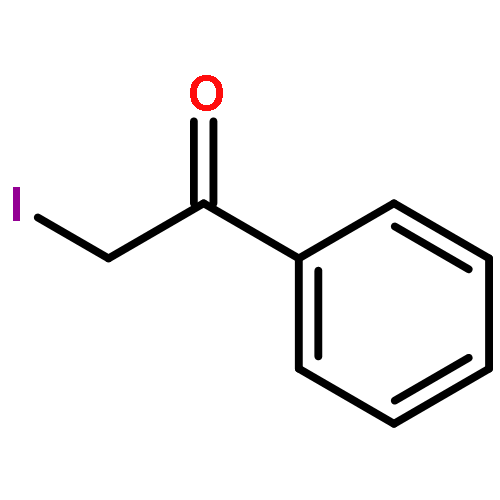
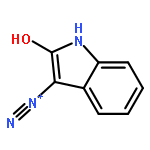
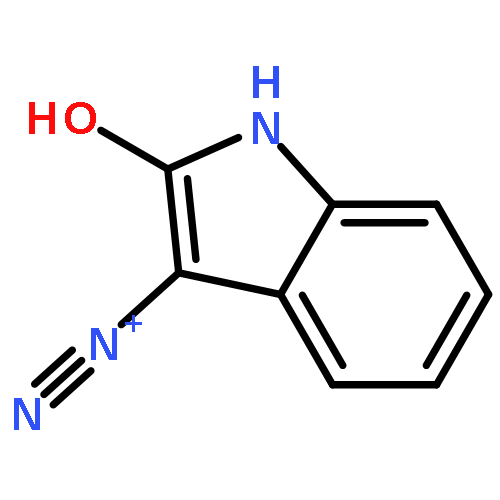
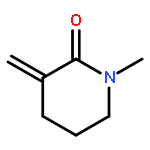
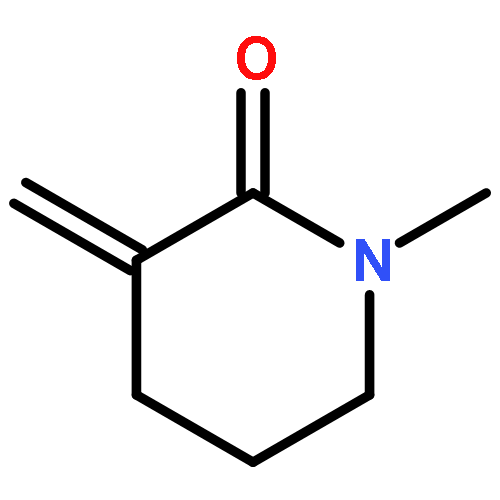
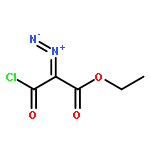
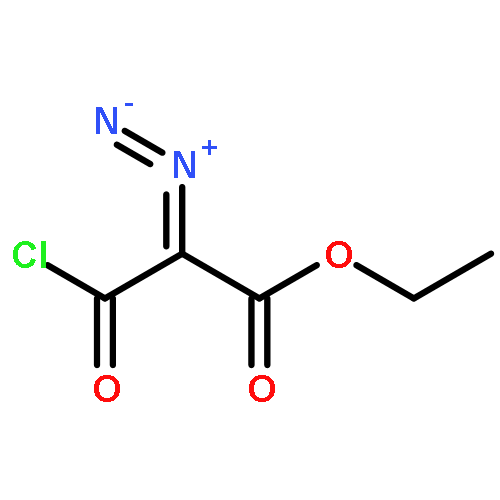
Co-reporter:Sara A. Bonderoff and Albert Padwa
The Journal of Organic Chemistry 2017 Volume 82(Issue 1) pp:642-651
Publication Date(Web):December 15, 2016
DOI:10.1021/acs.joc.6b02663
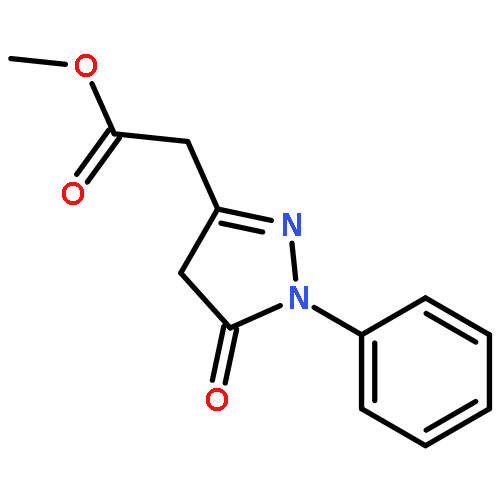
The chemoselective reaction of donor/acceptor (D/A) and acceptor/acceptor (A/A) diazo moieties in the same molecule was examined using 3-diazo-1-(ethyl 2-diazomalonyl)indolin-2-one under rhodium(II) catalysis. The metallo carbenoid derived from the D/A diazo group is preferentially formed and undergoes selective CH, NH, and OH insertion reactions, cyclopropanation, cyclopropenation, sulfur ylide formation/2,3-sigmatropic rearrangement, as well as nitrogen ylide formation followed by azetidine ring expansion. The initial reaction can be paired with a subsequent tandem cascade sequence involving dipole formation/cycloaddition in either an intra- or intermolecular sense to generate polycyclic N-heterocycles in one pot, with the formation up to three new rings in a single operation. Excellent diastereoselectivity was observed in the intramolecular cycloaddition reaction producing 5 to 7-membered rings.
Co-reporter:Carolyn A. Leverett, Gang Li, Stefan France, and Albert Padwa
The Journal of Organic Chemistry 2016 Volume 81(Issue 21) pp:10193-10203
Publication Date(Web):May 23, 2016
DOI:10.1021/acs.joc.6b00771
