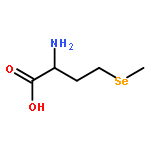
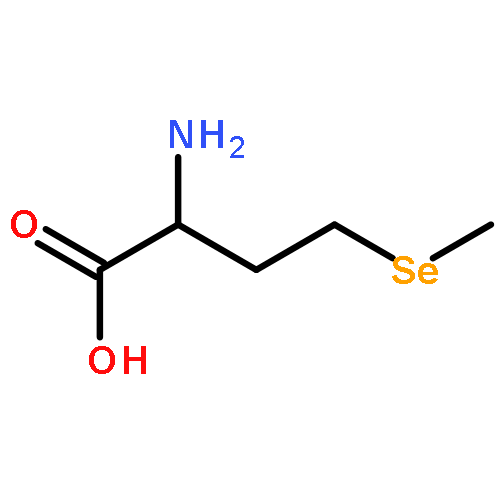
Co-reporter:Wenli Meng;Daniel P. Raleigh;Nicholas Lyle;Rohit V. Pappu;Bowu Luan
PNAS 2013 Volume 110 (Issue 6 ) pp:2123-2128
Publication Date(Web):2013-02-05
DOI:10.1073/pnas.1216979110
The sizes of unfolded proteins under highly denaturing conditions scale as N0.59 with chain length. This suggests that denaturing conditions mimic good solvents, whereby the preference for favorable chain–solvent
interactions causes intrachain interactions to be repulsive, on average. Beyond this generic inference, the broader implications
of N0.59 scaling for quantitative descriptions of denatured state ensembles (DSEs) remain unresolved. Of particular interest is the
degree to which N0.59 scaling can simultaneously accommodate intrachain attractions and detectable long-range contacts. Here we present data showing
that the DSE of the N-terminal domain of the L9 (NTL9) ribosomal protein in 8.3 M urea lacks detectable secondary structure
and forms expanded conformations in accord with the expected N0.59 scaling behavior. Paramagnetic relaxation enhancements, however, indicate the presence of detectable long-range contacts
in the denatured-state ensemble of NTL9. To explain these observations we used atomistic thermal unfolding simulations to
identify ensembles whose properties are consistent with all of the experimental observations, thus serving as useful proxies
for the DSE of NTL9 in 8.3 M urea. Analysis of these ensembles shows that residual attractions are present under mimics of
good solvent conditions, and for NTL9 they result from low-likelihood, medium/long-range contacts between hydrophobic residues.
Our analysis provides a quantitative framework for the simultaneous observation of N0.59 scaling and low-likelihood long-range contacts for the DSE of NTL9. We propose that such low-likelihood intramolecular hydrophobic
clusters might be a generic feature of DSEs that play a gatekeeping role to protect against aggregation during protein folding.
Co-reporter:Caitlin M. Davis ; Shifeng Xiao ; Daniel P. Raleigh ;R. Brian Dyer
Journal of the American Chemical Society 2012 Volume 134(Issue 35) pp:14476-14482
Publication Date(Web):August 8, 2012
DOI:10.1021/ja3046734
Understanding the folding of the β-hairpin is a crucial step in studying how β-rich proteins fold. We have studied CLN025, an optimized ten residue synthetic peptide, which adopts a compact, well-structured β-hairpin conformation. Formation of the component β-sheet and β-turn structures of CLN025 was probed independently using a combination of equilibrium Fourier transform infrared spectroscopy and laser-induced temperature jump coupled with time-resolved infrared and fluorescence spectroscopies. We find that CLN025 is an ultrafast folder due to its small free energy barrier to folding and that it exceeds the predicted speed limit for β-hairpin formation by an order of magnitude. We also find that the folding mechanism cannot be described by a simple two-state model, but rather is a heterogeneous process involving two independent parallel processes. Formation of stabilizing cross-strand hydrophobic interactions and turn alignment occur competitively, with relaxation lifetimes of 82 ± 10 and 124 ± 10 ns, respectively, at the highest probed temperature. The ultrafast and heterogeneous folding kinetics observed for CLN025 provide evidence for folding on a nearly barrierless free energy landscape, and recalibrate the speed limit for the formation of a β-hairpin.
Co-reporter:Ping Cao and Daniel P. Raleigh
Biochemistry 2012 Volume 51(Issue 13) pp:
Publication Date(Web):March 12, 2012
DOI:10.1021/bi2015162
Islet amyloid polypeptide (IAPP, amylin) is responsible for amyloid formation in type 2 diabetes and in transplanted islets. The flavanol (−)-epigallocatechin-3-gallate [EGCG; (2R,3R)-5,7-dihydroxy-2-(3,4,5-trihydroxyphenyl)-3,4-dihydro-2H-1-benzopyran-3-yl 3,4,5-trihydroxybenzoate] is an effective inhibitor of amyloid formation by IAPP; however, the interactions required for the inhibition of IAPP amyloid formation and for the remodeling of amyloid fibers are not known. A range of features have been proposed to be critical for EGCG protein interactions, including interactions with aromatic residues, interactions with amino groups, or sulfhydryls. Using a set of IAPP analogues, we show that none of these are required. Studies in which EGCG is added to the lag phase of amyloid formation shows that it interacts with intermediates as well as with monomers and amyloid. The features of EGCG required for effective inhibition were examined. The stereoisomer of EGCG, (−)-gallocatechin gallate (GCG), is an effective inhibitor, although less so than EGCG. Removing the gallate ester moiety leads to EGC which is a less effective inhibitor. Removing only the 3-hydroxyl group of the trihydroxyphenyl ring leads to a compound that has more pronounced effects on the lag phase than EGC but is less effective at reducing the amount of amyloid. Elimination of both the 3-hydroxy group and the gallate ester results in loss of activity. EGCG remodels IAPP amyloid fibers but does not fully resolubilize them to unstructured monomers, and the remodeling is not the reverse of amyloid assembly. The ability of the compounds to remodel IAPP amyloid closely follows their relative ability to inhibit amyloid formation.
Co-reporter:Peter J. Marek, Vadim Patsalo, David F. Green, and Daniel P. Raleigh
Biochemistry 2012 Volume 51(Issue 43) pp:
Publication Date(Web):September 27, 2012
DOI:10.1021/bi300574r
Amyloid formation plays a role in a wide range of human diseases. The rate and extent of amyloid formation depend on solution conditions, including pH and ionic strength. Amyloid fibrils often adopt structures with parallel, in-register β-sheets, which generate quasi-infinite arrays of aligned side chains. These arrangements can lead to significant electrostatic interactions between adjacent polypeptide chains. The effect of ionic strength and ion composition on the kinetics of amyloid formation by islet amyloid polypeptide (IAPP) is examined. IAPP is a basic 37-residue polypeptide responsible for islet amyloid formation in type 2 diabetes. Poisson–Boltzmann calculations revealed significant electrostatic repulsion in a model of the IAPP fibrillar state. The kinetics of IAPP amyloid formation are strongly dependent on ionic strength, varying by a factor of >10 over the range of 20–600 mM NaCl at pH 8.0, but the effect is not entirely due to Debye screening. At low ionic strengths, the rate depends strongly on the identity of the anion, varying by a factor of nearly 4, and scales with the electroselectivity series, implicating anion binding. At high ionic strengths, the rate varies by only 8% and scales with the Hofmeister series. At intermediate ionic strengths, no clear trend is detected, likely because of the convolution of different effects. The effects of salts on the growth phase and lag phase of IAPP amyloid formation are strongly correlated. At pH 5.5, where the net charge on IAPP is higher, the effect of different anions scales with the electroselectivity series at all salt concentrations.
