong Cheng, Rong-Shun Wang, Chang-Lu Shao, Shi-Lun Qiu
Journal of Molecular Structure: THEOCHEM 2002 Volume 578(1–3) pp:159-167
Publication Date(Web):14 February 2002
DOI:10.1016/S0166-1280(01)00687-X
Calculations of the stable structures and binding energies of [Si(O2Ph)n]m± (n=1–3, m=0,±2) have been carried out using ab initio HF and density functional theory B3LYP methods with different basis sets. The basis set superposition errors of the binding energies of the complexes were corrected by the counterpoise method. The composition characteristics of some frontier molecular orbitals in the complexes were discussed. All the complexes can stably exist in potential energy surfaces. The binding energies decrease accordingly with the increase in coordination number of silicon. In the tris(o-phenyl-enedioxy) silicon anion [Si(O2Ph)3]2−, the d orbitals of silicon contribute little to the stability of the complex, which is different from the transition metal hexacoordinated complexes with a d2sp3 hybridization of the central metal. The contribution to the stability of [Si(O2Ph)3]2− is from the formation of Si–O single bond, as well as the interaction of molecular orbitals among three PhO22− fragments.
Co-reporter:Xiao-Li Hu, Xin-Xin Yang, Xing-Quan He, Zhong-Min Su
Inorganic Chemistry Communications (March 2017) Volume 77() pp:
Publication Date(Web):March 2017
DOI:10.1016/j.inoche.2017.01.022
•The compounds 1–2 display novel and interesting structures based on two N-rich ligands and aromatic dicarboxylic acids.•1 and 2 are strongly luminescent with blue emission (λem = 440 nm and λem = 417 nm) in the solid-state.•The result reveals that 1 could be applied as a fluorescence sensor for NB with sensitivity and selectivity.•The possible quenching mechanism was investigated.Two novel Zn-based coordination polymer through employing of in-situ solvothermal techniques by using NO2-bdc (2-nitroterephthalic acid), HL1 = 1-(tetrazo-5-yl)-4-(triazo-1-yl)benzene, HL2 = 1-(tetrazo-5-yl)-4-(triazo-1-yl)benzene and Zn(NO3)2·6H2O to form [Zn2(HL1)2(NO2-bdc)]·0.5 (DMA)·CH3OH·H2O (1) and [Zn2(μ3-OH) (HL2)·(NO2-bdc)]·0.5 (DMA)·H2O (2). The structures have been determined by single-crystal X-ray diffraction analyses and further characterized by elemental analyses, IR spectra, powder X-ray diffraction (PXRD), and thermogravimetric (TG) analyses. In Compound 1, each Zn ion bridges one NO2-bdc and two HL1 ligands to yield infinite 2D layers, furthermore the aromatic π –π interactions link the adjacent layers yielding a 3D supramolecular structure. In compound 2, the HL2 ligands link Zn ions to form a 2D Zn(HL) sheet firstly, and further connected by double NO2-bdc pillars resulting in bi-pillared-layer type 3D frameworks. Moreover, 1 and 2 are luminescent with blue emission (λem = 440 nm and λem = 417 nm) in the solid-state. The fluorescent nature of 1and 2 inspired us to explore its potential application as a sensor for the detection of nitro explosives. The result reveals that 1 and 2 could be applied as a fluorescence sensor for NB (nitrobenzene) and 1,3-DNB (1,3-dinitrobenzene) with sensitivity and selectivity.Two novel Zn-based coordination polymer based on mixed ligands, [Zn2(HL1)2(NO2-bdc)(DMA)(MeOH)2]n (1) and [Zn2(μ3-OH) (HL2)·(NO2)]·0.5 (DMA)·H2O (2) were successfully synthesized under solvothermal condition, characterized by single-crystal X-ray diffraction, IR spectroscopy, thermogravimetric analysis (TGA). The fluorescent nature of 1 and 2 inspired us to explore its potential application as a sensor for the detection of nitro explosives.
Co-reporter:Yang Cui, Li-Li Wen, Guo-Gang Shan, Hai-Zhu Sun, Hui-Ting Mao, Min Zhang, Zhong-Min Su
Sensors and Actuators B: Chemical (June 2017) Volume 244() pp:314-322
Publication Date(Web):June 2017
DOI:10.1016/j.snb.2016.12.147
Co-reporter:Yang Jiang, Guangfu Li, Weilong Che, Yingjie Liu, Bin Xu, Guogang Shan, Dongxia Zhu, Zhongmin Su and Martin R. Bryce
Chemical Communications 2017 - vol. 53(Issue 21) pp:NaN3025-3025
Publication Date(Web):2017/02/27
DOI:10.1039/C7CC00769H
A neutral dinuclear Ir(III) Schiff base complex PIBIP has been synthesized and shown to exhibit both piezochromic luminescence (PCL) and aggregation induced emission (AIE) behaviour. An efficient second-level anti-counterfeit trademark and a data encryption device were fabricated using PIBIP as the active material.
Co-reporter:Yuan-Yuan Wang, Mi Zhang, Shun-Li Li, Shu-Ran Zhang, Wei Xie, Jun-Sheng Qin, Zhong-Min Su and Ya-Qian Lan
Chemical Communications 2017 - vol. 53(Issue 37) pp:NaN5207-5207
Publication Date(Web):2017/04/18
DOI:10.1039/C6CC10208E
Two novel isostructural polyoxometalate (POM)-based metal–organic frameworks (MOFs) with diamond topology, NENU-506 and NENU-507, were hydrothermally synthesized. They not only combine the advantages of both POMs and MOFs, but also show excellent chemical and thermal stability. Notably, NENU-507 exhibited a high reversible capacity of 640 mA h g−1 after 100 cycles when applied as an anode material in lithium-ion batteries.
Co-reporter:Tian-Tian Zheng, Jiao Zhao, Zhou-Wen Fang, Meng-Ting Li, Chun-Yi Sun, Xiao Li, Xin-Long Wang and Zhong-Min Su
Dalton Transactions 2017 - vol. 46(Issue 8) pp:NaN2461-2461
Publication Date(Web):2017/01/11
DOI:10.1039/C6DT04630D
The crystal structure of Cd-MOF-74 was obtained for the first time that possesses high sensitivity for the detection of copper ions from water and simulated biological fluids based on changes in luminescent intensity. Furthermore, Cd-MOF-74 could selectively remove Cu2+ from simulated biological fluids that contain Mg2+, Co2+, Zn2+, Fe2+, Ni2+, Na+, and K+. The adsorption capacity of this adsorbent for copper ions reached 189.5 mg g−1 and it quickly adsorbed copper ions within 10 minutes under 10 ppm Cu2+ in the simulated biological system. XPS, PXRD, and gas adsorption measurements revealed that this high sensitivity and selectivity of Cd-MOF-74 resulted from the partial substitution of Cd2+ by Cu2+ in the framework. Although many MOF materials have been employed for sensor or selective adsorption of Cu2+, Cd-MOF-74 is the first example of MOFs showing both capabilities in simulated biological fluids, which represents a pioneering work that extends the applications of MOF materials in the biological field.
Co-reporter:Guo-Gang Shan, Ling-Yu Zhang, Hai-Bin Li, Shuang Wang, Dong-Xia Zhu, Peng Li, Chun-Gang Wang, Zhong-Min Su and Yi Liao
Dalton Transactions 2012 - vol. 41(Issue 2) pp:NaN530-530
Publication Date(Web):2011/11/01
DOI:10.1039/C1DT11215E
We report the synthesis and characterization of two cationic iridium(III) complexes with dendritic carbazole ligands as ancillary ligands, namely, [Ir(ppy)2L3]PF6 (1) and [Ir(ppy)2L4]PF6 (2), where L3 and L4 represent 3,8-bis(3,6-di-tert-butyl-9H-carbazol-9-yl)-1,10-phenanthroline and 3,8-bis(3′,6′-di-tert-butyl-6-(3,6-di-tert-butyl-9H-carbazol-9-yl)-3,9′-bi(9H-carbazol)-9-yl)-1,10-phenanthroline, respectively. Their photophysical properties have been investigated and compared. The results have shown that complex 2 is aggregation-induced phosphorescent emission (AIPE) active and exhibits the highest photoluminescent quantum yield (PLQY) of 16.2% in neat film among the reported cationic Ir(III) complexes with AIPE activity. In addition, it also enjoys redox reversibility, good film-forming ability, excellent thermal stability as well as off/on luminescence switching properties, revealing its potential application as a candidate for light-emitting electrochemical cells and organic vapor sensing. To explore applications in biology, 2 was used to image cells.
Co-reporter:Liu Yang, Li Cao, Xiao Li, Chao Qin, Liang Zhao, Kui-Zhan Shao and Zhong-Min Su
Dalton Transactions 2017 - vol. 46(Issue 23) pp:NaN7576-7576
Publication Date(Web):2017/05/09
DOI:10.1039/C7DT01306J
Four metal–organic frameworks, namely [Cd2(tib)(btb)(H2O)2]·NO3·2.5DMF (1), [Cd(tib)(H2dhbqdc)0.5(NO3)]·6H2O (2), [Co(tib)(1,4-ndc)]·2DMF (3), and [Cu3(tib)2(2,6-ndc)2(H2O)2]·2NO3·2H2O (4), were synthesized based on 1,3,5-tris(1-imidazolyl)benzene and diverse carboxylic acid ligands. They have been characterized by elemental analysis, infrared spectroscopy (IR), powder X-ray diffraction (PXRD), thermogravimetric analyses (TGA) and single crystal X-ray diffraction. Compound 1 is a 3D framework constructed from a binuclear Cd cluster with (3,3,6)-connected (63)2(69·86) topology. Compound 2 exhibits a 2D wavy layered structure with (3,4)-connected topology, and compound 3 displays a two-fold interpenetrating network with (3,5)-connected topology. Compound 4 can be regarded as a three-fold interpenetrating framework. Moreover, compounds 1 and 2 can be used as fluorescent sensors sensing small molecules with high selectivity. In this context, we selected the typical toxic explosives, TNP, namely 2,4,6-trinitrophenol and NB, namely nitrobenzene, as examples to investigate the properties of sensing. Furthermore, the magnetic properties of compounds 3 and 4 are investigated.
Co-reporter:Wei Xie, Jun-Sheng Qin, Wen-Wen He, Kui-Zhan Shao, Zhong-Min Su, Dong-Ying Du, Shun-Li Li and Ya-Qian Lan
Inorganic Chemistry Frontiers 2017 - vol. 4(Issue 3) pp:NaN552-552
Publication Date(Web):2017/01/12
DOI:10.1039/C6QI00528D
We successfully synthesized a novel anionic luminescent metal–organic framework (MOF) (NENU-524) with a lonsdaleite topology. NENU-524 contains a trigonal prismatic unit {Zn8(btca)6(2-NH2-bdc)3} that can be regarded as a double secondary building unit with an unusual triply bound triangular frustum geometry. The prepared NENU-524 had a permanent porosity and excellent stability in air. NENU-524 was used as a platform to encapsulate yellow-emitting [Ir(ppy)2(bpy)]+ cations in the nanotube channels of the blue-emitting MOF via an ion-exchange process. The [Ir(ppy)2(bpy)]+@NENU-524 MOF ([Ir(ppy)2(bpy)]+ concentration 3.86 wt%) emitted a pure white light with CIE coordinates of (0.300, 0.336) and a high quantum yield of up to 15.2%. The white light-emitting diodes assembled using [Ir(ppy)2(bpy)]+@NENU-524 as a white phosphor emitted a bright white light, suggesting that the composite is a promising material for use in lighting. The assembled white light-emitting diodes continued to emit a bright white light for up to one month. This simple and feasible approach could be used to develop luminescent luminophor@MOFs composites for practical applications.
Co-reporter:Mei-Jie Wei, Jia-Qi Fu, Yi-Di Wang, Jing-Yang Gu, Bai-Ling Liu, Hong-Ying Zang, En-Long Zhou, Kui-Zhan Shao and Zhong-Min Su
Journal of Materials Chemistry A 2017 - vol. 5(Issue 3) pp:NaN1093-1093
Publication Date(Web):2016/11/29
DOI:10.1039/C6TA08581D
It is essential and vital to develop high-performance proton-conducting solid electrolyte materials for proton exchange membrane fuel cells (PEMFCs), but it remains challenging to design and synthesise such electrolytes with high proton conductivity which are also stable enough to be applied in PEMFCs. Herein, we employed the HCl steam-assisted conversion method to synthesize nonporous coordination complexes with a gradual increase of proton conductivity by stepwise protonation of sulfonated ligands and introduction of halide ions, including [Cu(Hsfpip)(H2O)2]·H2O (1), [CuH2(Hsfpip)2(H2O)] (2) and [CuH(Hsfpip)Cl(H2O)] (3) (where Hsfpip is 2-(2,4-disulfophenyl)imidazo(4,5-f)(1,10)-phenanthroline). We reveal the relationship between the nature of proton conduction and structural features. Three resulting coordination complexes showed high proton conductivity with a maximum value of 1.43 mS cm−1 for 1, 2.58 mS cm−1 for 2 and 15 mS cm−1 for 3 at 95 °C and 97% RH, and meanwhile, we proved their proton conduction nature and electron resistance using D2O-exchange experiments and the Hebb–Wagner polarization method. We believe that these nonporous solid electrolytes intrinsically possess proton carriers and may avoid fuel crossover, which makes them good candidates for PEMFCs in real-life applications.
Co-reporter:Yan-Qing Jiao, Hong-Ying Zang, Xin-Long Wang, En-Long Zhou, Bai-Qiao Song, Chun-Gang Wang, Kui-Zhan Shao and Zhong-Min Su
Chemical Communications 2015 - vol. 51(Issue 56) pp:NaN11316-11316
Publication Date(Web):2015/06/01
DOI:10.1039/C5CC03357H
The first polyoxometalate-based metal–organic nanotube constructed via covalent bonds has been synthesized. POM anions stick the metal–organic nanotubes to build 3D nanotubular arrays. The stability, magnetic and proton conducting properties are investigated.
Co-reporter:Kun Zhou, Yun Geng, Li-Kai Yan, Xin-Long Wang, Xian-Chun Liu, Guo-Gang Shan, Kui-Zhan Shao, Zhong-Min Su and Ying-Ning Yu
Chemical Communications 2014 - vol. 50(Issue 80) pp:NaN11937-11937
Publication Date(Web):2014/08/13
DOI:10.1039/C4CC05893C
An ultrastable [Ag55(MoO4)6]43+ ({Ag55Mo6} for short) nanocluster with a Ag-centered multishell structure in compound [Ag55(MoO4)6(CCtBu)24(CH3COO)18](OAc)·2H2O (1) has been obtained. The ultrastability of 1 was demonstrated by Mulliken population analysis. In addition, the potential wide gap semiconductor property and electrochemical properties of 1 were investigated.
Co-reporter:Wei-Chao Chen, Xin-Long Wang, Chao Qin, Kui-Zhan Shao, Zhong-Min Su and En-Bo Wang
Chemical Communications 2016 - vol. 52(Issue 61) pp:NaN9517-9517
Publication Date(Web):2016/06/21
DOI:10.1039/C6CC03763A
A carbon-free, stable, homogeneous water oxidation catalyst based on the unique hepta-nuclear cobalt–arsenic core (“fused” double-quasi-cubane) and polyoxometalate ligands, Na12[{CoII7AsIII6O9(OH)6}(A-α-SiW9O34)2]·8H2O (1), was synthesized, thoroughly characterized and employed to catalyze water oxidation under visible-light-driven conditions.
Co-reporter:Yu-Teng Zhang, Xin-Long Wang, Shuang-Bao Li, Ya-Ru Gong, Bai-Qiao Song, Kui-Zhan Shao and Zhong-Min Su
Chemical Communications 2016 - vol. 52(Issue 62) pp:NaN9635-9635
Publication Date(Web):2016/06/24
DOI:10.1039/C6CC04583A
Unprecedented Anderson-like alkoxo-polyoxovanadate [V6O6(OCH3)9(μ6-SO4)(COO)3]2− polyanions can serve as 3-connected second building units (SBUs) that assemble with dicarboxylate or tricarboxylate ligands to form a new family of metal organic tetrahedrons of V4E6 and V4F4 type (V = vertex, E = edge, and F = face). To our knowledge, this alkoxo-polyoxovanadate-based SBU is the first ever reported.
Co-reporter:Xiao-Li Hu, Chun-Yi Sun, Chao Qin, Xin-Long Wang, Hai-Ning Wang, En-Long Zhou, Wen-E Li and Zhong-Min Su
Chemical Communications 2013 - vol. 49(Issue 34) pp:NaN3566-3566
Publication Date(Web):2013/03/14
DOI:10.1039/C3CC39173F
Unprecedented 3d–4f MOFs encapsulating infinite linear polyiodide chains were firstly reported using iodine molecules as a versatile precursor template. They possess high framework stability in acid/base aqueous solutions. The kinetics of iodine molecule release/recovery and UV-light photocatalytic H2 evolution activities were investigated.
Co-reporter:Xue-Gang Hou, Yong Wu, Hong-Tao Cao, Hai-Zhu Sun, Hai-Bin Li, Guo-Gang Shan and Zhong-Min Su
Chemical Communications 2014 - vol. 50(Issue 45) pp:NaN6034-6034
Publication Date(Web):2014/04/22
DOI:10.1039/C3CC49395D
A new cationic Ir(III) complex with AIE characteristics was designed and synthesized with the help of density functional theory calculations, which exhibits highly sensitive and selective detection of explosives (2,4,6-trinitrophenol, TNP).
Co-reporter:Guang-Sheng Yang, Zhong-Ling Lang, Hong-Ying Zang, Ya-Qian Lan, Wen-Wen He, Xiao-Liang Zhao, Li-Kai Yan, Xin-Long Wang and Zhong-Min Su
Chemical Communications 2013 - vol. 49(Issue 11) pp:NaN1090-1090
Publication Date(Web):2012/12/10
DOI:10.1039/C2CC36894C
Two S-containing MOFs, interpenetrating IFMC-27 and non-interpenetrating IFMC-28, were synthesized by altering solvent size. The nanoporous IFMC-28 reveals high selective adsorption for Cu2+ ions and has been applied as a chromatographic column for separating transition metal ions for the first time.
Co-reporter:Peng Huang, Chao Qin, Xin-Long Wang, Chun-Yi Sun, Guang-Sheng Yang, Kui-Zhan Shao, Yan-Qing Jiao, Kun Zhou and Zhong-Min Su
Chemical Communications 2012 - vol. 48(Issue 1) pp:NaN105-105
Publication Date(Web):2011/11/04
DOI:10.1039/C1CC15684E
An unprecedented organic–inorganic hybrid {[Cu6L6(H2O)3][Nb10V4O40(OH)2]}2·13H2O (1) (L = 1,10-phenanthroline) containing the unreported {Nb10V4O40(OH)2}12− building blocks has been successfully synthesized and its photoluminescent properties, IR spectra, thermogravimetric analyses and single-crystal X-ray diffraction were investigated.
