Co-reporter:Viharika Bobba, Vishal Nanavaty, Nethrie D. Idippily, Anran Zhao, Bibo Li, Bin Su
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 12(Issue 12) pp:
Publication Date(Web):15 June 2017
DOI:10.1016/j.bmc.2017.04.009
•A series of sulfonamide tubulin inhibitors were synthesized.•The compounds were examined with mammalian cells and trypanosomal cells to identify selective drug candidates.•A new analog showed high selectivity and potency against the growth of trypanosome cells.•The compound decreased tubulin polymerization in trypanosome cells.African trypanosomiasis is still a threat to human health due to the severe side-effects of current drugs. We identified selective tubulin inhibitors that showed the promise to the treatment of this disease, which was based on the tubulin protein structural difference between mammalian and trypanosome cells. Further lead optimization was performed in the current study to improve the efficiency of the drug candidates. We used Trypanosoma brucei brucei cells as the parasite model, and human normal kidney cells and mouse macrophage cells as the host model to evaluate the compounds. One new analog showed great potency with an IC50 of 70 nM to inhibit the growth of trypanosome cells and did not affect the viability of mammalian cells. Western blot analyses reveal that the compound decreased tubulin polymerization in T. brucei cells. A detailed structure activity relationship (SAR) was summarized that will be used to guide future lead optimization.Download high-res image (100KB)Download full-size image
Co-reporter:Nethrie D. Idippily, Qiaoyun Zheng, Chunfang Gan, Aicha Quamine, Morgan M. Ashcraft, Bo Zhong, Bin Su
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 11(Issue 11) pp:
Publication Date(Web):1 June 2017
DOI:10.1016/j.bmcl.2017.04.046
Copalic acid, one of the diterpenoid acids in copaiba oil, inhibited the chaperone function of α-crystallin and heat shock protein 27 kD (HSP27). It also showed potent activity in decreasing an HSP27 client protein, androgen receptor (AR), which makes it useful in prostate cancer treatment or prevention. To develop potent drug candidates to decrease the AR level in prostate cancer cells, more copalic acid analogs were synthesized. Using the level of AR as the readout, 15 of the copalic acid analogs were screened and two compounds were much more potent than copalic acid. The compounds also dose-dependently inhibited AR positive prostate cancer cell growth. Furthermore, they inhibited the chaperone activity of α-crystallin as well.A series of copalic acid analogs was synthesized. Some analogs showed good potency to down-regulate androgen receptor in prostate cancer cells. They also showed small chaperone inhibitory activity in the in vitro assay. The results indicate that using small molecule chaperone inhibitor to down-regulate androgen receptor could be a potential approach for prostate cancer treatment or prevention.Download high-res image (150KB)Download full-size image
Co-reporter:Snigdha Chennamaneni, Chunfang Gan, Rati Lama, Bo Zhong, Bin Su
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 2) pp:277-285
Publication Date(Web):15 January 2016
DOI:10.1016/j.bmc.2015.12.016
Cyclooxygenase (COX) inhibitor Indomethacin analogs exhibited more potent cancer cell growth inhibition and apoptosis inducing activities than the parental compound. The anti-proliferative mechanism investigation of the analogs revealed that they inhibited tubulin polymerization at high concentrations whereas enhanced polymerization at low concentrations. The two opposite activities might antagonize each other and impaired the anti-proliferative activity of the derivatives eventually. In this study, we further performed lead optimization based on the structure activity relationship (SAR) generated. One of the new Indomethacin derivatives compound 11 {2-(4-(benzyloxy)phenyl)-N-(1-(4-bromobenzoyl)-3-(2-((2-(dimethylamino)ethyl)amino)-2-oxoethyl)-2-methyl-1H-indol-5-yl)acetamide} inhibited the proliferation of a panel of cancer cell lines with IC50s at the sub-micromole levels. Further study revealed that the compound only enhanced tubulin polymerization and was a tubulin stabilizer.