Co-reporter:Ping Cao ;Daniel P. Raleigh
Journal of the American Chemical Society 2010 Volume 132(Issue 12) pp:4052-4053
Publication Date(Web):March 4, 2010
DOI:10.1021/ja910763m
A major issue in studies of amyloid formation is the difficulty of preparing the polypeptide of interest in an initially monomeric state under physiologically relevant conditions. This is particularly problematic for polypeptides which are natively unfolded in their unaggregated state, and perhaps the most challenging such system is islet amyloid polypeptide (Amylin), the causative agent of amyloid formation in type-2 diabetes. Preparation of islet amyloid polypeptide with the Ser-19 Ser-20 amide bond replaced by an ester circumvents these problems. The modified peptide is unstructured and monomeric at slightly acidic pH’s as judged by analytical ultracentrifugation, gel filtration, dynamic light scattering, and CD. A rapid pH jump leads to deprotonation of the Ser-20 amide group, and a subsequent rapid O to N acyl shift regenerates normal human islet amyloid polypeptide. The half time, t1/2, for the conversion to normal islet amyloid polypeptide is 70 s at pH 7.4. The amyloid fibrils which are formed by the regenerated islet amyloid polypeptide are indistinguishable from those formed by the wild type polypeptide. The approach allows studies of amyloid formation by islet amyloid polypeptide to be carried out from a well-defined, physiologically relevant starting state in the absence of denaturants or organic cosolvents.
Co-reporter:Bing Shan ; Sebastian McClendon ; Carla Rospigliosi ; David Eliezer ;Daniel P. Raleigh
Journal of the American Chemical Society 2010 Volume 132(Issue 13) pp:4669-4677
Publication Date(Web):March 12, 2010
DOI:10.1021/ja908104s
Cold denaturation is a general property of globular proteins, and the process provides insight into the origins of the cooperativity of protein folding and the nature of partially folded states. Unfortunately, studies of protein cold denaturation have been hindered by the fact that the cold denatured state is normally difficult to access experimentally. Special conditions such as addition of high concentrations of denaturant, encapsulation into reverse micelles, the formation of emulsified solutions, high pressure, or extremes of pH have been applied, but these can perturb the unfolded state of proteins. The cold denatured state of the C-terminal domain of the ribosomal protein L9 can be populated under native-like conditions by taking advantage of a destabilizing point mutation which leads to cold denaturation at temperatures above 0 °C. This state is in slow exchange with the native state on the NMR time scale. Virtually complete backbone 15N, 13C, and 1H as well as side-chain 13Cβ and 1Hβ chemical shift assignments were obtained for the cold denatured state at pH 5.7, 12 °C. Chemical shift analysis, backbone N−H residual dipolar couplings, amide proton NOEs, and R2 relaxation rates all indicate that the cold denatured state of CTL9 (the C-terminal domain of the ribosomal protein L9) not only contains significant native-like secondary structure but also non-native structure. The regions corresponding to the two native α-helices show a strong tendency to populate helical Φ and Ψ angles. The segment which connects α-helix 2 and β-strand 2 (residues 107−124) in the native state exhibits a significant preference to form non-native helical structure in the cold denatured state. The structure observed in the cold denatured state of the I98A mutant is similar to that observed in the pH 3.8 unfolded state of wild type CTL9 at 25 °C, suggesting that it is a robust feature of the denatured state ensemble of this protein. The implications for protein folding and for studies of cold denatured states are discussed.
Co-reporter:Fanling Meng ; Daniel P. Raleigh ;Andisheh Abedini
Journal of the American Chemical Society 2010 Volume 132(Issue 41) pp:14340-14342
Publication Date(Web):September 28, 2010
DOI:10.1021/ja1046186
Amyloid formation plays a role in over 25 human disorders. A range of strategies have been applied to the problem of developing inhibitors of amyloid formation, but unfortunately, many inhibitors are effective only in molar excess and typically either lengthen the time to the onset of amyloid formation, (the lag time), while having modest effects on the total amount of amyloid fibrils produced, or decrease the amount of amyloid without significantly reducing the lag time. We demonstrate a general strategy whereby two moderate inhibitors of amyloid formation can be rationally selected via kinetic assays and combined in trans to yield a highly effective inhibitor which dramatically delays the time to the appearance of amyloid and drastically reduces the total amount of amyloid formed. A key feature is that the selection of the components of the mixture is based on their effect on the time course of amyloid formation rather than on just the amount of amyloid produced. The approach is validated using inhibitors of amyloid formation by islet amyloid polypeptide, the causative agent of amyloid formation in type 2 diabetes and the Alzheimer’s disease Aβ peptide.
Co-reporter:Peter Marek, Ann Marie Woys, Kelvin Sutton, Martin T. Zanni, and Daniel P. Raleigh
Organic Letters 2010 Volume 12(Issue 21) pp:4848-4851
Publication Date(Web):October 8, 2010
DOI:10.1021/ol101981b
A cost-efficient, time-reducing solid-phase synthesis of the amyloidogenic, 37 residue islet amyloid polypeptide (IAPP) is developed using two pseudoprolines (highlighted blue in sequence) in combination with microwave technology. A yield twice that obtained with conventional syntheses is realized. The utility of this protocol is demonstrated by the synthesis of a 13C18O-labeled Ser-20 IAPP variant, a prohibitively expensive and chemically challenging site to label via other protocols. TEM analysis shows the peptide forms normal amyloid (abstract image).
Co-reporter:Ping Cao, Fanling Meng, Andisheh Abedini and Daniel P. Raleigh
Biochemistry 2010 Volume 49(Issue 5) pp:
Publication Date(Web):December 22, 2009
DOI:10.1021/bi901751b
Islet amyloid polypeptide (IAPP) is a 37-residue polypeptide hormone that is responsible for islet amyloid formation in type II diabetes. Human IAPP is extremely amyloidogenic, while rat IAPP and mouse IAPP do not form amyloid in vitro or in vivo. Rat IAPP and mouse IAPP have identical primary sequences, but differ from the human polypeptide at six positions, five of which are localized between residues 20 and 29. The ability of rat IAPP to inhibit amyloid formation by human IAPP was tested, and the rat peptide was found to be an effective inhibitor. Thioflavin-T fluorescence-monitored kinetic experiments, transmission electron microscopy, and circular dichroism showed that rat IAPP lengthened the lag phase for amyloid formation by human IAPP, slowed the growth rate, reduced the amount of amyloid fibrils produced in a dose-dependent manner, and altered the morphology of the fibrils. The inhibition of human IAPP amyloid formation by rat IAPP can be rationalized by a model that postulates formation of an early helical intermediate during amyloid formation where the helical region is localized to the N-terminal region of IAPP. The model predicts that proline mutations in the putative helical region should lead to ineffective inhibitors as should mutations that alter the peptide−peptide interaction interface. We confirmed this by testing the ability of A13P and F15D point mutants of rat IAPP to inhibit amyloid formation by human IAPP. Both these mutants were noticeably less effective inhibitors than wild-type rat IAPP. The implications for inhibitor design are discussed.
