Co-reporter:Mengmeng Zhang;Wen Yang;Tingfeng Gong;Weiqun Zhou;Renyu Xue
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 32) pp:21672-21682
Publication Date(Web):2017/08/16
DOI:10.1039/C7CP03234J
Herein, tunable emissions in aggregation processes of triphenylamine derivatives (TPAQs) and their protonated cations, as well as protonated processes have been described. In this study, three triphenylamine-based compounds (TPAQs) were synthesized and their optical properties were investigated. Initially, the TPAQs displayed aggregation-induced emission enhancement (AIEE) properties via the restricted intramolecular charge transfer (ICT) state. Interestingly, the single-branched fluorophore (STPAQ) and its protonated cation emitted different color fluorescence in the solution and aggregation state. They emitted green fluorescence, which originated from the intramolecular charge transfer (ICT) state in a strong polar solvent, but the fluorescence bands turned blue, which was attributed to the LE state in the aggregated state. However, the cations of triple-branched fluorophores (TTPAQs) exhibited an inverse tunable emission process from bluish violet fluorescence of the LE state in a weak polar solvent (e.g., THF) to green fluorescence of the ICT state in the aggregated state. In a THF/water mixture solution (fw = 10%), the STPAQ could switch its emission between blue and green in the pH range of 10.0–0.5. This phenomenon enabled STPAQ to serve as a fluorescent pH sensor in solution. In the powder state, double-branched fluorophores (DTPAQs) could be used as a fluorescent sensor for the detection of acidic and basic organic vapors in the solid state.
Co-reporter:Wen Yang, Jing Li, Weiqun Zhou, Renyu Xue, Zhongqing Cheng, Mengying Li, Haili Liu, Youyong Li
Journal of Luminescence 2017 Volume 188(Volume 188) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.jlumin.2017.04.061
We synthesized 4, 4′-Di ((E)-2-(4-4-aniline) hydrazinyldiphenyl sulfones (DHS1-4) of D-π-A-π-D and characterized them using NMR, IR, and element analysis. The investigation of photophysical properties in THF and aggregation-induced emission (AIE) effects in THF/H2O mixture solvent of these fluorophores were carried out respectively. DHS-4 displayed the strongest AIE effect in THF/H2O mixture solvent, and the value of I/I0 reached to 136. The nano-size aggregate states in THF/H2O mixture solvent of these compounds were determined by Scanning Electronic Microscopy (SEM) technology and fluorescent microscope imaging. AIE mechanism was explained by molecular dynamics (MD) simulation methods. The special packing manners of V-shape molecules can avoid effectively face-to-face π-π stacking. The inter-molecular hydrogen bonding interactions affected the packing in the aggregate state and assisted their RIR (restricted intramolecular rotation) processes. Furthermore, living cell imaging of DHS-3 showed the bright blue fluorescence in A549 living cell. It indicates that DHS-3 can penetrate the cell membrane and aggregate in A549 living cell, which expands a new area to cell fluorescence staining.
Co-reporter:Jing Li, Wen Yang, Weiqun Zhou, Chunchun Li, Zhongqing Cheng, Mengying Li, Liqun Xie and Youyong Li
RSC Advances 2016 vol. 6(Issue 42) pp:35833-35841
Publication Date(Web):01 Apr 2016
DOI:10.1039/C6RA02209J
Four D–π–A–π–D structured 4,4′-di(E)-2-(4-4-alkoxyaniline)hydrazinyldiphenyl sulfones were synthesized and their structures were characterized using NMR, IR, and element analysis. These compounds displayed weak fluorescence emission in THF while their fluorescence was enhanced in a THF/water mixture solvent. Fluorescence microscope imaging showed the formation of the aggregate state. The mechanism of aggregation-induced emission (AIE) was supported by molecular dynamics calculations. The intermolecular hydrogen bonds between adjacent molecules inhibited the intramolecular rotations, preventing π–π stacking and causing AIE enhancement. Living cell imaging experiments opened up a new method for cell fluorescence staining.
Co-reporter:Xiang Chen, Jun-Qi Zhang, Shao-Jie Yin, Hai-Yan Li, Wei-Qun Zhou, and Xing-Wang Wang
Organic Letters 2015 Volume 17(Issue 17) pp:4188-4191
Publication Date(Web):August 19, 2015
DOI:10.1021/acs.orglett.5b01951
An enantioselective formal thio[3 + 3] spiroannulation reaction of indoline-2-thiones to 1-azadienes has been developed by the use of a quinine-derived bifunctional tertiary amine-thiourea catalyst, which furnished a series of optically active spiro[thiopyranoindole-benzoisothiazole] heterocycles with a spiro quaternary C–N/C–S stereogenic centers in high yields with good to excellent diastereo- and enantioselectivities.
