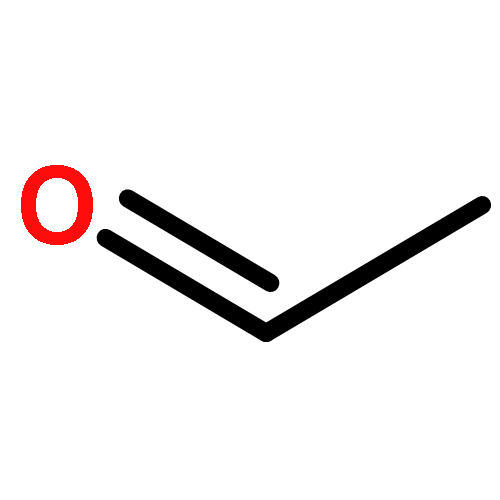
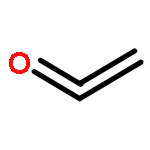
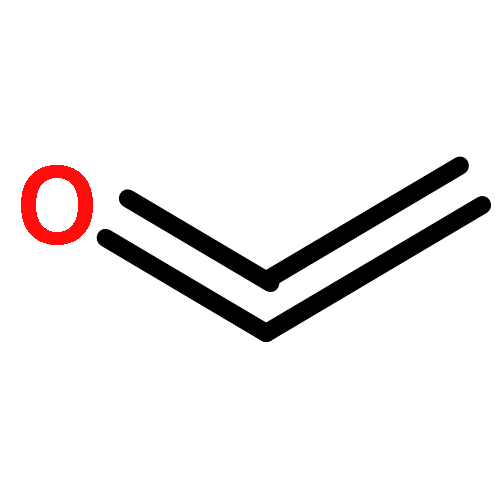
The graphdiyne family has attracted a high degree of concern because of its intriguing and promising properties. However, graphdiyne materials reported to date represent only a tiny fraction of the possible combinations. In this work, we demonstrate a computational approach to generate a series of conceivable graphdiyne-based frameworks (GDY-Rs and Li@GDY-Rs) by introducing a variety of functional groups (R = −NH2, −OH, −COOH, and −F) and doping metal (Li) in the molecular building blocks of graphdiyne without restriction of experimental conditions and rapidly screen the best candidates for the application of CO2 capture and sequestration (CCS). The pore topology and morphology and CO2 adsorption and separation properties of these frameworks are systematically investigated by combining density functional theory (DFT) and grand canonical Monte Carlo (GCMC) simulations. On the basis of our computer simulations, combining Li-doping and hydroxyl groups strategies offer an unexpected synergistic effect for efficient CO2 capture with an extremely CO2 uptake of 4.83 mmol/g at 298 K and 1 bar. Combined with its superior selectivity (13 at 298 K and 1 bar) for CO2 over CH4, Li@GDY-OH is verified to be one of the most promising materials for CO2 capture and separation.Keywords: CO2 adsorption; functional groups; graphdiyne; Li doping; porous carbon materials;
Journal of Photochemistry and Photobiology A: Chemistry 2017 Volume 332() pp:232-240
Publication Date(Web):1 January 2017
DOI:10.1016/j.jphotochem.2016.08.037
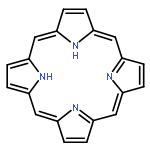
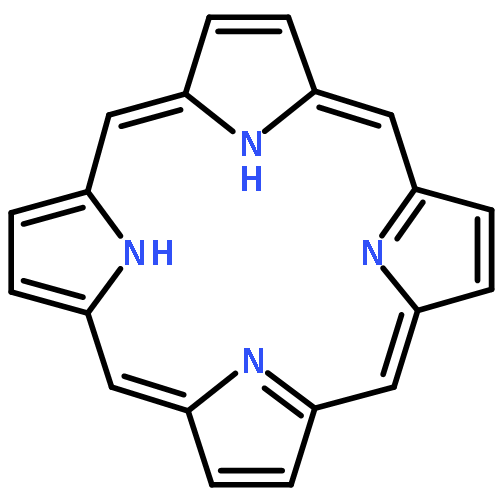
•The properties of new π-bridge modified porphyrin dyes based on GY50 prototpye are theoretically investigated.•A series of key parameters associating with the cell efficiency have been explored.•π-linker would greatly influence the light harvesting performance of dye.A series of new D-π-A porphyrin-based dyes, based on GY50 by utilizing 2-cyanoacrylic acid anchoring group and modifying the π bridge with thiophene, furan and/or vinyl groups, has been designed and investigated for screening efficient sensitizers in dye sensitized solar cells (DSSCs). The efficiency of the designed dye molecules is analyzed through various parameters. The absorption spectra of the designed dyes exhibit the dual Soret and Q bands as that of GY50 with increased molar extinction coefficients as well as red-shifted peaks for the Q band, and also present a modest hump between the Soret and Q bands (500–600 nm), yielding enhanced light harvesting efficiency. In terms of the absorption spectra together with the charge transfer characteristics, driving force for electron injection, dye regeneration as well as conduction band energy shift, the new sensitizers, especially with bithiophene and bithiophene plus vinyl on the π bridge, are found to be promising candidates for high-efficiency DSSCs.
•The brown coal shows super high selectivity of CO2 over CH4 at low pressures.•O-containing, pyridine, and thiophene groups promote the CO2/CH4 selectivity.•The preferential CO2 adsorption is strengthened by the electrostatic interactions.•Amine and sulfoxide groups display high efficient CO2 adsorption.The CO2/CH4 adsorption behaviors in brown coal at the temperatures of 298, 313, and 373 K and in the pressure range of 0.005–10 MPa were investigated by molecular dynamics (MD), density functional theory (DFT), and grand canonical Monte Carlo (GCMC) simulations. The absolute adsorption isotherms of single-component CH4 and CO2 exhibit type-I Langmuir adsorption behavior showing a negative influence of temperature. For the binary CO2/CH4 mixture, brown coal shows super high selectivity of CO2 over CH4 at pressures below 0.2 MPa, which then decreases quickly and finally tends to be constant when the pressure increases. The high competitive adsorption of CO2 originates from the effects of (i) the large electrostatic contributions, (ii) the conducive micropore environment with pore sizes below 0.56 nm, and (iii) the stronger adsorption of CO2 with respect to CH4. These effects are strengthened by the high-density oxygen-containing, pyridine, and thiophene functional groups contained in brown coal, which provide abundant and strong adsorption sites for CO2, but show weaker affinity to CH4. Furthermore, the influence of various nitrogen- and sulfur-containing functional groups on the CO2 adsorption capacity was also investigated. The results indicate that the basicity of the oxygen- and nitrogen-containing groups has a large influence on the CO2 adsorption, while for the sulfur functional groups the determining factor is the polarity.The preferential adsorption of CO2 than CH4 in brown coal is strengthened by the electrostatic interactions and high-density oxygen-containing, pyridine, and thiophene functional groups.Download high-res image (140KB)Download full-size image
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 26) pp:17449-17460
Publication Date(Web):2017/07/05
DOI:10.1039/C7CP01859B
The hydrodesulfurization (HDS) of thiophene on clean and S-modified MoP(010) is investigated to understand the HDS mechanism as well as the surface sulfur (S) atom effect using periodic density functional theory (DFT). The results show that thiophene prefers strongly flat adsorption on both the clean and S-modified surfaces, in either the molecular state or the dissociative state breaking simultaneously one C–S bond, and the adsorption of thiophene can be slightly weakened by the surface S atom. Thermodynamic and kinetic analysis indicates that the HDS of thiophene in both the molecular and dissociative adsorption states prefers to take place along the direct desulfurization (DDS) pathway rather than hydrogenation on both the clean and S-modified MoP(010) surfaces. Surface S shows a promotion effect on the HDS catalytic activity of MoP(010), because the energy barrier/rate constant of the rate-determining step on the DDS pathway is decreased/enlarged under the S modification. Compared with the situation of MoP(001), MoP(010) should have relatively low HDS activity, since a higher energy barrier as well as weaker exothermicity is involved in the reaction on the latter surface.
Co-reporter:Shuxian Wei, Yang Shao, Xiaofan Shi, Xiaoqing Lu, Ke Li, Zigang Zhao, Chen Guo, Houyu Zhu, Wenyue Guo
Organic Electronics 2016 Volume 29() pp:142-150
Publication Date(Web):February 2016
DOI:10.1016/j.orgel.2015.12.004
•Heteroleptic Cu(I) sensitizers designed for dye-sensitized solar cells.•Convenient photo-induced intramolecular electron transfer of Cu(I) sensitizers.•Transverse and longitudinal π-conjugation improve electronic and spectral property.A series of heteroleptic Cu(I) complexes integrating dicarboxylic acid dimethyl bipyridine/phenanthroline with functionalized chromophores have been investigated by density functional theory (DFT) and time-dependent DFT (TD-DFT). The molecular geometry, electronic structure, electronic excitation and absorption spectrum, light-harvesting efficiency (LHE), and intramolecular electron transfer (IET) are analyzed in dichloromethane solution. The four-coordinated heteroleptic Cu(I) complexes exhibit distorted trigonal-pyramidal geometries. Introducing the functionalized chromophores does not only disperse the electron distribution of HOMOs over the Cu(I) center and π-conjugated donors, but also downshift the HOMO levels and reduce the HOMO-LUMO gaps, and thus improve the light-harvesting efficiency significantly. The electronic excitations of all Cu(I) complexes at 300.0–375.0 and 475.0–575.0 nm are more favorable for the direct electron injection into semiconductor than those at 400.0–450.0 nm. The structural optimizations along the transverse direction of the Cu(I) complexes result in more transferred electrons, longer electron transfer distances, smaller IET rates, and less orbital overlaps than those along the longitudinal direction. Our results elucidate the detailed photon-to-electron conversion mechanisms of heteroleptic Cu(I) complexes, and provide a fresh insight into the design and screening of novel sensitizers for DSSCs.