Co-reporter:Bai-Qiao Song, Xin-Long Wang, Yu-Teng Zhang, Xue-Song Wu, Hong-Sheng Liu, Kui-Zhan Shao and Zhong-Min Su
Chemical Communications 2015 - vol. 51(Issue 46) pp:NaN9518-9518
Publication Date(Web):2015/04/30
DOI:10.1039/C5CC02639C
A unique cationic metal–organic framework was synthesized by connecting the neutral rod-shaped secondary building unit with a cationic dicarboxylate ligand. This framework showed a rare snub square tessellation pattern by the periodic tiling of triangular and square nanotubes. The charge- and size-dependent ion-exchange of anion dyes was investigated.
Co-reporter:Guo-Gang Shan, Hai-Bin Li, Hong-Tao Cao, Dong-Xia Zhu, Peng Li, Zhong-Min Su and Yi Liao
Chemical Communications 2012 - vol. 48(Issue 14) pp:NaN2002-2002
Publication Date(Web):2012/01/11
DOI:10.1039/C2CC15855H
We demonstrate that two new cationic Ir(III) complexes exhibit an interesting piezochromism, and their emission color can be smartly switched by grinding and heating. This is the first example that the Ir(III) complexes display piezochromic phosphorescence.
Co-reporter:Xiao-Li Hu, Chao Qin, Xin-Long Wang, Kui-Zhan Shao and Zhong-Min Su
Chemical Communications 2015 - vol. 51(Issue 99) pp:NaN17524-17524
Publication Date(Web):2015/09/23
DOI:10.1039/C5CC07004J
An anionic metal–organic framework (MOF) with 1D nanotube channels has been constructed. The charge and size dependent ion-exchange of cationic dyes was investigated. Rho@1 could be used as a dual-emitting fluorescent sensor for sensing explosives by self-referencing energy transfer behaviors.
Co-reporter:Yan-Qing Jiao, Chao Qin, Xin-Long Wang, Fu-Hong Liu, Peng Huang, Chun-Gang Wang, Kui-Zhan Shao and Zhong-Min Su
Chemical Communications 2014 - vol. 50(Issue 45) pp:NaN5963-5963
Publication Date(Web):2013/12/12
DOI:10.1039/C3CC47703G
A novel δ-Dawson [(WO5)3W14Mn2IIIO44Cl2]12− compound induced by the Jahn–Teller distortion of MnIII has been synthesized through utilizing {Mn12} as a reactant, which exhibits photocatalytic H2 evolution activity. Its electrochemical behavior and magnetic properties were investigated.
Co-reporter:Dong-Ying Du, Jun-Sheng Qin, Yang-Guang Li, Shun-Li Li, Ya-Qian Lan, Xin-Long Wang, Kui-Zhan Shao, Zhong-Min Su and En-Bo Wang
Chemical Communications 2011 - vol. 47(Issue 10) pp:NaN2834-2834
Publication Date(Web):2011/01/24
DOI:10.1039/C0CC04343E
The first 3D uninodal eight-connected {P4Mo6O31H6}-based pure inorganic framework linked by transition metal ions has been synthesized and its electrochemical behavior and diffuse reflectance UV-Vis spectrum were investigated.
Co-reporter:Hai-Ning Wang, Xing Meng, Guang-Sheng Yang, Xin-Long Wang, Kui-Zhan Shao, Zhong-Min Su and Chun-Gang Wang
Chemical Communications 2011 - vol. 47(Issue 25) pp:NaN7130-7130
Publication Date(Web):2011/05/26
DOI:10.1039/C1CC11932J
A 12-connected network with fcu topology was firstly reported focusing on using predesigned metal–organic polyhedron (MOP) as the precursor, and its adsorption and delivery of the drug 5-fluorouracil (5-FU) was also determined.
Co-reporter:Hai-jun Pang, Jun Peng, Chun-jing Zhang, Yang-guang Li, Peng-peng Zhang, Hui-yuan Ma and Zhong-min Su
Chemical Communications 2010 - vol. 46(Issue 28) pp:NaN5099-5099
Publication Date(Web):2010/06/15
DOI:10.1039/C003048A
A novel compound, [Cu(bimb)]2(HPW12O40)·3H2O (bimb = 1,4-bis(imidazol-1-ylmethyl)biphenyl) with a polyoxometalate-encapsulated 3D metal–organic pseudo-rotaxane framework that can be described as a 2-fold dianet topology showing well-defined 1D nano-scale tunnels, has been synthesized hydrothermally, and its de-/rehydration behavior has been investigated.
Co-reporter:Xiang-Rong Hao, Xin-Long Wang, Chao Qin, Zhong-Min Su, En-Bo Wang, Ya-Qian Lan and Kui-Zhan Shao
Chemical Communications 2007(Issue 44) pp:NaN4622-4622
Publication Date(Web):2007/09/20
DOI:10.1039/B711405B
A 3D chiral nanoporous coordination framework consisting of homochiral nanotubes assembled from octuple helices with a 19.4 Å by 22.4 Å aperture is formed by the parallel alignment of eight infinite helical chains.
Co-reporter:Ya-Qian Lan, Xin-Long Wang, Shun-Li Li, Zhong-Min Su, Kui-Zhan Shao and En-Bo Wang
Chemical Communications 2007(Issue 46) pp:NaN4865-4865
Publication Date(Web):2007/09/19
DOI:10.1039/B709835A
The first (6,8)-connected self-penetrating metal–organic framework has been constructed using an asymmetric neutral ligand based on dinuclear zinc clusters, as six-connected nodes, and trinuclear zinc clusters as eight-connected nodes, representing the highest-connected binodal network topology presently known for self-penetrating systems.
Co-reporter:Xin-Long Wang, Chao Qin, En-Bo Wang and Zhong-Min Su
Chemical Communications 2007(Issue 41) pp:NaN4247-4247
Publication Date(Web):2007/08/08
DOI:10.1039/B709563E
Threading molecular square “beads” on a twofold interpenetrated diamondoid skeleton gives a new type of 3D metal–organic polyrotaxane framework with large channels, in which nanosized Keggin anions as guests are encapsulated for the first time.
Co-reporter:Ronglin Zhong, Min Zhang, Hongliang Xu and Zhongmin Su
Chemical Science (2010-Present) 2016 - vol. 7(Issue 2) pp:NaN1032-1032
Publication Date(Web):2015/10/27
DOI:10.1039/C5SC03437J
Besides the classic double bond scheme, several novel schemes have been proposed to describe the nature of the chemical bond in dicarbon (C2), including a quadruple bond and a singlet diradical state. The results from a symmetry-broken CASSCF(8,8)/aug-cc-pVTZ study present a harmony between MO and VB theories, based on the orthogonal hybridization of the 3σg and 2σu orbitals together with the other six pristine valence orbitals. This scheme achieves the same bonding energy, RC–C, ωe and one electron density as that from the eight pristine valence orbitals. A quadruple bond scheme, identical to Prof. Shaik's result from VB theory, is achieved with the 4th bond energy in the range of 12.8–27.6 kcal mol−1. Meanwhile, the weight of a singlet open-shell configuration is the highest among all the possible configurations.
Co-reporter:Dong-Ying Du, Jun-Sheng Qin, Ting-Ting Wang, Shun-Li Li, Zhong-Min Su, Kui-Zhan Shao, Ya-Qian Lan, Xin-Long Wang and En-Bo Wang
Chemical Science (2010-Present) 2012 - vol. 3(Issue 3) pp:NaN710-710
Publication Date(Web):2011/12/19
DOI:10.1039/C2SC00586G
Here, we synthesize a novel polyoxometalate-based crystalline tubular inorganic–organic compound, Mn[Zn(im)]2{[Na(H2O)]2[Mn(H2O)2][Zn(im)2][P4Mo6O31H6]2}·8H2O (IFMC-100) (im and IFMC correspond to imidazole and Institute of Functional Material Chemistry, respectively). Au-anchored tubular microreactor, Au@IFMC-100, has been prepared by simple immersion of IFMC-100 in an ethanol solution of HAuCl4 without any extra reducing agents, photochemical and electrochemical auxiliaries. Furthermore, IFMC-100 and Au@IFMC-100 have been employed as catalysts for the reduction of K3Fe(CN)6 and 4-nitrophenol with NaBH4 in aqueous solution, respectively. The results indicate the as-prepared Au@IFMC-100 microtubes exhibit enhanced catalytic performance in redox catalysis.
Co-reporter:Dong-Ying Du, Jun-Sheng Qin, Shun-Li Li, Zhong-Min Su and Ya-Qian Lan
Chemical Society Reviews 2014 - vol. 43(Issue 13) pp:NaN4632-4632
Publication Date(Web):2014/03/27
DOI:10.1039/C3CS60404G
Polyoxometalate (POM)-based metal–organic framework (MOF) materials contain POM units and generally generate MOF materials with open networks. POM-based MOF materials, which utilize the advantages of both POMs and MOFs, have received increasing attention, and much effort has been devoted to their preparation and relevant applications over the past few decades. They have good prospects in catalysis owing to the electronic and physical properties of POMs that are tunable by varying constituent elements. In this review, we present recent developments in porous POM-based MOF materials, including their classification, synthesis strategies, and applications, especially in the field of catalysis.
Co-reporter:Jun-Sheng Qin, Dong-Ying Du, Wen-Liang Li, Jing-Ping Zhang, Shun-Li Li, Zhong-Min Su, Xin-Long Wang, Qiang Xu, Kui-Zhan Shao and Ya-Qian Lan
Chemical Science (2010-Present) 2012 - vol. 3(Issue 6) pp:NaN2118-2118
Publication Date(Web):2012/03/07
DOI:10.1039/C2SC00017B
A novel zeolite-like metal–organic framework (ZMOF) with sodalite topology, [Zn(HL)]·DMA (IFMC-1, L = 4,5-di(1H-tetrazol-5-yl)-2H-1,2,3-triazole and IFMC = Institute of Functional Material Chemistry), was solvothermally synthesized based on an N-rich aromatic ligand without a NH2 group. It exhibits high CO2 uptake and selective CO2/N2 adsorption capacity. For the first time, we investigated the influence of a large number of uncoordinated nitrogen atoms from aromatic rings for CO2 adsorption in ZMOFs. This result reveals that the high percentage of open N-donor sites leads to the high uptake capacity for CO2, even in the absence of any NH2 groups and open metal sites. In addition, it also exhibits efficient drug delivery capacity.
Co-reporter:Yu-Teng Zhang, Xin-Long Wang, En-Long Zhou, Xue-Song Wu, Bai-Qiao Song, Kui-Zhan Shao and Zhong-Min Su
Dalton Transactions 2016 - vol. 45(Issue 9) pp:NaN3701-3701
Publication Date(Web):2016/01/27
DOI:10.1039/C5DT04764A
Three polyoxovanadate-based metal–organic polyhedra (denoted as VMOP-1, -2, and -3), adopting isostructural discrete octahedral cage geometries, were successfully synthesized under solvothermal conditions. These structures are all built up from the same pentavanadate {V5O9Cl} cluster connected by linear bidentate ligands (H2L1 = H2BDC, H2L2 = H2BDC-NH2, H2L3 = H2BDC-Br), respectively.
Co-reporter:Fu-Hong Liu, Chao Qin, Yan Ding, Han Wu, Kui-Zhan Shao and Zhong-Min Su
Dalton Transactions 2015 - vol. 44(Issue 4) pp:NaN1760-1760
Publication Date(Web):2014/11/17
DOI:10.1039/C4DT02961E
Two novel pillared MOFs (metal organic frameworks) [Zn2(trz)2(tda)]·DMA CH3OH (1) and [Zn2(trz)2(bpdc)]·DMA (2) were obtained under solvothermal conditions. The resulting MOFs show similar structures but with different interlayer distances based on the different carboxylate ligands. 1 and 2 display a certain degree of framework stability in both acid/base solutions and water. The luminescence intensities of the activated phases 1a and 2a are sensitive to metal ions, particularly Fe3+ and Cd2+ ions. Furthermore, the luminescent properties of 1a and 2a well dispersed in different solvents have also been investigated systematically, which demonstrate distinct solvent-dependent luminescent spectra with emission intensities that are significantly quenched by acetone, nitrobenzene and trinitrotoluene.
Co-reporter:Bai-Qiao Song, Chao Qin, Yu-Teng Zhang, Li-Tao An, Kui-Zhan Shao and Zhong-Min Su
Dalton Transactions 2015 - vol. 44(Issue 6) pp:NaN2851-2851
Publication Date(Web):2014/10/22
DOI:10.1039/C4DT02808B
A novel interpenetrating metal–organic framework, namely [Zn3L2(oba)3(H2O)2]·4H2O (1), has been synthesized under hydrothermal conditions. Its structure was determined by single-crystal X-ray diffraction analysis and further characterized by elemental analysis, IR, and thermogravimetric (TG) analysis. In the structure of 1, the rigid and flexible V-shaped ligands link Zn(II) to form a 3D structure where two types of helices and four types of pseudo-helical chains containing three pairs of enantiomers and two pairs of conformational isomers have been characterized. One such 3D framework incorporates six identical networks to form a 7-fold interpenetrated 3D framework. From the topological analysis, the Zn(II) ions act as three- and four-connected nodes, and oba as well as L are linkers. The framework of compound 1 can be classified as a new (63)2(65·8) topology, which is a novel (3,3,4)-connected [4 + 3] 7-fold interpenetrating net showing 7-fold interlocking pseudo-helical chains and a unique catenane-like motif with Hopf links. In addition, the luminescence properties of the compound are discussed.
Co-reporter:Bai-Qiao Song, Chao Qin, Yu-Teng Zhang, Xue-Song Wu, Kui-Zhan Shao and Zhong-Min Su
Dalton Transactions 2015 - vol. 44(Issue 9) pp:NaN3958-3958
Publication Date(Web):2015/01/26
DOI:10.1039/C4DT03933E
A unique three-dimensional metal azolate framework containing a tetranuclear copper cluster constructed by six 1,2,4-triazole units was synthesized in which the 1,2,4-triazole units show unusual bridging “crevice” coordination mode with their 1- and 2-positioned sp2 N-atoms as symmetrically bridging centers. The photocatalytic activities of as-prepared compound were tested by degradation of rhodamine-B (RB) under different light irradiation.
Co-reporter:Xiao-Li Hu, Fu-Hong Liu, Chao Qin, Kui-Zhan Shao and Zhong-Min Su
Dalton Transactions 2015 - vol. 44(Issue 17) pp:NaN7827-7827
Publication Date(Web):2015/03/19
DOI:10.1039/C5DT00515A
A novel Cd-MOF (metal organic framework) [Cd3(NTB)2(DMA)3]·2DMA (H3NTB = 4,4′,4′′-nitrilotrisbenzoic acid; DMA = N,N-dimethylacetamide) (1) was obtained under solvothermal conditions. The resulting MOF exhibits a novel (2D→3D) interdigitated architecture that is obtained from a bilayered motif with hexagonal grids. Luminescence properties of the activated phase of 1a well dispersed in different solvents have also been investigated systematically, which demonstrate distinct solvent-dependent luminescence spectra with emission intensities significantly quenched toward nitrobenzene (NB) and 2,4,6-trinitrophenol (TNP). The results reveal that 1 can be applied as a fluorescent sensor for the detection of TNP with high sensitivity, selectivity, and recyclability.
Co-reporter:Hong-Tao Cao, Lei Ding, Guo-Gang Shan, Hai-Zhu Sun, Yong Wu and Zhong-Min Su
Dalton Transactions 2015 - vol. 44(Issue 46) pp:NaN20003-20003
Publication Date(Web):2015/10/19
DOI:10.1039/C5DT03129J
A sulfur-free iridium(III) complex (pbi)2Ir(mtpy) (1) was successfully prepared and adopted as a Hg(II)-chemosensor with high selectivity and sensitivity. Multi-signaling responses towards Hg(II) ions were observed by UV−vis absorption, phosphorescence and electrochemistry measurements. With addition of Hg(II) ions, complex 1 presented quenched emission in its phosphorescence spectrum and the detection limit was as low as 2.5 × 10−7 M. Additionally, its redox peak currents showed a broad linear relationship with the concentration of Hg(II) ions ranging from 0 to 500 μM, which was beneficial for the quantitative detection. Based on the 1H NMR and ESI-MS analyses, the probing mechanism was tentatively supposed to be the Hg2+-induced changes in the local environment of complex 1. Such a response process was useful for achieving simple and effective detection of Hg(II) ions as well as developing more chemosensors.
Co-reporter:Chun-Chen Yuan, Shi-Ming Wang, Wei-Lin Chen, Lin Liu, Chao Qin, Zhong-Min Su and En-Bo Wang
Dalton Transactions 2014 - vol. 43(Issue 4) pp:NaN1497-1497
Publication Date(Web):2013/11/12
DOI:10.1039/C3DT52676C
Two Anderson-type heteropolyanion-supported copper phenanthroline redox couples have been successfully introduced into dye-sensitized solar cells, which can significantly increase the short-circuit photocurrent, open-circuit voltage and the conversion efficiency by 2.2 times, 26.8% and 3.93 times respectively, compared to the pristine copper phenanthroline redox couple.
Co-reporter:Kun Zhou, Chao Qin, Xin-Long Wang, Kui-Zhan Shao, Li-Kai Yan and Zhong-Min Su
Dalton Transactions 2014 - vol. 43(Issue 28) pp:NaN10699-10699
Publication Date(Web):2014/04/17
DOI:10.1039/C4DT00762J
We report a rare all-thiol-stabilized [Ag28(StBu)23]5+ ({Ag28S23} for short) nanocluster with a “crab-like” shape in compound [Ag28(StBu)23](CF3COO)5·8CH3OH (1), which has been synthesized by the self-assembly of AgStBu with CF3COOH, Et3N and KBr/KI in methanol. The diffuse reflection spectrum and luminescence spectra of 1 were investigated.