Co-reporter:Juan Xie;Zhongqin Cheng;Wen Yang;Huanhuan Liu;Weiqun Zhou;Mengying Li;Yunlong Xu
Applied Organometallic Chemistry 2015 Volume 29( Issue 3) pp:157-164
Publication Date(Web):
DOI:10.1002/aoc.3262
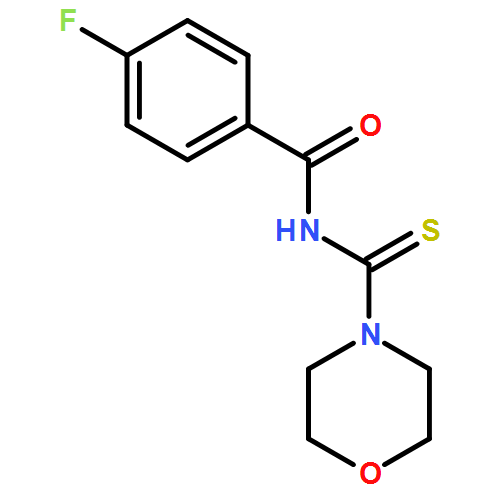
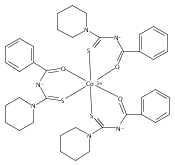
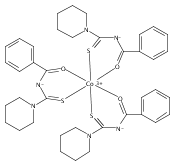
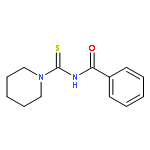
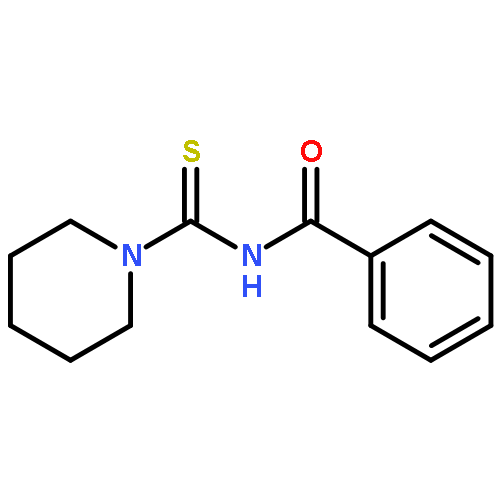
A new zinc(II) complex of N-(piperidylthiocarbonyl)benzamide, [ZnL2], has been synthesized and characterized using elemental analysis, Fourier transform infrared and 1H NMR spectroscopies. X-ray diffraction indicates that [ZnL2] presents a tetrahedral structure within an O2S2 donor set, which is different from analogous square planar [NiL2] and [CuL2] available in the literature. The antimicrobial activities of [ZnL2], [NiL2] and [CuL2] were evaluated against fungi and bacteria. The results show that [ZnL2] is the best for control of the studied fungi and bacteria, and its antimicrobial activity is close to that exhibited by commercial products. The relationship between the structures and antimicrobial activities of the complexes was further investigated using density functional theory calculations. It is elucidated that the increase of the polarity of carbonyl and thiocarbonyl groups determines antifungal and antibacterial activities. Moreover, the cytotoxicity of the complexes was tested against human cancer cells (hepatocellular carcinoma (SK-Hep-1) and breast carcinoma (MCF-7)). The [CuL2] complex is found to be the most cytotoxic. Copyright © 2015 John Wiley & Sons, Ltd.
Co-reporter:Yunlong Xu, Wen Yang, Jie Shao, Weiqun Zhou, Wei Zhu and Juan Xie
RSC Advances 2014 vol. 4(Issue 30) pp:15400-15405
Publication Date(Web):26 Mar 2014
DOI:10.1039/C4RA00034J
A simple probe (1, 1-(4-cyano-phenyl)-3-(4-dimethylamino-phenyl)-thiourea)) is prepared as a ligand for metal ions and characterized by elemental analysis, nuclear magnetic resonance (1H/13C NMR), mass spectroscopy and Fourier transform infrared spectrometry (FTIR). The probe exhibits fluorescence enhancement upon exposure to chromium(III) in THF–water (v/v = 9/1) buffer solution (trihydroxymethyl aminomethane–hydrochloric acid (Tris–HCl), pH = 7.4). A chelation photo-induced electronic transfer (PET) mechanism is introduced to explain the enhancement of the fluorescence and its high selectivity and sensitivity. It is found that the probe has a detection limit of 8.18 × 10−7 M. Furthermore, fluorescence imaging experiments of the exciplex (1@Cr3+) in living human glioma cells (U251) evidence its potential for practical application in biological systems.