Co-reporter:Yunjie Liu, Wenyue Guo, Xiaoqing Lu, Wei Gao, Guixia Li, Yahui Guo, Jun Zhu and Lanzhong Hao
RSC Advances 2016 vol. 6(Issue 8) pp:6289-6299
Publication Date(Web):04 Jan 2016
DOI:10.1039/C5RA20087C
In this work, the adsorption of S-containing species (S, HS, and H2S) and the hydrogenation of S on the Pt–Pd alloy were investigated by using the periodic density functional theory (DFT). The energy minimum of the adsorbed S, HS, and H2S were identified to bind preferentially on the fcc, bridge and top sites, respectively. Compared to single metal surfaces, the adsorption energies of adsorbates were calculated to be larger on the Pt–Pd alloy surfaces and adsorbed preferably on the sites with a majority of Pt atoms. The reaction pathways and energy profiles for the conversion of adsorbed S and H into gas phase H2S were determined. The results showed that both the S + H and HS + H reactions on Pt–Pd alloy surfaces were endothermic. The energy for the overall reaction on Pt–Pd alloy surfaces decreased significantly by 0.30–0.55 eV compared to pure Pt(111) surface. In addition, the energy barrier on Pt–1Pd(111) (one Pt atom was replaced by Pd atom on Pt(111) surface) was lower than that on other studied alloy surfaces. The above characteristics revealed that the hydrogenation of S to H2S was easier on Pt–1Pd(111) surface than on the other alloy surfaces. The partial density of states was utilized to illustrate the interaction mechanisms between S-containing species and surface atoms.
Co-reporter:Xiaoqing Lu, Weili Wang, Zhigang Deng, Houyu Zhu, Shuxian Wei, Siu-Pang Ng, Wenyue Guo and Chi-Man Lawrence Wu
RSC Advances 2016 vol. 6(Issue 3) pp:1729-1737
Publication Date(Web):21 Dec 2015
DOI:10.1039/C5RA21793H
The competitive oxidation reaction mechanism of methanol on the Ru(0001) surface has been investigated by periodic density functional theory (DFT). Stable adsorption configurations, elementary reaction energies and barriers, the potential energy surface (PES), and the electrochemical potential analysis were elucidated. The results showed that O–H bond activation was more competitive than C–H and C–O bond activation during the initial methanol oxidation. Competitive pathways occurred for CH3OH oxidation to CH2O via CH3OH → CH3O → CH2O versus CH3OH → CH2OH → CH2O, further to COOH via the CO pathway CH2O → CHO → CO → COOH versus the non-CO pathway CH2O → CH2OOH → CHOOH → COOH, and finally oxidation to CO2. Taking PES and the electrochemical potential analysis into account, CH3OH → CH2OH → CH2O → CH2OOH → CHOOH → COOH → CO2 appeared to be the preferred oxidation pathway. The OH group could inhibit CO formation by directly reacting with CH2O to yield CH2OOH but could not efficiently remove the CO that had already been produced by the reactions.
Co-reporter:Shuxian Wei, Ke Li, Xiaoqing Lu, Zigang Zhao, Yang Shao, Yong Dang, Shaoren Li, Wenyue Guo
Materials Chemistry and Physics 2016 Volume 173() pp:139-145
Publication Date(Web):15 April 2016
DOI:10.1016/j.matchemphys.2016.01.049
•Assessment of heteroleptic Cu(I) dyes for dye-sensitized solar cells.•Suitable energy levels render Cu(I) dyes ideal candidates for electron injection.•Heteroaromatic groups efficiently improve Cu(I) dyes light-harvesting properties.•Dye with dithiole group exhibits ideal photoelectronic property.A series of heteroleptic Cu(I)-based dyes were investigated by density functional theory (DFT) and time-dependent DFT (TD-DFT). Results showed that Cu(I)-based dyes were inclined to form distorted pseudo-trigonal pyramidal configurations with four-coordinated geometry index τ4 ranging from 0.905 to 0.914. The absorption spectra of Cu(I)-based dyes covered ∼300.0–600.0 nm region, and the lowest excitation states were crucial for efficient electron excitation and separation. Suitable energy levels of Cu(I)-based dyes rendered them thermodynamically favorable for efficient electron injection into semiconductor and regeneration from electrolyte. Relative to π-conjugation, heteroaromatic groups introduced into ancillary ligands could significantly improve the property of Cu(I)-based dyes by decreasing HOMO-LUMO gaps, red-shifting spectral range, strengthening absorption intensity, boosting light-harvesting efficiency, and promoting interfacial electron injection. Specifically, Cu(I)-based dye with dithiole-functionalized group exhibited outstanding optoelectronic property.
The Journal of Physical Chemistry C 2016 Volume 120(Issue 36) pp:20181-20191
Publication Date(Web):August 26, 2016
DOI:10.1021/acs.jpcc.6b07151
The water–gas shift (WGS) reaction plays a key role in hydrogen economy. Owing to the exothermic nature of the reaction, low-temperature WGS catalysts are highly desired. Zn-modified Pd-based catalysts are promising candidates for low-temperature WGS. Herein, the effect of Zn addition on the WGS catalysis is systematically studied by using the Pd(111) and PdZn(111) surface as models. Owing to the addition of Zn, the electron-accepting ability of the catalyst is weakened, while the electron-donating ability is increased. As a result, the adsorptions of electron-donor adsorbates, including H2O, CO, H, cis-COOH, trans-COOH, and H2, are weakened, while the adsorptions of electron-acceptor adsorbates, including O and OH, are strengthened. The same most favorable reaction path is found on Pd(111) and PdZn(111), which is the associative mechanism with the carboxyl dehydrogenation assisted by adsorbed OH. Although the most favorable path is the same, the weakening of CO adsorption makes the rate-determining step change from the association of CO and OH forming cis-COOH on Pd(111) to the dissociation of H2O on PdZn(111). The rate-determining step on PdZn(111) has an energy barrier lower than the rate-determining step on Pd(111). The promotion mechanism of the PdZn alloy for WGS is therefore attributed to the fact that the addition of Zn weakens the adsorption of CO and thereby alters the rate-determining step.
Co-reporter:Yuhua Chi;Lianming Zhao;Xiaoqing Lu;Changhua An
Journal of Materials Science 2016 Volume 51( Issue 11) pp:5046-5060
Publication Date(Web):2016 June
DOI:10.1007/s10853-016-9808-8
Density functional theory has been applied to study the geometric and electronic structures and the catalytic properties of Ag and AgAu clusters for CO oxidation. The calculated results suggest that the doping of Au atoms improve the stability of AgAu clusters. Correspondingly, the binding energy (BE) per atom of AgnAu is larger than that of pure Agn+1 cluster, due to strong hybridization between the d orbitals of Au and the s orbitals of Ag in AgnAu clusters. With the increasing Au concentration, the BE of Ag13−nAun(n = 1–8) clusters increase smoothly, while second-order difference of energies (Δ2E) and fragmentation energies (ΔE) show an even–odd oscillation. The AgAu clusters containing an odd n umber of gold atoms (Ag13−nAun, n = 3, 5, 7) are relatively stable compared to their neighbor. The CO and O2 adsorption properties on the Ag13, Ag10Au3, Ag8Au5, and Ag6Au7 clusters suggest that O2 is strongly activated by the clusters, while the activation of CO is much weak. Furthermore, the activation of O2 on AgAu cluster is stronger than that on pure Ag13 cluster, especially on Ag8Au5 cluster, due to the strengthened polarization of O–O bond. Compared to Ag13, Ag10Au3, and Ag6Au7 clusters, the lower energy barriers on Ag8Au5 cluster suggest a higher catalytic activity of Ag8Au5 cluster for O2 dissociation and CO oxidation reactions. The calculated results suggest that Ag8Au5 cluster could effectively reduce the carbon monoxide poisoning and exhibits the excellent catalytic performance for CO oxidation. Our study provides atomic-scale insights into the nature of the interfacial effects that determine CO oxidation on Ag–Au cluster catalysts.