The total synthesis of the Strychnos alkaloid (±)-minfiensine was achieved via an intramolecular amidofuran Diels–Alder cycloaddition/rearrangement followed by an iminium ion/cyclization cascade sequence. This domino process provides for a rapid access to the unique 1,2,3,4-tetrahydro-9a,4a-iminoethanocarbazole core structure found in the alkaloid minfiensine (2). In this paper, the full account of our synthetic study is described, highlighting the successful application of the cascade sequence to form the A/B/C/D rings of (±)-minfiensine (2) in high yield. A palladium-catalyzed enolate coupling reaction was then used to furnish the final E ring and complete the total synthesis of (±)-minfiensine (2).
Co-reporter:Albert Padwa;Scott Bur
Chemistry of Heterocyclic Compounds 2016 Volume 52( Issue 9) pp:616-626
Publication Date(Web):2016 September
DOI:10.1007/s10593-016-1942-3
The preparation of heterocyclic compounds using 1,3-dipolar cycloaddition chemistry is now well recognized in the fields of organic synthesis, drug discovery efforts, polymer chemistry, and materials science. As highlighted in this review, a growing area of interest in organic synthesis involves the enantioselectivity aspects of dipolar cycloaddition chemistry for the preparation of many different classes of natural products. Asymmetric synthesis of natural products using chiral substrates has been elegantly accomplished over the past decade using an assortment of dipole intermediates and represents the focus of this review article.
Co-reporter:Sara A. Bonderoff, Albert Padwa
Tetrahedron Letters 2015 Volume 56(Issue 23) pp:3127-3129
Publication Date(Web):3 June 2015
DOI:10.1016/j.tetlet.2014.12.020
Bis(diazo)piperidin-2-ones that are devoid of a donor substituent in the 4-position undergo a one-pot cyclopropanation/cascade dipole formation/cycloaddition reaction to produce novel azapolycycles.