Co-reporter:Wen Yang, Jie Shao, Yunlong Xu, Weiqun Zhou, Juan Xie
Journal of Photochemistry and Photobiology A: Chemistry 2014 Volume 292() pp:49-55
Publication Date(Web):15 October 2014
DOI:10.1016/j.jphotochem.2014.07.015
•One simple structure probe based on thiourea moiety was developed successfully.•I− can be discriminated according to the fluorescence quenching in THF/water (v/v = 9/1) Tris–HCl buffer solution.•The probe is applied to determine iodide in powdered milk and urine samples, and the result is satisfactory.•The mechanism of the fluorescence quenching is supported by quantum chemical calculation, UV and 1H NMR titration.1-(4-acetyl-phenyl)-3-(4-N,N-dimethylaminophenyl)-thiourea is synthesized and characterized by NMR, IR, MS and elemental analysis. The probe exhibits the “turn-off” fluorescence response to iodide anion in THF/H2O (v/v = 9/1) pH = 7.4 Tris–HCl buffer solution, which is attributed to the intramolecular charge transfer (ICT). It is found that the probe has a detection limit of 0.336 μM. Such interesting results could be further supported by quantum chemical calculation, UV and 1H NMR titration. Moreover, the probe is applied to determine iodide in powdered milk and urine samples. This experiment shows a good agreement between added and detected concentration of the iodide in real samples.
Co-reporter:Huanhuan Liu, Wen Yang, Weiqun Zhou, Yunlong Xu, Juan Xie, Mengying Li
Inorganica Chimica Acta 2013 Volume 405() pp:387-394
Publication Date(Web):24 August 2013
DOI:10.1016/j.ica.2013.06.029
•Crystal structures of three complexes were determined by X-ray diffraction.•The complexes have stronger antifungal ability than those of corresponding ligands.•The factors influencing the antibacterial abilities of complexes are analyzed.Cu(II) complexes with N-4-fluorobenzoylpiperidine-1-carbothioimidate (L1−), N-2-fluorobenzoylpiperidine-1-carbothioimidate (L2−) and 2-fluorobenzoate (L3−) have been synthesized and characterized by elemental analysis, Fourier Transform infrared (FT-IR), Ultraviolet (UV) and fluorescence spectroscopy methods. The crystal structures of [Cu(L1)2], [Cu(L2)2] and [Cu2(L3)4(DMF)2] (DMF: N,N-dimethylformamide) have been determined by X-ray diffraction method. The antimicrobial activities of the complexes against the bacteria (Escherichia coli, Staphylococcus aureus, Bacillus subtilis, Pseudomonas aeruginosa and Shewanella sp.) and the fungi (Botrytis cinerea, Trichoderma spp., Myrothecium and Verticillium spp.) have been investigated comparing with the corresponding ligands. The experiments showed that the complexes had the stronger antibacterial and antifungal activities than the corresponding ligands had.Three crystal structures of complexes with Cu(II) were determined by X-ray diffraction method and their antimicrobial activities against tested microorganism were investigated.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Wen Yang, Huanhuan Liu, Mengying Li, Fan Wang, Weiqun Zhou, Jianfen Fan
Journal of Inorganic Biochemistry 2012 Volume 116() pp:97-105
Publication Date(Web):November 2012
DOI:10.1016/j.jinorgbio.2012.08.001
Four new thiocarbonyl fluorobenzamides and their complexes with cobalt have been synthesized and characterized by elemental analysis, FTIR, and 1H NMR. Five crystal structures of the thioylbenzamides complexes of Co(PTCB)3, Co(2FPTCB)3, Co(4FPTCB)3, Co(2FMTCB)2 and Co(4FMTCB)3 have been determined by X-ray diffraction. The antibacterial properties of these compounds against the bacteria, E. coli, Staphylococcus aureus, B. subtilis, P. aeruginosa, and Shewanella sp. were investigated. The experiments showed that both compounds and the complexes had the antibacterial activities against all of the studied bacteria. The thioylbenzamides had stronger controls for the bacteria of E. coli, S. aureus, B. subtilis and P. aeruginosa than their corresponding cobalt complexes. There was the contrary result against the bacteria of Shewanella sp. The para‐substitution of fluorine atom increased antibacterial activities, while fluorine atom was substituted on ortho-benzoyl, the antibacterial activity weakened. The thioylbenzamides linked to piperidine instead of a morpholine group may increase the antibacterial activities.Synthesis, crystal structures and antibacterial activity of benzoylthiourea derivatives and their complexes with cobalt.Highlights► Four new thiocarbonyl fluorobenzamides and their complexes with cobalt have been synthesized and characterized. ► Five new crystal have been obtained and their structures have been determined by X-ray diffraction. ► The experiments showed that both ligands and the complexes had the antibacterial activity against the assayed bacteria. ► The para‐substitution of fluorine atom increased antibacterial activities. ► The thioylbenzamides linked to piperidine instead of a morpholine group may increase the antibacterial activity.