Co-reporter:Humeyra Taskent-Sezgin, Peter Marek, Rosanne Thomas, Daniel Goldberg, Juah Chung, Isaac Carrico and Daniel P. Raleigh
Biochemistry 2010 Volume 49(Issue 29) pp:
Publication Date(Web):June 21, 2010
DOI:10.1021/bi100932p
p-Cyanophenylalanine is an extremely useful fluorescence probe of protein structure that can be recombinantly and chemically incorporated into proteins. The probe has been used to study protein folding, protein−membrane interactions, protein−peptide interactions, and amyloid formation; however, the factors that control its fluorescence are not fully understood. Hydrogen bonding to the cyano group is known to play a major role in modulating the fluorescence quantum yield, but the role of potential side-chain quenchers has not yet been elucidated. A systematic study of the effects of different side chains on p-cyanophenylalanine fluorescence is reported. Tyr is found to have the largest effect followed by deprotonated His, Met, Cys, protonated His, Asn, Arg, and protonated Lys. Deprotonated amino groups are much more effective fluorescence quenchers than protonated amino groups. Free neutral imidazole and hydroxide ion are also effective quenchers of p-cyanophenylalanine fluorescence with Stern−Volmer constants of 39.8 and 22.1 M−1, respectively. The quenching of p-cyanophenylalanine fluorescence by specific side chains is exploited in developing specific, high-sensitivity, fluorescence probes of helix formation. The approach is demonstrated with Ala-based peptides that contain a p-cyanophenylalanine-His or a p-cyanophenylalanine-Tyr pair located at positions i and i + 4. The p-cyanophenylalanine-His pair is most useful when the His side chain is deprotonated and is, thus, complementary to the Trp-His pair which is most sensitive when the His side chain is protonated.
Co-reporter:Fanling Meng, Andisheh Abedini, Annette Plesner, C. Bruce Verchere, and Daniel P. Raleigh
Biochemistry 2010 Volume 49(Issue 37) pp:
Publication Date(Web):August 13, 2010
DOI:10.1021/bi100939a
Islet amyloid polypeptide (IAPP, amylin) is the major protein component of the islet amyloid deposits associated with type 2 diabetes. The polypeptide lacks a well-defined structure in its monomeric state but readily assembles to form amyloid. Amyloid fibrils formed from IAPP, intermediates generated in the assembly of IAPP amyloid, or both are toxic to β-cells, suggesting that islet amyloid formation may contribute to the pathology of type 2 diabetes. There are relatively few reported inhibitors of amyloid formation by IAPP. Here we show that the tea-derived flavanol, (−)-epigallocatechin 3-gallate [(2R,3R)-5,7-dihydroxy-2-(3,4,5-trihydroxyphenyl)-3,4-dihydro-2H-1-benzopyran-3-yl 3,4,5-trihydroxybenzoate] (EGCG), is an effective inhibitor of in vitro IAPP amyloid formation and disaggregates preformed amyloid fibrils derived from IAPP. The compound is thus one of a very small set of molecules which have been shown to disaggregate IAPP amyloid fibrils. Fluorescence-detected thioflavin-T binding assays and transmission electron microscopy confirm that the compound inhibits unseeded amyloid fibril formation as well as disaggregates IAPP amyloid. Seeding studies show that the complex formed by IAPP and EGCG does not seed amyloid formation by IAPP. In this regard, the behavior of IAPP is similar to the reported interactions of Aβ and α-synuclein with EGCG. Alamar blue assays and light microscopy indicate that the compound protects cultured rat INS-1 cells against IAPP-induced toxicity. Thus, EGCG offers an interesting lead structure for further development of inhibitors of IAPP amyloid formation and compounds that disaggregate IAPP amyloid.
Co-reporter:Humeyra Taskent-Sezgin;Juah Chung;Partha S. Banerjee;Dr. Sureshbabu Nagarajan; R. Brian Dyer; Isaac Carrico; Daniel P. Raleigh
Angewandte Chemie International Edition 2010 Volume 49( Issue 41) pp:7473-7475
Publication Date(Web):
DOI:10.1002/anie.201003325
Co-reporter:K. J. Potter;A. Abedini;P. Marek;A. M. Klimek;S. Butterworth;M. Driscoll;R. Baker;G. L. Warnock;M. R. Nilsson;J. Oberholzer;S. Bertera;M. Trucco;G. S. Korbutt;P. E. Fraser;D. P. Raleigh;C. B. Verchere
PNAS 2010 Volume 107 (Issue 9 ) pp:4305-4310
Publication Date(Web):2010-03-02
DOI:10.1073/pnas.0909024107
Islet transplantation is a promising treatment for diabetes but long-term success is limited by progressive graft loss. Aggregates
of the beta cell peptide islet amyloid polypeptide (IAPP) promote beta cell apoptosis and rapid amyloid formation occurs in
transplanted islets. Porcine islets are an attractive alternative islet source as they demonstrate long-term graft survival.
We compared the capacity of transplanted human and porcine islets to form amyloid as an explanation for differences in graft
survival. Human islets were transplanted into streptozotocin-diabetic immune-deficient mice. Amyloid deposition was detectable
at 4 weeks posttransplantation and was associated with islet graft failure. More extensive amyloid deposition was observed
after 8 weeks. By contrast, no amyloid was detected in transplanted neonatal or adult porcine islets that had maintained normoglycemia
for up to 195 days. To determine whether differences in IAPP sequence between humans and pigs could explain differences in
amyloid formation and transplant viability, we sequenced porcine IAPP. Porcine IAPP differs from the human sequence at 10
positions and includes substitutions predicted to reduce its amyloidogenicity. Synthetic porcine IAPP was considerably less
amyloidogenic than human IAPP as determined by transmission electron microscopy, circular dichroism, and thioflavin T binding.
Viability assays indicated that porcine IAPP is significantly less toxic to INS-1 beta cells than human IAPP. Our findings
demonstrate that species differences in IAPP sequence can explain the lack of amyloid formation and improved survival of transplanted
porcine islets. These data highlight the potential of porcine islet transplantation as a therapeutic approach for human diabetes.