Co-reporter:Yuhua Chi;Lianming Zhao;Xiaoqing Lu;Changhua An
Journal of Nanoparticle Research 2016 Volume 18( Issue 3) pp:
Publication Date(Web):2016 March
DOI:10.1007/s11051-016-3386-1
Density functional theory (DFT) has been applied to study the geometrical and electronic structures and the catalytic properties for NO oxidation of pure Pt and PtAu clusters. The calculated results suggest that Pt10 clusters shows the most stable structure among the pure Ptn (n = 2–13) clusters with the local maximum Δ2E value. The doping of Au atoms reduces the stability of the clusters, and Pt6Au4 cluster has the most stable structure among Pt10−nAun (n = 1–7) clusters, due to the closest band centers between Pt and Au atoms (0.83 eV) and the obvious s–p resonance peaks near the Fermi level. Pt6Au4 cluster displays the strongest activation of O2 molecules among Pt10−nAun (n = 0–7) clusters, owing to the clear overlap between O 2p and Pt 6 s and Au 6 s near the Fermi level, and the more positive d band center than the others. The interaction between NO and metals changes slightly in NO/Pt10-nAun (n = 2–7) systems, which is weaker than that in NO/Pt9Au system, as a result of the decreasing resonance peaks of sp hybridization near the Fermi level. Compared to pure Pt10 cluster, the lower energy barriers and larger reaction energies on Pt6Au4 cluster suggest a higher catalytic activity of PtAu cluster for the O2 dissociation and NO oxidation reactions. Our study provides atomic-scale insights into the nature of the interfacial effect that determines NO oxidation on PtAu cluster catalysts.
Co-reporter:Guixia Li, Houyu Zhu, Lianming Zhao, Wenyue Guo, Huifang Ma, Yanchen Yu, Xiaoqing Lu, and Yunjie Liu
The Journal of Physical Chemistry C 2016 Volume 120(Issue 40) pp:23009-23023
Publication Date(Web):September 14, 2016
DOI:10.1021/acs.jpcc.6b07103
Molybdenum phosphide (MoP) has been extensively experimentally shown to possess high and surprisingly increasing hydrodesulfurization (HDS) activities during the HDS process. In order to understand the HDS mechanism, we investigate the HDS of thiophene on clean and single-sulfur-atom-modified MoP(001) using self-consistent periodic density functional theory (DFT). Thiophene strongly prefers flat adsorption, which is slightly weakened in the presence of a surface S atom. Thermodynamic and kinetic analyses of the elementary steps show that the HDS of thiophene takes place along the direct desulfurization (DDS) pathway on both clean and S-modified MoP(001), because of the very low C–S bond activation barriers as well as very high exothermicities involved. More importantly, the surface S atom does not elevate the C–S bond activation barriers but opens a new concerted pathway for the simultaneous rupture of both C–S bonds in thiophene. These results indicate that the presence of a surface S atom could be helpful for thiophene desulfurization. For comparison, we also investigate the influence of a surface S atom on the HDS of thiophene on Pt(111). The results show clearly a negative effect of the surface S atom, in accordance with the lower sulfur resistance of noble metals.
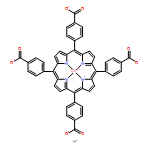
The effect of edge-functionalization on the competitive adsorption of a binary CO2–CH4 mixture in nanoporous carbons (NPCs) has been investigated for the first time by combining density functional theory (DFT) and grand canonical Monte Carlo (GCMC) simulation. Our results show that edge-functionalization has a more positive effect on the single-component adsorption of CO2 than CH4, therefore significantly enhancing the selectivity of CO2 over CH4, in the order of NH2–NPC > COOH–NPC > OH–NPC > H–NPC > NPC at low pressure. The enhanced adsorption originates essentially from the effects of (1) the conducive environment with a large pore size and an effective accessible surface area, (2) the high electronegativity/electropositivity, (3) the strong adsorption energy, and (4) the large electrostatic contribution, due to the inductive effect/direct interaction of the embedded edge-functionalized groups. The larger difference from these effects results in the higher competitive adsorption advantage of CO2 in the binary CO2–CH4 mixture. Temperature has a negative effect on the gas adsorption, but no obvious influence on the electrostatic contribution on selectivity. With the increase of pressure, the selectivity of CO2 over CH4 first decreases sharply and subsequently flattens out to a constant value. This work highlights the potential of edge-functionalized NPCs in competitive adsorption, capture, and separation for the binary CO2–CH4 mixture, and provides an effective and superior alternative strategy in the design and screening of adsorbent materials for carbon capture and storage.
Co-reporter:Xiaoqing Lu, Yang Shao, Shuxian Wei, Zigang Zhao, Ke Li, Chen Guo, Weili Wang, Mingmin Zhang and Wenyue Guo
Journal of Materials Chemistry A 2015 vol. 3(Issue 39) pp:10129-10139
Publication Date(Web):02 Sep 2015
DOI:10.1039/C5TC02286J
A series of porphyrin sensitizers for dye-sensitized solar cells (DSSCs) have been systematically investigated by density functional theory (DFT) and time-dependent DFT (TD-DFT) in tetrahydrofuran (THF) solution. The effects of π-bridge length, heteroaromatic unit, longitudinal conjugation, and relative position of functionalized groups on the optical and electrical properties are elucidated by analyzing the geometry, electronic structure, electron excitation, spectrum, photo-induced intramolecular electron transfer (IET), and light-harvesting efficiency (LHE). Our results show that the increase in π-bridge length by the addition of phenyl groups distances the electron distribution of LUMO away from the anchoring group and sharply decreases the effective electron excitation at the long wavelength region. The introduction of heteroaromatics in the π-bridge, especially electron-deficient units, stabilizes LUMO levels and improves the light-harvesting capability and donor-to-acceptor IET characteristics significantly. The extension of longitudinal π-conjugation in the π-bridge broadens the B band and slightly strengthens and redshifts the Q band but results in undesired orbital overlap. Repositioning the phenyl/thiophene group away from carboxylic acid increases the energy gap but leads to more effective long-range IET processes with more electrons, longer distance, lower orbital overlap, and moderate transfer rate. Our results highlight the significant effect of the functionalized π-bridge on porphyrin sensitizers, and provide a new insight into the design and screening of sensitizers for DSSCs.
Co-reporter:Xiaoqing Lu, Dongliang Jin, Shuxian Wei, Zhaojie Wang, Changhua An and Wenyue Guo
Journal of Materials Chemistry A 2015 vol. 3(Issue 23) pp:12118-12132
Publication Date(Web):19 Mar 2015
DOI:10.1039/C4TA06829G
Uncontrolled massive CO2 emission into the atmosphere is becoming a huge threat to our global climate and environment. Carbon capture and storage (CCS), starting with the crucial step of CO2 capture and separation, provides a promising approach to alleviate this issue. The major challenge for CO2 capture and separation is exploring efficient adsorbent materials with high storage capacity and selectivity. This review firstly summarized the significant advancement in a variety of state-of-the-art adsorbent materials. Then, particular attention was focused on the practical strategies to enhance CO2 capture and separation based on current adsorbent materials by topological structure design, chemical doping, chemical functionalization, open metal sites, and electric fields. These strategies paved constructive ways for the design and synthesis of novel adsorbent materials. Finally, we gave a perspective view on future directions in this rapidly growing field.
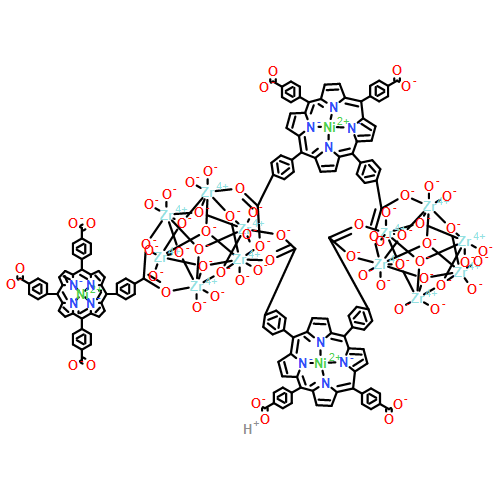
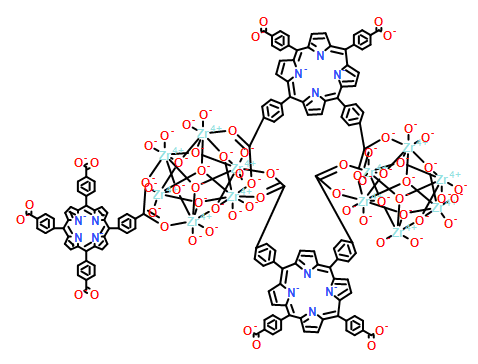
Co-reporter:Fuling Liu, Yuwen Xu, Lianming Zhao, Liangliang Zhang, Wenyue Guo, Rongming Wang and Daofeng Sun
Journal of Materials Chemistry A 2015 vol. 3(Issue 43) pp:21545-21552
Publication Date(Web):04 Aug 2015
DOI:10.1039/C5TA03680A
The current study describes the first barium–organic framework with permanent porosity, efficient catalytic capacity, and highly selective luminescence sensing of DMSO molecules and metal ions. Single-crystal-to-single-crystal transformations (from complex 1 to complexes 2 and 3) were used to thermally generate coordinatively unsaturated metal sites (CUSs) as catalytically active sites (CASs). Complex 3 exhibits efficient catalytic capacity for the cyanosilylation of aldehydes and ketones, and the cycloaddition of CO2 and epoxides. To the best of our knowledge, complex 3 keeps a record among the MOF-based catalysts for the cyanosilylation of aldehydes and ketones. The generation of Ba2+ CUSs with high catalytic activity in a SCSC fashion is responsible for the excellent properties of 3, which is further confirmed by the theoretical calculation. Besides, complex 2 can highly sense DMSO molecules through fluorescence enhancement.