Co-reporter:Albert Padwa and Yan Zou
The Journal of Organic Chemistry 2015 Volume 80(Issue 3) pp:1802-1808
Publication Date(Web):January 5, 2015
DOI:10.1021/jo502725d
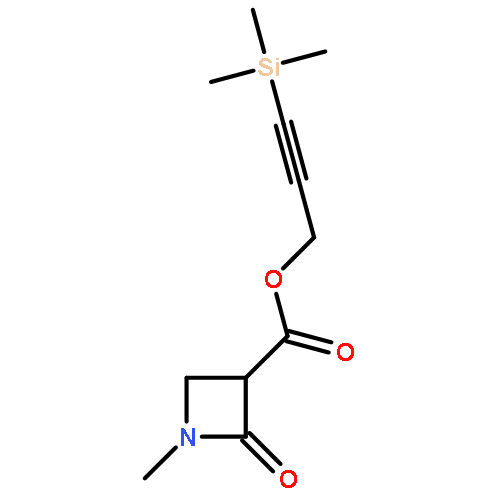
We report a detailed investigation into the Rh(II)-catalyzed reactions of 2-alkynyl 2-diazo amido-substituted esters. The distribution of products was found to be dependent on the substituent group on the nitrogen atom, the ligand on the Rh(II) center, and the solvent used. The dominant product obtained from the reaction of 3-(trimethylsilyl)prop-2-ynyl-2-(dibenzylcarbamoyl)-2-diazoacetate (34) with Rh2(OAc)4 in hexane corresponds to an azetidinone derived by CH-insertion of the carbenoid into the neighboring benzyl group. In contrast, the Rh2(esp)2-catalyzed reaction of 34 in CH2Cl2 afforded a 3-oxocyclohepta[c]pyrrole formed by cyclopropanation of the rhodium carbenoid across the aromatic π-bond. Related systems were studied, and CH-insertion into an adjacent alkyl group was found to be the dominant or exclusive pathway. In none of the cases studied was it possible to detect products derived from a carbenoid/alkyne cascade sequence as had previously been found with a series of 2-alkynyl-2-diazo-3-oxobutanoates.
Co-reporter:Hao Li, Sara A. Bonderoff, Bo Cheng, and Albert Padwa
The Journal of Organic Chemistry 2014 Volume 79(Issue 1) pp:392-400
Publication Date(Web):December 13, 2013
DOI:10.1021/jo4026622
Model studies dealing with the rhodium(II)-catalyzed carbenoid insertion/cyclization/cycloaddition cascade of several α-diazo dihydroindolinones have been carried out as an approach to the alkaloid mersicarpine. The cascade reaction of α-diazo dihydroindolinone 21 proceeded in high yield with excellent diastereoselectivity to give cycloadduct 22, which possesses the required stereochemistry of the two adjacent quaternary carbon centers present in mersicarpine. The overall reaction enabled the rapid assemblage of a polycyclic ring system that contains three new stereocenters and three continuous quaternary carbons in a single operation in high yield with excellent diastereoselectivity. The 3-indolinone derivative 36 was eventually formed from cycloadduct 22 by an acid-induced hydrolysis of 22 to give 23, which was subsequently converted in several steps to 36. The synthesis of this compound constitutes a successful construction of the tricyclic core of mersicarpine. Reduction of the nitrile group of 36 followed by a subsequent reductive cyclization/ring-opening aromatization cascade, as was found to occur with the related compound 29, will be employed for an eventual synthesis of demethylmersicarpine.
Co-reporter:Albert Padwa, Yan Zou, Bo Cheng, Hao Li, Nadale Downer-Riley, and Christopher S. Straub
The Journal of Organic Chemistry 2014 Volume 79(Issue 7) pp:3173-3184
Publication Date(Web):March 11, 2014
DOI:10.1021/jo500331j
Model studies dealing with the Cu(II)- or Rh(II)-catalyzed carbenoid cyclization/cycloaddition cascade of several α-diazo indolo amido esters have been carried out as an approach to the alkaloid scandine. The Cu(II)-catalyzed reaction of an α-diazo indolo diester that contains a tethered oxa-pentenyl side chain was found to give rise to a reactive benzo[c]furan which undergoes a subsequent [4 + 2]-cycloaddition across the tethered π-bond. The reaction proceeds by the initial generation of a copper carbenoid intermediate which cyclizes onto the adjacent carbonyl group to give a reactive benzo[c]furan which in certain cases can be isolated. Disappointingly, the analogous reaction with the related amido indolo ester failed to take place, even when the tethered π-bond contained an electron-withdrawing carbomethoxy group. It would seem that the geometric requirements for the intramolecular cycloaddition of the furo[3,4-b]indole system with the tethered π-bond imposes distinct restrictions upon the bond angles of the reacting centers to prevent the cycloaddition reaction from occurring. However, the incorporation of another carbonyl group on the nitrogen atom of the tethered alkenyl diazo amido indolo ester seemingly provides better orbital overlap between the reacting π-systems and allows the desired cycloaddition reaction to occur.