Co-reporter:Jae-Hyun Cho ; Nichole O’Connell ; Daniel P. Raleigh ;Arthur G. Palmer ; III
Journal of the American Chemical Society 2009 Volume 132(Issue 2) pp:450-451
Publication Date(Web):December 22, 2009
DOI:10.1021/ja909052h
Proteins that fold rapidly, on the (sub-) microsecond time scale, offer the prospect of direct comparison between experimental data and molecular dynamics simulations. However, experimental studies for such proteins frequently are hindered because folding rates are too fast to measure using conventional stopped-flow methods. To overcome this impediment, NMR spin relaxation dispersion experiments are used to quantify mutational effects on kinetics (ΔΔG°), stability (ΔΔG°), and ϕ-values (ΔΔG†/ΔΔG°) for proteins exhibiting chemical exchange line broadening that is fast on the NMR chemical shift time scale. The accuracy of ϕ-value analysis is enhanced because mutational effects on denatured or intermediate states can be detected through changes in line broadening. The transition and intermediate states of the villin headpiece domain, HP67, are characterized in varying solvent conditions to validate the method.
Co-reporter:Shifeng Xiao, Yuan Bi, Bing Shan and Daniel P. Raleigh
Biochemistry 2009 Volume 48(Issue 21) pp:
Publication Date(Web):April 8, 2009
DOI:10.1021/bi8021763
The helical subdomain of the villin headpiece is the smallest naturally occurring cooperatively folded protein. Its small size, simple three-helix topology, and very rapid folding have made it an extremely popular model system for computational and theoretical studies of protein folding. The domain has a well-packed hydrophobic core comprised in part of an unusual set of three closely packed phenylalanine residues, F47, F51, and F58 (denoted using the numbering of the larger headpiece protein). Aromatic−aromatic interactions have been thought to play a critical role in specifying the subdomain fold and have been proposed to play a general role in stabilizing small proteins. The modest stability of the subdomain has hindered studies of core packing since multiple mutations can lead to constructs which fail to fold, and even single mutants can result in poorly folded variants. Using a previously characterized hyperstable mutant of the domain, generated by targeting surface residues, a complete set of single, double, and triple core Phe to Leu mutants were characterized. A highly conserved surface Trp which is part of a Trp−Pro interaction was also examined. All mutants are well-folded as judged by CD and NMR, and all exhibit sigmoidal urea and thermally induced unfolding transitions, thus proving that aromatic−aromatic, aromatic−proline, or aromatic−hydrophobic interactions are not required for specifying the subdomain fold. Double mutant cycle analysis demonstrates that F47 and F51 make the strongest pairwise interaction. Mutations which lack F58 are the most destabilized, although even the triple mutant is folded. Interestingly, mutation of the central Phe, F51, has the smallest effect on stability even though it makes contact with both F47 and F58 and appears to form the strongest pairwise interaction.
Co-reporter:Bing Shan, David Eliezer and Daniel P. Raleigh
Biochemistry 2009 Volume 48(Issue 22) pp:
Publication Date(Web):March 20, 2009
DOI:10.1021/bi802299j
Interest in the structural and dynamic properties of unfolded proteins has increased in recent years owing to continued interest in protein folding and misfolding. Knowledge of the unfolded state under native conditions is particularly important for obtaining a complete picture of the protein folding process. The C-terminal domain of protein L9 is a globular α, β protein with an unusual mixed parallel and antiparallel β-strand structure. The folding kinetics and equilibrium unfolding of CTL9 strongly depend on pH, and follow a simple two state model. Both the native and the unfolded state can be significantly populated at pH 3.8 in the absence of denaturant, allowing the native state and the unfolded state to be characterized under identical conditions. Backbone 15N, 13C, 1H and side-chain 13Cβ, 1Hβ chemical shifts, amide proton NOEs, and 15N R2 relaxation rates were obtained for the two conformational states at pH 3.8. All the data indicate that the pH 3.8 native state is well folded and is similar to the native state at neutral pH. There is significant residual structure in the pH 3.8 unfolded state. The regions corresponding to the two native state α-helices show strong preference to populate helical φ and ψ angles. The segment that connects α-helix 2 and β-strand 2 has a significant tendency to form non-native α-helical structure. Comparison with the pH 2.0 unfolded state and the urea unfolded state indicates that the tendency to adopt both native and non-native helical structure is stronger at pH 3.8, demonstrating that the unfolded state of CTL9 under native-like conditions is more structured. The implications for the folding of CTL9 are discussed.
Co-reporter:Humeyra Taskent-Sezgin, Juah Chung, Vadim Patsalo, Shigeki J. Miyake-Stoner, Andrew M. Miller, Scott H. Brewer, Ryan A. Mehl, David F. Green, Daniel P. Raleigh and Isaac Carrico
Biochemistry 2009 Volume 48(Issue 38) pp:
Publication Date(Web):August 6, 2009
DOI:10.1021/bi900938z
The use of noncoded amino acids as spectroscopic probes of protein folding and function is growing rapidly, in large part because of advances in the methodology for their incorporation. Recently p-cyanophenylalanine has been employed as a fluorescence and IR probe, as well as a FRET probe to study protein folding, protein−membrane interactions, protein−protein interactions and amyloid formation. The probe has been shown to be exquisitely sensitive to hydrogen bonding interactions involving the cyano group, and its fluorescence quantum yield increases dramatically when it is hydrogen bonded. However, a detailed understanding of the factors which influence its fluorescence is required to be able to use this popular probe accurately. Here we demonstrate the recombinant incorporation of p-cyanophenylalanine in the N-terminal domain of the ribosomal protein L9. Native state fluorescence is very low, which suggests that the group is sequestered from solvent; however, IR measurements and molecular dynamics simulations show that the cyano group is exposed to solvent and forms hydrogen bonds to water. Analysis of mutant proteins and model peptides demonstrates that the reduced native state fluorescence is caused by the effective quenching of p-cyanophenylalanine fluorescence via FRET to tyrosine side-chains. The implications for the interpretation of p-cyanophenylalanine fluorescence measurements and FRET studies are discussed.
Co-reporter:David B. Strasfeld, Yun L. Ling, Ruchi Gupta, Daniel P. Raleigh and Martin T. Zanni
The Journal of Physical Chemistry B 2009 Volume 113(Issue 47) pp:15679-15691
Publication Date(Web):November 2, 2009
DOI:10.1021/jp9072203
The 37-residue human islet amyloid polypeptide (hIAPP or amylin) self-assembles into fibers, the assembly of which has been associated with the disease mechanism of type II diabetes. Infrared spectroscopy in conjunction with isotope labeling is proving to be a powerful tool for studying the aggregation process of hIAPP and other amyloid forming proteins with residue specific structure and kinetic information, but the relationship between the spectroscopic observables and the structure is not fully established. We report a detailed analysis of the linear and 2D IR spectra of hIAPP fibers isotope labeled at seven different residue positions. The features of the 2D IR spectra, including the frequencies, linewidths, intensities, and polarization dependence of the diagonal and cross-peaks, rely heavily on the position of the isotope labeled residue. In order to understand how these measured parameters depend on fiber secondary and tertiary structure, we have simulated 1D and 2D IR spectra utilizing idealized structural models in addition to a recently published solid-state NMR based model of the amyloid fibril. The analysis provides a more rigorous foundation for interpreting the infrared spectra of amyloids. In addition, we demonstrate that 2D IR spectra can be employed to distinguish between residues in β-sheets versus those in turn regions, and that transitional residues between secondary structures can be identified by the suppression of their cross-peaks in 2D IR spectra. This latter approach is not limited to amyloid fibrils and will be generally useful in identifying regions of secondary structure in proteins using 2D IR spectroscopy and isotope labeling.