The geometrical and electronic structures and photocatalytic performance of subnanometer Agn clusters (n = 2–6) deposited on AgBr(110) are studied under the framework of density functional theory (DFT) plus Hubbard U contributions. The most stable adsorption is facilitated by AgBr(110) interacting with the most stable structure of Agn and results in a new metal-induced gap band (MIGB) located between the valence (VB) and the conduction bands (CB). Both the MIGB and CB are mainly contributed by the sp hybridization states from the metal clusters, while the VB is composed primarily of the 4p states of the surface Br and the 4d states of Ag from both the adsorbate and the surface. The variety of the electronic structures favors visible and infrared light absorption, which strengthens substantially as the cluster size is enlarged. The dominant localization of the photo-excited electrons on the Agn clusters facilitates the oxidation–reduction reactions occurring on the surface and reduces effectively the photolysis of AgBr under sunlight irradiation. The overpotentials of the CB and VB edges indicate that photocatalytic conversion of CO2 with H2O to methanol is possible on AgBr(110) deposited with the Agn clusters which has been realized experimentally.
First-principle calculations were performed to explore the initial reduction of CO2 on perfect and O-defective CeO2 (111) surfaces via direct dissociation and hydrogenation, to elucidate the product selectivity towards CO, COOH, or HCOO. The results showed that CO2 prefers a bent configuration with the C atom of CO2 occupying the oxygen vacancy site. Reductive hydrogenation CO2 + H → COOH* was more competitive than CO2 + H → HCOO* on both perfect and O-defective CeO2 (111) surfaces. Comparatively, CO2 hydrogenation towards COOH was slightly more favorable on the perfect surface, whereas reductive dissociation of CO2 was predominant on the O-defective CeO2 (111) surface. Electronic localization function, charge density difference, and density of states were utilized to analyze the effect of charge accumulation and redistribution on the adsorption and reductive dissociation of CO2 caused by the presence of O vacancies. The results of this study provided detailed insight into the initial reduction mechanisms of CO2 towards different products on perfect and O-defective CeO2 (111) surfaces.
Materials Chemistry and Physics 2015 Volume 162() pp:6-10
Publication Date(Web):15 July 2015
DOI:10.1016/j.matchemphys.2015.05.041
•Photo-induced intramolecular electron transfer of heterodinuclear Ru–Co complex.•New donors improve the spectral range and intensity in the visible region.•Fine-tuned donors boost the Ru → Co IET rate at least one order of magnitude.Photo-induced intramolecular electron transfer (IET) of a series of heterodinuclear Ru(II)–Co(III) complexes has been theoretically investigated by density functional theory (DFT) and time-dependent DFT (TD-DFT). The Ru(II)–Co(III) complexes display Ru → Co metal-to-metal electron transfer (MMET) in the visible region. The photosensitivity involving spectral response range and absorption intensity, and IET rate, are improved by introducing (Z)-N-(1H-isoindol-1-ylidene)-2H-isoindol-1-amine as donor ligands. The Ru → Co IET rate in the newly designed complexes shows at least one order of magnitude larger than that in [(bpy)2-Ru(pytp)Co(tren)]5+. These superior performances indicate that heterodinuclear Ru(II)–Co(III) complexes could be promising as the effective ligand release carriers for the selective cancer treatment.
The Journal of Physical Chemistry C 2015 Volume 119(Issue 35) pp:20389-20400
Publication Date(Web):August 12, 2015
DOI:10.1021/acs.jpcc.5b03951
Self-consistent periodic density functional theory (PW91-GGA) calculations are employed to study the oxidation of methanol on PtRu(111). Geometries and energies for all the intermediates involved are analyzed, and the oxidation network is mapped out to illustrate the reaction mechanism. On PtRu(111), the Ru atoms with less electronegativity are more favorable to binding the adsorbates than the Pt atoms. Alloying Pt with Ru weakens the bond of CO to Pt, but strengthens the bond of CO to Ru. All possible pathways through initial C–H, O–H, and C–O bond scissions are considered. The initial O–H bond scission is found to be the most favorable and bears an energy barrier comparable to that for methanol desorption. The further oxidation occurs preferentially via the non-CO path from species CHO. The most possible reaction pathway of methanol on PtRu(111) is CH3OH → CH3O → CH2O → CHO → CHOOH → COOH → CO2. Furthermore, the activation of H2O on PtRu(111) is more favorable than that on the pure Pt(111) surface. The enhancement of methanol oxidation catalytic activity of the PtRu alloy is due primarily to altering the major reaction pathways from the CO path on pure Pt to the non-CO path on the alloy surface as well as promoting adsorption of methanol and formation of active OH species from H2O.
The Journal of Physical Chemistry C 2015 Volume 119(Issue 38) pp:21943-21951
Publication Date(Web):September 3, 2015
DOI:10.1021/acs.jpcc.5b04641
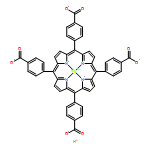
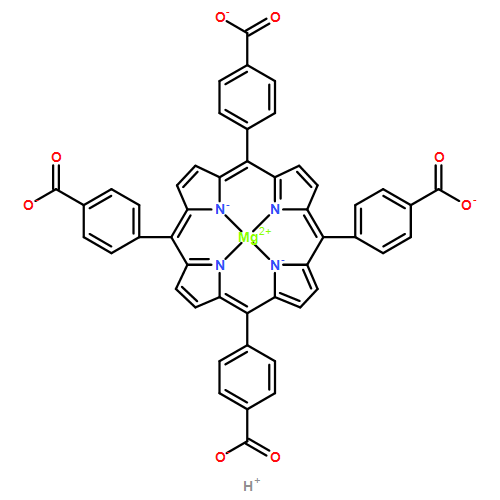
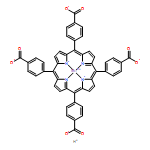
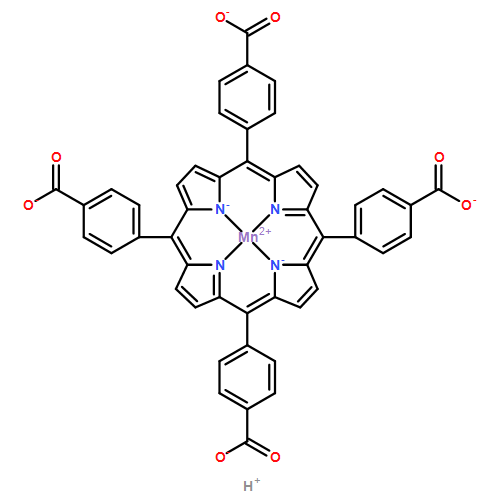
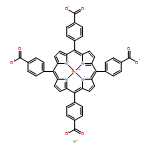
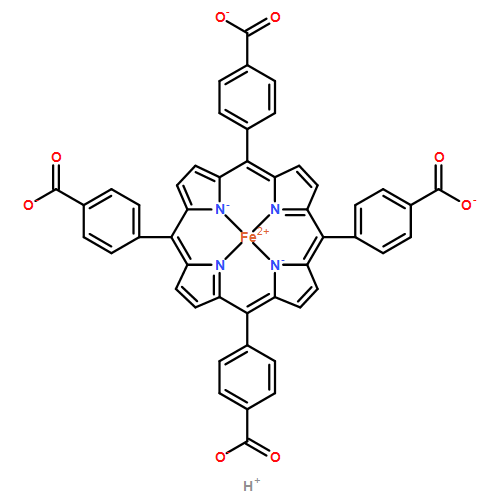
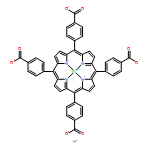
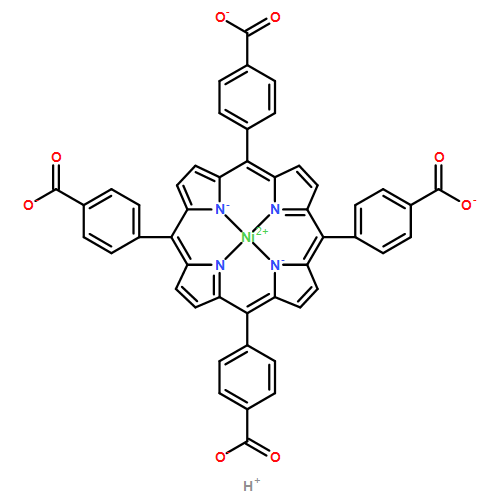
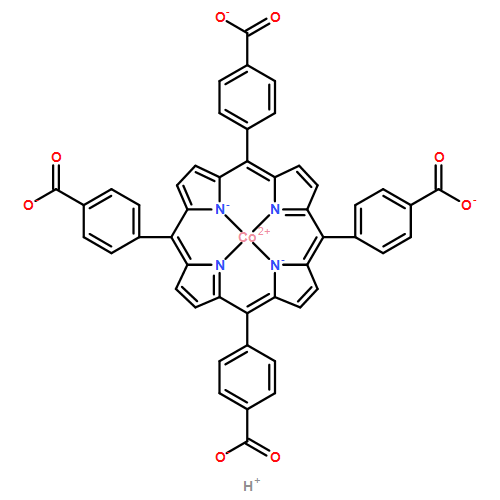
A combination of grand canonical Monte Carlo (GCMC) and density functional theory (DFT) simulations was used to investigate the effect of modified metal center in ligand for CO2 capture in novel Zr-based porphyrinic metal–organic frameworks (PCN-224-Ms, M = Mg, Fe, Co, Ni, Mn, and Zr). The results show that the MTCPP ligands (TCPP = tetrakis(4-carboxyphenyl)porphyrin) provide more favorable adsorption sites than the inorganic Zr6 nodes for CO2 molecules. The modification of metal center in MTCPP ligand has a remarkable effect on the single-component adsorption of CO2 compared to CH4 and thus enhances the adsorption of CO2 and the selectivity of CO2 over CH4. Furthermore, Coulomb interaction between adsorbate and framework plays a dominant role compared with non-Coulomb interaction in the process of adsorption and separation. Among various modified metal centers, the Zr-MTCPP is found to be the best for enhancing the adsorption and selectivity of CO2. In addition, a small amount of water has a negative effect on the selectivity of CO2/CH4, and its influence follows the order PCN-224-Zr > PCN-224-Mn > PCN-224-Ni, depending on the strength of Coulomb interaction between H2O molecules and frameworks.