Co-reporter:Wen Yang, Wei Zhu, Weiqun Zhou, Huanhuan Liu, Yunlong Xu and Jianfen Fan
RSC Advances 2012 vol. 2(Issue 24) pp:8998-9004
Publication Date(Web):22 Aug 2012
DOI:10.1039/C2RA20997G
Three donor–acceptor benzoylthiourea derivatives were characterized structurally by FTIR and NMR methods. Electronic spectra were investigated by UV absorption and steady fluorescence methods. Quadruple fluorescence bands on the fluorescence spectra were observed and re-assigned. Differently from the previous assignment for benzanilide (BA), the fluorescence bands with emission maxima of 480–524 nm were attributed to ESIPT (excited state intermolecular proton transfer). The fluorescence bands in the region 420–460 nm were explained by the TICT (twisted intramolecular charge transfer) model. Two short-wavelength fluorescence emissions at about 300 and 360 nm were assigned to the S2 state of two rotating isomers characteristic of the nature of the local excited (LE) state. The proposed mechanism for the emission was quantitatively supported by MP2 and CASSCF calculations.
Co-reporter:Wen Yang, Wei Zhu, Weiqun Zhou, Huanhuan Liu, Jianfen Fan
Journal of Fluorine Chemistry 2012 Volume 144() pp:38-44
Publication Date(Web):December 2012
DOI:10.1016/j.jfluchem.2012.09.005
Crystal structures of two benzoylthiourea isomers, named N-4-fluorobenzoyl-N′-4-tolylthiourea (FBTT4) and N-2-fluorobenzoyl-N′-2-tolylthiourea (FBTT2) were determined by X-ray diffraction method. It was found that intra- and intermolecular hydrogen bonds played an essential role in determining their conformations. Electronic spectra of these two isomers (FBTTs) were investigated by UV absorption spectra. The UV absorption bands indicating n → π* transition become widened due to intramolecular hydrogen bonds, while intermolecular hydrogen bonds between FBTTs and the solvent molecules led to unusual blue shifts of the UV absorption band for π → π* transition. The intensity of UV absorption decreased as increasing the solvent polarity and the formation of intermolecular hydrogen bonds. The absorption spectra of two isomers have been calculated with the TDDFT formalism. The calculated absorption curves by the CPCM model considering the first solvent shell match well with the experimental results.Graphical abstractWe have studied the hydrogen bonding interactions in two isomers of fluorobenzoylthioureas and their absorption spectra by theoretical and experimental methods.Highlights► Crystal structures of two benzoylthiourea isomers were determined by X-ray diffraction method. ► Intra- and intermolecular hydrogen bonds affect each other in the crystals. ► The intramolecular hydrogen bonds broaden the UV absorption bands of n → π* transition. ► The intermolecular hydrogen bonds cause to unusual blue shifts of the UV absorption band for π → π* transition.
Co-reporter:Wen Yang;Wei Zhu;Weiqun Zhou;Huanhuan Liu;Yunlong Xu
Journal of Fluorescence 2012 Volume 22( Issue 5) pp:1383-1393
Publication Date(Web):2012 September
DOI:10.1007/s10895-012-1077-6
Two benzoylthiourea isomers, N-2-flurobenzoy-N’-4- (N,N-dimethyl)amidophenylthiourea (2FBDAPT) and N-4-fluro-benzoy-N’-4- (N,N-dimethyl)amidophenylthiourea (4FBDAPT) were determined by fourier transform infrared spectroscopy (FTIR), nuclear magnetic resonance (NMR) and X-ray diffraction. It was found that intra- and intermolecular hydrogen bonds played an important role in determining their conformations. Electronic spectra of the two compounds were investigated by UV absorption and steady-state fluorescence methods. The intermolecular hydrogen bond between the title compounds and methanol molecules caused the long wavelength absorption bands in methanol to weaken and vanish indeed. Quadruple fluorescence bands in ultraviolet and visible region were observed in the studied solvents upon the variable excitation wavelength. As same as Azumaya’s suggestions for benzanilide (BA), F4 fluorescence bands with the maximum wavelength (λmax) between 546 nm and 622 nm were characteristic of TICT fluorescence. F3 bands of λmax from 434 nm to 483 nm were explained by the ESIPT model of the S1 state of the thiol tautomer to the S1 state of the keto tautomer. The new proposition was that F2 bands with λmax at about 365 nm were attributed to ESIPT from the S1 state of the thiol tautomer to the S0 state of the enol tautomer. And F1 fluorescence emissions with λmax at about 310 nm originated from the local S1 transitions of the enol tautomer. All experimental results were supported by MP2, CASSCF and CASPT2 quantum chemical calculations.