Co-reporter:Yun L. Ling, David B. Strasfeld, Sang-Hee Shim, Daniel P. Raleigh and Martin T. Zanni
The Journal of Physical Chemistry B 2009 Volume 113(Issue 8) pp:2498-2505
Publication Date(Web):January 30, 2009
DOI:10.1021/jp810261x
Islet amyloid polypeptide (IAPP, also known as amylin) is responsible for pancreatic amyloid deposits in type 2 diabetes. The deposits, as well as intermediates in their assembly, are cytotoxic to pancreatic β-cells and contribute to the loss of β-cell mass associated with type 2 diabetes. The factors that trigger islet amyloid deposition in vivo are not well understood, but peptide membrane interactions have been postulated to play an important role in islet amyloid formation. To better understand the role of membrane interactions in amyloid formation, two-dimensional infrared (2D IR) spectroscopy was used to compare the kinetics of amyloid formation for human IAPP both in the presence and in the absence of negatively charged lipid vesicles. Comparison of spectral features and kinetic traces from the two sets of experiments provides evidence for the formation of an ordered intermediate during the membrane-mediated assembly of IAPP amyloid. A characteristic transient spectral feature is detected during amyloid formation in the presence of vesicles that is not observed in the absence of vesicles. The spectral feature associated with the intermediate raises in intensity during the self-assembly process and subsequently decays in intensity in the classic manner of a kinetic intermediate. Studies with rat IAPP, a variant that is known to interact with membranes but does not form amyloid, confirm the presence of an intermediate. The analysis of 2D IR spectra in terms of specific structural features is discussed. The unique combination of time and secondary structure resolution of 2D IR spectroscopy has enabled the time-evolution of a hIAPP intermediate to be directly monitored for the first time. The data presented here demonstrates the utility of 2D IR spectroscopy for studying membrane-catalyzed amyloid formation.
Co-reporter:Sang-Hee Shim;David B. Strasfeld;Daniel P. Raleigh;Ruchi Gupta;Yun L. Ling;Martin T. Zanni
PNAS 2009 Volume 106 (Issue 16 ) pp:6614-6619
Publication Date(Web):2009-04-21
DOI:10.1073/pnas.0805957106
There is considerable interest in uncovering the pathway of amyloid formation because the toxic properties of amyloid likely
stems from prefibril intermediates and not the fully formed fibrils. Using a recently invented method of collecting 2-dimensional
infrared spectra and site-specific isotope labeling, we have measured the development of secondary structures for 6 residues
during the aggregation process of the 37-residue polypeptide associated with type 2 diabetes, the human islet amyloid polypeptide
(hIAPP). By monitoring the kinetics at 6 different labeled sites, we find that the peptides initially develop well-ordered
structure in the region of the chain that is close to the ordered loop of the fibrils, followed by formation of the 2 parallel
β-sheets with the N-terminal β-sheet likely forming before the C-terminal sheet. This experimental approach provides a detailed
view of the aggregation pathway of hIAPP fibril formation as well as a general methodology for studying other amyloid forming
proteins without the use of structure-perturbing labels.
Co-reporter:Bing Shan, Shibani Bhattacharya, David Eliezer and Daniel P. Raleigh
Biochemistry 2008 Volume 47(Issue 36) pp:
Publication Date(Web):August 16, 2008
DOI:10.1021/bi8006862
There is considerable interest in the properties of the unfolded states of proteins, particularly unfolded states which can be populated in the absence of high concentrations of denaturants. Interest in the unfolded state ensemble reflects the fact that it is the starting point for protein folding as well as the reference state for protein stability studies and can be the starting state for pathological aggregation. The unfolded state of the C-terminal domain (residues 58−149) of the ribosomal protein L9 (CTL9) can be populated in the absence of denaturant at low pH. CTL9 is a 92-residue globular α, β protein. The low-pH unfolded state contains more secondary structure than the low-pH urea unfolded state, but it is not a molten globule. Backbone (1H, 13C, and 15N) NMR assignments as well as side chain 13Cβ and 1Hβ assignments and 15N R2 values were obtained for the pH 2.0 unfolded form of CTL9 and for the urea unfolded state at pH 2.5. Analysis of the deviations of the chemical shifts from random coil values indicates that residues that comprise the two helices in the native state show a clear preference for adopting helical φ and ψ angles in the pH 2.0 unfolded state. There is a less pronounced but nevertheless clear tendency for residues 107−124 to preferentially populate helical φ and ψ values in the unfolded state. The urea unfolded state has no detectable tendency to populate any type of secondary structure even though it is as compact as the pH 2.0 unfolded state. Comparison of the two unfolded forms of CTL9 provides direct experimental evidence that states which differ significantly in their secondary structure can have identical hydrodynamic properties. This in turn demonstrates that global parameters such as Rh or Rg are very poor indicators of “random coil” behavior.
Co-reporter:Fanling Meng, Peter Marek, Kathryn J. Potter, C. Bruce Verchere and Daniel P. Raleigh
Biochemistry 2008 Volume 47(Issue 22) pp:
Publication Date(Web):May 6, 2008
DOI:10.1021/bi702518m
Amyloid formation has been implicated in more than 20 different human diseases, including Alzheimer’s disease, Parkinson’s disease, and type 2 diabetes. The development of inhibitors of amyloid is a topic of considerable interest, both because of their potential therapeutic applications and because they are useful mechanistic probes. Recent studies have highlighted the potential use of rifampicin as an inhibitor of amyloid formation by a variety of polypeptides; however, there are conflicting reports on its ability to inhibit amyloid formation by islet amyloid polypeptide (IAPP). IAPP is the cause of islet amyloid in type 2 diabetes. We show that rifampicin does not prevent amyloid formation by IAPP and does not disaggregate preformed IAPP amyloid fibrils;, instead, it interferes with standard fluorescence-based assays of amyloid formation. Rifampicin is unstable in aqueous solution and is readily oxidized. However, the effects of oxidized and reduced rifampicin are similar, in that neither prevents amyloid formation by IAPP. Furthermore, use of a novel p-cyanoPhe analogue of IAPP shows that rifampicin does not significantly affect the kinetics of IAPP amyloid formation. The implications for the development of amyloid inhibitors are discussed as are the implications for studies of the toxicity of islet amyloid. The work also demonstrates the utility of p-cyanoPhe IAPP for the screening of inhibitors. The data indicate that rifampicin cannot be used to test the relative toxicity of IAPP fibrils and prefibril aggregates of IAPP.