Co-reporter:Bo Zhong, Sergei Vatolin, Nethrie D. Idippily, Rati Lama, Laila A. Alhadad, Frederic J. Reu, Bin Su
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 4) pp:1272-1275
Publication Date(Web):15 February 2016
DOI:10.1016/j.bmcl.2016.01.020
Inhibition of DNA methyltransferase 1 (DNMT1) can reverse the malignant behavior of cancer cells by restoring expression of aberrantly silenced genes that are required for differentiation, senescence, and apoptosis. Clinically used DNMT1 inhibitors decitabine and azacitidine inhibit their target by covalent trapping after incorporation into DNA as azacytidine analogs. These nucleoside compounds are prone to rapid enzymatic inactivation in blood, posing challenges to the development of purely epigenetic dosing schedules. Non-nucleoside compounds that suppress expression or function of DNMT1 may overcome this problem. Using a high-throughput PCR-based site specific chromatin condensation assay, we identified a compound that reactivated Cyclin-Dependent Kinase Inhibitor 2A (CDKN2A) in myeloma cells and suppressed expression of DNMT1 from a library of 5120 chemically diverse small molecules. Lead optimization was performed to generate 26 new analogs with lung cancer proliferation and DNMT1 expression as activity readout. Two of the new derivatives showed 2 fold improvement of growth inhibiting potency and also decreased DNMT1 protein levels in lung cancer cells.A series of non-nucleoside DNA methyltransferase 1 (DNMT1) inhibitors was designed and synthesized. The potency of the compounds on cancer cell growth was determined with lung cancer cells. A new analog showed improved activity and also downregulated DNMT1 protein level.Download high-res image (92KB)Download full-size image
Co-reporter:Bo Zhong, Rati Lama, Wannarasmi Ketchart, Monica M. Montano, Bin Su
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 5) pp:1410-1413
Publication Date(Web):1 March 2014
DOI:10.1016/j.bmcl.2014.01.025
Co-reporter:Bo Zhong, Rati Lama, Daniel G. Kulman, Bibo Li, Bin Su
European Journal of Medicinal Chemistry 2014 80() pp: 243-253
Publication Date(Web):
DOI:10.1016/j.ejmech.2014.04.038
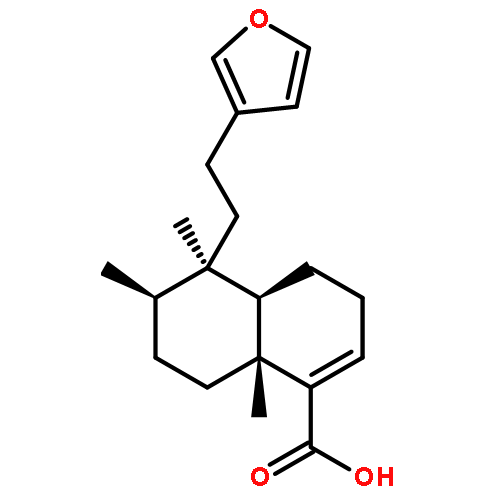
Co-reporter:Rati Lama, Bo Zhong, Daniel G. Kulman, Bin Su
Phytochemistry Letters 2014 10() pp: 65-75
Publication Date(Web):
DOI:10.1016/j.phytol.2014.08.006
Co-reporter:Bo Zhong ; Snigdha Chennamaneni ; Rati Lama ; Xin Yi ; Werner J. Geldenhuys ; John J. Pink ; Afshin Dowlati ; Yan Xu ; Aimin Zhou
Journal of Medicinal Chemistry 2013 Volume 56(Issue 13) pp:5306-5320
Publication Date(Web):June 16, 2013
DOI:10.1021/jm4004736
Heat shock protein 27 (Hsp27) is a chaperone protein, and its expression is increased in response to various stress stimuli including anticancer chemotherapy, which allows the cells to survive and causes drug resistance. We previously identified lead compounds that bound to Hsp27 and tubulin via proteomic approaches. Systematic ligand based optimization in the current study significantly increased the cell growth inhibition and apoptosis inducing activities of the compounds. Compared to the lead compounds, one of the new derivatives exhibited much better potency to inhibit tubulin polymerization but a decreased activity to inhibit Hsp27 chaperone function, suggesting that the structural modification dissected the dual targeting effects of the compound. The most potent compounds 20 and 22 exhibited strong cell proliferation inhibitory activities at subnanomolar concentration against 60 human cancer cell lines conducted by Developmental Therapeutic Program at the National Cancer Institute and represented promising candidates for anticancer drug development.