Co-reporter:Wei Zhu, Wen Yang, Weiqun Zhou, Huanhuan Liu, Shuanghua Wei, Jianfen Fan
Journal of Molecular Structure 2011 Volume 1004(1–3) pp:74-81
Publication Date(Web):12 October 2011
DOI:10.1016/j.molstruc.2011.07.028
A new crystal of N-2-flurobenzoyl-N′-4-tolylthiourea (FBTT) was obtained from slow evaporation of solvent benzene and the crystal structure of FBTT was determined by X-ray diffraction method. The packing mode of FBTT molecules was affected by crystallization conditions. The new crystal of FBTT crystallizes in the monoclinic with the space group C2/c, and it was stabilized by two pairs of intermolecular interactions. The first pair of intermolecular hydrogen bonds are N(1#)–H(1#)⋯S(1) and N(1)–H(1)⋯S(1#1), the second pair of intermolecular hydrogen bonds, are C(11#)–H(11#)⋯O(1) and C(11)–H(11)⋯O(1#). There also have other unpaired intermolecular hydrogen bonds C(15)–H(15C)⋯O(1#1) and C(15#2)–H(15#2)⋯O(1). Double fluorescence bands are observed in both non-polar and polar solvents. The fluorescence emission at 350–360 nm originates from the transitions of π∗ → π state and is assigned to S2 fluorescence for the nature of LE state. The long wavelength fluorescence emission with large Stokes shift is the characteristic of ESIPT state. All experimental results are supported by MP2 and CASSCF calculations.Highlights► A new crystal structure of thiourea is determined by X-ray diffraction method. ► The crystal is stabilized by some intra- and intermolecular hydrogen bonding interactions. ► The fluorescence bands with large Stokes shift present an ESIPT fluorescence characteristic. ► The fluorescence emission at 350–360nm is assigned to S2 fluorescence for the nature of LE state. ► The experimental results are supported by MP2 and CASSCF quantum chemical calculations.
Co-reporter:Wen Yang, Weiqun Zhou, Ke Peng
Journal of Molecular Structure: THEOCHEM 2008 Volume 858(1–3) pp:101-106
Publication Date(Web):15 June 2008
DOI:10.1016/j.theochem.2008.02.024
Structure and stability of thioureate anion with the water clusters CSNH2NH−-(H2O)n = 1–7, are studied, using density functional theory. Molecular structures and the stability of the clusters are discussed based on the calculation results of the stable conformers and their relative energies. All the clusters are stable thermodynamically in gas phase with respect to separate monomers. The clusters are stabilized progressively with an increasing number of water molecules, as indicated by the increasing of the binding energies. The binding energies of CSNH2NH− and a water molecule are 14.34 and 16.36 kcal/mol for cis CSNH2NH− and trans CSNH2NH−, respectively. As the reaction in aqueous solution progresses, the CS bond distance increases monotonically, indicating that the CS bond of the thioureate anion unit in the clusters is de-stabilized with an increasing number of water molecules.
Co-reporter:Weiqun Zhou, Ke Peng, Wen yang, Minfeng Wang
Journal of Molecular Structure: THEOCHEM 2008 Volume 850(1–3) pp:121-126
Publication Date(Web):15 February 2008
DOI:10.1016/j.theochem.2007.10.042
This article presents a theoretical study on the oxidation reaction of thiourea by hydrogen peroxide in water or alkaline solutions using density functional and ab initio theories. This work also focuses on the analysis of the thermodynamic and kinetic properties of the predicted oxidation mechanism of thiourea using density functional and ab initio theories. The calculated results show that the activation energies, activation enthalpies, and activation Gibbs free energies of the reaction decreased and the releasable reaction energies, enthalpies and Gibbs free energies increased with the cooperation of water or hydroxyl anion. We conclude that the oxidation reaction of thiourea by hydrogen peroxide in water or alkaline solutions was easier and more completed than that in the gas state. The calculated results are consistent with the experiments.
Co-reporter:Qiu Li-Hua;Zhou Wei-Qun;Shen Zong-Xuan
Journal of Chemical Crystallography 2007 Volume 37( Issue 3) pp:181-186
Publication Date(Web):2007 March
DOI:10.1007/s10870-006-9130-4
The title compound (C23H30N2O), a chiral ligand, has been successfully synthesized from a natural proline. FTIR spectrum has been discussed. DFT (B3LYP) method has been used to determine the structure. With the basis set of the 6–311G* quality, the DFT calculated bond parameters and harmonic vibrations are predicted in very good agreement with experimental data.