Co-reporter:Bo Zhu, Li-Kai Yan, Wei Guan and Zhong-Min Su
Dalton Transactions 2015 - vol. 44(Issue 19) pp:NaN9070-9070
Publication Date(Web):2015/04/10
DOI:10.1039/C5DT00318K
A thorough theoretical analysis was carried out on the sulfoxidation with H2O2 catalyzed by a tetranuclear peroxotungstate [SiO4{WO(O2)2}4]4−. The active species is the [SiO4{WO(O2)2}4(H2O2)]4− (SiW4(H2O2)) complex rather than [SiO4{WO(O2)2}4]4− (SiW4). The catalytic cycle consists of three elementary processes: oxygen transfer, sulfoxide dissociation, and catalyst regeneration. The oxygen transfer occurs from the peroxo oxygen atom O1 of SiW4(H2O2) to the sulfur center of dimethyl sulfide with a moderate Gibbs activation energy (ΔG°‡) of 17.1 kcal mol−1. By comparing potential energy surfaces and condensed Fukui functions (ƒ+), the electrophilicity of the outer peroxo atoms in SiW4(H2O2) determines which oxygen transfers to the dimethyl sulfide. Then, the sulfoxide dissociation proceeds with a small ΔG°‡ value of 2.3 kcal mol−1 by elongation of the peroxo O1–O4 distance and elimination of the product dimethylsulfoxide. Finally, the catalyst regeneration is found to occur via two successive proton transfers from H2O2 to the oxygen atoms of peroxotungstates with the ΔG°‡ values of 15.9 and 15.3 kcal mol−1, which has been firstly examined in the present study. All of these steps occur easily with moderate ΔG°‡ values, but the oxygen transfer is the rate-determining step of this catalytic reaction. In addition, the catalytic activity of peroxotungstates can be effectively tuned by changing the heteroatom X of [XO4{WO(O2)2}4(H2O2)]n− in the order: SeVI ≈ SVI > AsV ≈ PV > SiIV.
Co-reporter:Xin-Long Wang, Chao Qin, Ya-Qian Lan, Kui-Zhan Shao, Zhong-Min Su and En-Bo Wang
Chemical Communications 2009(Issue 4) pp:NaN412-412
Publication Date(Web):2008/11/14
DOI:10.1039/B815629H
The first uninodal 10-connected metal–organic framework, based on pentanuclear cadmium cluster building blocks, exhibits an unprecedented γ-Pu topology, which adds a new member to the series of metal–organic analogues which have a natural materials topology.
Co-reporter:Kun Zhou, Chao Qin, Hai-Bin Li, Li-Kai Yan, Xin-Long Wang, Guo-Gang Shan, Zhong-Min Su, Chuang Xu and Xiu-Li Wang
Chemical Communications 2012 - vol. 48(Issue 47) pp:NaN5846-5846
Publication Date(Web):2012/04/23
DOI:10.1039/C2CC32321D
Bridging between silver clusters and polyoxoanion clusters, the first 1D assembly, [Ag34(StBu)26(W6O21)(CF3COO)](CF3COO)·Et3N·20CH3OH (1), based on POM-templated silver-thiolate nanoclusters featuring a [Ag34(StBu)26(CF3COO)]7+ shell and a [W6O21]6− core is reported. This novel core–shell nanocluster possesses nanoscopic morphology, displays intense deep-blue emission in solution under ambient conditions and also shows special electrochemical properties.
Co-reporter:Lei Chen, Ke Tan, Ya-Qian Lan, Shun-Li Li, Kui-Zhan Shao and Zhong-Min Su
Chemical Communications 2012 - vol. 48(Issue 47) pp:NaN5921-5921
Publication Date(Web):2012/04/18
DOI:10.1039/C2CC31257C
Two isostructural 2D → 2D parallel → 3D inclined interpenetrating polycatenane-like metal–organic frameworks were successfully constructed based on length-adjusted tricarboxylate ligands. With the merit of being microporous, IFMC-10 can serve as host for encapsulating lanthanide cations and I2 to exhibit luminescent sensing and rapid adsorption of iodine.
Co-reporter:Wen-Wen He, Shun-Li Li, Guang-Sheng Yang, Ya-Qian Lan, Zhong-Min Su and Qiang Fu
Chemical Communications 2012 - vol. 48(Issue 80) pp:NaN10003-10003
Publication Date(Web):2012/08/13
DOI:10.1039/C2CC34196D
A novel non-interpenetrating metal–organic framework IFMC-15 was successfully constructed based on octahedral cage-like building units and its outstanding performance in reversible adsorption of iodine was investigated.
Co-reporter:Xiao Li, Liu Yang, Tan Su, Xinlong Wang, Chunyi Sun and Zhongmin Su
Journal of Materials Chemistry A 2017 - vol. 5(Issue 10) pp:NaN5006-5006
Publication Date(Web):2017/01/24
DOI:10.1039/C6TA10405C
In this study, we synthesized a low-cost electrocatalyst, Ni/Mo2C nanoparticles coated with graphene shells (denoted as NiMo2C@C), via a facile carburization process of porous bimetallic metal–organic frameworks (NiMo-MOF). This is the first example of a Ni and Mo2C nanocomposite derived from a bimetallic MOF that demonstrates excellent electrocatalytic activity and remarkable durability as long as 10 h under acidic and basic conditions. The overpotentials are 169 mV and 181 mV to reach the current density of 10 mA cm−2, respectively. The favorable performance can be ascribed to the synergistic effect between Mo2C and Ni as well as the homogeneous distribution, graphene coating and mesoporous structure which is in favor of the charge transfer in the HER. This work may provide some guidelines for fabricating nanostructured hybrids composed of versatile transition metal carbides and graphene with high performance and stability in different media based on designed MOFs.
Co-reporter:Wei-Chao Chen, Chao Qin, Xin-Long Wang, Yang-Guang Li, Hong-Ying Zang, Kui-Zhan Shao, Zhong-Min Su and En-Bo Wang
Dalton Transactions 2015 - vol. 44(Issue 25) pp:NaN11293-11293
Publication Date(Web):2015/05/20
DOI:10.1039/C5DT01711D
A large lanthanide-containing tungstotellurites(IV) nanocluster Na18[Ce10Te8W88O298(OH)12(H2O)40]·54H2O (1) was synthesized by combining cerium linkers and TeO32− heteroanion templates. The macroanion in 1 consists of two identical [Te4W42O144(OH)6Ce4(H2O)13]14− subunits and two triangle-shaped {W2O5Ce(H2O)7} linkers.
Co-reporter:Bai-Qiao Song, Xin-Long Wang, Chun-Yi Sun, Yu-Teng Zhang, Xue-Song Wu, Liu Yang, Kui-Zhan Shao, Liang Zhao and Zhong-Min Su
Dalton Transactions 2015 - vol. 44(Issue 31) pp:NaN13822-13822
Publication Date(Web):2015/07/06
DOI:10.1039/C5DT01560J
A novel 3D organic–inorganic hybrid framework constructed from tetra-CoII-substituted sandwich-type phosphotungstates with a rare 8-connected bcu topology is reported, which exhibited highly efficient photocatalysis activity under visible light and could be used for 5 cycles without any obvious decrease in activity.
Co-reporter:Guo-Gang Shan, Hai-Bin Li, Hai-Zhu Sun, Dong-Xia Zhu, Hong-Tao Cao and Zhong-Min Su
Journal of Materials Chemistry A 2013 - vol. 1(Issue 7) pp:NaN1449-1449
Publication Date(Web):2012/12/14
DOI:10.1039/C2TC00558A
Three multifunctional cationic iridium(III)-based materials with aggregation-induced emission (AIE) and piezochromic luminescence (PCL) behavior have been rationally designed with the help of the theoretical calculations and successfully synthesized. All complexes contain the same cyclometalated ligand, 1-(2,4-difluorophenyl)-1H-pyrazole (dfppz), while functionalized ancillary ligands with different substitution are used to control their photophysical properties. Complex 1 and 2 with ancillary ligands modified with aliphatic chains and carbazole end-capped alkyl groups, respectively, undergo remarkable and reversible changes in emission color in the solid state upon grinding or heating. In addition, 2, characterized as having the 3ILCT excited-state feature, simultaneously exhibits an interesting AIE behavior, showing almost non-emission in good solution but enhanced emission in its solid state. Further modification of 2 by attachment of a tert-butyl group on the ligand obtains complex 3, an amorphous material, which only displays AIE activity. More importantly, with the merits of reversible PCL and AIE properties of 2, the rare multi-channel color change and temperature-dependent emission behavior of the iridium(III) complex have been observed. Furthermore, the emissive nanoaggregates of 2 can be efficiently quenched by picric acid, making it a highly sensitive chemosensor for explosives, which is demonstrated in iridium(III)-based luminescent materials for the first time.
Co-reporter:Guo-Gang Shan, Hai-Bin Li, Dong-Xia Zhu, Zhong-Min Su and Yi Liao
Journal of Materials Chemistry A 2012 - vol. 22(Issue 25) pp:NaN12744-12744
Publication Date(Web):2012/04/13
DOI:10.1039/C2JM30480E
To make Ir(III)-based complexes potentially multifunctional materials, two new cationic Ir(III) complexes with a 2-(5-phenyl-2-phenyl-2H-1,2,4-triazol-3-yl)pyridine (Phtz) ancillary ligand were designed and synthesized. By introducing the pendant phenyl ring into the ancillary ligand, the two complexes possess desired intramolecular π–π stacking between the pendant phenyl ring of the Phtz ligand and one of the phenyl rings of the cyclometalated ligand, which renders the complexes more stable. Density functional theory calculation indicates that the intramolecular π–π interactions in both complexes can reduce the degradation reaction in metal-centered (3MC) states to some extent, which further implies their stability. With these results in combination with their reversible oxidation and reduction processes as well as excellent photophysical properties, the stable light-emitting cells (LECs) would be expected. Furthermore, the two synthesized complexes exhibit reversible piezochromism. Their emission color can be smartly switched by grinding and heating, which is visible to the naked eye. In light of our experimental results, the present piezochromic behavior is due to interconversion between crystalline and amorphous states.
Co-reporter:Xueqing Zhang, Hong Ren, Tingting Wang, Lingyu Zhang, Lu Li, Chungang Wang and Zhongmin Su
Journal of Materials Chemistry A 2012 - vol. 22(Issue 26) pp:
Publication Date(Web):
DOI:10.1039/C2JM31141K
Co-reporter:Yun Geng, Hai-Bin Li, Shui-Xing Wu and Zhong-Min Su
Journal of Materials Chemistry A 2012 - vol. 22(Issue 39) pp:NaN20851-20851
Publication Date(Web):2012/06/25
DOI:10.1039/C2JM33369D
Perylene diimide (PDI) and its derivatives hold great promise, since they are undeniably considered as an important family of organic n-type semiconductors with both high carrier mobilities and air stabilities comparable to p-type ones, although they traditionally stand out as a class of high-performance dyes and pigments. In this feature article, we summarize the influences of substituents on different positions (imide, ortho, bay) of PDI on their electronic and morphological (packing) properties, which are in close connection with the ability for carrier transport. Then representative molecular packing motifs for PDIs are also classified, with an emphasis on the intricate interplay of intermolecular interactions, packing motifs and electron transport properties of perylene imide related carrier transport materials from a theoretical point of view, towards paving the way for boosting and improving the electron transport mobilities and air stabilities of PDIs-based materials.
Co-reporter:Dong-Ying Du, Jun-Sheng Qin, Chun-Gang Wang, Xian-Chun Liu, Shun-Li Li, Zhong-Min Su, Xin-Long Wang, Ya-Qian Lan and En-Bo Wang
Journal of Materials Chemistry A 2012 - vol. 22(Issue 39) pp:NaN21044-21044
Publication Date(Web):2012/08/17
DOI:10.1039/C2JM33959E
A 3D eight-connected redox-active polyoxometalate (POM)-based crystalline material, IFMC-101, has been synthesized based on reduced {P4Mo6O31H6}-based tetrameric clusters. IFMC-101 was used as a reducing agent and stabilizer to prepare Au and Pt nanoparticles (NPs) by auto-redox reactions without extra reduction assistance. For the first time, we have demonstrated a new strategy to prepare noble metal NP-loaded POM-based crystalline catalysts. These crystalline catalysts were employed toward the reduction of 4-nitrophenol by NaBH4 and the efficiency of the Au NP-loaded material, Au@IFMC-101, was nearly 9 times higher than that of the POM-based crystalline material, IFMC-101. This result reveals that Au@IFMC-101 exhibits enhanced activity owing to the synergistic catalysis of noble metal NPs and crystalline POM components.
Co-reporter:Guang-Sheng Yang, Mei-Na Li, Shun-Li Li, Ya-Qian Lan, Wen-Wen He, Xin-Long Wang, Jun-Sheng Qin and Zhong-Min Su
Journal of Materials Chemistry A 2012 - vol. 22(Issue 34) pp:NaN17953-17953
Publication Date(Web):2012/06/13
DOI:10.1039/C2JM32990E
Three metal–organic frameworks (MOFs) comprising micropore, mesocage and nanotube structures have been prepared based on the same ligands and binuclear zinc secondary building units. We have successfully achieved flexible modulation of pore size from micropore to mesopore in a binary system by adjusting the reactant ratio. With the merit of a nanoscale channel, IFMC-8 can be applied as the host material for encapsulating Alq3 chromophores to exhibit tunable luminescence. The fluorescence emission of composite material Alq3@IFMC-8 has changed from green to blue. More importantly, the inclusion of IFMC-8 effectively prolonged the excited-state lifetime of Alq3 in ethanol. To the best of our knowledge, it is the first time that a MOF was studied as the host material for modulating the fluorescence properties of Alq3 chromophores.
Co-reporter:Tingting Wang, Fang Chai, Qin Fu, Lingyu Zhang, Haiyan Liu, Lu Li, Yi Liao, Zhongmin Su, Chungang Wang, Beiye Duan and Dongxue Ren
Journal of Materials Chemistry A 2011 - vol. 21(Issue 14) pp:NaN5306-5306
Publication Date(Web):2011/02/18
DOI:10.1039/C0JM04115G
In this article, we report the fabrication of monodisperse hollow mesoporous silica (HMS) nanocages with uniform size possessing a hollow cubic core and mesoporous shell with penetrating pore channels based on a template-coating-etching process. It is worthwhile noting the obtained HMS nanocages with cubic void space and highly porous shell endow the structures with much higher storage capacity and sustained release of anticancer drugs. More importantly, the therapeutic efficacy of doxorubicin-loaded HMS nanocages was evaluated in vitro and in vivo for liver cancer therapy. The results show that the doxorubicin-loaded HMS nanocages have good cell uptake and can induce efficient cell deathinvitro. Taken together, this study demonstrates that the biocompatible HMS nanocages can effectively deliver drugs to the tumors and suppress tumor growth compared to free doxorubicinin vivo. Additionally, the synthetic strategy has also extended to fabrication of the uniform and monodisperse HMS nanocapsules with spherical shape.
Co-reporter:Dong-Ying Du, Jun-Sheng Qin, Cheng-Xin Sun, Xin-Long Wang, Shu-Ran Zhang, Ping Shen, Shun-Li Li, Zhong-Min Su and Ya-Qian Lan
Journal of Materials Chemistry A 2012 - vol. 22(Issue 37) pp:NaN19678-19678
Publication Date(Web):2012/07/31
DOI:10.1039/C2JM34661C
Five heterometallic luminescent crystalline materials with the metalloligand, [Zn(HL)EuxTby(H2O)2][ZnBr4]·H2O (x = 1, y = 0, IFMC-21; x = 0.75, y = 0.25, IFMC-22; x = 0.5, y = 0.5, IFMC-23; x = 0.25, y = 0.75, IFMC-24; x = 0, y = 1, IFMC-25; H4L = 4,4′,4′′,4′′′-(2,2′,2′′,2′′′-(ethane-1,2-diylbis(azanetriyl))tetrakis(methylene)-tetrakis-(1H-benzo[d]imidazole-2,1-diyl))tetrakis(methylene)-tetrabenzoic acid; IFMC = Institute of Functional Material Chemistry), were prepared by the combination of hydrothermal and ionothermal methods for the first time. IFMC-21–25 can be obtained by introducing the desired Eu(III) and Tb(III) in the initial experiments. In these crystalline materials, the metalloligand Zn(HL) was connected by bi-lanthanide cores leading to a 2D sheet-structure and [ZnBr4]2− ions were distributed in the interspaces of the sheet. The luminescent properties of IFMC-21 to 25 were investigated and the results reveal that they exhibit characteristic Eu(III) and Tb(III) ion emissions, and the intensities of red and green arising from Eu(III) and Tb(III) emissions are shifted correspondingly by tuning the ratios of Eu(III):Tb(III).
Co-reporter:Yun Geng, Jianping Wang, Shuixing Wu, Haibin Li, Fei Yu, Guochun Yang, Hongze Gao and Zhongmin Su
Journal of Materials Chemistry A 2011 - vol. 21(Issue 1) pp:NaN143-143
Publication Date(Web):2010/10/19
DOI:10.1039/C0JM02119A
Seven perylene bisimide derivatives with different molecular packings and intermolecular interactions were investigated in detail within Marcus-Levich-Jortner formalism at the level of density functional theory (DFT). In theory, we further proved the report that different halogen substitutions in the core position of perylene bisimide lead to different molecular packings in their single crystals and thus obviously different electron transport properties. Here, insight into the geometries, the character of the frontier molecular orbitals, the decompositions of reorganization energies and transfer integrals in different directions was provided to shed light on the relationship between structures and properties. The molecular dynamics (MD) simulations and band structures calculations were also employed to give a multiscale understanding of their transport properties. The results show that there are small discrepancies of the intramolecular electron reorganization energies among these compounds and the transfer integrals determine their electron transport properties. Compounds 1a, 3a and 3b, with typical “brick” packing, π-stacked face-to-face packing and “herringbone” packing, respectively, have larger electron mobilities among these systems and possess different transport dimensionalities. Moreover, we also find there is close relationship between the intermolecular interaction energy and the transfer integral.
Co-reporter:Yun Geng, Shui-Xing Wu, Hai-Bin Li, Xiao-Dan Tang, Yong Wu, Zhong-Min Su and Yi Liao
Journal of Materials Chemistry A 2011 - vol. 21(Issue 39) pp:NaN15566-15566
Publication Date(Web):2011/08/26
DOI:10.1039/C1JM12483H
Three naphthalene tetracarboxylic diimide derivatives 1–3 with high electron mobilities and long-term ambient stabilities were investigated employing Marcus–Levich–Jortner formalism at the density functional theory (DFT) level. The complicated relationships among molecular packings, intermolecular interactions, and transport properties for these compounds were focused on and analyzed through investigating the sensitivities of transfer integrals to intermolecular relative orientations, the optimizations of the major transport pathways and the calculations of intermolecular interaction energies by using dispersion-corrected DFT. The results show that the transfer integrals are sensitive to the subtle changes of relative orientations of molecules, especially for core-chlorinated compounds, and there is an interplay between intermolecular interaction and molecular packing. It is found that the transfer integrals associated with the molecular packing motifs of these systems determine their electron mobilities. Interestingly, further discussions on band structures, the anisotropies and temperature dependences of mobilities, and the comparisons of mobilities before and after optimization indicate that the intermolecular packing motifs in the film state may be different from those in the crystalline state for 2. Finally, we hope that our conjecture would facilitate the future design and preparation of high-performance charge-transport materials.