Co-reporter:Sara A. Bonderoff and Albert Padwa
Organic Letters 2013 Volume 15(Issue 16) pp:4114-4117
Publication Date(Web):July 26, 2013
DOI:10.1021/ol4017468
The chemoselective reaction of donor/acceptor (D/A) and acceptor/acceptor (A/A) diazo moieties in the same molecule was examined using 3-diazo-1-(ethyl 2-diazomalonyl)indolin-2-one under rhodium(II) catalysis. The D/A diazo group undergoes selective cyclopropanation as well as XH-insertion, leaving behind the second diazo group for a further intramolecular dipolar cycloaddition reaction.
Co-reporter:Gang Li and Albert Padwa
Organic Letters 2011 Volume 13(Issue 15) pp:3767-3769
Publication Date(Web):June 22, 2011
DOI:10.1021/ol201320v
An efficient synthesis of (±)-minfiensine has been accomplished employing an intramolecular Diels–Alder cycloaddition/rearrangement cascade of an amidofuran derivative. Thermal reorganization of the initially formed [4 + 2]-cycloadduct affords the critical tetrahydroiminoethanocarbazole skeleton of the alkaloid in high yield.
Co-reporter:Hao Li, Nawong Boonnak, Albert Padwa
Tetrahedron Letters 2011 Volume 52(Issue 17) pp:2062-2064
Publication Date(Web):27 April 2011
DOI:10.1016/j.tetlet.2010.10.072
A mild cross-coupling reaction has been used to access several N-vinyl substituted indoles. When treated with acid, these unique enamines produce novel dimeric and trimeric products derived from a preferred protonation reaction at the enamine π-bond.
Co-reporter:Albert Padwa
Tetrahedron 2011 67(42) pp: 8057-8072
Publication Date(Web):
DOI:10.1016/j.tet.2011.07.009
Co-reporter:Hao Li, Bo Cheng, Nawong Boonnak, Albert Padwa
Tetrahedron 2011 67(51) pp: 9829-9836
Publication Date(Web):
DOI:10.1016/j.tet.2011.09.118
Co-reporter:Jutatip Boonsompat and Albert Padwa
The Journal of Organic Chemistry 2011 Volume 76(Issue 8) pp:2753-2761
Publication Date(Web):March 10, 2011
DOI:10.1021/jo200125c
Using an intramolecular [4 + 2] cycloaddition/rearrangement cascade of 3-(1,4-dioxaspiro[4.4]non-7-en-7-yl)-N-furan-2-ylpropionamide (23) as the key step, the BCD core of the lycopodium alkaloid fawcettidine was constructed. Heating the initially formed Diels−Alder cycloadduct at 180 °C results in a nitrogen-assisted ring opening followed by a deprotonation/reprotonation of the ensuing zwitterion to give a rearranged hexahydroindolinone. Our attempts to induce a related intramolecular furan Diels−Alder reaction (IMDAF) from the corresponding ketone of 23 failed to give any cycloaddition product. Instead, the only product obtained corresponded to a cyclopentenone derivative derived by isomerization of the double bond into the thermodynamically more stable α,β-position. Efforts toward construction of the final skeleton of fawcettidine by ring A closure of the rearranged cycloadduct derived from furanyl amide 23 are discussed.