Co-reporter:Weiqun Zhou, Ke Peng, Fu-Ming Tao
Journal of Molecular Structure: THEOCHEM 2007 Volume 821(1–3) pp:116-124
Publication Date(Web):1 November 2007
DOI:10.1016/j.theochem.2007.06.031
This article presents a theoretical reaction mechanism for the oxidation of thiourea by hydrogen peroxide using density functional theory and ab initio methods. This work also focuses on the analysis of the potential energy surface (PES), thermodynamic and kinetic properties for the designed oxidation mechanism of thiourea using BH&HLYP and B3LYP methods. Both DFT calculations lead to similar reaction paths. The results of the thermodynamic analysis for the designed oxidation mechanism of thiourea are reasonable and feasible. Based on the research on the activity energies, activity enthalpies and activity Gibbs free energies obtained from the determined reaction pathways, we conclude that the oxidation of thiourea by hydrogen peroxide in vacuum was well completed. Because the active relative energies, the relative enthalpies and the relative Gibbs free energies of Reaction 4 predicted by Path II are higher than those predicted by Path I. Kinetics calculations suggest that the designed Path I is more favorable than Path II. The basis sets and calculation methods have strong effects on the thermodynamics and kinetics calculations.
Co-reporter:Zhou Weiqun, Li Baolong, Cao Yang, Zhang Yong, Lu Lude Yang Xujie
Journal of Molecular Structure: THEOCHEM 2005 Volume 715(1–3) pp:117-124
Publication Date(Web):28 February 2005
DOI:10.1016/j.theochem.2004.09.058
The structure of the N-2-fluorobenzoyl-N′-2-methoxyphenyl thiourea (C15H13FN2O2S, Mr=304.34) derived from X-ray diffraction on single crystal has been presented. The crystal is triclinic, space group: P−1 (#2), a=7.058(4), b=9.067(5), c=11.907(8) Å, α=89.00(2), β=86.59(2), γ=69.02(1)°, V=710.2(8) Å3, Z=2. FTIR, NMR and X-ray diffraction determination give the existence of hydrogen bond interactions. Density functional theory (DFT) using Becke's three-parameter hybrid method with the Lee, Yang, and Parr correlation functional (B3LYP) methods have been used to determine the structure and energies of stable conformers. The optimized geometry corresponding to crystal structure is the most stable conformation. This has partly been attributed to multi six-tempered and five-tempered ring formation resulting from the N(2)–H(2)⋯(1), N–(1) H(1)⋯F(1), N(2)–H(2)⋯O(2) and C(6)–H(6)⋯O(2) intra-molecular hydrogen bonds, respectively. With the basis sets of the 6-311G** quality, the DFT calculated bond parameters, harmonic vibrations and nuclear magnetic resonance values are in a very good agreement with experimental results.
Co-reporter:Weiqun Zhou, Jianmei Lu, Zhenjiang Zhang, Yong Zhang, Yang Cao, Lude Lu, Xujie Yang
Vibrational Spectroscopy 2004 Volume 34(Issue 2) pp:199-204
Publication Date(Web):31 March 2004
DOI:10.1016/j.vibspec.2003.09.006
The structure of the N-4-chlorobenzoyl–N′-4-methoxylphenylthiourea (C15H13N2OClS, Mr=320.79) derived from X-ray diffraction on single crystal has been presented. The crystals are monoclinic, space group: P21/n (#14), a=12.125(2), b=6.3650(6), c=19.755(3) Å, β=76.800(6)°, V=1484.3(3) Å3, Z=4. FT-IR spectrum has been determined. The interactions of intramolecular and intermolecular hydrogen bonds have been discussed. Density functional theory (DFT) (B3LYP) methods have been used to determine the structure and energies of stable conformers. Minimum energy conformations are calculated as a function of the torsion angle θ (C7N1C8N2) varied every 30°. The optimized geometry corresponding to crystal structure is the most stable conformation. This has partly been attributed to intramolecular hydrogen bonds. With the basis sets of the 6-311G∗ quality, the DFT calculated bond parameters and harmonic vibrations are predicted in a very good agreement with experimental data.
Co-reporter:Weiqun Zhou, Liming Zhu, Yong Zhang, Zhengfeng Yu, Lude Lu, Xujie Yang
Vibrational Spectroscopy 2004 Volume 36(Issue 1) pp:73-78
Publication Date(Web):18 October 2004
DOI:10.1016/j.vibspec.2004.03.002
The complex Ni(mtcb)2 (mtcb = N-(morpholinothiocarbonyl)benzamide) has been synthesized and characterized by elemental analysis, FTIR. The crystal structure was determined by X-ray diffraction analysis. Ni(mtcb)2·(1/2) toluene crystallized is in the triclinic space group , with Z=2. Unit cell parameters were as a=8.246(4) Å, b=11.594(5) Å, c=15.712(6) Å, α=103.885(5)°, β=97.358(5)°, γ=104.543(4)°, V=1382.9(10) Å3. Intra-molecular hydrogen bonded interaction have been discussed. DFT (B3LYP) methods have been used as predicted N-(morpholinothiocarbonyl)benzamide molecular structure and vibrational spectra. There is good agreement between theoretical vibrational spectra and FTIR.