Co-reporter:Rongming Wang, Qingguo Meng, Liangliang Zhang, Haifeng Wang, Fangna Dai, Wenyue Guo, Lianming Zhao and Daofeng Sun
Chemical Communications 2014 vol. 50(Issue 38) pp:4911-4914
Publication Date(Web):24 Mar 2014
DOI:10.1039/C4CC00477A
Two porous metal–organic frameworks (1 and 2) with a fsc topology based on mixed ligands have been assembled and characterized. The different pillared ligands (pyrazine for 1 and 4,4′-bipyridine for 2) significantly influence the pore size of the frameworks. Gas uptake measurements reveal that complex 1 possesses higher H2, CO2, and CH4 uptake capacities than 2, although the surface area of 1 is lower than that of complex 2. These results further experimentally prove that the pore size plays an important role in gas uptake in porous MOFs, and the slit pore with a size of ∼6 Å exhibits stronger interactions with gas molecules.
Owing to far-ranging industrial applications and theoretical researches, tailored synthesis of well-defined nanocrystals has attracted substantial research interest. Herein, β-AgI nanoplates have been synthesized through a facile polyvinylpyrrolidone (PVP)-assisted-aqueous-solution (PAAS) method under mild conditions. The parametric studies on the effect of ratio of reactants, solvents and surfactants were performed, revealing that a molar ratio of I− to Ag+ of 1.2 in deionized water and the presence of appropriate PVP as stabilizing agent can stimulate the preferred orientation growth of AgI nanoplates. The as-synthesized AgI nanoplates exhibit excellent photocatalytic activity and enhanced durability towards the degradation of organics, i.e., rhodamine B (RhB), under visible light illumination in comparison with corresponding bulk nanoparticles. A possible photocatalytic reaction mechanism was discussed, revealing O2˙− and h+ are main reactive species and free ˙OH radicals in solution also contribute to the degradation reaction. The superior photocatalytic performance renders the as-achieved AgI nanoplates promising candidates for applications in the fields of environmental purification or water disinfection. The present work opens an avenue to the synthesis of other shaped silver halide nanophotocatalysts.
Co-reporter:Dongliang Jin, Xiaoqing Lu, Mingmin Zhang, Shuxian Wei, Qing Zhu, Xiaofan Shi, Yang Shao, Weili Wang and Wenyue Guo
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 22) pp:11037-11046
Publication Date(Web):03 Apr 2014
DOI:10.1039/C3CP55107E
The effects of chemical and structural surface heterogeneity on the CH4 adsorption behaviour on microporous carbons have been investigated using a hybrid theoretical approach, including the use of density functional theory (DFT), molecular dynamics (MD), and grand canonical Monte Carlo (GCMC) simulations. Bader charge analysis is first performed to analyze the surface atomic partial charges. The CH4 adsorption densities in defective and functionalized graphite slit pores are lower than that in the perfect pore according to the MD simulations. Finally, the CH4 adsorption isotherms for the perfect, defective and functionalized slit pores are analyzed using the GCMC simulations in combination with the DFT and MD results. For pores with a defective surface, the adsorption capacities decrease; the embedded functional groups decrease the adsorption capacity at low pressure and enhance it at high pressure. Our results demonstrate the significant effects of chemical and structural surface heterogeneity on the CH4 adsorption and provide a systematic approach to understand the gas adsorption behaviour.
Co-reporter:Zhigang Deng, Xiaoqing Lu, Zengqiang Wen, Shuxian Wei, Qing Zhu, Dongliang Jin, Xiaofan Shi and Wenyue Guo
RSC Advances 2014 vol. 4(Issue 24) pp:12266-12274
Publication Date(Web):06 Jan 2014
DOI:10.1039/C3RA46544F
Periodic density functional theory (DFT) calculations were performed to systematically investigate the decomposition mechanism of methylamine (CH3NH2) to hydrogen cyanide (HCN) on Pt(111). The geometries and energies for all species involved are analyzed, and the decomposition network is mapped out to elaborate the reaction mechanism. Our results show that the CH3NH2, methanimine (CH2NH) and HCN prefer to desorb, while the other species prefer to decompose; the decomposition pathway prefers the successive N–H bond scissions followed by the C–H bond scissions, that is, CH3NH2 → CH3NH → CH3N → CH2N → HCN. The electronic structure and energy barrier analysis are used to identify the initial competitive scissions of C–H, N–H and C–N bonds. The interaction between fragments and surface in the TS plays a decisive role in controlling the energy barrier of initial CH3NH2 decomposition on Pt(111). Finally, the Brønsted–Evans–Polanyi (BEP) relation identifies that the C–H and N–H bond scissions stay competitive, but the C–N bond scission is not facile to occur.
Co-reporter:Dianling Fu, Wenyue Guo, Ming Li, Houyu Zhu, Yunjie Liu
Journal of Molecular Structure 2014 1062() pp: 68-76
Publication Date(Web):24 March 2014
DOI:10.1016/j.molstruc.2014.01.035
•Adsorption of reactants, intermediates, and products involved were investigated.•The Mulliken charge and projected density of states were analyzed.•The reactions of SO2 on Rh(111) were discussed.•The reaction mechanism was investigated.The adsorption and decomposition reaction mechanisms of SO2 on Rh(111) has been systematically studied using self-consistent periodic density functional theory (DFT). All possible adsorptions of reactants, intermediates, and products involved are investigated. Two stable adsorption configurations of SO2 on Rh(111) were confirmed: chair mode (η2(S)–η1(O)) and parallel mode ((η1(S)–η1(O)–η1(O)). The decomposition reaction of SO2 takes place along SO2 → SO + O → S + 2O, in which the second step is the rate-determining step. For the high energy barrier and low rate constant, the disproportionation reaction is not facile among all the possible reactions.
Co-reporter:Guangwu Yang, Chengcheng Miao, Zhongheng Bu, Qing Wang, Wenyue Guo
Journal of Power Sources 2013 Volume 233() pp:74-78
Publication Date(Web):1 July 2013
DOI:10.1016/j.jpowsour.2013.01.087
We report in this paper a cheap and easy method for the preparation of ZnO nanowire/TiO2 nanoparticle hybrid films for applications in dye-sensitized solar cells (DSCs), which is achieved by post-embedding ZnO nanowires in mesoporous TiO2 film via a seed free low-temperature hydrothermal process. The electron transport and recombination properties in the as-fabricated DSCs are studied by using electrochemical impedance spectroscopy. Results indicate that the electron transport, electron lifetime, effective diffusion length and the electron collection efficiency are increased, while the charge recombination is reduced, resulting in the remarkably enhanced power conversion efficiency of 7.46%, higher than 6.09% of standard TiO2 nanoparticle based DSCs.Graphical abstractWe report in this paper a cheap and easy method for the preparation of ZnO nanowire/TiO2 nanoparticle hybrid films for applications in dye-sensitized solar cells (DSCs), which is achieved by post-embedding ZnO nanowires in mesoporous TiO2 film via a seed free low-temperature hydrothermal process. The electron transport and recombination properties in the as-fabricated DSCs are studied by using electrochemical impedance spectroscopy. Results indicate that the electron transport, electron lifetime, effective diffusion length and the electron collection efficiency are increased, while the charge recombination is reduced, resulting in the remarkably enhanced power conversion efficiency of 7.46%, higher than 6.09% of standard TiO2 nanoparticle based DSCs.Highlights► ZnO nanowires are grown in TiO2 film via a seed free and low temperature process. ► The hybrid film based DSCs exhibit significantly enhanced photovoltaic performance. ► The electron transport and recombination properties are studied by using EIS. ► The effects of ZnO nanowires in the hybrid film are highlighted.