Co-reporter:Rati Lama, Lin Zhang, Janine M. Naim, Jennifer Williams, Aimin Zhou, Bin Su
Bioorganic & Medicinal Chemistry 2013 21(4) pp: 922-931
Publication Date(Web):
DOI:10.1016/j.bmc.2012.12.007
Co-reporter:Xin Yi ; Bo Zhong ; Kerri M. Smith ; Werner J. Geldenhuys ; Ye Feng ; John J. Pink ; Afshin Dowlati ; Yan Xu ; Aimin Zhou
Journal of Medicinal Chemistry 2012 Volume 55(Issue 7) pp:3425-3435
Publication Date(Web):March 21, 2012
DOI:10.1021/jm300100d
We previously developed a series of anticancer agents based on cyclooxygenase-2 (COX-2) inhibitor nimesulide as a lead compound. However, the molecular targets of these agents still remain unclear. In this study, we synthesized a biotinylated probe based on a representative molecule of the compound library and performed protein pull-down assays to purify the anticancer targets of the compound. Via proteomic approaches, the major proteins bound to the probe were identified to be tubulin and heat shock protein 27 (Hsp27), and the compound inhibited tubulin polymerization by binding at the colchicine domain. However, the tubulin inhibitory effect of the compound activated the Hsp27 phosphorylation and possibly overrode the direct Hsp27 inhibitory effects, which made it difficult to solely validate the Hsp27 target. Taken together, the compound was a dual ligand of tubulin and Hsp27, inhibited tubulin polymerization, and had the potential to be a class of new chemotherapeutic agents.
Co-reporter:Bo Zhong, Xiaohan Cai, Snigdha Chennamaneni, Xin Yi, Lili Liu, John J. Pink, Afshin Dowlati, Yan Xu, Aimin Zhou, Bin Su
European Journal of Medicinal Chemistry 2012 Volume 47() pp:432-444
Publication Date(Web):January 2012
DOI:10.1016/j.ejmech.2011.11.012
Cyclooxygenase-2 (COX-2) inhibitor nimesulide inhibits the proliferation of various types of cancer cells mainly via COX-2 independent mechanisms, which makes it a good lead compound for anti-cancer drug development. In the presented study, a series of new nimesulide analogs were synthesized based on the structure–function analysis generated previously. Some of them displayed very potent anti-cancer activity with IC50s around 100 nM–200 nM to inhibit SKBR-3 breast cancer cell growth. CSUOH0901 (NSC751382) from the compound library also inhibits the growth of the 60 cancer cell lines used at National Cancer Institute Developmental therapeutics Program (NCIDTP) with IC50s around 100 nM–500 nM. Intraperitoneal injection with a dosage of 5 mg/kg/d of CSUOH0901 to nude mice suppresses HT29 colorectal xenograft growth. Pharmacokinetic studies demonstrate the good bioavailability of the compound.CSUOH0901, a non-COX-2 inhibitory derivative of COX-2 inhibitor nimesulide, significantly suppresses the growth of multiple cancer cell lines with IC50s around 100–500 nM.Highlights► COX-2 inhibitor nimesulide inhibited cancer cell growth independent of COX-2. ► A set of non-COX-2 active nimesulide anti-cancer derivatives were synthesized. ► The potential hepatotoxicity of nimesulide derivatives was also abolished. ► New analogs showed very promising in vitro and vivo anti-cancer activity.