Co-reporter:W.Q. Zhou, L.M. Zhu, Zh.B. Cao, Y. Zhang, W.D. Lu, L.D. Lu
Journal of Molecular Structure 2003 Volume 655(Issue 3) pp:405-411
Publication Date(Web):13 August 2003
DOI:10.1016/S0022-2860(03)00283-7
4,4′-Carbonyl-di-morpholine was synthesized and characterized by X-ray diffraction, the FTIR and NMR spectra. The extended MO calculations using density functional theory (DFT) and self-consistent field molecular orbital Hartree–Fock theory were carried out. The results of the calculations were compared with experimental data. The experimental and calculated results were supported each other. The performance of a hybrid B3LYP density functional was compared with the ab initio restricted Hartree–Fock method. With the basis sets of the 6-311G** quality, the DFT calculated bond lengths, dipole moments and harmonic vibrations were predicted in a very good agreement with available experimental data.
Co-reporter:Zhou Weiqun, Leng Kuisheng, Zhang Yong, Lude Lu
Journal of Molecular Structure 2003 Volume 657(1–3) pp:215-223
Publication Date(Web):September 2003
DOI:10.1016/S0022-2860(03)00371-5
The crystal and molecular structure of N-(4-chloro)benzoyl-N′-2-tolylthiourea (C15H13N2OSCl, Mr=304.79) is determined by X-ray diffraction. The crystal structure is monoclinic, space group P21/n with a=3.9898(3), b=22.922(2), β=104.367(4)°, Z=4, The results of extended MO calculations using density functional theory (DFT) with a hybrid B3LYP density functional approximation and FTIR and NMR studies on the title compound are reported. With the basis sets of the 6-311G* quality, the DFT calculated bond parameters, harmonic vibrations and chemical shifts are in a very good agreement with experimental data. The intramolecular and intermolecular hydrogen bonds are discussed. Non-linear optical properties of the molecule are predicted.

![[4-(phenylsulphonyl)phenyl]hydrazine](http://img.cochemist.com/ccimg/70800/70714-83-9.png)
![[4-(phenylsulphonyl)phenyl]hydrazine](http://img.cochemist.com/ccimg/70800/70714-83-9_b.png)
![(2e)-3-[4-(dimethylamino)phenyl]-1-(4-fluorophenyl)-2-propen-1-on E](http://img.cochemist.com/ccimg/28100/28081-19-8.png)
![(2e)-3-[4-(dimethylamino)phenyl]-1-(4-fluorophenyl)-2-propen-1-on E](http://img.cochemist.com/ccimg/28100/28081-19-8_b.png)
![[4-(4-hydrazinylphenyl)sulfonylphenyl]hydrazine](http://img.cochemist.com/ccimg/14100/14052-65-4.png)
![[4-(4-hydrazinylphenyl)sulfonylphenyl]hydrazine](http://img.cochemist.com/ccimg/14100/14052-65-4_b.png)






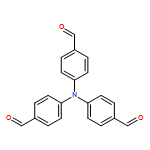

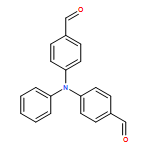
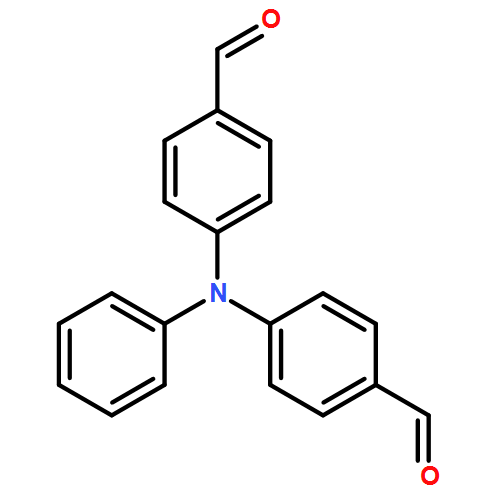
![Benzamide, 2-fluoro-N-[[(2-methylphenyl)amino]thioxomethyl]-](/data/chemimg/135700/536722-20-0.png)
![Benzamide, 2-fluoro-N-[[(2-methylphenyl)amino]thioxomethyl]-](/data/chemimg/135700/536722-20-0_b.png)
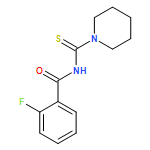
![Benzamide, 4-fluoro-N-[[(4-methylphenyl)amino]thioxomethyl]-](/data/chemimg/135700/446831-62-5.png)