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 38) pp:16172-16182
Publication Date(Web):07 Aug 2013
DOI:10.1039/C3CP51948A
Periodic density functional theory (DFT) calculations have been performed to systematically investigate the effect of reaction intermediate on catalytic activity for hydrazine (N2H4) decomposition on Rh(111). Reaction mechanisms via intramolecular and NH2-assisted N2H4 decompositions are comparatively analyzed, including adsorption configuration, reaction energy and barrier of elementary step, and reaction network. Our results show that the most favorable N2H4 decomposition pathway starts with the initial N–N bond scission to the NH2 intermediate, followed by stepwise H stripping from adsorbed N2Hx (x = 1–4) species, and finally forms the N2 and NH3 products. Comparatively, the stepwise intramolecular dehydrogenation via N2H4 → N2H3 → N2H2 → N2H → N2, and N2H4 → NH2 → NH → N with or without NH2 promotion effect, are unfavorable due to higher energy barriers encountered. Energy barrier analysis, reaction rate constants, and electronic structures are used to identify the crucial competitive route. The promotion effect of the NH2 intermediate is structurally reflected in the weakening of the N–H bond and strengthening of the N–N bond in N2Hx in the coadsorption system; it results intrinsically from the less structural deformation of the adsorbate, and weakening of the interaction between dehydrogenated fragment and departing H in transition state. Our results highlight the crucial effect of reaction intermediate on catalytic activity and provide a theoretical approach to analyze the effect.
Ethanol decomposition on Pd(110) is comprehensively investigated using self-consistent periodic density functional theory. Geometries and energies for all the intermediates involved are analyzed, and the decomposition network is mapped out to illustrate the reaction mechanism. On Pd(110), the most stable adsorption of the involved species tends to follow the gas-phase bond order rules, wherein C is tetravalent and O is divalent with the missing H atoms replaced by metal atoms. The most likely decomposition pathway of ethanol on Pd(110) is CH3CH2OH → CH3CH2O → CH3CHO → CH3CO → CH3 + CO → CO + H + CH4 + C, in which the initial dehydrogenation is the rate-limited step. No C–O scission pathway is identified. Comparing with ethanol decomposition on Pd(111) [Langmuir, 2010, 26, 1879–1888], Pd(110) characterizes relatively high activity and different selectivity. Two crucial factors controlling the variations of reactivity and selectivity from Pd(111) to Pd(110), i.e., the local electronic effect of the metals and the geometrical effect of the relevant transition states, are identified. Four distinct Brønsted-Evans-Polanyi (BEP) relations are identified for the three types of bond scission (C–H, C–O, and C–C) if we consider Pd(111) and Pd(110) as a whole, one for C–H bond scission, one for C–O bond scission, and two for C–C bond scission.
Energy & Fuels 2013 Volume 27(Issue 4) pp:2010-2017
Publication Date(Web):March 26, 2013
DOI:10.1021/ef3018207
Laser-induced acoustic desorption coupled with tunable synchrotron vacuum ultraviolet photoionization mass spectrometry (LIAD/SVUVPI-MS) is employed to analyze aromatics prepared under different conditions from Lungu atmospheric residue (LGAR), i.e., the primary aromatics separated directly from LGAR, and the secondary aromatics after hydrogenation of LGAR and its resins. The mass spectra of the primary aromatics present a bimodal normal distribution in the range of 200–900 Da, in which the relative intensity of the two peaks changes significantly with the SVUV photon energies (9.0, 11.0, and 14.0 eV), indicating that at least two categories of compounds with different ionization energies (IEs) are included, i.e., polycyclic aromatics (IEs < 10.0 eV) in the mass range of 400–900 Da, and aliphatic and alicyclic compounds (IEs close to 11.0 eV) in 200–400 Da. Also detected in the aromatics are metalloporphyrins. Furthermore, the mass spectra of the secondary aromatics separated from LGAR and its resins at different hydrogenation temperatures (390, 400, 410, and 420 °C) are also recorded. The results indicate that the hydrogenation process, especially at higher temperatures, results in removal of alkyl-side and bridge chains in the aromatics, and the secondary aromatics from LGAR resins contain more alkyl side and bridge chains and metal compounds than those from LGAR.
Computational and Theoretical Chemistry 2013 Volume 1023() pp:29-37
Publication Date(Web):1 November 2013
DOI:10.1016/j.comptc.2013.09.001
•DFT study of the reactivity of ethanol with Ni+(2D).•Five different O and ethyl-H attached Ni+–ethanol isomers are found.•The most favorable mechanism is a new one-step syn-elimination pathway.The bond activation of ethanol by the ground-state Ni+(2D) in the gas phase has been theoretically investigated using density functional theory. The approach of Ni+ towards ethanol could form five different Ni+–ethanol adducts, which correspond to Ni+ interaction through the O atom and C–H bonds, respectively. The O attached complexes are much more stable than that with Ni+ interaction through the C–H bonds. Extensive conversions into each other could occur readily for these encounter complexes. The loss of H2O and C2H4 proceeds via three pathways, i.e., Ni+ insertion into the polar C–O bond through the O attached Ni+–ethanol complexes, initial Cβ–H activation via the Ni+–ethanol complex with Ni+ interaction through the methyl C–H bonds, and electrophile-inducted one-step syn-elimination through the Ni+–gauche-ethanol complex, and the last one is the most favorable. The loss of H2 is less favorable, because all mechanisms involve high activation barriers, while the initial Cα–H bond activation of ethanol by Ni+ through the O attached Ni+–ethanol complexes is the most favorable.Graphical abstract
Co-reporter:Lianming Zhao, Min Tan, Juan Chen, Qiuyue Ding, Xiaoqing Lu, Yuhua Chi, Guangwu Yang, Wenyue Guo, and Qingtao Fu
The Journal of Physical Chemistry A 2013 Volume 117(Issue 24) pp:5161-5170
Publication Date(Web):May 23, 2013
DOI:10.1021/jp4021454
The activation of ethanol and methanol by VO2+ in gas phase has been theoretically investigated by using density functional theory (DFT). For the VO2+/ethanol system, the activation energy (ΔE) is found to follow the order of ΔE(Cβ–H) < ΔE(Cα–H) ≈ ΔE(O–H). Loss of methyl and glycol occurs respectively via O–H and Cβ–H activation, while acetaldehyde elimination proceeds through two comparable O–H and Cα–H activations yielding both VO(H2O)+ and V(OH)2+. Loss of water not only gives rise to VO(CH3CHO)+ via both O–H and Cα–H activation but also forms VO2(C2H4)+ via Cβ–H activation. The major product of ethylene is formed via both O–H and Cβ–H activation for yielding VO(OH)2+ and VO2(H2O)+. In the methanol reaction, both initial O–H and Cα–H activation accounts for formaldehyde and water elimination, but the former pathway is preferred.
Periodic DFT calculations have been performed to systematically investigate the catalytic mechanisms of ammonia (NH3) decomposition on clean, O- and OH-assisted Ir(1 1 1) surfaces. The adsorption configurations, reaction energies and barriers, and elementary steps were elaborated. Our results show that the NHx (x=1–3) decomposition prefers to proceed by means of the O- and OH-assisted reaction mechanisms, NH3+ONH2+OH, NH2+OHNH+H2O, and NH+OHN+H2O, rather than the direct NHx decomposition of NH3NH2NHN as a result of the high energy barriers involved. The promotion effect of the O- and OH-oxidizing agents are then discussed using energy barrier analysis. The relationships between the selectivity toward the final product and coverage, O to N coverage, and reaction temperature are elucidated. Finally, we compare our results with analogous investigations of NH3 decomposition on Pt, Rh, and Ir surfaces.
Co-reporter:Xiaoqing Lu, Li Liu, Yang Li, Wenyue Guo, Lianming Zhao and Honghong Shan
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 16) pp:5642-5650
Publication Date(Web):17 Feb 2012
DOI:10.1039/C2CP40149E
The conversion of acetylene to ethylidyne on Pt(111) has been comprehensively investigated using self-consistent periodic density functional theory. Geometries and energies for all of the intermediates involved as well as the conversion mechanism were analyzed. On Pt(111), the carbon atoms in the majority of stable C2Hx (x = 1–4) intermediates prefer saturated sp3 configurations with the missing H atoms substituted by the adjacent metal atoms. The most favorable conversion pathway for acetylene to ethylidyne is via a three-step reaction mechanism, acetylene → vinyl → vinylidene → ethylidyne. The first step, acetylene → vinyl, depends on the availability of surface H atoms: without preadsorbed H the reaction occurs via the initial disproportionation of acetylene, which resulted in adsorbed vinyl; with an abundance of preadsorbed H, acetylene could transform to vinyl via both the disproportionation and hydrogenation reactions. Conversions through initial dehydrogenation of acetylene and isomerizations of acetylene and vinyl are unfavorable due to high energy barriers along the relevant pathways. The conversion rate involving vinylidene as an intermediate is at least 100 times larger than that involving ethylidene.