Co-reporter:Peter Marek;Ruchi Gupta ;Daniel P. Raleigh
ChemBioChem 2008 Volume 9( Issue 9) pp:1372-1374
Publication Date(Web):
DOI:10.1002/cbic.200800052
Co-reporter:Andisheh Abedini, Gagandeep Singh, Daniel P. Raleigh
Analytical Biochemistry 2006 Volume 351(Issue 2) pp:181-186
Publication Date(Web):15 April 2006
DOI:10.1016/j.ab.2005.11.029
Islet amyloid polypeptide (IAPP) is a 37-residue pancreatic hormone. It is responsible for the formation of islet amyloid in vivo and is very insoluble and aggregation-prone in vitro, particularly at basic pH. The peptide contains a disulfide bridge between residues two and seven and an amidated C terminus. There is no reported expression system for the production of amidated IAPP. The peptide is difficult to synthesize and formation of the disulfide by traditional methods is problematic. We have found that the use of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) or dimethyl sulfoxide (DMSO) significantly improves disulfide formation and purification of highly aggregation-prone IAPP sequences. The use of these organic solvents increases the solubility of the hydrophobic peptides, avoids the use of aqueous basic solutions, and eliminates the need for continuous stirring during oxidation to form the Cys-2 to Cys-7 disulfide bridge. Elimination of the stirring step and basic solution helps to reduce aggregation and allows for more consistent high-performance liquid chromatography (HPLC) retention times. Formation of the intramolecular disulfide using DMSO was found to be the most effective method for IAPP oxidation, reducing the reaction time from 24 to 5 h. Aggregated IAPP can be resolubilized by HFIP or DMSO and recovered by HPLC with very good yield.
Co-reporter:Scott H. Brewer;Dung M. Vu;Yuefeng Tang;Ying Li;Stefan Franzen;Daniel P. Raleigh;R. Brian Dyer
PNAS 2005 102 (46 ) pp:16662-16667
Publication Date(Web):2005-11-15
DOI:10.1073/pnas.0505432102
Equilibrium Fourier transform infrared (FTIR) and temperature-jump (T-jump) IR spectroscopic techniques were used to study
the thermodynamics and kinetics of the unfolding and folding of the villin headpiece helical subdomain (HP36), a small three-helix
protein. A double phenylalanine mutant (HP36 F47L, F51L) that destabilizes the hydrophobic core of this protein also was studied.
The double mutant is less stable than wild type (WT) and has been shown to contain less residual secondary structure and tertiary
contacts in its unfolded state. The relaxation kinetics after a T-jump perturbation were studied for both HP36 and HP36 F47L,
F51L. Both proteins exhibited biphasic relaxation kinetics in response to a T-jump. The folding times for the WT (3.23 μs
at 60.2°C) and double phenylalanine mutant (3.01 μs at 49.9°C) at the approximate midpoints of their thermal unfolding transitions
were found to be similar. The folding time for the WT was determined to be 3.34 μs at 49.9°C, similar to the folding time
of the double phenylalanine mutant at that temperature. The double phenylalanine mutant, however, unfolds faster with an unfolding
time of 3.01 μs compared with 6.97 μs for the WT at 49.9°C.
Co-reporter:Satoshi Sato, Daniel P. Raleigh
Journal of Molecular Biology (6 July 2007) Volume 370(Issue 2) pp:349-355
Publication Date(Web):6 July 2007
DOI:10.1016/j.jmb.2007.02.084
Our present understanding of the nature of the transition state for protein folding depends predominantly on studies where individual side-chain contributions are mapped out by mutational analysis (ϕ value analysis). This approach, although extremely powerful, does not in general provide direct information about the formation of backbone hydrogen bonds. Here, we report the results of amide H/D isotope effect studies that probe the development of hydrogen bonded interactions in the transition state for the folding of a small α−β protein, the N-terminal domain of L9. Replacement of amide protons by deuterons in a solvent of constant isotopic composition destabilized the domain, decreasing both its Tm and ΔG0 of unfolding. The folding rate also decreased. The parameter ΦH/D, defined as the ratio of the effect of isotopic substitution upon the activation free energy to the equilibrium free energy was determined to be 0.6 in a D2O background and 0.75 in a H2O background, indicating that significant intraprotein hydrogen bond interactions are developed in the transition state for the folding of NTL9. The value is in remarkably good agreement with more traditional measures of the position of the transition state, which report on the relative burial of surface area. The results provide a picture of a compact folding transition state containing significant secondary structure. Indirect analysis argues that the bulk of the kinetic isotope effect arises from the β-sheet-rich region of the protein, and suggests that the development of intraprotein hydrogen bonds in this region plays a critical role in the folding of NTL9.
Co-reporter:Fanling Meng, Daniel P. Raleigh
Journal of Molecular Biology (25 February 2011) Volume 406(Issue 3) pp:491-502
Publication Date(Web):25 February 2011
DOI:10.1016/j.jmb.2010.12.028
Islet amyloid polypeptide (IAPP; also known as amylin) is responsible for islet amyloid formation in type 2 diabetes, and IAPP-induced toxicity is believed to contribute to the loss of β-cell mass associated with the late stages of type 2 diabetes. Islet amyloid formation may also play a role in graft failure after transplantation. IAPP is produced as a prohormone, pro-islet amyloid polypeptide (proIAPP), and processed in the secretory granules of the pancreatic β-cells. Partially processed forms of proIAPP are found in amyloid deposits; most notable is a 48-residue intermediate, proIAPP1–48, which includes the N-terminal pro-extension, but which has been properly processed at the C-terminus. Incomplete processing may play a role in islet amyloid formation by promoting interactions with sulfated proteoglycans of the extracellular matrix, which, in turn, promote amyloid formation. We show that acid fuchsin (3-(1-(4-amino-3-methyl-5-sulphonatophenyl)-1-(4-amino-3-sulphonatophenyl)methylene)cyclohexa-1,4-dienesulphonic acid), a simple sulfonated triphenyl methyl derivative, is a potent inhibitor of amyloid formation by the proIAPP1–48 intermediate. The more complicated triphenyl methane derivative fast green FCF {ethyl-[4-[[4-[ethyl-[(3-sulfophenyl)methyl]amino]phenyl]-(4-hydroxy-2-sulfophenyl)methylidene]-1-cyclohexa-2,5-dienylidene]-[(3-sulfophenyl)methyl]azanium} also inhibits amyloid formation by IAPP and the proIAPP processing intermediate. Both compounds inhibit amyloid formation by mixtures of the proIAPP intermediate and the model glycosaminoglycan heparan sulfate. Acid fuchsin also inhibits glycosaminoglycan-mediated amyloid formation by mature IAPP. The ability to inhibit amyloid formation is not simply due to the compounds being sulfonated, since the sulfonated inhibitor of amyloid-β, tramiprosate, is not an inhibitor of amyloid formation by proIAPP1–48.