Co-reporter:Hao Li, Nawong Boonnak, and Albert Padwa
The Journal of Organic Chemistry 2011 Volume 76(Issue 22) pp:9488-9496
Publication Date(Web):October 18, 2011
DOI:10.1021/jo201955c
A mild cross-coupling reaction to access several N-alkenyl-substituted indoles has been developed. The coupling procedure involves treating a NH-indole with various alkenyl bromides using a combination of 10 mol % of copper(I) iodide and 20 mol % of ethylenediamine as the catalyst in dioxane at 110 °C in the presence of K3PO4 as the base. When treated with acid, these unique enamines produce a dimeric product derived from a preferred protonation reaction at the enamine π-bond. A cationic cyclization reaction of the readily available 2-(2-(1H-indol-1-yl)allyl)cyclopentanol was utilized to construct tetracyclic indole derivatives with a quaternary stereocenter attached to the C2-position of the indole ring. An alternative strategy for selective functionalization at the C2-position of a N-alkenyl-substituted indole derivative that was also studied involves a radical cyclization of a xanthate derivative. The work described provides an attractive route to the tetracyclic core of some vinca alkaloids, including the tetrahydroisoquinocarbazole RS-2135.
Co-reporter:Andrew C. Flick, Maria José Arevalo Caballero, Hyoung Ik Lee and Albert Padwa
The Journal of Organic Chemistry 2010 Volume 75(Issue 6) pp:1992-1996
Publication Date(Web):February 12, 2010
DOI:10.1021/jo100055u
An efficient stereocontrolled route to the azatricyclic core of an advanced halichlorine intermediate is described. Reaction of the oxime derived from 2-(oxo-cyclopentyl)acetic acid ethyl ester with 2,3-bis(phenylsulfonyl)-1,3-butadiene gives rise to a 7-oxa-1-azanorbornane cycloadduct in high yield. The formation of the bicyclic isoxazolidine arises from conjugate addition of the oxime onto the diene to afford a transient nitrone that then undergoes an intramolecular dipolar cycloaddition. Treatment of the cycloadduct with 5% Na/Hg results in reductive nitrogen−oxygen bond cleavage to furnish a spirocyclic piperidinone, which was further elaborated to an advanced intermediate employed in an earlier synthesis of halichlorine.
Co-reporter:Andrew C. Flick, Maria José Arevalo Caballero, Albert Padwa
Tetrahedron 2010 66(21) pp: 3643-3650
Publication Date(Web):
DOI:10.1016/j.tet.2010.03.088
Co-reporter:Guido Verniest, Dylan England, Norbert De Kimpe, Albert Padwa
Tetrahedron 2010 66(7) pp: 1496-1502
Publication Date(Web):
DOI:10.1016/j.tet.2009.10.033
Co-reporter:Albert Padwa
Chemical Society Reviews 2009 vol. 38(Issue 11) pp:3072-3081
Publication Date(Web):03 Apr 2009
DOI:10.1039/B816701J
In this tutorial review, the rhodium(II)-catalyzed domino reactions of α-diazo carbonyl compounds are summarized. The article will emphasize some of the more recent synthetic applications of the rhodium carbenoid cyclization/cycloaddition domino cascade for alkaloid synthesis. The many structurally diverse and highly successful examples of both oxa and azapolycyclic ring formation cited in this tutorial review clearly demonstrate that the domino cyclization/cycloaddition cascade of metallo carbenoids has evolved as an important strategy for the synthesis of nitrogen containing natural products.
Co-reporter:Sezgin Kiren and Albert Padwa
The Journal of Organic Chemistry 2009 Volume 74(Issue 20) pp:7781-7789
Publication Date(Web):September 24, 2009
DOI:10.1021/jo9017793
A practical benzannulation method to prepare variously substituted aryl amines and sulfides was developed. The approach involves a Michael-aldol reaction of β-keto sulfones with enones followed by a subsequent condensation of the initial adduct with various amines. The base-induced Michael-aldol cascade proceeds smoothly with a number of different β-keto sulfones, affording the adducts as single diastereomers. Heating the resulting Michael-aldol product with an amine in toluene at 120 °C results in the formation of a transient enamine, which then undergoes loss of phenyl sulfenic acid to furnish the aromatized amine in good yield. A related reaction also occurred when the Michael-aldol product was heated with thiols or alcohols, giving rise to aryl-substituted sulfides or ethers.