Co-reporter:Yang Li, Wenyue Guo, Houyu Zhu, Lianming Zhao, Ming Li, Shaoren Li, Dianling Fu, Xiaoqing Lu, and Honghong Shan
Langmuir 2012 Volume 28(Issue 6) pp:3129-3137
Publication Date(Web):January 18, 2012
DOI:10.1021/la2051004
The initial hydrogenations of pyridine on MoP(001) with various hydrogen species are studied using self-consistent periodic density functional theory (DFT). The possible surface hydrogen species are examined by studying interaction of H2 and H2S with the surface, and the results suggest that the rational hydrogen source for pyridine hydrogenations should be surface hydrogen atoms, followed by adsorbed H2S and SH. On MoP(001), pyridine has two types of adsorption modes, i.e., side-on and end-on; and the most stable η5(N,Cα,Cβ,Cβ,Cα) configuration of the side-on mode facilitates the hydrogenation of pyridine. The optimal hydrogenation path of pyridine with surface hydrogen atoms in the Langmuir–Hinshelwood mechanism is the formation of 3-monohydropyridine, followed by producing 3,5-dihydropyridine, in which the two-step hydrogenations take place on the Cβ atoms. When adsorbed H2S is considered as the source of hydrogen, slightly higher hydrogenation barriers are always involved, while the energy barriers for hydrogenations involving adsorbed SH are much lower. However, the hydrogenation of pyridine should be suppressed by the adsorption of H2S, and the promotion effect of adsorbed SH is limited.
Journal of Solid State Electrochemistry 2012 Volume 16( Issue 12) pp:3761-3767
Publication Date(Web):2012 December
DOI:10.1007/s10008-012-1818-0
In this paper, we report 3D nickel (II) hydroxide thin films with porous nanostructures prepared on Ni foam by direct current electrodeposition from aqueous solution of Ni(NO3)2 through basic chemicals. The effect of deposition temperature on Ni(OH)2 thin film morphology is examined by field emission scanning electron microscopy, which is found to have significant influence on capacitance performance of Ni(OH)2 thin films. Moreover, the effect of annealing temperature on electrochemical capacitance and long-time stability of Ni(OH)2 thin films is investigated. An optimum-specific capacitance value of 2,447 farads g−1 is obtained for Ni(OH)2 thin film deposited at 20 °C and annealed at 100 °C.
Journal of Molecular Catalysis A: Chemical 2012 Volumes 363–364() pp:18-25
Publication Date(Web):November 2012
DOI:10.1016/j.molcata.2012.05.011
Adsorption and desulfurization of thiophene on Pt(1 1 0) are investigated using self-consistent periodic density functional theory (DFT), and the reaction network is mapped out for elaborating the possible desulfurization processes. Thiophene has two types of adsorption geometries on Pt(1 1 0), Upright and Flat adsorptions. The S atom inclines to sp3 nonequivalent hybridization in all the adsorption structures of thiophene, whereas the C atom alters between sp2 and sp3 hybridization determined by the interaction modes between the adsorbate and the surface Pt atoms. The extent of molecular distortion of thiophene on Pt(1 1 0) is correlated to the HOMO–LUMO gap of the molecule and the interaction with the surface. Thermodynamic and kinetic analysis of the elementary steps suggest that direct desulfurization pathway is most favorable for the hydrodesulfurization of thiophene on Pt(1 1 0). This desulfurization process exhibits lower CS bond activation energy but higher conversion barrier compared to the situation of thiophene on Pt(1 1 1).Graphical abstractHighlights► Upright and Flat adsorption modes were observed for thiophene on Pt(1 1 0). ► C4H4S distortion was linked with HOMO–LUMO gap and interaction with Pt(1 1 0). ► Direct desulfurization was more favorable than hydrodesulfurization. ► Rate-limiting step of thiophene desulfurization was different on Pt(1 1 0) and Pt(1 1 1).
The Journal of Physical Chemistry A 2012 Volume 116(Issue 1) pp:512-519
Publication Date(Web):December 7, 2011
DOI:10.1021/jp206894y
The potential energy surfaces of Mn+ reaction with ethylene oxide in both the septet and quintet states are investigated at the B3LYP/DZVP level of theory. The reaction paths leading to the products of MnO+, MnO, MnCH2+, MnCH3, and MnH+ are described in detail. Two types of encounter complexes of Mn+ with ethylene oxide are formed because of attachments of the metal at different sites of ethylene oxide, i.e., the O atom and the CC bond. Mn+ would insert into a C–O bond or the C–C bond of ethylene oxide to form two different intermediates prior to forming various products. MnO+/MnO and MnH+ are formed in the C–O activation mechanism, while both C–O and C–C activations account for the MnCH2+/MnCH3 formation. Products MnO+, MnCH2+, and MnH+ could be formed adiabatically on the quintet surface, while formation of MnO and MnCH3 is endothermic on the PESs with both spins. In agreement with the experimental observations, the excited state a5D is calculated to be more reactive than the ground state a7S. This theoretical work sheds new light on the experimental observations and provides fundamental understanding of the reaction mechanism of ethylene oxide with transition metal cations.
Co-reporter:Lianming Zhao, Xiaoqing Lu, Yuanyuan Li, Juan Chen, and Wenyue Guo
The Journal of Physical Chemistry A 2012 Volume 116(Issue 12) pp:3282-3289
Publication Date(Web):March 1, 2012
DOI:10.1021/jp300211v
The potential energy surface (PES) corresponding to the Co+-mediated oxidation of ethane by N2O has been investigated by using density functional theory (DFT). After initial N2O reduction by Co+ to CoO+, ethane oxidation by the nascent oxide involves C–H activation followed by two possible pathways, i.e., C–O coupling accounting for ethanol, Co+-mediated β–H shift giving the energetically favorable product of CoC2H4+ + H2O, with minor CoOH2+ + C2H4. CoC2H4+ could react with another N2O to yield (C2H4)Co+O, which could subsequently undergo a cyclization mechanism accounting for acetaldehyde and oxirane and/or a direct H-abstraction mechansim for ethenol. Loss of oxirane and ethenol is hampered by respective endothermicity and high kinetics barrier, whereas acetaldehyde elimination is much energetically favorable. CoOH2+ could facilely react with N2O to form OCoOH2+, rather than Co(OH)2+ or CoO+.
Co-reporter:Houyu Zhu, Wenyue Guo, Ming Li, Lianming Zhao, Shaoren Li, Yang Li, Xiaoqing Lu, and Honghong Shan
ACS Catalysis 2011 Volume 1(Issue 11) pp:1498
Publication Date(Web):September 23, 2011
DOI:10.1021/cs2002548
Desulfurization of thiophene and its hydrogenated derivatives on Pt(111) are studied using self-consistent periodic density functional theory (DFT), and the hydrodesulfurization network is mapped out. On Pt(111), thiophene has two types of adsorption configurations (parallel cross-bridge and partially tilted bridge-hollow), and for its hydrogenated derivates, the molecule is gradually lifted up from the surface with the addition of hydrogen atoms. In all the adsorbed thiophenic compounds, the S atom is always sp3 hybridized; the C atom in the methylene group is always sp3 hybridized, whereas it is either sp2 or sp3 hybridized in the methyne group, depending on how the group interacts with the surface Pt atoms. On the basis of the thermodynamic and kinetic analysis of the elementary steps, a direct desulfurization pathway is proposed for the hydrodesulfurization of thiophene on Pt(111). In contrast to the common thought that hydrogenation toward aromatic organosulfur compounds would make desulfurization easier, the present work clearly demonstrates that hydrogenations of thiophene on Pt(111) do not reduce the energy barrier for the C–S bond cleavage.Keywords: C−S bond; desulfurization; hydrodesulfurization on Pt(111); hydrogenation; organosulfur compounds; thiophene;
We report herein a comprehensive study of the gas-phase Fe+-mediated oxidation of ethane by N2O on both the sextet and quartet potential energy surfaces (PESs) using density functional theory. The geometries and energies of all the relevant stationary points are located. Initial oxygen-atom transfer from N2O to iron yields FeO+. Then, ethane oxidation by the nascent oxide involves C–H activation forming the key intermediate of (C2H5)Fe+(OH), which can either undergo C–O coupling to Fe+ + ethanol or experience β-H shift giving the energetically favorable product of FeC2H4+ + H2O. Reaction of FeC2H4+ with another N2O constitutes the third step of the oxidation. N2O coordinates to FeC2H4+ and gets activated by the metal ion to yield (C2H4)Fe+O(N2). After releasing N2 through the direct H abstraction and/or cyclization pathways, the system would be oxidized to ethenol, acetaldehyde, and oxirane, regenerating Fe+. Oxidation to acetaldehyde along the cyclization –C–to–C hydrogen shift pathway is the most energetically favored channel.