Co-reporter:Rati Lama, Ranjodh Sandhu, Bo Zhong, Bibo Li, Bin Su
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 17) pp:5508-5516
Publication Date(Web):1 September 2012
DOI:10.1016/j.bmcl.2012.07.023
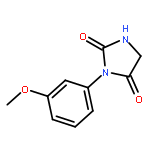
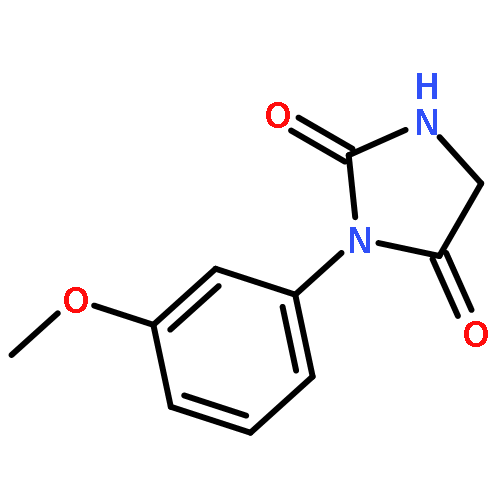
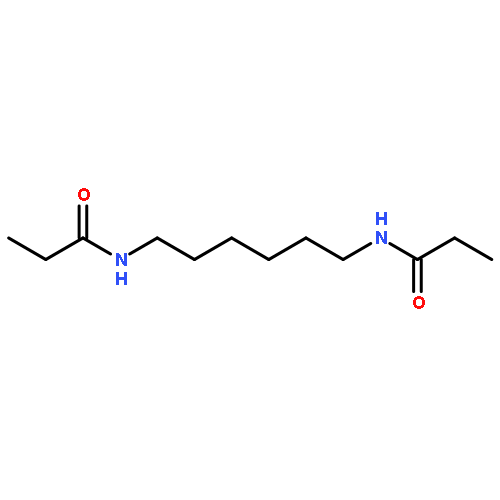
The potency of a series of sulfonamide tubulin inhibitors against the growth of Trypanosoma brucei (T. brucei), as well as human cancer and primary fibroblast cells were evaluated with the aim of determining whether compounds that selectively inhibit parasite proliferation could be identified. Several compounds showed excellent selectivity against T. brucei growth, and have the potential to be used for the treatment of Human African trypanosomiasis. A T. brucei tubulin protein homology model was built based on the crystal structure of the bovine tubulin. The colchicine-binding domain, which is also the binding site of the tested sulfonamide tubulin inhibitors, showed clear differences between the tubulin structures and presumably explained the selectivity of the compounds.A series of sulfonamide tubulin inhibitors were tested with Trypanosoma brucei and human cancer and normal cells, and several of them selectively inhibited the growth of the parasite cells without affecting mammalian cell proliferation.
Co-reporter:Bo Zhong, Rati Lama, Kerri M. Smith, Yan Xu, Bin Su
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 18) pp:5324-5327
Publication Date(Web):15 September 2011
DOI:10.1016/j.bmcl.2011.07.025
JCC76 is a derivative of cyclooxygenase-2(COX-2) selective inhibitor nimesulide and exhibits potent anti-breast cancer activity. It selectively induces apoptosis of Her2 positive breast cancer cells. However, the specific molecular targets of JCC76 still remain unclear, which significantly withdraw the further drug development of JCC76. To identify the molecular targets of JCC76, a six carbon linker and biotin conjugated JCC76 probe was designed and synthesized. The anti-proliferation activity of the probe and its analogs was evaluated.JCC76 is a derivative of COX-2 inhibitor nimesulide. It significantly inhibits cancer cell growth. Biotinylated JCC76 as a probe was designed and synthesized and exhibited similar anti-cancer activity as JCC76.