Co-reporter:Guochun Yang, Yanling Si and Zhongmin Su
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 42) pp:NaN8425-8425
Publication Date(Web):2012/09/10
DOI:10.1039/C2OB26374B
We have investigated the chiroptical, linear, and second-order nonlinear optical (NLO) properties of seven binaphthol derivatives and elucidated structure–property relationships from the micromechanism for the first time. The excitation energies, oscillator strengths, and rotational strengths of the 150 lowest energy electron excitations for the most stable conformers have been calculated at TDB3LYP/cc-pVDZ level of theory. The experimental UV–vis absorption energies were reproduced well by our calculations. The simulated circular dichroism (CD) spectra and calculated optical rotation (OR) values are in reasonable agreement with experimental ones. These results demonstrate that TDDFT calculations can not only describe the electron transition property but also can be used to assign the absolute configurations (ACs) of binaphthol derivatives with high confidence. Whereas OR values are more sensitive to the molecular structures than CD spectra. The electron transition property and chiroptical origin have been assigned and analyzed. These derivatives possess remarkably large molecular first hyperpolarizabilities, especially compound 7 which has a value of 241.65 × 10−30 esu. This value is about 60 times as large as that of highly π-delocalized phenyliminomethyl ferrocene complex. Moreover, compound 6 exhibits pronounced different second-order NLO response values from neutral state to the two cationic states (62+2+2+ and 64+4+4+), which indicates that this compound could act as a potential NLO switch material. The cooperativity of intramolecular charge transfer of the studied compounds was also discussed.
Co-reporter:Rong-Lin Zhong, Hong-Liang Xu and Zhong-Min Su
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 20) pp:NaN13959-13959
Publication Date(Web):2016/04/20
DOI:10.1039/C6CP00647G
Carbon–boron–nitride heteronanotubes (BNCNT) have attracted a lot of attention because of their adjustable properties and potential applications in many fields. In this work, a series of CA, PA and HA armchair BNCNT models were designed to explore their nonlinear optical (NLO) properties and provide physical insight into the structure–property relationships; CA, PA and HA represent the models that are obtained by doping the carbon segment into pristine boron nitride nanotube (BNNT) fragments circularly around the tube axis, parallel to the tube axis and helically to the tube axis, respectively. Results show that the first hyperpolarizability (β0) of an armchair BNCNT model is dramatically dependent on the connecting patterns of carbon with the boron nitride fragment. Significantly, the β0 value of PA-6 is 2.00 × 104 au, which is almost two orders of magnitude larger than those (6.07 × 102 and 1.55 × 102 au) of HA-6 and CA-6. In addition, the β0 values of PA and CA models increase with the increase in carbon proportion, whereas those of HA models show a different tendency. Further investigations on transition properties show that the curved charge transfer from N-connecting carbon atoms to B-connecting carbon atoms of PA models is essentially the origin of the big difference among these models. This new knowledge about armchair BNCNTs may provide important information for the design and preparation of advanced NLO nano-materials.
Co-reporter:Yong Wu, Guo-Gang Shan, Hai-Bin Li, Shui-Xing Wu, Xin-Yao Ren, Yun Geng and Zhong-Min Su
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 4) pp:NaN2446-2446
Publication Date(Web):2014/12/01
DOI:10.1039/C4CP04919E
The geometries, electronic structures, photophysical properties and spin–orbit coupling (SOC) effects in the radiative process for the recently synthesized complexes (Bppy)Pt(acac) (1) and (BNppy)Pt(acac) (2) as well as the designed complexes 3–6 were investigated by DFT and TD-DFT calculations, to reveal the influences of the functional ligands on charge injection ability and phosphorescence efficiency of emitters. It is found that compared with electron acceptor complex 1, complexes 2–6 have lower ionization potentials and comparable high electronic affinities, which are suited for bipolar luminescent materials. The results also demonstrated that Bppy complexes 1, 5 and 6 have more 3MLCT compositions in T1 emitting states compared with BNppy complexes 2–4, which results in strong SOC and fast kr. Thus, the phosphorescence efficiency of 1 is higher than that of 2. In addition, 5 and 6 have the balanced charge transport and better hole injection ability when the hole-transporting ligand is incorporated to 1. Therefore, 5 and 6 can server as promising candidates for efficient multifunctional phosphorescent OLED emitters owing to their ambipolar characters, balanced charge carrier injection/transport features and high phosphorescence quantum efficiency.
Co-reporter:Caixia Wu, Tengying Ma, Likai Yan, Ting Zhang and Zhongmin Su
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 17) pp:NaN11526-11526
Publication Date(Web):2015/03/30
DOI:10.1039/C4CP06042C
Quantum chemical calculations were performed to explore the structural and electronic properties of the two polyoxoaurates, [AuIII4AsV4O20]8− (Au4As4) and [AuIII4SeIV4O16]4− (Au4Se4), known to date, and a number of hypothetical polyoxoaurate derivatives comprising heteroatoms different from arsenic and selenium (namely, Si, Ge and P). In addition, the interactions of [AuIII4X4Om]n− (X = As, Se) with alkali-metal cations (Li+, Na+, K+ and Rb+) are also analysed. The studies suggest that the geometry structure, electronic properties and nucleophilicity of oxygen atoms of these polyoxoaurates are tuned by the size or electronegativity of the heteroatoms (Si, Ge, P, As and Se). Then, the geometry of [AuIII4X4Om]n− (X = As and Se) coordinating with alkali cations from Li+ to Rb+ and the complexation energy between [AuIII4X4Om]n− and alkali cations were compared. The results show that the stability and electronic structure of heteropolyoxoaurates depend on the entrapped cations. On the basis of the complexation energy, it can be concluded that the ion-pairing effect in arsenate-capped oxoaurate is stronger than that in selenite-capped oxoaurate. These heteropolyoxoaurates are expected to play a role in aqueous behaviour, self-assembly characteristics of polyoxoaurates, ion recognition, selectivity studies and may exhibit potential guest-switchable redox properties.
Co-reporter:Ting Zhang, Wei Guan, Likai Yan, Tengying Ma, Jing Wang and Zhongmin Su
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 7) pp:NaN5465-5465
Publication Date(Web):2014/12/22
DOI:10.1039/C4CP04890C
Polyoxometalate (POM)-based organic–inorganic hybrid systems II1–II7 are designed as p-type dyes containing double D–A1–π–A2 chains. The A1 spacers are thiophene, 1,2,3-triazole, 1,3,4-oxadlazole, thienothiadiazole units or their combinations and the A2 spacer is hexamolybdate. The electronic structures, absorption spectra, and electronic transition characteristics of systems were systematically studied on the basis of density functional theory (DFT) and time-dependent DFT (TDDFT). The highest occupied molecular orbital (HOMO) levels of systems II1–II7 were below the valence bond (VB) of NiO and the lowest unoccupied molecular orbital (LUMO) levels of studied systems were higher than the I2/I3− redox level, which benefit hole injection and dye regeneration. The HOMOs of systems II1–II4 were predominantly delocalized over the organic groups and MoN, which are more helpful to hole injection than systems II5–II7. Introduction of thienothiadiazole units is an effective way to improve the light absorption ability of dyes, and inserting thiophene and 1,2,3-triazole as A1 spacers can increase the efficiency of dye in dye-sensitized solar cells (DSSC).
Co-reporter:Bo Zhu, Zhong-Ling Lang, Na-Na Ma, Li-Kai Yan and Zhong-Min Su
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 33) pp:NaN18022-18022
Publication Date(Web):2014/07/14
DOI:10.1039/C4CP01389A
Density functional theory (DFT) calculations and natural bond orbital (NBO) analysis were carried out to investigate the electronic structures and bonding features between the ruthenium(II) atom and the SO2 molecule in two ruthenium–sulfur dioxide (SO2) adducts, trans-Ru(NH3)4(SO2)Cl+ and [{SiW11O39}RuII(SO2)]6−. In addition, the bonding interactions between SO2 and the metal-ruthenium fragment were determined by binding energy (ΔEabs) calculation and electronic structures. The results indicate that the η1-S-planar model in both trans-Ru(NH3)4(SO2)Cl+ and [{SiW11O39}RuII(SO2)]6− are more favorable. NBO analysis of the bonding interaction between ruthenium and sulfur centers in the [{SiW11O39}RuII(SO2)]6− complex shows that it possesses a σ and a π bond. It predicts that the polyoxometalate [SiW11O39Ru]6− can serve as a potential adsorbent for the SO2 molecule because of the strong Ru–S bond relative to Ru(NH3)4Cl+.
Co-reporter:Ji Zhang;Jian-Zhao Zhang;Hai-Bin Li;Yong Wu;Yun Geng
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 45) pp:
Publication Date(Web):2014/10/30
DOI:10.1039/C4CP03355H
Ten porphyrin sensitizers with different electron-withdrawing groups derived from the best sensitizer SM315 were investigated by means of the density functional theory (DFT) and time-dependent DFT calculations. To this end, major factors affecting the performance of the cell, including light harvesting, electron injection, dye regeneration, and conduction band energy shift are taken into consideration. Especially, the calculated distance (r) from the electron recapture center to the semiconductor surface is used to probe the charge recombination process. In addition, considering the complexity of the porphyrin sensitizers' absorption, the maximum short circuit current density (Jmaxsc) is determined for investigating the light harvesting ability quantitatively. We find that when compared to SM315 with 2,1,3-benzothiadiazole, 1 with naphtho[1,2-c:5,6-c]bis[1,2,5]thiadiazole shows better performance due to both larger Jmaxsc and r, and 7 with diketopyrrolopyrrole could also be a promising candidate due to the much larger Jmaxsc and comparable r.
Co-reporter:Shuang-Bao Li, Yu-Ai Duan, Yun Geng, Hai-Bin Li, Jian-Zhao Zhang, Hong-Liang Xu, Min Zhang and Zhong-Min Su
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 47) pp:NaN25808-25808
Publication Date(Web):2014/09/03
DOI:10.1039/C4CP03022B
In the current work, a series of bithiopheneimide (BTI)-based D–A copolymers were investigated based on the reported PDTSBTI (1) to screen excellent molecules toward organic photovoltaic (OPV) donor materials. It is found that the PCE based on the proposed derivative 4, where the silicon atom is replaced with vinyl and cyano groups on the DTS unit, shows a 70 percent improvement by Scharber diagrams compared with its prototype 1. Then, the charge transfer dynamics of 1/PC71BM and 4/PC71BM were investigated, including the intermolecular charge transfer (inter-CT) and recombination (inter-CR) rates. The theoretical data demonstrate that the ratio kinter-CT/kinter-CR of 4/PC71BM heterojunction is about 1 × 105 times higher than that of 1/PC71BM. These results clearly reveal that the designed donor molecule 4 will be a promising candidate for high-performance OPV device. We expect that this work from electron processing at the D/A interface may provide a theoretical guideline for further optimization and design of organic copolymer donor materials.
Co-reporter:Shizheng Wen, Wei Guan, Yuhe Kan, Guochun Yang, Nana Ma, Likai Yan, Zhongmin Su and Guanhua Chen
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 23) pp:NaN9185-9185
Publication Date(Web):2013/04/11
DOI:10.1039/C3CP51380G
Nano-hybrid materials based on a combination of polyoxometalate (POM) clusters and single-walled carbon nanotubes (SWNT) exhibit a great interesting application in molecular cluster batteries. The interactions between POM and SWNT and their detailed electronic properties have been investigated by employing first-principles calculations. Various models were constructed to study the geometries, interactions (binding sites and energies), and charge transfer behavior. Analysis of charge distributions reveals two different charge transfer characteristic depending on the type of POM interaction with SWNT. The simulation provides insight into the optimal structures in lieu of interfacial stability. Finally, the implications of these results for understanding the properties of molecular cluster batteries are discussed.
Co-reporter:Dong-Lai Wang, Hong-Liang Xu, Zhong-Min Su and Guang Xin
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 43) pp:NaN15105-15105
Publication Date(Web):2012/09/17
DOI:10.1039/C2CP42669B
Very recently, two novel Sc3NC-based cluster fullerenes Sc3NC@C80 (Wang et. al. J. Am. Chem. Soc. 2010, 132, 16362) and Sc3NC@C78 (Wu et. al. J. Phys. Chem. C 2011, 115, 23755) were prepared and characterized, respectively. Inspired by these findings, the possibility of encapsulating Sc3NC cluster in the C84 fullerene is performed using density functional theory (DFT). Firstly, the isolated pentagon rule (IPR) D2d (23) C84 fullerene is employed to encase the Sc3NC cluster: four possible endohedral metallofullerene isomers a–d are designed. The large binding energies (ranging from 163.7 to 210.0 kcal mol−1) indicate that the planar quinary cluster Sc3NC can be stably encapsulated in the C84 (isomer 23) cage. Further, we consider the incorporation of Sc3NC into the non-IPR Cs (51365) C84 cage leading to isomer e and show the high stability of isomer e, which has a larger binding energy, larger HOMO-LUMO gap, higher adiabatic (vertical) ionization potential, and lower adiabatic (vertical) electron affinity than the former four Sc3NC@C84 (isomer 23). Significantly, the predicted binding energy (294.2 kcal mol−1) of isomer e is even larger than that (289.2 and 277.7 kcal mol−1, respectively) of the synthesized Sc3NC@C80 and Sc3NC@C78, suggesting a considerable possibility for experimental realization. The 13C NMR chemical shifts and Raman spectra of this a new endofullerene have been explored to assist future experimental characterization.
Co-reporter:Min Zhang, ZhongMin Su and GuanHua Chen
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 14) pp:NaN4702-4702
Publication Date(Web):2012/02/02
DOI:10.1039/C2CP23164F
The electron excitations of Single-Walled Silicon Nanotubes (SWSiNTs), with sp2 and sp3 hybridization, were studied using the localized-density-matrix (LDM) method with INDO/S parameters. Strong anisotropic characteristics of the dynamic polarizabilities were found for all the nanotubes. The transitional intensity along the tubular axis is much larger than that perpendicular to the axis for all the nanotubes. The optical gaps of sp3-hybridized infinitely-long pentagonal SWSiNTs are near 3.0 eV and 4.7 eV owing to σ–σ* transitions along the direction of the tubular axis. The optical gaps of sp2-hybridized infinitely-long armchair SWSiNTs along the tube axis direction are about 0.7 eV and 2.4 eV for Si(3,3) SWSiNTs and 0.7 eV and 2.7 eV for Si(4,4) SWSiNTs. The former peak at 0.7 eV originated from π–π* electron transitions and the latter peak at 2.4 eV or 2.7 eV originated from σ–σ* electron transitions. Meanwhile, the intensities of π–π* electron transitions are stronger than those of σ–σ* electron transitions in SWSiNTs. The low sp2 transition energy derived from the weak overlap of unpaired pz orbitals of silicon atoms. Moreover, the electronic excitations of zigzag SWSiNTs are similar to those of armchair structures. This indicates that sp2-hybridized silicon nanotubes possess much greater potential for application in optical fields.
Co-reporter:Nana Ma, Likai Yan, Wei Guan, Yongqing Qiu and Zhongmin Su
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 16) pp:NaN5612-5612
Publication Date(Web):2012/02/27
DOI:10.1039/C2CP00054G
We report a theoretical study based on density functional theory (DFT) on the geometric and electronic structure, linear optical and second-order nonlinear optical properties of a series of new inorganic–organic hybrid hexamolybdate–organoimido–(car)boranes. By the incorporation of borane/carborane at the end of the phenyl ring of the organoimido segment, the studied systems show excellent nonlinear optical (NLO) response than the organoimido-substituted hexamolybdate. The computed static first hyperpolarizability βvec value of [Mo6O18(NC8H8)(B12H11)]4− (II) is largest, −167.2 × 10−30 esu, and a higher βvec value of [Mo6O18(NC8H8)(C2B10H11)]2− (III-2p) is 58.6 × 10−30 esu. Moreover, the time-dependent (TD)DFT calculation illustrates that the maximum absorption, which is helpful for the large NLO responses, is mainly assigned to the charge transfer (CT) from (car)borane and organoimido segment to the hexamolybdate cluster. The density of density (DOS) calculations further illustrate the excitation from valence orbitals of boron atoms to that of Mo and O atoms in hexamolybdate can be responsible for larger NLO responses. The linear and nonlinear optical properties of species III both vary with the position of the vertex on the carborane. Furthermore, the order of the βvec values is consistent with the bathochromic shift of the maximum absorption for our studied systems, and the studied systems show a wider transparency range extending into the entire visible and infrared (IR) region.
Co-reporter:Shabbir Muhammad, Hongliang Xu, Muhammad Ramzan Saeed Ashraf Janjua, Zhongmin Su and Muhammad Nadeem
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 18) pp:NaN4799-4799
Publication Date(Web):2010/03/24
DOI:10.1039/B924241D
A novel sequence for reversible second-order nonlinear optical (NLO) molecular switching with protonation/deprotonation has been achieved and tuned as well. The NLO switching with first hyperpolarizabilities (β0) as low as 14 × 10−30 esu (Off-phase) and as large as 1189 × 10−30 esu (On-phase) have been computed by using density functional theory (DFT). Remarkably large differences between the β0 values of benzimidazole containing chromophores and their deprotonated anions have shown their significant potential for a new type of NLO molecular switching, as (1-(4-methoxyphenyl)-2-(2-thienyl)pyrrolyl)-1,3-benzimidazole anion (1−) has a β0 value computed to be 61 × 10−30 esu, which is 4 times larger than its neutral molecule 1. This β0 value has been tuned up to 2028 × 10−30 esu by effective substitutions in the derivatives of 1− (1a−, 1b−, 1c−, and 1d−). Interestingly, the substituted compounds have illustrated robustly large off–on NLO switching with a difference in β0 values of 7, 63, 85 and 75 times larger than their neutral counterparts, respectively. TD-DFT calculations along with natural bond orbital (NBO), frontier molecular orbitals (FMOs) and molecular electrostatic potential (MEP) analyses show that the abstraction of an imido proton brings about a change in push–pull configurations resulting in a red shift for both absorption and emission spectra which subsequently leads to a high performance second-order NLO molecular switching. A similar trend of NLO switching in F− compounds of these chromophores has also been observed with significantly large β0 values having analogous electro-optical properties like deprotonated anions. Furthermore, gas-phase acidity (GPA) calculations for the neutral molecule 1 and its derivatives (1a, 1b, 1c, and 1d) have also revealed that these are rationally potent nitrogen acids and can easily be dissociated to produce stable deprotonated anions.