Co-reporter:Sezgin Kiren, Xuechuan Hong, Carolyn A. Leverett, Albert Padwa
Tetrahedron 2009 65(33) pp: 6720-6729
Publication Date(Web):
DOI:10.1016/j.tet.2009.03.011
Co-reporter:Drew R. Bobeck, Stefan France, Carolyn A. Leverett, Fernando Sánchez-Cantalejo, Albert Padwa
Tetrahedron Letters 2009 50(26) pp: 3145-3147
Publication Date(Web):
DOI:10.1016/j.tetlet.2008.12.088
Co-reporter:Chad J. Stearman, Michael Wilson and Albert Padwa
The Journal of Organic Chemistry 2009 Volume 74(Issue 9) pp:3491-3499
Publication Date(Web):April 1, 2009
DOI:10.1021/jo9003579
A highly efficient total synthesis of (±)-yohimbenone and a formal synthesis of (±)-emetine is described. The key element of the synthesis consists of a conjugate addition−dipolar cycloaddition of 2,3-bis(phenylsulfonyl)-1,3-butadiene with an appropriate oxime. The resulting cycloadducts are cleaved reductively to provide azapolycyclic scaffolds with strategically placed functionality for further manipulation to the target compounds.
Co-reporter:Drew R. Bobeck, Hyoung Ik Lee, Andrew C. Flick and Albert Padwa
The Journal of Organic Chemistry 2009 Volume 74(Issue 19) pp:7389-7402
Publication Date(Web):August 27, 2009
DOI:10.1021/jo901336z
An efficient stereocontrolled route to the isoschizozygane alkaloid core has been developed utilizing an intramolecular 1,4-dipolar cycloaddition of a cross-conjugated heteroaromatic betaine. The resulting cycloadduct undergoes loss of COS, and further reduction delivers a 5a-azaacenaphthylene intermediate that was transformed into the isoschizozygane skeleton upon treatment with acid. A variation of this tactic was then employed for a synthesis of the hexacyclic framework of the shizozygane alkaloid (±)-strempeliopine. The key step of the synthesis corresponds to an intramolecular 1,4-dipolar cycloaddition of a heteroaromatic betaine across a tethered 4-((2-nitrophenyl)but-3-enyl) side chain. Catalytic reduction of the nitro group followed by reaction with NBS resulted in the formation of the required pentacyclic indoline framework of the target alkaloid. Closure of the final ring of the shizozygane skeleton was carried using an oxidative cyclization.
Co-reporter:JoséM. Mejía-Oneto
Helvetica Chimica Acta 2008 Volume 91( Issue 2) pp:285-302
Publication Date(Web):
DOI:10.1002/hlca.200890034
Abstract
The RhII-catalyzed cycloaddition cascade of an indolyl-substituted α-diazo imide was used for the total synthesis of the complex pentacyclic alkaloid (±)-aspidophytine. Treatment of the resulting dipolar cycloadduct with BF3⋅OEt2 induces a domino fragmentation cascade. The reaction proceeds by an initial cleavage of the oxabicyclic ring and formation of a transient N-acyl iminium ion which reacts further with the adjacent tert-butyl ester and sets the required lactone ring present in aspidophytine. A three-step sequence was then used to remove both the ester and OH groups. Subsequent functional group manipulations allowed for the high-yielding conversion to (±)-aspidophytine.