![Benzamide, 4-fluoro-N-[[(4-methylphenyl)amino]thioxomethyl]-](/data/chemimg/135700/446831-62-5_b.png)
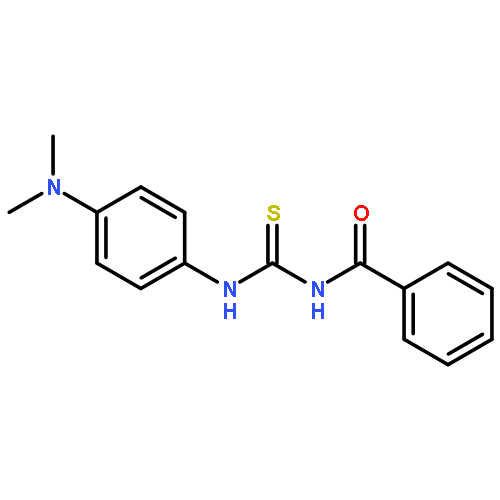
![Benzamide, N-[[[4-(dimethylamino)phenyl]amino]thioxomethyl]-2-fluoro-](/data/chemimg/135700/425682-41-3.png)
![Benzamide, N-[[[4-(dimethylamino)phenyl]amino]thioxomethyl]-2-fluoro-](/data/chemimg/135700/425682-41-3_b.png)
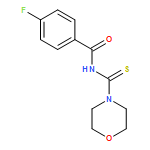
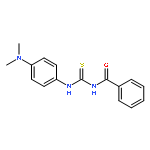
![Benzamide, N-[[[4-(dimethylamino)phenyl]amino]thioxomethyl]-4-fluoro-](/data/chemimg/135700/335212-90-3.png)
![Benzamide, N-[[[4-(dimethylamino)phenyl]amino]thioxomethyl]-4-fluoro-](/data/chemimg/135700/335212-90-3_b.png)
![Benzamide, 2-fluoro-N-[[(4-methylphenyl)amino]thioxomethyl]-](/data/chemimg/135700/156005-06-0.png)
![Benzamide, 2-fluoro-N-[[(4-methylphenyl)amino]thioxomethyl]-](/data/chemimg/135700/156005-06-0_b.png)


![Propanedinitrile,2-[(3-methylphenyl)methylene]-](http://img.cochemist.com/ccimg/15800/15728-26-4.png)
![Propanedinitrile,2-[(3-methylphenyl)methylene]-](http://img.cochemist.com/ccimg/15800/15728-26-4_b.png)


![Propanedinitrile,2-[(3-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/3000/2972-72-7.png)
![Propanedinitrile,2-[(3-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/3000/2972-72-7_b.png)
![Propanedinitrile,2-[(3-nitrophenyl)methylene]-](http://img.cochemist.com/ccimg/2900/2826-32-6.png)
![Propanedinitrile,2-[(3-nitrophenyl)methylene]-](http://img.cochemist.com/ccimg/2900/2826-32-6_b.png)
![Propanedinitrile,2-[(4-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/2900/2826-26-8.png)
![Propanedinitrile,2-[(4-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/2900/2826-26-8_b.png)
![2-[(4-methylphenyl)methylidene]propanedinitrile](http://img.cochemist.com/ccimg/2900/2826-25-7.png)
![2-[(4-methylphenyl)methylidene]propanedinitrile](http://img.cochemist.com/ccimg/2900/2826-25-7_b.png)
![Propanedinitrile,2-[(2-bromophenyl)methylene]-](http://img.cochemist.com/ccimg/2700/2698-42-2.png)
![Propanedinitrile,2-[(2-bromophenyl)methylene]-](http://img.cochemist.com/ccimg/2700/2698-42-2_b.png)
![Propanedinitrile,2-[(4-chlorophenyl)methylene]-](http://img.cochemist.com/ccimg/1900/1867-38-5.png)
![Propanedinitrile,2-[(4-chlorophenyl)methylene]-](http://img.cochemist.com/ccimg/1900/1867-38-5_b.png)
![9-Octadecenoic acid(9Z)-,(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-2-[(1-oxohexadecyl)oxy]ethylester](http://img.cochemist.com/ccimg/26700/26662-94-2.png)
![9-Octadecenoic acid(9Z)-,(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-2-[(1-oxohexadecyl)oxy]ethylester](http://img.cochemist.com/ccimg/26700/26662-94-2_b.png)


![9-Octadecenoic acid(9Z)-,1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-2-[(1-oxohexadecyl)oxy]ethylester](http://img.cochemist.com/ccimg/10100/10015-88-0.png)
![9-Octadecenoic acid(9Z)-,1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-2-[(1-oxohexadecyl)oxy]ethylester](http://img.cochemist.com/ccimg/10100/10015-88-0_b.png)