Co-reporter:Ting Gao, Li-Li Shi, Hai-Bin Li, Shan-Shan Zhao, Hui Li, Shi-Ling Sun, Zhong-Min Su and Ying-Hua Lu
Physical Chemistry Chemical Physics 2009 - vol. 11(Issue 25) pp:NaN5129-5129
Publication Date(Web):2009/03/23
DOI:10.1039/B812492B
The combination of genetic algorithm and back-propagation neural network correction approaches (GABP) has successfully improved the calculation accuracy of absorption energies. In this paper, the absorption energies of 160 organic molecules are corrected to test this method. Firstly, the GABP1 is introduced to determine the quantitative relationship between the experimental results and calculations obtained by using quantum chemical methods. After GABP1 correction, the root-mean-square (RMS) deviations of the calculated absorption energies reduce from 0.32, 0.95 and 0.46 eV to 0.14, 0.19 and 0.18 eV for B3LYP/6-31G(d), B3LYP/STO-3G and ZINDO methods, respectively. The corrected results of B3LYP/6-31G(d)-GABP1 are in good agreement with experimental results. Then, the GABP2 is introduced to determine the quantitative relationship between the results of B3LYP/6-31G(d)-GABP1 method and calculations of the low accuracy methods (B3LYP/STO-3G and ZINDO). After GABP2 correction, the RMS deviations of the calculated absorption energies reduce to 0.20 and 0.19 eV for B3LYP/STO-3G and ZINDO methods, respectively. The results show that the RMS deviations after GABP1 and GABP2 correction are similar for B3LYP/STO-3G and ZINDO methods. Thus, the B3LYP/6-31G(d)-GABP1 is a better method to predict absorption energies and can be used as the approximation of experimental results where the experimental results are unknown or uncertain by experimental method. This method may be used for predicting absorption energies of larger organic molecules that are unavailable by experimental methods and by high-accuracy theoretical methods with larger basis sets. The performance of this method was demonstrated by application to the absorption energy of the aldehyde carbazole precursor.
Co-reporter:Yong Li, Hong-Liang Xu, Heng-Qing Wu, Rong-Lin Zhong, Shi-Ling Sun and Zhong-Min Su
Dalton Transactions 2014 - vol. 43(Issue 6) pp:NaN2660-2660
Publication Date(Web):2013/11/08
DOI:10.1039/C3DT52923A
Recently, two isomeric thiophene-fused benzocarborane derivatives Tb1 and Tb2 with different locations of sulfur atoms, labeled as 1, 4 and 6, 9 of the thiophene were synthesized by Morisaki (Chem.–Eur. J., 2012, 18, 11251–11257) and Barrere (Macromolecules, 2009, 42, 2981–2987), respectively. In the present work, natural bond orbital (NBO) analysis shows that after doping one lithium atom into the isomeric structures Tb1 and Tb2, the electrons transfer to different regions in Tb1 and Tb2. For Tb1-Li, the transferred electrons mainly locate at S1, C2, C3, and S4, but for Tb2-Li, the transferred electrons mainly locate at C2, C3, C7, and C8. Significantly, the charge distribution is a crucial factor influencing the static first hyperpolarizabilities for Tb1-Li and Tb2-Li. Furthermore, the βtot value of Tb1-Li is 6222 au, which is about 160 times larger than that of Tb1 (39 au). However, the βtot value of Tb2-Li (498 au) is only about 5 times larger than that of the corresponding Tb2 (91 au). It is our expectation that this work could provide useful information for the development of nonlinear optical materials based on carboranes.
Co-reporter:Na-Na Ma, Shu-Jun Li, Li-Kai Yan, Yong-Qing Qiu and Zhong-Min Su
Dalton Transactions 2014 - vol. 43(Issue 13) pp:NaN5075-5075
Publication Date(Web):2013/12/13
DOI:10.1039/C3DT53298D
The rotary motion based on metallacarboranes around a molecular axis can be controlled by simple electron transfer processes, which provides a basis for the structure–property relationship for the nonlinear optical (NLO) switching. However, this phenomenon has not been previously reported in the development of NLO properties of metallacarboranes. In this work, the metallacarboranes [NiIII/IV(C2B9H11)2]−/0 and their C-,B-functionalized derivatives are studied by the density functional theory (DFT) method. By calculating relative energies, we obtained the stable states before and after rotation controlled by simple electron transfer. Then, the static and frequency-dependent second-order NLO properties were calculated by several DFT functionals. According to the TDDFT results, the large NLO responses of the studied compounds are mainly caused by substituent group-to-carborane cage charge transfer (L′LCT) and substituent group-to-metal charge transfer (L′MCT) processes. The order of first hyperpolarizabilities (β values) illustrates that the NLO response can be enhanced by introducing a strong electron-donating group. Significantly, the geometric interconversions resulting from the redox reaction of 1C/1T–6C/6T allow the NLO responses to be switched “ON” or “OFF”. The B(9,9′)-methoxyphenyl-functionalized derivative of nickelacarborane, having low energetic cost and large different NLO responses between two states (from 0 to 20998 a.u.), can be an excellent switchable NLO material.
Co-reporter:Li-Jie Wang, Rong-Lin Zhong, Shi-Ling Sun, Hong-Liang Xu, Xiu-Mei Pan and Zhong-Min Su
Dalton Transactions 2014 - vol. 43(Issue 25) pp:NaN9660-9660
Publication Date(Web):2014/03/10
DOI:10.1039/C3DT53329H
Recently, a new sulfide cluster fullerene, Sc2S@Cs (10528)-C72 containing two pairs of fused pentagons has been isolated and characterized (Chen et al., J. Am. Chem. Soc., 2012, 134, 7851). Inspired by this investigation, we propose a question: what properties will be influenced by the interaction between the encapsulated V-shaped polar molecule and C72? To answer this question, four encapsulated metallic fullerenes (EMFs) M2N@C72 (M = Sc or Y, N = S or O) along with pristine Cs-C72 (10528) were investigated by quantum chemistry methods. The results show that the Egap (3.01–3.14 eV) of M2N@C72 are significantly greater than that of pristine Cs-C72 (10528) (2.34 eV). This indicates that the stabilities of these EMFs increase by encapsulating the V-shaped polar molecule into the fullerene. Furthermore, the natural bond orbital (NBO) charge analysis indicates electron transfer from M2N to C72 cage, which plays a crucial role in enhancing first hyperpolarizability (βtot). The βtot follows the order of 1174 au (Y2O@C72) ≈ 1179 au (Sc2O@C72) > 886 au (Y2S@C72) ≈ 864 au (Sc2S@C72) > 355 au (C72). This indicates that the βtot of M2N@C72 is more remarkable than that of pristine Cs-C72 (10528) due to the induction effect of the encapsulated molecule. Compared with sulfide cluster fullerenes (Y2S@C72 and Sc2S@C72), oxide cluster fullerenes (Sc2O@C72 and Y2O@C72) show much larger βtot due to the small ionic radius and the large electronegativity of oxygen. In contrast, the metal element (scandium and yttrium) has a slight influence on the βtot. Thus, oxide cluster fullerenes are candidates to become promising nonlinear optical materials with higher performance.
Co-reporter:Sa Chen, Shi-Ling Sun, Heng-Qing Wu, Hong-Liang Xu, Liang Zhao and Zhong-Min Su
Dalton Transactions 2014 - vol. 43(Issue 33) pp:NaN12662-12662
Publication Date(Web):2014/06/02
DOI:10.1039/C4DT01240B
Recently, the well-known phenalenyl radical π-dimer with its fascinating 2-electron/12-center (2e/12c) bond has attracted our attention. In this work, we designed two molecules, Li3O⋯C13H9 (1a) and BeF3⋯C13H9 (1b). Interestingly, owing to the inductive effect of superatoms, an electron is transferred from Li3O to phenalenyl in 1a, while an electron is transferred from phenalenyl to BeF3 in 1b. Further, we employed 1a and 1b as building blocks to assemble two novel molecules with 2e/12c bonds: Li3O⋯(C13H9)2⋯BeF3 (2a) and Li3O⋯(C13H9)2⋯BeF3 (2b). Remarkably, 2a and 2b with novel 2e/12c bonds exhibit a dramatic interlayer charge-transfer character, which results in a significant difference of dipole moments (Δμ: 2.6804 for 2a and 3.8019 Debye for 2b) between the ground state and the crucial excited state. As a result, the static first hyperpolarizabilities (β0: 5154 for 2a and 12500 au for 2b) are considerably larger than the values of 347 for 1a and 328 au for 1b. It is our expectation that the results of the present work might provide beneficial information for further theoretical and experimental studies on the fascinating properties of molecules with interlayer charge-transfer character.
Co-reporter:Hai-Ning Wang, Xing Meng, Chao Qin, Xin-Long Wang, Guang-Sheng Yang and Zhong-Min Su
Dalton Transactions 2012 - vol. 41(Issue 3) pp:NaN1053-1053
Publication Date(Web):2011/11/22
DOI:10.1039/C1DT11304F
Four new compounds, [Cd(5-aip)(bpy)]·1.5DMA (1), [Cu(5-aip)(bpy)]·1.3DMA (2), [Co(5-aip)(bpy)]·1.6DMA (3), and [Cd(5-aip)(bpy)0.5(H2O)]·1.3DMA (4), based on 5-aminoisophthalic acid and 4,4′-bipyridine, have been synthesized by the solvothermal method and structurally determined using single crystal X-ray diffraction. Compounds 1–3 are structurally similar and show non-interpenetrating three-dimensional (3D) pillar-layer frameworks, while compound 4 displays a two-dimensional (2D) (3,4)-connected parallel non-interpenetrating architecture. In all these compounds, 1D rectangular channels are observed and the ligand 5-aminoisophthalic acid exhibits three kinds of coordination modes. Furthermore, 1 displays a single-crystal-to-single-crystal transformation when immersed in a methanol solution. More significantly, 1 can absorb and deliver I2 molecules by means of its channels, and could induce a reversible luminescent transformation from quenching to the initial state. The luminescent properties of 1 and 4 have also been studied.
Co-reporter:Kun Zhou, Xin-Long Wang, Chao Qin, Hai-Ning Wang, Guang-Sheng Yang, Yan-Qing Jiao, Peng Huang, Kui-Zhan Shao and Zhong-Min Su
Dalton Transactions 2013 - vol. 42(Issue 5) pp:NaN1355-1355
Publication Date(Web):2012/11/15
DOI:10.1039/C2DT32145A
The first macrocycle-based high-nuclearity nanocluster [Ag20(StBu)10(CF3COO)2]8+ in complex 1, with a sandwich-like structure constructed by bilevel inversion Ag5S5 pentagrams and an interlayer Ag10 ring, has been obtained. Structure analysis indicates that Cl− acts as an anion template to direct the formation of 1. In addition, the photoluminescent property of complex 1 is also investigated.
Co-reporter:Hai-Ning Wang, Guang-Sheng Yang, Xin-Long Wang and Zhong-Min Su
Dalton Transactions 2013 - vol. 42(Issue 18) pp:NaN6297-6297
Publication Date(Web):2013/03/07
DOI:10.1039/C3DT32958E
pH-induced different crystalline behaviors based on the same reactants and reaction conditions are illustrated by our present study. Compound 2 has been used for the adsorption and delivery of 5-FU.
Co-reporter:Guo-Gang Shan, Hai-Bin Li, Hai-Zhu Sun, Hong-Tao Cao, Dong-Xia Zhu and Zhong-Min Su
Dalton Transactions 2013 - vol. 42(Issue 31) pp:NaN11065-11065
Publication Date(Web):2013/03/19
DOI:10.1039/C3DT50358E
Herein we designed and synthesized a series of cationic iridium(III) complexes with a phenylbenzoimidazole-based cyclometalated ligand, containing different numbers of carbazole moieties from zero to three (complexes 1–4). The photophysical and electrochemical properties of this series have been systematically investigated. The complexes exhibit strong luminescence in both solution and in neat films, as well as excellent redox reversibility. Introducing carbazole groups into the complexes is found to lead to substantially enhanced photoluminescence quantum efficiency in the neat film, but has little effect on the emitting color and excited-state characteristics as supported by density functional theory (DFT) results. DFT calculations also suggest that functionalized complexes 2–4 reveal better hole-transporting properties than 1. More importantly, all complexes effectively reduce the degradation reaction to some extent in metal-centered (3MC) excited-states, demonstrating their stability. Further studies indicate that restriction of opening of the structures in the 3MC state is caused by the unique molecular conformation of the phenylbenzoimidazole ligand, which is first demonstrated here in cationic iridium(III) complexes without intramolecular π–π stacking. These results presented here would provide valuable information for designing and synthesizing highly efficient and stable cationic iridium(III) complexes suitable for the optical devices.
Co-reporter:Zhong-Ling Lang, Wei Guan, Li-Kai Yan, Shi-Zheng Wen, Zhong-Min Su and Li-Zhu Hao
Dalton Transactions 2012 - vol. 41(Issue 37) pp:NaN11368-11368
Publication Date(Web):2012/07/18
DOI:10.1039/C2DT31166F
The formation mechanism is always a fundamental and confused issue for polyoxometalate chemistry. Two formation mechanisms (M1 and M2) of the Lindqvist anion [W6O19]2− have been adopted to investigate it's self-assembly reaction pathways at a density functional theory (DFT) level. The potential energy surfaces reveal that both the mechanisms are thermodynamically favorable and overall barrierless at room temperature, but M2 is slightly dominant to M1. The formation of the pentanuclear species [W5O16]2− and [W5O15(OH)]− are recognized as the rate-determining steps in the whole assembly polymerization processes. These two steps involve the highest energy barriers with 30.48 kcal mol−1 and 28.90 kcal mol−1, respectively, for M1 and M2. [W4O13]2− and [W4O12(OH)]− are proved to be the most stable building blocks. In addition, DFT results reveal that the formation of [W3O10]2− experiences a lower barrier along the chain channel.
Co-reporter:Bai-Qiao Song, Chao Qin, Yu-Teng Zhang, Xue-Song Wu, Liu Yang, Kui-Zhan Shao and Zhong-Min Su
Dalton Transactions 2015 - vol. 44(Issue 42) pp:NaN18394-18394
Publication Date(Web):2015/09/09
DOI:10.1039/C5DT03218K
Two unprecedented homochiral enantiomers based on two different kinds of rigid ligands, namely [Cd(NDC)L]2·H2O (1R and 1L), have been synthesized under hydrothermal conditions through spontaneous resolution. Their structures were determined by single-crystal X-ray diffraction analysis and further characterized by elemental analysis, IR, and thermogravimetric (TG) analysis. The resulting framework 1, constructed by four kinds of homo-handed helical chains represents the first 3D self-penetrating framework formed by decoration of single (10,3)-a net with helical chains. The single (10,3)-a net in 1 formed by three kinds of different homo-handed helical chains is different from the standard one, which should be ascribed to the usage of V-shaped ligand L. A unique self-penetration motif can be discovered in 1 where one helical chain alternately passes through 10-membered shortest circuits linked to each other and in contrary, the corresponding circuits are bound to the helical chain. Interestingly, 1 exhibits fluorescent emission in both the solid and solution phase. The uncoordinated nitrogen atom and amino group from the triazole core on the crystal surface make it suitable to detect picric acid in water. The luminescence intensity of 1 in water can be efficiently quenched by the addition of picric acid (PA). The sensitive detection of PA can be continuously performed for at least five cycles without diminishing the fluorescence intensity and destroying the framework structure of 1. The possible quenching mechanisms for PA are also investigated.
Co-reporter:Si-Quan Jiang, Zi-Yan Zhou, Shu-Ping Zhuo, Guo-Gang Shan, Ling-Bao Xing, Hai-Ning Wang and Zhong-Min Su
Dalton Transactions 2015 - vol. 44(Issue 48) pp:NaN20833-20833
Publication Date(Web):2015/11/09
DOI:10.1039/C5DT03814F
An efficient turn-on fluorescent sensor for PO43− has been developed by rationally designing an in situ-generated iron(III) complex with a 1,8-naphthalene-based Schiff base unit. The sensor exhibits high sensitivity and selectivity in both solution and solid-state film, even in the presence of other phosphate anions such as HPO42− and H2PO4−.
Co-reporter:Hai-Ning Wang, Xing Meng, Xin-Long Wang, Guang-Sheng Yang and Zhong-Min Su
Dalton Transactions 2012 - vol. 41(Issue 8) pp:NaN2233-2233
Publication Date(Web):2011/12/22
DOI:10.1039/C2DT11872F
Three allomorphs with the same stoichiometry, [Mg3(H2O)4(5-aip)2(5-Haip)2]·4DMA, were solvothermally synthesized in the presence of different additives and represented the first 8-connected nanotubular networks in Mg-based metal–organic frameworks. The adsorption and delivery of drugs were also determined.
Co-reporter:Guo-Gang Shan, Dong-Xia Zhu, Hai-Bin Li, Peng Li, Zhong-Min Su and Yi Liao
Dalton Transactions 2011 - vol. 40(Issue 12) pp:NaN2953-2953
Publication Date(Web):2011/02/15
DOI:10.1039/C0DT01559H
Three cationic iridium complexes containing 4,7-bis(3,6-di-tert-butyl-9H-carbazol-9-yl)-1,10-phenanthroline (L1) and 4,7-bis(3′,6′-di-tert-butyl-6-(3,6-di-tert-butyl-9H-carbazol-9-yl)-3,9′-bi(9H-carbazol)-9-yl)-1,10-phenanthroline (L2) as the ancillary ligands, namely, [Ir(ppy)2(L1)]PF6 (1), [Ir(ppy)2(L2)]PF6 (2) and [Ir(oxd)2(L2)]PF6 (3) (ppy is 2-phenylpyridine, oxd is 2,5-diphenyl-1,3,4-oxadiazole), have been designed and prepared. With more intramolecular rotational units on the ancillary ligand (L2), 2 and 3 possess a unique aggregation-induced phosphorescent emission (AIPE) property. This phenomenon was unprecedentedly observed in the cationic iridium(III) complexes. In order to investigate the underlying mechanism of this AIPE behavior, their photophysical, temperature-dependent aggregation properties as well as theoretical calculations, were performed. The results suggest that restricted intramolecular rotation is responsible for the AIPE of cationic complexes. Moreover, photoluminescent quantum yields in the neat film, thermal stabilities and off/on luminescence switching of 2 were investigated, revealing its potential application as a candidate for LECs and organic vapor sensing.
Co-reporter:Chun-Guang Liu, Wei Guan, Li-Kai Yan and Zhong-Min Su
Dalton Transactions 2011 - vol. 40(Issue 12) pp:NaN2974-2974
Publication Date(Web):2011/02/15
DOI:10.1039/C0DT01085E
High-valent transition-metal-substituted Keggin-type polyoxometalates (POMs) are active and robust oxidation catalyst. The important oxidized intermediates of these POM complexes are very difficult to be characterized by using the experimental method, and thus no detail information is available on such species. In the present paper, density functional theory (DFT) calculations have been carried out to characterize the electronic structures of a series of mono-ruthenium-substituted Keggin-type POMs. We find that the aquaruthenium(II/III/IV) species possess dxy2dxz2dyz2, dxy2dxz2dyz1, and dxy2dxz1dyz1 electronic configuration, respectively, and hydroxyl/oxoruthenium(IV/V/VI) species possess dxy2dxz1π*yz1, dxy2π*xz1π*yz1, dxy1π*xz1π*yz1, and dxy1π*xz1π*yz0 electronic configuration, respectively. Mulliken spin population shows that spin density is localized on the ruthenium center in aquaruthenium(II/III/IV) POM complexes, and the RuOa unit in hydroxyl/oxoruthenium(IV/V/VI) POM complexes. The Oa atom has substantial radical character in oxoruthenium(IV/V) species, and the radical character of the Oa atom are significantly weakened in the oxoruthenium(VI) species. The relevant energy of the important Ru–Oa π*-antibonding unoccupied orbitals with high RuOa compositions of oxoruthenium(IV/V/VI) POM complexes decrease in the order: oxoruthenium(IV) > oxoruthenium(V) > oxoruthenium(VI). The pH-independent multiple reduction energies for Ru(III/II), Ru(V/IV), and Ru(VI/V) couples are calculated, which is in agreement with the experimental data.