Co-reporter:Albert Padwa
Journal of Organometallic Chemistry 2005 Volume 690(24–25) pp:5533-5540
Publication Date(Web):1 December 2005
DOI:10.1016/j.jorganchem.2005.06.010
The transition metal catalyzed reaction of α-diazo carbonyl compounds has found numerous applications in organic synthesis, and its use in either heterocyclic or carbocyclic ring formation is well precedented. In contrast to other catalysts that are suitable for carbenoid reactions of diazo compounds, those constructed with the dirhodium(II) framework are most amenable to ligand modification that, in turn, can influence reaction selectivity. The reaction of rhodium carbenoids with carbonyl groups represents a very efficient method for generating carbonyl ylide dipoles. Rhodium-mediated carbenoid–carbonyl cyclization reactions have been extensively utilized as a powerful method for the construction of a variety of novel polycyclic ring systems. This article will emphasize some of the more recent synthetic applications of the tandem rhodium carbenoid cyclization/cycloaddition cascade for natural product synthesis. Discussion centers on the chemical behavior of the rhodium metal carbenoid complex that is often affected by the nature of the ligand groups attached to the metal center.This mini-review article emphasizes some of the more recent synthetic applications of the tandem rhodium carbenoid cyclization/cycloaddition cascade for natural product synthesis. Discussion centers on the chemical behavior of the rhodium metal carbenoid complex that is often affected by the nature of the ligand groups attached to the metal center.
Co-reporter:Albert Padwa
Helvetica Chimica Acta 2005 Volume 88(Issue 6) pp:1357-1374
Publication Date(Web):21 JUN 2005
DOI:10.1002/hlca.200590109
The transition metal catalyzed reaction of α-diazo carbonyl compounds has found numerous applications in organic synthesis, and its use in either heterocyclic or carbocyclic ring formation is well-precedented. In contrast to other catalysts that are suitable for carbenoid reactions of diazo compounds, those constructed with the dirhodium(II) framework are most amenable to ligand modification that, in turn, can influence reaction selectivity. The reaction of rhodium carbenoids with carbonyl groups represents a very efficient method for generating carbonyl ylide dipoles. Rhodium-mediated carbenoid–carbonyl cyclization reactions have been extensively utilized as a powerful method for the construction of a variety of novel polycyclic ring systems. This article will emphasize some of the more recent synthetic applications of the tandem cyclization/cycloaddition cascade for natural product synthesis. Discussion centers on the chemical behavior of the rhodium metal–carbenoid complex that is often affected by the nature of the ligand groups attached to the metal center.
Co-reporter:Albert Padwa
Journal of Organometallic Chemistry 2001 Volumes 617–618() pp:3-16
Publication Date(Web):15 January 2001
DOI:10.1016/S0022-328X(00)00764-6
The rhodium(II)-catalyzed reaction of α-diazo ketones bearing tethered alkyne units represents a new and useful method for the construction of a variety of substituted cyclopentenones. The process proceeds by addition of the rhodium-stabilized carbenoid onto the acetylenic π-bond to give a vinyl carbenoid intermediate. The resulting rhodium complex undergoes a wide assortment of reactions including cyclopropanation, 1,2-hydrogen migration, CH-insertion, addition to tethered alkynes and ylide formation. The exact pathway followed is dependent on the specific metal/ligand employed and is also influenced by the nature of the solvent. Sulfonium ylide formation occurred both intra and intermolecularly when the reaction was carried out in the presence of a sulfide. In the case where an ether oxygen was present on the backbone of the vinyl carbenoid, cyclization afforded an oxonium ylide which underwent a [1,2] or [2,3]-sigmatropic shift to give a rearranged product. These cyclic metallocarbenoids were also found to interact with a neighboring carbonyl π-bond to produce carbonyl ylide dipoles that could be trapped with added dipolarophiles. The domino transformation was also performed intramolecularly by attaching an alkene directly to the carbonyl group. When 2-alkynyl-2-diazo-3-oxobutanoates were treated with a Rh(II)-catalyst, furo[3,4-c]furans were formed in excellent yield. The 1,5-electrocyclization process involved in furan formation has also been utilized to produce indeno[1,2-c]furans. Rotamer population was found to play a significant role in the cyclization of α-diazo amide systems containing tethered alkynes. In this account, an overview of our work in this area is presented.