Co-reporter:Peter Marek, Sudipta Mukherjee, Martin T. Zanni, Daniel P. Raleigh
Journal of Molecular Biology (23 July 2010) Volume 400(Issue 4) pp:878-888
Publication Date(Web):23 July 2010
DOI:10.1016/j.jmb.2010.05.041
Amyloid formation normally exhibits a lag phase followed by a growth phase, which leads to amyloid fibrils. Characterization of the species populated during the lag phase is experimentally challenging, but is critical since the most toxic entities may be pre-fibrillar species. p-Cyanophenylalanine (FC≡N) fluorescence is used to probe the nature of lag-phase species populated during the formation of amyloid by human islet amyloid polypeptide. The polypeptide contains two phenylalanines at positions 15 and 23 and a single tyrosine located at the C-terminus. Each aromatic residue was separately replaced by FC≡N. The substitutions do not perturb amyloid formation relative to wild-type islet amyloid polypeptide as detected using thioflavin T fluorescence and electron microscopy. FC≡N fluorescence is high when the cyano group is hydrogen bonded and low when it is not. It can also be quenched via Förster resonance energy transfer to tyrosine. Fluorescence intensity was monitored in real time and revealed that all three positions remained exposed to solvent during the lag phase but less exposed than unstructured model peptides. The time course of amyloid formation as monitored by thioflavin T fluorescence and FC≡N fluorescence is virtually identical. Fluorescence quenching experiments confirmed that each residue remains exposed during the lag phase. These results place significant constraints on the nature of intermediates that are populated during the lag phase and indicate that significant sequestering of the aromatic side chains does not occur until β-structure sufficient to bind thioflavin T has developed. Seeding studies and analysis of maximum rates confirm that sequestering of the cyano groups occurs concomitantly with the development of thioflavin T binding capability. Overall, the process of amyloid formation and growth appears to be remarkably homogenous in terms of side-chain ordering. FC≡N also provides information about fibril structure. Fluorescence emission measurements, infrared measurements, and quenching studies indicate that the aromatic residues are differentially exposed in the fibril state with Phe15 being the most exposed. FC≡N is readily accommodated into proteins; thus, the approach should be broadly applicable.
Co-reporter:Ying Li, Bing Shan, Daniel P. Raleigh
Journal of Molecular Biology (20 April 2007) Volume 368(Issue 1) pp:256-262
Publication Date(Web):20 April 2007
DOI:10.1016/j.jmb.2007.02.011
A point mutation of a small globular protein, the C-terminal domain of L9 destabilizes the protein and leads to observable cold-denaturation at temperatures above zero. The cold denatured state is in slow exchange with the native state on the NMR time scale, and this allows the hydrodynamic properties of the cold unfolded state and the native state to be measured under identical conditions using pulsed-field gradient NMR diffusion measurements. This provides the first experimental measurement of the hydrodynamic properties of a cold unfolded protein and its folded form under identical conditions. Hydrodynamic radii of the cold-induced unfolded states were measured for a set of temperatures ranging from 2 °C to 25 °C at pD 6.6 in the absence of denaturant. The cold unfolded state is compact compared to the urea or acid unfolded state and a trend of increasing radii of hydration is observed as the temperature is lowered. These observations are confirmed by experiments on the same protein at pD 8.0, where it is more stable, in the presence of a modest concentration of urea. The expansion of the cold-denatured state at lower temperatures is consistent with the temperature dependence of hydrophobic interactions.
Co-reporter:Humeyra Taskent, Jae-Hyun Cho, Daniel P. Raleigh
Journal of Molecular Biology (2 May 2008) Volume 378(Issue 3) pp:699-706
Publication Date(Web):2 May 2008
DOI:10.1016/j.jmb.2008.02.024
Characterization of the transition-state ensemble and the nature of the free-energy barrier for protein folding are areas of intense activity and some controversy. A key issue that has emerged in recent years is the width of the free-energy barrier and the susceptibility of the transition state to movement. Here we report denaturant-induced and temperature-dependent folding studies of a small mixed α–β protein, the N-terminal domain of L9 (NTL9). The folding of NTL9 was determined using fluorescence-detected stopped-flow fluorescence measurements conducted at seven different temperatures between 11 and 40 °C. Plots of the log of the observed first-order rate constant versus denaturant concentration, “chevron plots,” displayed the characteristic V shape expected for two-state folding. There was no hint of deviation from linearity even at the lowest denaturant concentrations. The relative position of the transition state, as judged by the Tanford β parameter, βT, shifts towards the native state as the temperature is increased. Analysis of the temperature dependence of the kinetic and equilibrium m values indicates that the effect is due to significant movement of the transition state and also includes a contribution from temperature-dependent ground-state effects. Analysis of the Leffler plots, plots of ΔG‡ versus ΔG°, and their cross-interaction parameters confirms the transition-state movement. Since the protein is destabilized at high temperature, the shift represents a temperature-dependent Hammond effect. This provides independent confirmation of a recent theoretical prediction. The magnitude of the temperature-denaturant cross-interaction parameter is larger for NTL9 than has been reported for the few other cases studied. The implications for temperature-dependent studies of protein folding are discussed.
Co-reporter:Fanling Meng, Andisheh Abedini, Annette Plesner, Chris T. Middleton, ... Daniel P. Raleigh
Journal of Molecular Biology (16 July 2010) Volume 400(Issue 3) pp:555-566
Publication Date(Web):16 July 2010
DOI:10.1016/j.jmb.2010.05.001
Islet amyloid polypeptide (IAPP), also known as amylin, is responsible for amyloid formation in type 2 diabetes. The formation of islet amyloid is believed to contribute to the pathology of the disease by killing β-cells, and it may also contribute to islet transplant failure. The design of inhibitors of amyloid formation is an active area of research, but comparatively little attention has been paid to inhibitors of IAPP in contrast to the large body of work on β-amyloid, and most small-molecule inhibitors of IAPP amyloid are generally effective only when used at a significant molar excess. Here we show that the simple sulfonated triphenyl methane derivative acid fuchsin, 3-(1-(4-amino-3-methyl-5-sulfonatophenyl)-1-(4-amino-3-sulfonatophenyl) methylene) cyclohexa-1,4-dienesulfonic acid, is a potent inhibitor of in vitro amyloid formation by IAPP at substoichiometric levels and protects cultured rat INS-1 cells against the toxic effects of human IAPP. Fluorescence-detected thioflavin-T binding assays, light-scattering, circular dichroism, two-dimensional IR, and transmission electron microscopy measurements confirm that the compound prevents amyloid fibril formation. Ionic-strength-dependent studies show that the effects are mediated in part by electrostatic interactions. Experiments in which the compound is added at different time points during the lag phase after amyloid formation has commenced reveal that it arrests amyloid formation by trapping intermediate species. The compound is less effective against the β-amyloid peptide, indicating specificity in its ability to inhibit amyloid formation by IAPP. The work reported here provides a new structural class of IAPP amyloid inhibitors and demonstrates the power of two-dimensional infrared spectroscopy for characterizing amyloid inhibitor interactions.