Co-reporter:Shizheng Wen, Wei Guan, Jianping Wang, Zhongling Lang, Likai Yan and Zhongmin Su
Dalton Transactions 2012 - vol. 41(Issue 15) pp:NaN4607-4607
Publication Date(Web):2012/02/01
DOI:10.1039/C2DT12465C
The structural and electronic properties of [PW12O40]3− (PW12) anion deposited on a graphene layer are investigated by using periodic density functional theory. The equilibrium geometries of graphene–PW12 (G–PW12) are examined based on six different configurations. The adsorption energy and charge transfer between PW12 and graphene are calculated and analyzed. We found that the interaction between PW12 and graphene are noncovalent. The formation of G–PW12 complex is theoretically predicted to be feasible from an energetic perspective with electron transfer from the PW12 to graphene.
Co-reporter:Peng Huang, Chao Qin, Xin-Long Wang, Chun-Yi Sun, Yan Xing, Hai-Ning Wang, Kui-Zhan Shao and Zhong-Min Su
Dalton Transactions 2012 - vol. 41(Issue 20) pp:NaN6077-6077
Publication Date(Web):2012/04/12
DOI:10.1039/C2DT30265A
A new organic–inorganic hybrid, [Cu(en)2]3{[Cu(en)2][H6SiNb18O54]}·22H2O (1, en = ethylenediamine) containing the crescent-shaped polyoxoanion [H6SiNb18O54]8− and copper–organic cations has been successfully synthesized, and elemental analyses, IR spectra, thermogravimetric analyses and single-crystal X-ray diffraction were investigated.
Co-reporter:Guo-Gang Shan, Hai-Bin Li, Jun-Sheng Qin, Dong-Xia Zhu, Yi Liao and Zhong-Min Su
Dalton Transactions 2012 - vol. 41(Issue 32) pp:NaN9593-9593
Publication Date(Web):2012/06/01
DOI:10.1039/C2DT31013A
A new cationic Ir(III) complex based on a dendritic ancillary ligand has been designed and synthesized, which simultaneously exhibits piezochromic luminescent (PCL) behavior and aggregation-induced emission (AIE) property for the first time.
Co-reporter:Ping Song, Wei Guan, Likai Yan, Chunguang Liu, Chan Yao and Zhongmin Su
Dalton Transactions 2010 - vol. 39(Issue 15) pp:NaN3713-3713
Publication Date(Web):2010/03/08
DOI:10.1039/B922876D
The polarizability and redox properties for the tetranuclear vanadium–oxide–carboxylates have been investigated using density functional theory (DFT). These so-called inorganic crown ethers possess large polarizabilities, which are equivalent to fullerene and generate large polarization on the guest anions into the host. The dipole-induced dipole interaction between the host and the guest anions induces charge transfer from the guest to the host, which is enhanced with the increasing polarization. Moreover, the redox potentials are sensitive to different guests inside the host bowl, and they shift negatively as compared to the isolated host bowl. In contrast, the modification of the methyl group by CH2tBu on the rim of the bowl has evoked higher polarizability (over 560 a.u.) with larger polarization on the guest anions and more negative redox potentials. The weak interaction energies in accord with the organic crown ether incorporating alkali metal ions indicate that the guest anions can move freely inside and outside the host bowl, so this kind of inorganic crown ether may exhibit potential guest-switchable redox properties based on reversible complexation–decomplexation and will be expected to find applications in ion recognition and selectivity studies based on the sensitivity to different guests.
Co-reporter:Hong-Ying Zang, Ya-Qian Lan, Shun-Li Li, Guang-Sheng Yang, Kui-Zhan Shao, Xin-Long Wang, Li-Kai Yan and Zhong-Min Su
Dalton Transactions 2011 - vol. 40(Issue 13) pp:NaN3182-3182
Publication Date(Web):2011/02/21
DOI:10.1039/C0DT00656D
With the bottom-up design principle, we use metal-ions to bridge the predesigned tectons (1 [(H2L1)2(Mo8O26)]·4H2O and 3 [(H2L2)(L2)0.5(Mo8O26)0.5]·H2O) so that two higher dimensional γ-octamolybdate based inorganic–organic hybrid compounds 2 [CuI2(L1)3(Mo8O26)0.5] and 4 [Ni(L2)2(HL2)2(Mo8O26)]·4H2O are successfully obtained.
Co-reporter:Ya-Qian Lan, Shun-Li Li, Kui-Zhan Shao, Xin-Long Wang, Xiang-Rong Hao and Zhong-Min Su
Dalton Transactions 2009(Issue 6) pp:NaN947-947
Publication Date(Web):2008/12/16
DOI:10.1039/B810287B
Two POM-pillared 3D porous compounds, [CuICuII(CuIIfcz)2(H2O)5(PMoVI10MoV2O40)]·6H2O (1) and [CuI2(CuIIfcz)2(H2O)2(PMoVI8VV3VIV3O42)]·6H2O (2) (Hfcz = fluconazole, (1-(2,4-difluorophenyl)-1,1-bis[(1H-1,2,4-triazol-1-yl)methyl]benzyl alcohol) have been constructed based on different polyanions, (Cufcz)22+ macrocations and copper cations by the hydrothermal method. The (Cufcz)22+ macrocations link Cu cations to generate a 2D wavelike cationic sheet. Then the POM anions act as pillars to the cationic sheet to form different POM-pillared 3D frameworks. In compound 1, the polyanion exhibits a rare coordination mode and acts as a penta-dentate ligand, which acts as to pillars to the cationic sheet to form an unprecedented 3D (3,4,5,6)-connected open framework with (3·6·7)(32·6·73)(33·4·62·73·8)(34·42·62·76·8)(32·62·76·84·10) topology. In compound 2, polyanions covalently link cationic sheets to extend to an unusual 3D (3,4,6)-connected framework with the (52·6)(52·62·7·9)(54·64·74·93) topology. To the best of our knowledge, it is the first time that POM-pillared 3D metal–organic frameworks have been realized by combining POMs with deliberately designed macrocations and transition-metal ions, using a rational approach for synthesis of POM-based open metal–organic frameworks. In addition, the electrochemical behaviors of compounds 1 and 2 have been investigated.
Co-reporter:Chun-Guang Liu, Wei Guan, Li-Kai Yan, Ping Song and Zhong-Min Su
Dalton Transactions 2009(Issue 31) pp:NaN6213-6213
Publication Date(Web):2009/06/24
DOI:10.1039/B902837D
In the present paper, the electronic structures and bonding features between the ruthenium atom and nitrido ligand of known nitrido ruthenium (VI) porphyrin and hexavalent ruthenium nitrido derivative of Keggin typical polyoxometalate (POM) have been investigated by using density functional theory (DFT) calculations and natural bond orbital (NBO) analysis. The results show that the Keggin POM complex and porphyrin complex have the RuN triple bond, the NBO calculations yield a nitrido ligand charge close to zero in both complexes because of the charge transfer from the rich-electron nitrido ligand to the ruthenium center. The molecular orbitals predict that the ruthenium porphyrin complex would generate oxidized species with no change in the oxidation state of the metal center; however, the ruthenium center of the POM complex would be oxidized in a one-electron-oxidation process because of the different HOMO character, and moreover, the spin unrestricted calculations confirm this prediction made by molecular orbital analysis. On the other hand, both complexes have the same reduced center, the RuN unit, and the one-electron-reduction process weakens the bonding interaction between the ruthenium atom and nitrido ligand for both complexes because the antibonding LUMO is occupied. Compared with the porphyrin complex, the POM complex possesses a higher RuN composition and lower energy π* antibonding orbital according to gas-phase and solvent calculations.
Co-reporter:Chan Yao, Li-Kai Yan, Wei Guan, Chun-Guang Liu, Ping Song and Zhong-Min Su
Dalton Transactions 2010 - vol. 39(Issue 33) pp:NaN7649-7649
Publication Date(Web):2010/04/16
DOI:10.1039/C002547J
The second-order nonlinear optical (NLO) properties of porphyrin–metal–polyoxometalate (por–metal–POM) sandwich structures [(por)M(PW11O39)]5− (por = TPP, TPyP, TPPF20, M = Hf; por = TPP, M = Zr) and [(TPP)Hf(XW11O36)]6− (X = Si, Ge) are investigated by time-dependent density functional theory (TDDFT). The character of charge-transfer transition indicates that the porphyrin ligand acts as an electron acceptor and the lacunary Keggin-type POM acts as an electron donor. Our results show that this kind of organic–inorganic hybrid compound possesses remarkably large molecular second-order NLO polarizability, ∼100 × 10−30 esu, and might be an excellent second-order NLO material. Furthermore, the NLO response can be tuned by the element substituents. The computed β0 values increase with the auxiliary electron-accepting group on the porphyrin ring (TPPF20 > TPyP > TPP) and a heavy central heteroatom (Ge > Si > P). The present investigation provides important insight into the NLO properties of this class of por–metal–POM sandwich compound.
Co-reporter:Li-Li Shi, Yun Geng, Hong-Ze Gao, Zhong-Min Su and Zhi-Jian Wu
Dalton Transactions 2010 - vol. 39(Issue 33) pp:NaN7740-7740
Publication Date(Web):2010/07/23
DOI:10.1039/C0DT00146E
The complexes AlQ3 and Ir(ppy)3 (Q = 8-hydroxyquinolate; ppy = 2-phenylpyridyl) are typical green emitting fluorescence and phosphorescence materials, respectively. Here we hybridize Ir(ppy)3 with AlQ3 to investigate the optoelectronic properties of the Ir(III)-centred derivatives including (ppy)2IrQ, (ppy)IrQ2 and IrQ3 by using density functional methods. Our calculations show that the derivative Ir(III) complexes are red emitting phosphorescence materials. The characters of the lowest triplet excited states for these Ir(III) complexes are mainly dominated by the 8-hydroxyquinolate ligand. IrQ3 and (ppy)2IrQ possess good electron transfer performance, while (ppy)IrQ2 might have hole transport properties.
Co-reporter:Ya-Qian Lan, Shun-Li Li, Kui-Zhan Shao, Xin-Long Wang and Zhong-Min Su
Dalton Transactions 2008(Issue 29) pp:NaN3835-3835
Publication Date(Web):2008/06/13
DOI:10.1039/B802626B
Five POM-based hybrid materials have been designed and synthesized based on different metal ions under hydrothermal conditions, namely, [Zn(Hfcz)(H2O)3](H3fcz)(SiMo12O40)·3H2O (1), [Cd2(Hfcz)6(H2O)2](SiMo12O40)·H2O (2), [Co2(Hfcz)2(SiW12O40)](H3fcz)2(SiW12O40)·10H2O (3), [Ni2(Hfcz)4(H2O)2](SiW12O40)·5H2O (4) and [Ag4(Hfcz)2(SiMo12O40)] (5), where Hfcz is fluconazole [2-(2,4-difluorophenyl)-1,3-di(1H-1,2,4-triazol-1-yl)propan-2-ol]. Their crystal structures have been determined by X-ray diffraction, elemental analyses, IR spectra, and thermogravimetric analyses (TGA). There are 1D mono and double chain-like metal–organic units in compounds 1 and 2, respectively. Polyoxometalates and metal–organic units co-crystallize through hydrogen bonds. In compound 3, metal–organic sheets are pillared by one kind of polyanion through covalent connections to generate a sandwich double-sheet. The other kind of polyanion acts as a counter-ion and lies in two adjacent sandwich double-sheets through non-covalent interactions. Polyanions covalently link metal–organic sheets to extend to an unusual 3D 5-connected framework with the (44·66) topology in 4. In compound 5, polyanions link metal–organic chains to form a sheet through covalent connections. It is interesting that compound 5 shows an intricate (4,5,10)-connected framework with (44·62)4(48·62)2(414·619·812) topology based on two kinds of Ag cations as four-connected and five-connected nodes, and polyanions as ten-connected nodes, when Ag⋯O interactions are considered. It represents the highest connected network topology presently known for polyoxometalate systems. The structural differences among 1–5 indicate the importance of different metal–organic units, coordination modes of polyanions for framework formation, and the interactions between polyanions and metal–organic units. In addition, the luminescent properties of compounds 1, 2 and 5, and electrochemical behaviours of compounds 1–5 have been investigated.
Co-reporter:Ya-Qian Lan, Shun-Li Li, Yao-Mei Fu, Yan-Hong Xu, Lu Li, Zhong-Min Su and Qiang Fu
Dalton Transactions 2008(Issue 47) pp:NaN6807-6807
Publication Date(Web):2008/10/22
DOI:10.1039/B809336A
A series of mixed-ligand coordination complexes, namely [Zn(L1)(oba)] (1), [Cd(L1)(oba)] (2), [Zn2(L2)(oba)2]·8H2O (3), [Cd2(L2)(oba)2]·2H2O (4), [Zn3(L3)(oba)3] (5), [Cd2(L3)(oba)2]·(L3) (6), [Cd(L4)(oba)]·H2O (7) and [Cd(L5)(oba)]·3H2O (8), where L1 = 2-(2-pyridyl)imidazole, L2 = 1,4-bis[2-(2-pyridyl)imidazol-1-yl]butane, L3 = 1,4-bis[2-(2-pyridyl)imidazol-1-ylmethyl]benzene, L4 = 1,3-bis[2-(2-pyridyl)imidazol-1-ylmethyl]benzene, L5 = 1,2-bis[2-(2-pyridyl)imidazol-1-ylmethyl]benzene and H2oba = 4,4′-oxydibenzoic acid, have been synthesized under hydrothermal conditions. Their structures have been determined by single crystal X-ray diffraction analyses and further characterized by elemental analyses, IR spectra, and thermogravimetric (TG) analyses. In compounds 1 and 2, oba2−, L1 ligand and ZnII or CdII ions assemble to form the parallel chains or parallel sheets which are linked by the weak hydrogen bonding and π⋯π stacking interactions to give the 2D supramolecular sheet or 3D supramolecular net, respectively. For 3, L2 ligands connect [Zn(oba)] chains to generate a unusual (10,3)-b topological structure which is the first example for eight-fold interpenetrating framework based on the (10,3)-b net. In 4, L2 ligands link [Cd(oba)] double-chains to give a 2D sheet which is assembled by π⋯π stacking interactions to obtain a 3D supramolecular net. In 5, L3 ligands link ZnII ions from α-Po net formed by ZnII ions and oba2− anions to show a novel 3D 8-connected self-penetrating framework with the unreported (416·611·8) topological structure. In 6, the double-chains constructed by CdII and oba2− anions are linked by one kind of L3 ligand to form a layer-like structure which is assembled by π⋯π stacking interactions to show a 3D supramolecular structure. In 7, oba2− anions coordinate to CdII cations to form chains which are connected by L4 to form a four-fold interpenetrating diamond network. In 8, the weak hydrogen bonding and π⋯π stacking interactions connect the [Cd(L5)(oba)] chains to give a 2D supramolecular sheet. By careful inspections of the structures of 1–8, we believe that the different flexible and angular neutral ligands, coordination geometries of metal centers and weak interactions (hydrogen bonds and π⋯π stacking interactions) are crucial factors for the formation of the different structures. The photoluminescent properties of 1–8 have been studied in the solid state at room temperature.
Co-reporter:Dashu Chen, Hongzhu Xing, Chungang Wang and Zhongmin Su
Journal of Materials Chemistry A 2016 - vol. 4(Issue 7) pp:NaN2662-2662
Publication Date(Web):2016/02/01
DOI:10.1039/C6TA00429F
A new microporous robust zirconium metal–organic framework (Zr-MOF), NNU-28, has been synthesized and employed as a visible-light photocatalyst for carbon dioxide (CO2) reduction to produce formate. NNU-28 is constructed by using a visible light responsive organic ligand derived from an anthracene group. Studies reveal that the as-prepared Zr-MOF shows desirable characteristics including excellent chemical and thermal stability, high CO2 uptake, broad-band visible light absorption and efficient photoinduced charge generation. Remarkably, NNU-28 is highly efficient for visible-light-driven CO2 reduction with a formate formation rate of 183.3 μmol h−1 mmolMOF−1, which is among the highest performances of Zr-MOFs. Both photocatalytic experiments and electron paramagnetic resonance (EPR) studies reveal that both the inorganic building unit Zr6 oxo cluster and the anthracene-based ligand contribute to the highly efficient photocatalysis of CO2 reduction. The dual photocatalytic routes are demonstrated here to be more efficient for visible-light-driven CO2 photoreduction than that typically relying on a ligand-to-metal charge transfer process, illustrating a new strategy to design and synthesize novel visible-light photocatalysts for CO2 reduction with high efficiency.
Co-reporter:Xianchun Liu, Renxi Jin, Dashu Chen, Lin Chen, Shuangxi Xing, Hongzhu Xing, Yan Xing and Zhongmin Su
Journal of Materials Chemistry A 2015 - vol. 3(Issue 8) pp:NaN4313-4313
Publication Date(Web):2014/12/09
DOI:10.1039/C4TA06049K
In this study, we report a simple in situ auto-reduction strategy for the fabrication of Ag@FDU-15 nanocomposites, in which the small sized Ag nanoparticles are monodispersed in the channels of FDU-15 ordered mesopolymers. The as-prepared Ag@FDU-15 nanocomposites were characterized by small and wide angle X-ray diffraction (XRD), nitrogen sorption, thermo-gravimetric analysis (TGA), energy dispersive X-ray spectroscopy (EDS), X-ray photoelectron spectroscopy (XPS), transmission electron microscopy (TEM) and scanning transmission electron microscopy (STEM). TEM results show that the silver nanoparticles are uniformly dispersed with a mean diameter of 5.6 ± 0.5 nm. When used as a hydrogenation catalyst, the Ag@FDU-15 nanocomposites exhibit excellent catalytic performance and reusability for the reduction of 4-nitrophenol (4-NP) in the presence of NaBH4.
Co-reporter:En-Long Zhou, Peng Huang, Chao Qin, Kui-Zhan Shao and Zhong-Min Su
Journal of Materials Chemistry A 2015 - vol. 3(Issue 14) pp:NaN7228-7228
Publication Date(Web):2015/03/02
DOI:10.1039/C5TA00231A
A stable luminescent anionic metal–organic framework with unprecedented topological structure was constructed, which showed a certain degree of framework stability in water and acid/base solutions. Furthermore, the compound showed moderate CO2 adsorption and extremely selective detection of 2,4,6-trinitrophenol (TNP) in both an organic solvent and aqueous solution.