Co-reporter:Albert Padwa
Journal of Heterocyclic Chemistry 1999 Volume 36(Issue 6) pp:1349-1364
Publication Date(Web):12 MAR 2009
DOI:10.1002/jhet.5570360601
Co-reporter:Albert Padwa
Chemical Society Reviews 2009 - vol. 38(Issue 11) pp:NaN3081-3081
Publication Date(Web):2009/04/03
DOI:10.1039/B816701J
In this tutorial review, the rhodium(II)-catalyzed domino reactions of α-diazo carbonyl compounds are summarized. The article will emphasize some of the more recent synthetic applications of the rhodium carbenoid cyclization/cycloaddition domino cascade for alkaloid synthesis. The many structurally diverse and highly successful examples of both oxa and azapolycyclic ring formation cited in this tutorial review clearly demonstrate that the domino cyclization/cycloaddition cascade of metallo carbenoids has evolved as an important strategy for the synthesis of nitrogen containing natural products.

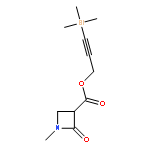
![Propanedioic acid, mono[3-(trimethylsilyl)-2-propynyl] ester](http://img.cochemist.com/ccimg/497100/497070-59-4.png)
![Propanedioic acid, mono[3-(trimethylsilyl)-2-propynyl] ester](http://img.cochemist.com/ccimg/497100/497070-59-4_b.png)
![1H,3H-Furo[3,4-c]furan-1-one, 6-(1-pyrrolidinyl)-4-(trimethylsilyl)-](http://img.cochemist.com/ccimg/289700/289673-86-5.png)
![1H,3H-Furo[3,4-c]furan-1-one, 6-(1-pyrrolidinyl)-4-(trimethylsilyl)-](http://img.cochemist.com/ccimg/289700/289673-86-5_b.png)
![1H,3H-Furo[3,4-c]furan-1-one, 6-(dimethylamino)-4-(trimethylsilyl)-](http://img.cochemist.com/ccimg/289700/289673-85-4.png)
![1H,3H-Furo[3,4-c]furan-1-one, 6-(dimethylamino)-4-(trimethylsilyl)-](http://img.cochemist.com/ccimg/289700/289673-85-4_b.png)


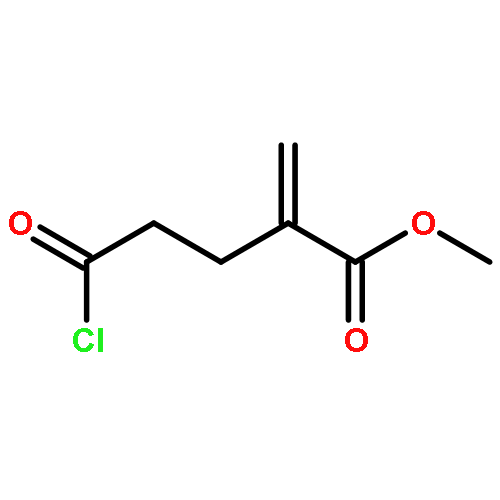
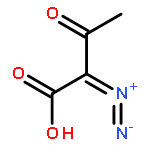
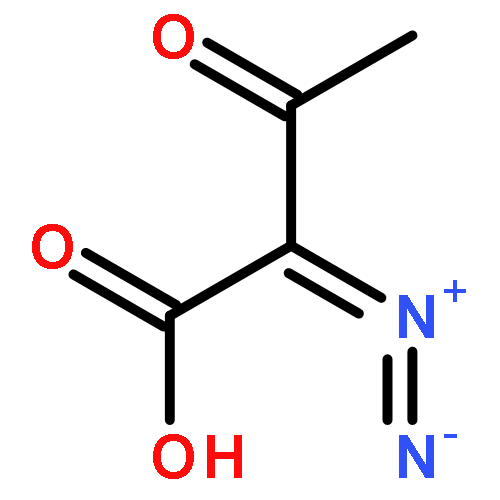
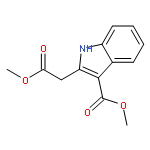
dimethyl-](http://img.cochemist.com/ccimg/82600/82511-02-2.png)
dimethyl-](http://img.cochemist.com/ccimg/82600/82511-02-2_b.png)