Co-reporter:Shifeng Xiao, Daniel P. Raleigh
Journal of Molecular Biology (13 August 2010) Volume 401(Issue 2) pp:274-285
Publication Date(Web):13 August 2010
DOI:10.1016/j.jmb.2010.05.070
The helical subdomain of the villin headpiece (HP36) is one of the smallest naturally occurring proteins that folds cooperatively. Its small size, rapid folding, and simple three-helix topology have made it an extraordinary popular model system for computational, theoretical, and experimental studies of protein folding. Aromatic–proline interactions involving Trp64 and Pro62 have been proposed to play a critical role in specifying the subdomain fold by acting as gatekeeper residues. Note that the numbering corresponds to full-length headpiece. Mutation of Pro62 has been shown to lead to a protein that does not fold, but this may arise for two different reasons: The residue may make interactions that are critical for the specificity of the fold or the mutation may simply destabilize the domain. In the first case, the protein cannot fold, while in the second, the small fraction of molecules that do fold adopt the correct structure. The modest stability of the wild type prevents a critical analysis of these interactions because even moderately destabilizing mutations lead to a very small folded state population. Using a hyperstable variant of HP36, denoted DM HP36, as our new wild type, we characterized a set of mutants designed to assess the role of the putative gatekeeper interactions. Four single mutants, DM Pro62Ala, DM Trp64Leu, DM Trp64Lys, and DM Trp64Ala, and a double mutant, DM Pro62Ala Trp64Leu, were prepared. All mutants are less stable than DM HP36, but all are well folded as judged by CD and 1H NMR. All of the mutants display sigmoidal thermal unfolding and urea-induced unfolding curves. Double-mutant cycle analysis shows that the interactions between Pro62 and Trp64 are weak but favorable. Interactions involving Pro62 and proline–aromatic interactions are, thus, not required for specifying the subdomain fold. The implications for the design and thermodynamics of miniature proteins are discussed.
Co-reporter:Ruchi Gupta, Neeraj Kapoor, Daniel P. Raleigh, Thomas P. Sakmar
Journal of Molecular Biology (10 August 2012) Volume 421(Issues 2–3) pp:378-389
Publication Date(Web):10 August 2012
DOI:10.1016/j.jmb.2012.04.017
Many human diseases are associated with amyloid fibril deposition, including type 2 diabetes mellitus where human islet amyloid polypeptide (hIAPP) forms fibrils in the pancreas. We report here that engineered, soluble forms of the human Ca2+-binding protein nucleobindin 1 (NUCB1) prevent hIAPP fibril formation and disaggregate preexisting hIAPP fibrils. Scanning transmission electron microscopy (STEM) and atomic force microscopy indicate that NUCB1 binds to and stabilizes heterogeneous prefibrillar hIAPP species. The NUCB1-stabilized prefibrillar species were isolated by size-exclusion chromatography and analyzed by STEM, dynamic light scattering, and multi-angle light scattering. The stabilized prefibrillar species show a size range of 2–6 million Da and have other similarities to hIAPP protofibrils, but they do not progress to become mature fibrils. The effects of NUCB1 are absent in the presence of Ca2+. We postulate that the engineered forms of NUCB1 prevent hIAPP fibril formation by a mechanism where protofibril-like species are “capped” to prevent further fibril assembly and maturation. This mode of action appears to be different from other protein-based inhibitors, suggesting that NUCB1 may offer a new approach to inhibiting amyloid formation and disaggregating amyloid fibrils.Download high-res image (322KB)Download full-size imageHighlights► Soluble Nucleobindin 1 (NUCB1) prevents fibrillization of hIAPP (human islet amyloid polypeptide). ► NUCB1 inhibits fibril formation through a novel “capping” mechanism. ► NUCB1 anti-amyloidogenic activity is independent of the stage of fibril formation.
Co-reporter:Ping Cao, Ling-Hsien Tu, Andisheh Abedini, Olesya Levsh, ... Daniel P. Raleigh
Journal of Molecular Biology (10 August 2012) Volume 421(Issues 2–3) pp:282-295
Publication Date(Web):10 August 2012
DOI:10.1016/j.jmb.2011.12.032
Islet amyloid polypeptide (IAPP, amylin) is responsible for amyloid formation in type 2 diabetes and in islet cell transplants. The only known natural mutation found in mature human IAPP is a Ser20-to-Gly missense mutation, found with small frequency in Chinese and Japanese populations. The mutation appears to be associated with increased risk of early-onset type 2 diabetes. Early measurements in the presence of organic co-solvents showed that S20G-IAPP formed amyloid more quickly than the wild type. We confirm that the mutant accelerates amyloid formation under a range of conditions including in the absence of co-solvents. Ser20 adopts a normal backbone geometry, and the side chain makes no steric clashes in models of IAPP amyloid fibers, suggesting that the increased rate of amyloid formation by the mutant does not result from the relief of steric incompatibility in the fiber state. Transmission electronic microscopy, circular dichroism, and seeding studies were used to probe the structure of the resulting fibers. The S20G-IAPP peptide is toxic to cultured rat INS-1 (transformed rat insulinoma-1) β-cells. The sensitivity of amyloid formation to the identity of residue 20 was exploited to design a variant that is much slower to aggregate and that inhibits amyloid formation by wild-type IAPP. An S20K mutant forms amyloid with an 18-fold longer lag phase in homogeneous solution. Thioflavin T binding assays, together with experiments using a p-cyanophenylalanine (p-cyanoPhe) variant of human IAPP, show that the designed S20K mutant inhibits amyloid formation by human IAPP. The experiments illustrate how p-cyanoPhe can be exploited to monitor amyloid formation even in the presence of other amyloidogenic proteins.Download high-res image (97KB)Download full-size imageResearch Highlights► The Ser20-to-Gly mutation enhances amyloid formation by IAPP. ► The Ser20-to-Lys mutant forms amyloid 18 times more slowly than wild-type IAPP. ► The Lys20 variant inhibits amyloid formation by wild-type IAPP. ► p-CyanoPhe can be used to follow amyloid formation in a mixture of proteins.

![Pentanoic acid,5-[4-[[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]methyl]-3,5-dimethoxyphenoxy]-](http://img.cochemist.com/ccimg/115200/115109-65-4.png)
![Pentanoic acid,5-[4-[[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]methyl]-3,5-dimethoxyphenoxy]-](http://img.cochemist.com/ccimg/115200/115109-65-4_b.png)
![9,9'-[(2R,3R,3aS,7aR,9R,10R,10aS,14aR)-3,5,10,12-tetrahydroxy-5,12-dioxidooctahydro-2H,7H-difuro[3,2-d:3',2'-j][1,3,7,9,2,8]tetraoxadiphosphacyclododecine-2,9-diyl]bis(2-amino-3,9-dihydro-6H-purin-6-one)](http://img.cochemist.com/ccimg/61100/61093-23-0.png)
![9,9'-[(2R,3R,3aS,7aR,9R,10R,10aS,14aR)-3,5,10,12-tetrahydroxy-5,12-dioxidooctahydro-2H,7H-difuro[3,2-d:3',2'-j][1,3,7,9,2,8]tetraoxadiphosphacyclododecine-2,9-diyl]bis(2-amino-3,9-dihydro-6H-purin-6-one)](http://img.cochemist.com/ccimg/61100/61093-23-0_b.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)