Co-reporter:Bin Su, Xiaohan Cai, Yanyan Hong, Shiuan Chen
The Journal of Steroid Biochemistry and Molecular Biology (October 2010) Volume 122(Issue 4) pp:232-238
Publication Date(Web):1 October 2010
DOI:10.1016/j.jsbmb.2010.06.004
Cyclooxygenase-2 (COX-2) inhibitor nimesulide derivatives compounds A and B decreased aromatase activity in breast cancer cells via a novel mechanism different to aromatase inhibitors (AIs), and were defined as “aromatase suppressors”. Breast carcinoma cells (MCF-7aro and T47Daro) transfected with aromatase full gene were used to explore the mechanisms of the two compounds. They dose and time-dependently suppressed aromatase activity in MCF-7aro and T47Daro cells in the nanomole range. However, they neither directly inhibited aromatase, nor improved aromatase degradation even at much higher concentrations. They could also suppress androgen stimulated cell growth, but did not affect estrogen enhanced cell proliferation. These results suggest that compounds A and B selectively interfere with aromatase in breast cancer cells, but not estrogen receptor (ER) downstream to disrupt androgen mediated cell growth. Interestingly, compound B effectively inhibited LTED (long-term estrogen deprived MCF-7aro cell) cell growth, which is a model for AIs resistance, with an IC50 of 4.68 ± 0.54 μM. The results indicate that compound B could potentially overcome AI resistance in breast cancer cell and could be used as a lead to design more potent derivatives.
Co-reporter:Bin Su, Cynthie Wong, Yanyan Hong, Shiuan Chen
The Journal of Steroid Biochemistry and Molecular Biology (February 2011) Volume 123(Issues 3–5) pp:101-108
Publication Date(Web):1 February 2011
DOI:10.1016/j.jsbmb.2010.11.012
It has been demonstrated that growth factors produced by breast cancer cells stimulate aromatase expression in both breast cancer and adjacent adipose fibroblasts and stromal cells. However, whether these growth factors affect aromatase activity by other mechanisms still remain unclear. In the current study, MCF-7aro and T47Daro aromatase transfected breast carcinoma cells were used to explore the mechanisms of post-transcriptional regulation of aromatase activity by growth factor pathways. Our study reveals that PI3K/Akt and MAPK inhibitors suppressed aromatase activity in MCF-7aro cells. However, PI3K/Akt pathway inhibitors stimulated aromatase activity in T47Daro cells. This is due to enhanced MAPK phosphorylation as compensation after the PI3K/Akt pathway has been blocked. IGF-1 treatment increased aromatase activity in both breast cancer cell lines. In addition, LTEDaro cells (long-term estrogen deprived MCF-7aro cells) which have enhanced MAPK activity, show higher aromatase activity compared to parental MCF-7aro cells, but the aromatase protein level remains the same. These results suggest that aromatase activity could be enhanced by growth factor signaling pathways via post-transcriptional mechanisms.Research highlights▶ Growth factors secreted by breast cancer cells stimulate aromatase expression in breast cancer cells. ▶ Although there are some reports regarding the non-transcriptional regulation of aromatase by these factors, there is no systematic study about this phenomenon. ▶ Our study addressed this mechanism and reveals that aromatase activity could be increased by these signaling via post transcriptional mechanisms. ▶ This is an important supplement of the current understanding that growth factors only up-regulate aromatase expression in breast cancer tissue.