Co-reporter:Shao-Juan Bao, Rajamani Krishna, Ya-Bing He, Jun-Sheng Qin, Zhong-Min Su, Shun-Li Li, Wei Xie, Dong-Ying Du, Wen-Wen He, Shu-Ran Zhang and Ya-Qian Lan
Journal of Materials Chemistry A 2015 - vol. 3(Issue 14) pp:NaN7367-7367
Publication Date(Web):2015/01/30
DOI:10.1039/C5TA00256G
An air-stable tetrazolate-containing framework, [Zn2L2]·2DMF (NENU-520, H2L = 4-(1H-tetrazole-5-yl)biphenyl-4-carboxylic acid), with uncoordinated N atoms on its internal surface was solvothermally synthesized and structurally characterized. This metal–organic framework (MOF) exhibited high CO2 uptake of 79.9 cm3 cm−3 at 298 K and 100 kPa, as well as excellent adsorption selectivity for CO2 over CH4 and N2. Particularly, its exceptionally high selectivity of CO2 over N2 at 298 K has ranked NENU-520 among the highest MOFs for selective CO2 separation. Furthermore, the potential application of NENU-520 for the fixed bed pressure swing adsorption (PSA) separation of CO2 from CH4 and N2 has been validated via simulated breakthrough experiments. The small channel with the size of 3.6 Å, combined with CO2-accessible free nitrogen atoms directed toward the inner surface, is believed to contribute to its high CO2 uptake capacity and selectivity. Thus, this work represents a unique way to target MOF materials for highly selective CO2 separation by incorporating CO2-philic functional sites on pore surfaces, and at the same time optimizing pore sizes.
Co-reporter:Shu-Ran Zhang, Jing Li, Dong-Ying Du, Jun-Sheng Qin, Shun-Li Li, Wen-Wen He, Zhong-Min Su and Ya-Qian Lan
Journal of Materials Chemistry A 2015 - vol. 3(Issue 46) pp:NaN23434-23434
Publication Date(Web):2015/10/12
DOI:10.1039/C5TA07427D
In this work, a novel microporous anionic metal–organic framework (MOF), [Zn(ABTC)0.5(NO3)][(CH3)2NH2]·DMA·3H2O (NENU-505; NENU = Northeast Normal University; H4ABTC = 3,3′,5,5′-azobenzenetetracarboxylic acid; DMA = N,N-dimethylacetamide), has been rationally synthesized under solvothermal conditions. Single-crystal X-ray analysis reveals that NENU-505 is a (4,4)-connected 3D network with pts topology. Charge neutrality is achieved by [(CH3)2NH2]+ ions. It is noteworthy that NENU-505 displays high stability in air for more than two months. In particular, the adsorption ability of NENU-505 toward ionic dyes has been also investigated. According to the UV/vis spectroscopy analysis and the colour variance of NENU-505, we found that the cationic dyes could be efficiently adsorbed over a period of time, while the neutral and anionic dyes could not be adsorbed. Therefore, NENU-505 exhibits selective adsorption toward cationic dyes and can potentially serve as a column-chromatographic filler for the separation of dye molecules. Furthermore, the cationic dyes can be gradually released in the presence of NaCl. More interestingly, when NENU-505 was immersed in different metal ion DMA solutions, it performs as a rare example of a highly selective and sensitive sensor for Cr3+ ions. In connection to this, the probable sensing mechanism was also further investigated in detail in this paper. Remarkably, this is the first MOF to exhibit an excellent ability for the detection and adsorption of Cr3+ ions in a convenient, economical, and environmentally friendly manner.
Co-reporter:Wen-Wen He, Shun-Li Li, Wen-Liang Li, Ji-Sen Li, Guang-Sheng Yang, Shu-Ran Zhang, Ya-Qian Lan, Ping Shen and Zhong-Min Su
Journal of Materials Chemistry A 2013 - vol. 1(Issue 37) pp:NaN11116-11116
Publication Date(Web):2013/07/25
DOI:10.1039/C3TA12662E
A novel microporous metal–organic framework, IFMC-16, has been successfully constructed by using mixed ligands. IFMC-16 has multipoint interaction sites and exhibits high hydrogen storage capacity not only at ambient pressure, but also at lower pressure in a high pressure region, and displays high adsorptive efficiency in the removal of organosulfur compounds.
Co-reporter:Hai-Ning Wang, Fu-Hong Liu, Xin-Long Wang, Kui-Zhan Shao and Zhong-Min Su
Journal of Materials Chemistry A 2013 - vol. 1(Issue 42) pp:NaN13063-13063
Publication Date(Web):2013/09/30
DOI:10.1039/C3TA13242K
By means of a stepwise synthesis strategy, three 12-connected MOFs with fcu topology have been obtained, which are rare examples of 3D frameworks constructed from a MOP precursor. In addition, the adsorption and separation of dye molecules have been investigated.
Co-reporter:Shabbir Muhammad, Hong-Liang Xu, Rong-Lin Zhong, Zhong-Min Su, Abdullah G. Al-Sehemi and Ahmad Irfan
Journal of Materials Chemistry A 2013 - vol. 1(Issue 35) pp:NaN5449-5449
Publication Date(Web):2013/08/06
DOI:10.1039/C3TC31183J
Nonlinear optical (NLO) materials are the smartest materials of the era, and have the ability to generate new electromagnetic fields with changed frequencies, phases, and other physical properties. Recently, many cutting edge research reports have been focused on NLO materials especially on those which are composed of sp2 hybridized carbon nanostructures. As the carbon nanostructures are composed of abundant π-electrons and have significant delocalization, these are potential candidates for modern NLO materials. Generally, sp2 hybridized carbon nanostructures can be divided into zero-dimensional fullerenes, one-dimensional nanotubes and two-dimensional graphene nanoribbons and quantum dots etc. These dimensionally different carbon nanomaterials are promising candidates for a wide range of applications in next-generation nanotechnologies. In present feature article, we first briefly explain a theoretical structure–NLO property relationship based on perturbation theory and then elucidate the crucial factors to control the NLO responses. We put together the different random investigations of sp2 hybridized carbon nanostructures for NLO application by highlighting the importance of their several structural designs to tune NLO amplitudes. Furthermore, we make a comparative and updated analysis of the NLO properties of dimensionally different sp2 hybridized carbon nanomaterials i.e. fullerenes, carbon nanotubes, and graphene nanoribbons and quantum dots. Finally, we make a brief discussion about different aspects and opportunities to use the sp2 hybridized carbon nanomaterials as high performance NLO materials of the future. This review is a focused perspective based on different updated quantum chemical investigations about fullerenes, nanotubes and graphene nanoribbons and quantum dots for their possible use in nonlinear optical applications.
Co-reporter:Shunrui Luo, Fang Chai, Lingyu Zhang, Chungang Wang, Lu Li, Xianchun Liu and Zhongmin Su
Journal of Materials Chemistry A 2012 - vol. 22(Issue 11) pp:NaN4836-4836
Publication Date(Web):2012/01/30
DOI:10.1039/C2JM16476K
In this work, we have developed a facile and rapid synthesis of urchin-shaped Fe3O4@Bi2S3 core-shell hierarchical structures through a sonochemical method. The as-prepared Fe3O4@Bi2S3 hierarchical core-shell structures show excellent photocatalytic efficiency for the degradation of rhodamine B (RhB) and retained this photocatalytic activity after being recycled five times with the help of an external magnetic field. The possible mechanism for the formation of the urchin-shaped Fe3O4@Bi2S3 core-shell structures is also proposed. These novel urchin-shaped Fe3O4@Bi2S3 core-shell structures may have potential applications in water treatment, sensors, and energy storage.
Co-reporter:Xiao-Dan Tang, Yi Liao, Hong-Ze Gao, Yun Geng and Zhong-Min Su
Journal of Materials Chemistry A 2012 - vol. 22(Issue 14) pp:
Publication Date(Web):
DOI:10.1039/C2JM14871D
Co-reporter:Li Wang, Yong Wu, Guo-Gang Shan, Yun Geng, Jian-Zhao Zhang, Dong-Mei Wang, Guo-Chun Yang and Zhong-Min Su
Journal of Materials Chemistry A 2014 - vol. 2(Issue 16) pp:NaN2868-2868
Publication Date(Web):2014/01/14
DOI:10.1039/C3TC31831A
A series of heteroleptic iridium(III) complexes were investigated by using the density functional theory/time-dependent density functional theory (DFT/TD-DFT) approach to determine the influence of the diphenylphosphoryl (Ph2PO) moiety on the electronic structures, phosphorescent properties and the organic light-emitting diode (OLED) performance. The results reveal that the introduction of the Ph2PO group could not only dramatically change the electron density distributions of the LUMO and cause red shifts of the emission wavelengths, but also increase the oscillator strengths and the metal character, thus leading to larger radiative decay rates. Additionally, compared with FIrpic (a widely used kind of blue guest material in OLED devices), those complexes with Ph2PO substituents could improve the electron injection/balance ability, increase the Förster energy transfer rate and confine the triplet excitons to the guest phosphors, hence resulting in better OLED performance. Interestingly, further analysis indicates that, compared to IrpicPO with the Ph2PO group sited at the phenyl ring of the phenylpyridine (ppy) ligands, IrpicPOpy with the Ph2PO group sited at the pyridine ring of the ppy ligands performs better in the hole-trapping and hole-injection ability. Finally, we hope our investigations will facilitate the future design of high efficient phosphorescent materials.
Co-reporter:Yanling Si, Guochun Yang and Zhongmin Su
Journal of Materials Chemistry A 2013 - vol. 1(Issue 7) pp:NaN1406-1406
Publication Date(Web):2012/12/13
DOI:10.1039/C2TC00413E
Time-dependent density functional theory (TDDFT) calculations have been used to investigate chiroptical, linear, and second-order nonlinear optical (NLO) properties of the novel tetrathiafulvalenylallene in both neutral and two cationic states for the first time. The calculated UV-Vis/ECD spectra of the studied compound are in good agreement with the experimental ones, which can be used to assign its absolute configuration (AC) with high confidence. From neutral state to the two cationic states, the studied compound exhibits pronounced different chiroptical effects and second-order NLO response values. For example, the calculated β0 value of 12+ is 10.36 times as large as that of 1, while the β0 value of 14+ is 46.51 times as large as that of 1. These effects mainly result from the structural modifications of TTF units in the redox process. It is found that charge transfer between the tetrathiafulvalene (TTF) unit and the allene framework plays a key role in determining the chiroptical properties and electronic transition properties. It is interesting to find that the two benzene rings have vanishingly small effects on the chiroptical properties. The studied compound could act as both a chiroptical switch and NLO switch material from the standpoint of different chiroptical and NLO responses, reversible redox processes, and high stability. The effects of different functionals and basis sets, including solvent effects on the UV-Vis/ECD spectra were also considered.
Co-reporter:Chun-Yi Sun, Chao Qin, Xin-Long Wang, Guang-Sheng Yang, Kui-Zhan Shao, Ya-Qian Lan, Zhong-Min Su, Peng Huang, Chun-Gang Wang and En-Bo Wang
Dalton Transactions 2012 - vol. 41(Issue 23) pp:NaN6909-6909
Publication Date(Web):2012/04/24
DOI:10.1039/C2DT30357D
Zeolitic Imidazolate Framework-8 (ZIF-8), for the first time for ZIFs, exhibits a remarkable capacity for the anticancer drug 5-fluorouracil (5-FU), around 660 mg of 5-FU/g of ZIF-8, and presents a pH-triggered controlled drug release property. These prove ZIF-8 to be a valuable candidate for delivery of anticancer agents and reveal its potential applications in the treatment of cancer.
Co-reporter:Shanshan Zhao, Fei Yu, Guochun Yang, Hongyu Zhang, Zhongmin Su and Yue Wang
Dalton Transactions 2012 - vol. 41(Issue 24) pp:NaN7277-7277
Publication Date(Web):2012/04/20
DOI:10.1039/C2DT00009A
To deeply understand the charge-transporting nature of Pt(CNtBu)2(CN)2 nanowires induced by intermolecular Pt⋯Pt interactions, calculations based on first-principle band structure and Marcus theory have been performed. The calculated bandwidths of the valence band, conducting band, and the effective masses of hole and electron are almost equal. This suggests that this complex has ambipolar transport characteristics, in agreement with experimental results. Density of states analysis revealed that the hole transport resulted mainly from the Pt⋯Pt interactions, while the electron transport was derived mainly from the CN groups. The character of the frontier molecular orbitals, reorganization energies and transfer integrals in different directions also supports the calculated first-principle band structure. Moreover, an investigation into the intermolecular interaction energy of neighbors revealed that there is a remarkable relationship between the intermolecular interaction energy and the transfer integral.
Co-reporter:Zhong-Ling Lang, Guo-Chun Yang, Na-Na Ma, Shi-Zheng Wen, Li-Kai Yan, Wei Guan and Zhong-Min Su
Dalton Transactions 2013 - vol. 42(Issue 29) pp:NaN10625-10625
Publication Date(Web):2013/05/14
DOI:10.1039/C3DT50666E
Water oxidation is a key half reaction in the energy conversion scheme. The reaction mechanism for the oxidation of H2O to O2 catalyzed by single-Ru-substituted polyoxometalates, [RuIII(H2O)XW11O39]n− (X = SiIV, GeIV), was investigated by means of density functional calculations. The electronic structure of the pre-activation intermediates indicates that the aqua ligand is prone to accommodate the proton coupled electron transfer (PCET) process to achieve the active group [RuVOa], and the high valent oxo-ruthenium(V) species are responsible for the O–O forming event. Three possible proton acceptors were designed for the rate-determining step (Ob, Oa, and H2O), the calculated results support that the bridge Ob atom of the polytungstate ligand will act as the most favorable proton acceptor in the O–O bond formation, with an energy barrier of 28.43 kcal mol−1. A detailed information of the peroxidic intermediates in the oxidation process was also characterized, both the peroxo-species [RuIV(OO)SiW11O39]6− and [RuV(OO)SiW11O39]5− show the six-coordinate isomer with an open terminal geometry is more favorable than the close seven-coordinate ones. In addition, the replacement of the heteroatom in XO4n− can effectively tune the catalytic activity of polyoxometalates, in the order of GeIV > SiIV.
Co-reporter:Yu-Teng Zhang, Peng Huang, Chao Qin, Li-Kai Yan, Bai-Qiao Song, Zhao-Xia Yang, Kui-Zhan Shao and Zhong-Min Su
Dalton Transactions 2014 - vol. 43(Issue 26) pp:NaN9850-9850
Publication Date(Web):2014/04/14
DOI:10.1039/C4DT00507D
A new organic–inorganic hybrid titaniobate compound, [Cu(en)2][Cu(en)2(H2O)2]3[Ti2Nb8O28]·8H2O (1) (en = ethylenediamine), was successfully synthesized, characterized by single-crystal X-ray diffraction, IR spectroscopy, thermogravimetric analysis (TGA) and UV-Vis diffuse reflectance spectroscopy and its photoluminescence studied.
Co-reporter:Wei Guan, Zhijian Wu and Zhongmin Su
Dalton Transactions 2012 - vol. 41(Issue 9) pp:NaN2803-2803
Publication Date(Web):2012/01/20
DOI:10.1039/C2DT12068B
DFT investigations have been carried out on encapsulation of Lindqvist-type W6O192− anion inside hydrogenated (n,n) armchair single-walled carbon nanotubes (h-CNTs) with n = 8, 9, 10 to understand the confinement effect of the CNTs on the rotation of W6O192−. The energy-decomposition analysis (EDA) of interaction between W6O192− and CNTs shows that with the increase of confinement effect from n = 8, 9, to 10, the destabilizing ΔEPauli plays a more important role in the relative orientation of W6O192− inside CNTs. For W6O192−@(9,9) h-CNT, the most stable orientation appears at the y/z angle 45°/36°. The confinement effect reduces significantly the energy gap of W6O192−@(n,n) h-CNT (n = 8, 9, 10) compared with free W6O192−. Electron transfer from the W6O192− to CNT is observed.
Co-reporter:Yong Wu, Shui-Xing Wu, Hai-Bin Li, Yun Geng and Zhong-Min Su
Dalton Transactions 2011 - vol. 40(Issue 17) pp:NaN4488-4488
Publication Date(Web):2011/03/15
DOI:10.1039/C0DT01299H
The electronic structures and photophysical properties of eight Pt-complexes with different N-heterocyclic carbene ligands and potential to serve as light emitting diode materials were investigated by density functional theory and time-dependent density functional theory, employing the BP86 functional for geometry optimisations, SAOP potential for excited state calculations and all-electron TZ2P basis set throughout. Non-radiative and radiative decay rate constants were determined for each system through analyses of the geometric relaxations, d-orbital splitting and spin–orbit couplings at the optimised S0 and T1 geometries. Three Pt-systems bound to two N-heterocyclic carbenes were shown to be nonemissive, while a fourth was shown to be emissive from the T1 excited state. Similar T1-initated emission was observed for three other Pt-systems investigated, each bound to four N-heterocyclic carbenes, while a fourth similarly tetra-ligated system showed T2-initation of emission. The results highlight the coupling of ligand-identity to photophysical properties and more importantly, the potential for rational optimisation and tuning of emission wavelengths and phosphorescent efficiencies. Encouragingly, two of the tetra-N-heterocyclic carbene ligated systems show strong potential to serve as highly-efficient blue and green light emitting materials, respectively.
Co-reporter:Ya-Qian Lan, Shun-Li Li, Zhong-Min Su, Kui-Zhan Shao, Jian-Fang Ma, Xin-Long Wang and En-Bo Wang
Chemical Communications 2008(Issue 1) pp:
Publication Date(Web):
DOI:10.1039/B714413J
Co-reporter:Xianchun Liu, Yan Xing, Xinlong Wang, Hongbin Xu, Xizheng Liu, Kuizhan Shao and Zhongmin Su
Chemical Communications 2010 - vol. 46(Issue 15) pp:NaN2616-2616
Publication Date(Web):2010/02/24
DOI:10.1039/B923028A
A new chiral open-framework cobalt phosphite [C6N2H14][Co(HPO3)2] (1) with helical channels has been synthesized under solvothermal conditions in the presence of achiral organic amine. The circular dichroism (CD) spectrum indicates that the bulk crystals have optical activity. Magnetic measurements reveal that 1 is a weak ferromagnet.
Co-reporter:Ping Shen, Wen-Wen He, Dong-Ying Du, Hai-Long Jiang, Shun-Li Li, Zhong-Ling Lang, Zhong-Min Su, Qiang Fu and Ya-Qian Lan
Chemical Science (2010-Present) 2014 - vol. 5(Issue 4) pp:NaN1374-1374
Publication Date(Web):2013/12/02
DOI:10.1039/C3SC52666F
In this work, we have demonstrated an unprecedented single-crystal-to-single-crystal (SCSC) transformation between two 3D metal-organic frameworks (MOFs). The centrosymmetric IFMC-68 ([(Zn4O)2(L)3]·10H2O·46DMA) transforms into a chiral IFMC-69 ([(Zn4O)2(L)3H2O]·H2O·4DMA) doubly triggered by reaction temperature and time simultaneously in the presence or absence of solvent. To our knowledge, this is the first representative that the non-interpenetrated structure transforms into self-penetrated structure in MOFs. For the first time, we have studied the influence of reaction temperature and time on SCSC transformation, simultaneously, and get the transformation relationship among IFMC-68, IFMC-69 and the intermediate coming from the direct synthesis method and stepwise synthesis method at different temperatures and for different times. Meanwhile, we have achieved the conversion from an air-unstable to air-stable structure. Air-stable IFMC-69 exhibits the selective CO2 uptake over N2 and more excellent gas adsorption ability than IFMC-68. In addition, IFMC-69 shows an efficient capability in reversible adsorption of iodine. The electrical conductivity value (σ) of I2@IFMC-69 is much higher than the pristine MOF and thus is promising for potential semiconductor materials in the future.
Co-reporter:Yinghui Wang, Bin Li, Yanhong Liu, Liming Zhang, Qinghui Zuo, Linfang Shi and Zhongmin Su
Chemical Communications 2009(Issue 39) pp:
Publication Date(Web):
DOI:10.1039/B910305H
Co-reporter:Bai-Qiao Song, Da-Qin Chen, Zhenguo Ji, Junhong Tang, Xin-Long Wang, Hong-Ying Zang and Zhong-Min Su
Chemical Communications 2017 - vol. 53(Issue 11) pp:NaN1895-1895
Publication Date(Web):2017/01/03
DOI:10.1039/C6CC07729C
We have demonstrated for the first time that isostructural homochiral metal–organic frameworks (MOFs) can be synthesized directly from chiral ligands and indirectly from achiral ligands via spontaneous resolution combined with cooperative chirality induction, respectively. Moreover, the usage of different ligands leads to distinct proton conduction behaviors.
Co-reporter:Hong Ren, Lingyu Zhang, Tingting Wang, Lu Li, Zhongmin Su and Chungang Wang
Chemical Communications 2013 - vol. 49(Issue 54) pp:NaN6038-6038
Publication Date(Web):2013/04/30
DOI:10.1039/C3CC41284A
A novel, fast and simple method was developed to fabricate poly(acrylic acid sodium salt) microspheres (PAAS MSs). The resulting PAAS MSs were utilized as active templates to universally synthesize the mesoporous lanthanide-doped gadolinium oxide hollow nanospheres with multicolored upconversion emissions under mild conditions.
Co-reporter:Jun Liang, Xin-Long Wang, Yan-Qing Jiao, Chao Qin, Kui-Zhan Shao, Zhong-Min Su and Qing-Yin Wu
Chemical Communications 2013 - vol. 49(Issue 76) pp:NaN8557-8557
Publication Date(Web):2013/07/31
DOI:10.1039/C3CC43990A
Three novel self-catenated 4-connected uninodal (65·8)-mok metal–organic rotaxane frameworks (MORFs) containing cucurbit[6]uril were constructed from the in situ trans/cis-configuration (1:1) of rotaxanes by taking advantage of a d10 metal ion directed synthesis. It was revealed that the effect of hydrogen bonds and π–π stacking interactions play significant roles in the self-assembly process.
Co-reporter:Wei-Chao Chen, Chao Qin, Xin-Long Wang, Yang-Guang Li, Hong-Ying Zang, Yan-Qing Jiao, Peng Huang, Kui-Zhan Shao, Zhong-Min Su and En-Bo Wang
Chemical Communications 2014 - vol. 50(Issue 87) pp:NaN13267-13267
Publication Date(Web):2014/09/10
DOI:10.1039/C4CC06447J
Two Fe-substituted Dawson-type nanoscale selenotungstate clusters, {Fe6Se6W34} and {Fe10Se8W62} involving {α-Se2W14} and {γ-Se2W14} building blocks, have been isolated, which exhibit photocatalytic H2 evolution activity. Their electrochemical behaviours and magnetic properties were also investigated.
Co-reporter:Ji Zhang, Hai-Bin Li, Shi-Ling Sun, Yun Geng, Yong Wu and Zhong-Min Su
Journal of Materials Chemistry A 2012 - vol. 22(Issue 2) pp:NaN576-576
Publication Date(Web):2011/11/03
DOI:10.1039/C1JM13028E
To rationalize the marked difference in the energy conversion efficiency of dye sensitized solar cells (DSSCs) based on organic dyes 1 and 2 different only in their π spacer, density functional theory (DFT) and time-dependent DFT calculations of the geometries, electronic structures and absorption spectra of the organic dyes before and after binding to titanium oxide were carried out. These enable us to determine factors such as dipole moments associated with the open-circuit photovoltage (Voc), and to quantify parameters such as the light harvesting efficiency, the electron injection efficiency associated with the short-circuit photocurrent density (Jsc). The results reveal that compared to 2 with a thiazole spacer, 1 with a thiophene spacer could cause a red shift of the absorption spectrum, increase the oscillator strength and improve the driving force for electron injection, thus leading to the larger Jsc, in good agreement with experimental data. As for Voc, our results stress that apart from the generally emphasized vertical dipole moment of the dyes pointing outward from the semiconductor surface, the number of photoinjected electrons from the dye to the semiconductor is also crucial to obtain high performance dyes with high Voc. After justifying the reliability of the quantum-chemical methods, we designed another four dyes with different π spacers to screen more efficient organic dyes. Fortunately, taking 1 as reference, we find that dye 4 with a thienothiophene spacer displays an enhanced Jsc and Voc, indicating that it will be a more efficient diarylamine-fluorene-based organic dye used in DSSCs, which will play a theoretical guiding role in the design and synthesis of new organic dyes.
Co-reporter:Rong-Lin Zhong, Hong-Liang Xu, Shabbir Muhammad, Ji Zhang and Zhong-Min Su
Journal of Materials Chemistry A 2012 - vol. 22(Issue 5) pp:NaN2202-2202
Publication Date(Web):2011/12/15
DOI:10.1039/C1JM14358A
Excess electron compounds have been proposed to be novel candidates of high-performance nonlinear optical (NLO) materials because of their large static first hyperpolarizabilities (β0). To enhance the stability of an unstable excess electron compound (LiCN⋯Li) with an extremely large β0 value (310196 a.u.), we designed a boron nitride nanotube (BNNT) as a protective shield molecule to encapsulate it (in theory). The stability of LiCN⋯Li was enhanced: the vertical ionization potentials (VIP) of LiCN⋯Li increased after encapsulating. Therefore, by comparison with LiCN⋯Li, the encapsulated complexes are more difficult to oxidize. Significantly, the BNNT encapsulated LiCN⋯Li complex exhibits a considerable β0 value (10645 a.u.), which is significantly (almost 380 times) larger than 28 a.u. of BNNT. Our further investigations into the intrinsic hyperpolarizabilites (βint) of these compounds show that there are clearly dependencies of the NLO response on the transition energy. Furthermore, it is easy to encapsulate LiCN⋯Li from the B-rich edge rather than N-rich edge of BNNT due to the lower energy barrier, which makes our calculations more useful to experimentalists who may try to synthesize these compounds. Knowledge of the encapsulation process of LiCN⋯Li within BNNT provides a new strategy for the design and synthesis of stable high-performance NLO materials.
Co-reporter:Tengying Ma, Shizheng Wen, Caixia Wu, Likai Yan, Min Zhang, Yuhe Kan and Zhongmin Su
Journal of Materials Chemistry A 2015 - vol. 3(Issue 39) pp:NaN10090-10090
Publication Date(Web):2015/07/29
DOI:10.1039/C5TC00792E
The electronic and transport properties of a series of 11-ASiNRs (armchair silicene nanoribbons) at different torsion angles were studied by using density functional theory combined with nonequilibrium Green's function method. Several key factors determining the transport properties, such as the electron transmission coefficient and band structure, have been discussed. The interesting results suggest that the transport properties of ASiNRs are insensitive to the torsional silicene nanoribbon configuration in the scattering region. With the increase of the torsion angle, the transmission coefficient is still well maintained within the limits of the torsion angle. Although the torsion angle is increased to 120°, the current dropped by just 22% compared to the initial 11-ASiNRs at a torsion angle of 0°. Furthermore, all the configurations of 11-ASiNRs in this study behave as conventional conductors with nearly linear current–voltage dependence. On the basis of these distinctive transport properties with metabolic structure, ASiNRs present potential promising applications in silicon-based electronic nanodevices.
Co-reporter:Xing Meng, Chao Qin, Xin-Long Wang, Zhong-Min Su, Bo Li and Qi-Hua Yang
Dalton Transactions 2011 - vol. 40(Issue 39) pp:NaN9966-9966
Publication Date(Web):2011/09/09
DOI:10.1039/C1DT11227A
Immobilization of the chiral salen-metal complex [MnIII(salen)(H2O)2ClO4] on the Keggin-type polyoxometalate (POM) skeletons leads to the isolation of POM derivatives functionalized with chiral salen-metal complexes, which represent the first examples of introducing chiral salen-metal complexes into the POM systems.
Co-reporter:Jian-Ping Wang, Guo-Chun Yang, Li-Kai Yan, Wei Guan, Shi-Zheng Wen and Zhong-Min Su
Dalton Transactions 2012 - vol. 41(Issue 33) pp:NaN10104-10104
Publication Date(Web):2012/05/28
DOI:10.1039/C2DT30449J
We theoretically investigate a novel switching phenomenon based on the divacant Keggin-type polyoxotungstate bearing chiral organophosphonate [{NH2CH(CH3)PO}2(γ-SiW10O36)]4−, that is the synchronous chiroptical and nonlinear optical (NLO) switch triggered by redox. The ECD calculations on the Boltzmann weighted conformations of the three oxidation states of this chiral polyoxometalate (POM) clearly present a chiroptical switching process. The electronic transition and the bond-length alternation studies show that the chirality transfer from chiral carbon atom to POM cage increases as the polyanion is reduced. Simultaneously, the static first hyperpolarizability of studied chiral POM quadrupled from the oxidized state to the 1e-reduced state, and is further doubled to the 2e-reduced state, which is mainly due to the increasing electronic-dipole-allowed d–d charge transfer transitions in the POM cage. This work firstly reproduces the ECD spectrum of chiral POM with high accuracy and proves the possibility for confirming the molecular conformations of flexible chiral POMs in solution by the aid of ECD calculations. Most importantly, a sensitive diplex switch based on a chiral POM is predicted in theory, which may aid the design of novel POM-based switches.
Co-reporter:Xiang-Rong Hao, Xin-Long Wang, Zhong-Min Su, Kui-Zhan Shao, Ya-Hui Zhao, Ya-Qian Lan and Yao-Mei Fu
Dalton Transactions 2009(Issue 40) pp:NaN8566-8566
Publication Date(Web):2009/09/01
DOI:10.1039/B906728K
Two unprecedented 3D porous anionic metal–organic frameworks, [Me2NH2]2[Cd2(bpdc)3]·4dma 1 and [Me2NH2]2[Cd2(NH2bdc)3]·4dma 2 (dma = N,N′-dimethylacetamide, bpdc = 4,4′-biphenyldicarboxylate, NH2bdc = 2-amino-1,4-benzenedicarboxylate) have been solvothermally synthesized with a dimethylammonium cations template. Both 1 and 2 are constructed from low-symmetry SBUs. 1 has a chiral framework with helical nanotube-like channels, and 2 has a MOF-5-like motif. The fluorescence, N2 adsorption and ion-exchange properties for 1 have been examined.
Co-reporter:Hong Ren, Lingyu Zhang, Jiping An, Tingting Wang, Lu Li, Xiaoyan Si, Liu He, Xiaotong Wu, Chungang Wang and Zhongmin Su
Chemical Communications 2014 - vol. 50(Issue 8) pp:NaN1002-1002
Publication Date(Web):2013/11/11
DOI:10.1039/C3CC47666A
The polyacrylic acid@zeolitic imidazolate framework-8 (PAA@ZIF-8) nanoparticles (NPs) were first fabricated using a facile and simple route. It is worthwhile noting that the as-fabricated PAA@ZIF-8 NPs possessed ultrahigh doxorubicin (DOX) loading capability (1.9 g DOX g−1 NPs), which were employed as pH-dependent drug delivery vehicles.
Co-reporter:Wei Xie, Wen-Wen He, Dong-Ying Du, Shun-Li Li, Jun-Sheng Qin, Zhong-Min Su, Chun-Yi Sun and Ya-Qian Lan
Chemical Communications 2016 - vol. 52(Issue 16) pp:NaN3291-3291
Publication Date(Web):2015/12/16
DOI:10.1039/C5CC08703A
A stable mesoporous blue-emitting MOF NENU-521 was successfully constructed. NENU-521 can serve as a host for encapsulating Alq3 to obtain tunable and efficient white-light emission. The Alq3@NENU-521 composite possesses excellent stability and can be used as a promising white phosphor in WLEDs.
Co-reporter:Cai Xia Wu, Shi Zheng Wen, Li Kai Yan, Min Zhang, Teng Ying Ma, Yu He Kan and Zhong Min Su
Journal of Materials Chemistry A 2017 - vol. 5(Issue 16) pp:NaN4062-4062
Publication Date(Web):2017/03/28
DOI:10.1039/C6TC05545A
Using density functional theory (DFT) in combination with non-equilibrium Green's functions, we have investigated the electronic structures, magnetization, and quantum transport properties of zigzag graphene nanoribbons (ZGNRs) functionalized with conventional conductive metal adatoms (Al, Cu, Ag and Au). On the basis of the adsorption energies, our simulation demonstrates that Al and Cu adatoms are chemically bonded with ZGNRs, while the adsorptions for Ag and Au are between weak chemisorption and strong physisorption. The properties of charge transfer and magnetic moment are in reasonable agreement with the previous calculations. The adsorption of metal adatoms induce a net magnetic moment of −1 μB in 6ZGNR–metal systems. On the other hand, the transport studies of metal adatoms adsorbed ZGNRs suggest that the metal adatoms play an important role in the transport properties of devices and exhibit different effects on the transport properties of 6ZGNR-based and 7ZGNR-based devices. The 7ZGNR-based devices show the opposite conductive order in 6ZGNR-based devices. For 6ZGNR-based devices, the transport current in 6ZGNRs can be enhanced effectively by the adsorption of metal adatoms. However, the currents in 7ZGNR functionalized with conductive metal atoms are obviously smaller than that in pristine 7ZGNR, implying that metal adsorptions reduce the electrical conductivity of 7ZGNR-based devices. In contrast to the properties of the bulk materials, the conductivity of 6ZGNR–Al is highest among 6ZGNR–metal systems, which is in agreement with that of single atomic wires of Ag, Al, Au, and Cu.
Co-reporter:Cai Xia Wu, Shi Zheng Wen, Li Kai Yan, Min Zhang, Teng Ying Ma, Yu He Kan and Zhong Min Su
Journal of Materials Chemistry A 2017 - vol. 5(Issue 16) pp:NaN4062-4062
Publication Date(Web):2017/03/28
DOI:10.1039/C6TC05545A
Using density functional theory (DFT) in combination with non-equilibrium Green's functions, we have investigated the electronic structures, magnetization, and quantum transport properties of zigzag graphene nanoribbons (ZGNRs) functionalized with conventional conductive metal adatoms (Al, Cu, Ag and Au). On the basis of the adsorption energies, our simulation demonstrates that Al and Cu adatoms are chemically bonded with ZGNRs, while the adsorptions for Ag and Au are between weak chemisorption and strong physisorption. The properties of charge transfer and magnetic moment are in reasonable agreement with the previous calculations. The adsorption of metal adatoms induce a net magnetic moment of −1 μB in 6ZGNR–metal systems. On the other hand, the transport studies of metal adatoms adsorbed ZGNRs suggest that the metal adatoms play an important role in the transport properties of devices and exhibit different effects on the transport properties of 6ZGNR-based and 7ZGNR-based devices. The 7ZGNR-based devices show the opposite conductive order in 6ZGNR-based devices. For 6ZGNR-based devices, the transport current in 6ZGNRs can be enhanced effectively by the adsorption of metal adatoms. However, the currents in 7ZGNR functionalized with conductive metal atoms are obviously smaller than that in pristine 7ZGNR, implying that metal adsorptions reduce the electrical conductivity of 7ZGNR-based devices. In contrast to the properties of the bulk materials, the conductivity of 6ZGNR–Al is highest among 6ZGNR–metal systems, which is in agreement with that of single atomic wires of Ag, Al, Au, and Cu.



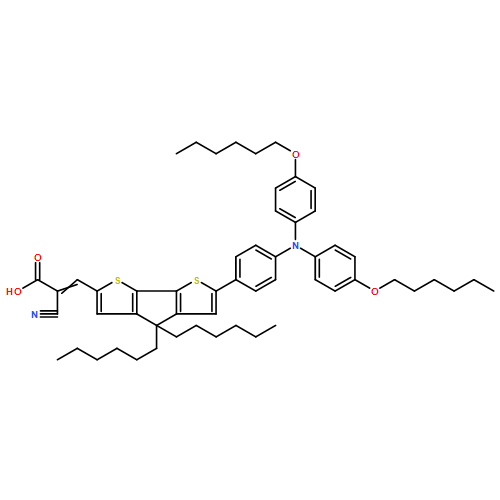















![6-[[4-[(6-oxocyclohexa-2,4-dien-1-ylidene)methylamino]anilino]methylidene]cyclohexa-2,4-dien-1-one](http://img.cochemist.com/ccimg/5400/5344-68-3.png)
![6-[[4-[(6-oxocyclohexa-2,4-dien-1-ylidene)methylamino]anilino]methylidene]cyclohexa-2,4-dien-1-one](http://img.cochemist.com/ccimg/5400/5344-68-3_b.png)
![2-(1H-Benzo[d]imidazol-2-yl)phenol](http://img.cochemist.com/ccimg/3000/2963-66-8.png)
![2-(1H-Benzo[d]imidazol-2-yl)phenol](http://img.cochemist.com/ccimg/3000/2963-66-8_b.png)


![Tungstate(3-),tetracosa-m-oxododecaoxo[m12-[phosphato(3-)-kO:kO:kO:kO':kO':kO':kO'':kO'':kO'':kO''':kO''':kO''']]dodeca-,hydrogen (1:3)](http://img.cochemist.com/ccimg/1400/1343-93-7.png)
![Tungstate(3-),tetracosa-m-oxododecaoxo[m12-[phosphato(3-)-kO:kO:kO:kO':kO':kO':kO'':kO'':kO'':kO''':kO''':kO''']]dodeca-,hydrogen (1:3)](http://img.cochemist.com/ccimg/1400/1343-93-7_b.png)