Co-reporter:Bo Zhong, Xiaohan Cai, Xin Yi, Aimin Zhou, Shiuan Chen, Bin Su
The Journal of Steroid Biochemistry and Molecular Biology (August 2011) Volume 126(Issues 1–2) pp:10-18
Publication Date(Web):1 August 2011
DOI:10.1016/j.jsbmb.2011.03.018
Third generation aromatase inhibitors (AIs) are more effective than tamoxifen in the treatment of estrogen receptor (ER) positive breast cancer. However, long-term use of AIs commonly results in resistance. We examined whether compound JCC76{Cyclohexanecarboxylic acid [3-(2,5-dimethyl-benzyloxy)-4-(methanesulfonyl-methyl-amino)-phenyl]-amide}, an analog of Cyclooxygenase-2 (COX-2) inhibitor nimesulide, can inhibit the growth of AI-insensitive breast cancer cells and the mechanisms by which the compound affects cell proliferation. LTEDaro (long term estrogen deprived MCF-7aro cell) cells, which are a model for AI resistance, were used in this study. JCC76 effectively inhibited LTEDaro cell proliferation with an IC50 of 2.75 ± 0.31 μM. Further investigations reveal that the compound significantly induced apoptosis in LTEDaro cells by decreasing pAKT, BCL-2 and pBad protein levels, which were all up regulated in the cells after long term estrogen deprivation. LTEDaro tumor size and weight were decreased in ovariectomized nude mice treated with the compound, and cell apoptosis in the tumor tissue was increased compared to the control. The animal weight remained almost unchanged which indicated the low toxicity of the compound. These results suggest that JCC76 overcame AI resistance by inducing cell apoptosis as illustrated in the in vitro and in vivo models. Collectively, results from this study provide data to support that nimesulide analog JCC76 may be a new drug candidate to treat AI-resistant breast cancers. It could be also used as a lead to design and synthesize more potent derivatives.Highlights► Nimesulide analog JCC 76 inhibits aromatase inhibitor resistant breast cancer cell growth. ► Further studies reveal that the compound significantly induces apoptosis in the resistant cells. ► The inhibition and apoptosis inducing activity are also observed in the in vivo studies. ► JCC76 is a promising drug candidate for aromatase inhibitor resistant breast cancer.
Co-reporter:Nethrie D. Idippily, Chunfang Gan, Paul Orefice, Jane Peterson, Bin Su
Bioorganic & Medicinal Chemistry Letters (15 February 2017) Volume 27(Issue 4) pp:
Publication Date(Web):15 February 2017
DOI:10.1016/j.bmcl.2017.01.025
Histone deacetylase (HDAC) inhibitors modulate various cellular functions including proliferation, differentiation, and apoptosis. Vorinostat (SuberAniloHydroxamic Acid, SAHA) is the first HDAC inhibitor approved by FDA for cancer treatment. However, SAHA distributes in cancer tissue and normal tissue in similar levels. It will be ideal to selectively deliver SAHA into cancer cells. Rapidly growing cancer cells have a great need of cholesterol. Low-density lipoprotein (LDL) is the major cholesterol carrier in plasma and its uptake is mediated by LDL-receptor (LDL-R), a glycoprotein overexpressed on the surface of cancer cells. Herein, we designed and synthesized a SAHA cholesterol conjugate, and further formed the conjugate containing particles with LDL as the carrier. The diameters of the particles were determined. The inhibitory activity of the particles carrying the conjugate was determined with cancer cell proliferation assay, and the hydrolysis of the conjugate by the enzymes in cancer cells was confirmed with LC–MS/MS.A SAHA and cholesterol conjugate was designed and synthesized. The conjugate was loaded into low-density lipoprotein (LDL) particles and the biological activity was examined with cancer cell proliferation assay. The results indicate that the conjugate can be transported into cancer cells via LDL receptor and inhibits cancer cell growth.



![Carbamic acid, [6-(acetylamino)hexyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/145200/145118-23-6.png)
![Carbamic acid, [6-(acetylamino)hexyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/145200/145118-23-6_b.png)
![(5E)-3-(4-chlorophenyl)-5-[(4-chlorophenyl)methylidene]imidazolidine-2,4-dione](http://img.cochemist.com/ccimg/97400/97310-74-2.png)
![(5E)-3-(4-chlorophenyl)-5-[(4-chlorophenyl)methylidene]imidazolidine-2,4-dione](http://img.cochemist.com/ccimg/97400/97310-74-2_b.png)