Co-reporter:Xiaoqing Lu ; Shuxian Wei ; Chi-Man Lawrence Wu ; Shaoren Li
The Journal of Physical Chemistry C 2011 Volume 115(Issue 9) pp:3753-3761
Publication Date(Web):February 14, 2011
DOI:10.1021/jp111325y
Design of light-absorbent dyes with cheaper, safer, and more sustainable materials is one of the key issues for the future development of dye-sensitized solar cells (DSSCs). We report herein a theoretical investigation on a series of polypyridyl Cu(I)-based complexes with general formula [CuLL′]+ (L and L′ represent bipyridyl ligands) by density functional theory (DFT) and time-dependent DFT. Molecular geometries, electronic structures, and optical absorption spectra are predicted in both the gas phase and methyl cyanide solution. Our results show that all the [CuLL′]+ derivatives display Cu → bipyridine metal-to-ligand charge transfer absorption spectra in the range of 350−700 nm. Structural optimizations by enhancing π-conjugation and introducing heteroaromatic groups on ancillary ligands lead to upshift of molecular orbital energies, increase in oscillator strength, and red shift of absorption spectra. Compared with Ru(II) sensitizers, polypridyl Cu(I)-based complexes show similar optical properties and improving trend of the DSSCs performance along with the optimizations of structures. The results of this work highlight the point that polypyridyl Cu(I)-based complexes could provide promising sensitizers for efficient next-generation DSSCs.
Co-reporter:Ruibin Jiang, Wenyue Guo, Ming Li, Xiaoqing Lu, Jianye Yuan and Honghong Shan
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 28) pp:7794-7803
Publication Date(Web):19 May 2010
DOI:10.1039/B927050G
Dehydrogenation of methanol on Pd(100) is systematically investigated using self-consistent periodic density functional theory. The theoretical results are compared with those of the same reaction on Pd(111) published very recently [J. Phys. Chem. C, 2009, 113, 4188–4197]. Switching from (111) to (100), adsorptions are strengthened for most species except for CHO, CO and H at hollow sites. Moreover, Pd(100) affords relatively low energy barriers and higher rate constants for most elementary dehydrogenation steps as well as smaller desorption rates for the saturated adsorbates (methanol and formaldehyde), suggesting that the more open Pd surface indeed possesses the higher activity and selectivity for the complete dehydrogenation of methanol. At lower temperatures (e.g., 250 K), Pd(100) affords the same dehydrogenation path as Pd(111) for methanol, which is unchanged on the latter surface at both lower and higher temperatures; whereas at the typical steam re-forming (MSR) temperature (500 K), the path on Pd(100), i.e., CH3OH → CH3O and/or CH2OH → CH2O → CHO → CO, is different from the situation of Pd(111). In both cases, the initial bond scission process constitutes the rate-determining step.
Co-reporter:Xiaoqing Lu, Shuxian Wei, Wenyue Guo, and Chi-Man Lawrence Wu
The Journal of Physical Chemistry A 2010 Volume 114(Issue 47) pp:12490-12497
Publication Date(Web):November 4, 2010
DOI:10.1021/jp106397g
The gas-phase reaction mechanisms of methylamine (MA) with the ground-state Co+(3F) and Ni+(2D) are theoretically investigated using density functional theory at both the B3LYP/6-311++G(d,p) and B3LYP/6-311++G(3df,2p) levels. The reactions for hydride abstraction and dehydrogenation are analyzed in terms of the topology of potential energy surfaces (PESs). Co+ and Ni+ perform similar roles along the isomerization processes to the final products. Hydride abstraction takes place via the key species of metal cation-methyl-H intermediate, followed by a charge transfer process before the direct dissociation of CH2NH2+···MH (M = Co, Ni). The enthalpies of reaction, stability of metal cation-methyl-H species, and competition between different channels account for the sequence of the hydride abstraction products: CoH < NiH < CuH. The most competitive dehydrogenation route occurs through a stepwise reaction, consisting of initial C−H activation, amino-H shift, and direct dissociation of the precursor CH2NHM+···H2. This theoretical work sheds new light on the experimental observations and provides fundamental understanding of the reaction mechanisms of amine prototype with late first-row transition metal cations.
Ethanol decomposition over Pd(111) has been systematically investigated using self-consistent periodic density functional theory, and the decomposition network has been mapped out. The most stable adsorption of the involved species tends to follow the gas-phase bond order rules, wherein C is tetravalent and O is divalent with the missing H atoms replaced by metal atoms. Desorption is preferable for adsorbed ethanol, methane, and CO, while for the other species decomposition is preferred. For intermediates going along the decomposition pathways, energy barriers for the C−C, Cα−H, and O−H scissions are decreased, while it is increased for the C−O path or changes less for the Cβ−H path. For each of the C−C, C−O, and C−H paths, the Bro̷nsted−Evans−Polanyi relation holds roughly. The most likely decomposition path is CH3CH2OH → CH3CHOH → CH3CHO → CH3CO → CH2CO → CHCO → CH + CO → CO + H + CH4 + C.
Co-reporter:Houyu Zhu, Wenyue Guo, Ruibin Jiang, Lianming Zhao, Xiaoqing Lu, Ming Li, Dianling Fu and Honghong Shan
Langmuir 2010 Volume 26(Issue 14) pp:12017-12025
Publication Date(Web):June 28, 2010
DOI:10.1021/la101678d
Decomposition of methanthiol on Pt(111) is systematically investigated using self-consistent periodic density functional theory (DFT), and the decomposition network has been mapped out. The most stable adsorption of the involved species tends to form the sp3 hybridized configuration of both C and S atoms, in which C is almost tetrahedral and S has the tendency to bond to three atoms. Spontaneous dissociation rather than desorption is preferred for adsorbed methanthiol. Based on the harmonic transition state theory calculations, the decomposition rate constants of the thiolmethoxy and thioformaldehyde intermediates are found to be much lower than those for their formation, leading to long lifetimes of the intermediates for observation. Under the ultrahigh vacuum (UHV) condition, the most possible decomposition pathway for methanthiol on Pt(111) is found as CH3SH → CH3S → CH2S → CHS → CH + S → C + S, in which the C−S bond cleavage mainly occurs at the CHS species. However, the decomposition pathway is CH3SH → CH3S → CH3 + S under the hydrogenation condition; the C−S bond scission mainly occurs at CH3S. The Brønsted−Evans−Polanyi relation holds for each of the S−H, C−H, and C−S bond scission reactions.
Co-reporter:Xiaoqing Lu, Chi-Man Lawrence Wu, Shuxian Wei and Wenyue Guo
The Journal of Physical Chemistry A 2010 Volume 114(Issue 2) pp:1178-1184
Publication Date(Web):December 10, 2009
DOI:10.1021/jp909731t
Molecular geometries, electronic structures, and optical absorption spectra were investigated using density functional theory (DFT) at the B3LYP/6-31G(d) and B3LYP/DZVP levels for [CuL2]+ and [CuL2][PF6] (L = 6,6′-dimethyl-2,2′-bipyridine-4,4′-dimethylformate), both in the gas phase and in methyl cyanide (MeCN) solution. The vertical excitation energies were calculated within the framework of the time-dependent DFT (TD-DFT) approach, whereas the solvent effects were taken into account using the polarizable continuum model (C-PCM). Our results show that the five highest occupied molecular orbitals (HOMOs) are composed of a set of distorted degenerate Cu 3d orbitals, whereas the four lowest unoccupied molecular orbitals (LUMOs) are the bipyridine ligand π*C═N orbitals. The spectra in the range of 400−600 nm were found to originate from metal-to-ligand charge-transfer (MLCT) transitions, whereas the spectra in the range of 350−400 nm are excitations mainly from the metal Cu 3d orbitals to the carboxyl π* orbitals. The solvent effects lead to changes in both the geometries and the absorption spectra. The results of this work suggest that copper-based complexes might be effective sensitizers for next-generation dye-sensitized solar cells.
Co-reporter:Ming Li, Wenyue Guo, Ruibin Jiang, Lianming Zhao, Xiaoqing Lu, Houyu Zhu, Dianling Fu and Honghong Shan
The Journal of Physical Chemistry C 2010 Volume 114(Issue 18) pp:8440-8448
Publication Date(Web):April 14, 2010
DOI:10.1021/jp100970c
The conversion of ethylene to ethylidyne on Rh(111) is examined using self-consistent periodic density function theory. The adsorptions of the reactants, intermediates, and products involved as well as the thermodynamics and kinetics of the conversion are characterized. Ethylene could form two adsorption configurations designated as di-σ and π adsorptions on Rh(111); ethyl, vinyl, vinylidene, ethylidyne, and ethylidene prefer the saturated sp3 configuration of both carbon atoms with the lost H atoms replaced by the metal atoms. The three-step conversion path on Rh(111), i.e., ethylene → vinyl → vinylidene → ethylidyne, is the most feasible, in which the vinylidene hydrogenation is the rate-limiting step. The pathway through ethylidene intermediate, ethylene → vinyl → ethylidene → ethylidyne, is impractical because it has a conversion rate at least 104 times lower than the most favorable path at the real reaction conditions. The mechanism via ethyl intermediate, ethylene → ethyl → ethylidene → ethylidyne, is impossible because of the high dehydrogenation barrier of ethyl to ethylidene as well as the low barriers for the conversions of ethyl to ethane and/or ethylene. Conversion involving direct isomerizations is also unlikely to be important due to the very high energy barriers involved.
Co-reporter:Zhaochun Liu, Wenyue Guo, Lianming Zhao and Honghong Shan
The Journal of Physical Chemistry A 2010 Volume 114(Issue 7) pp:2701-2709
Publication Date(Web):February 3, 2010
DOI:10.1021/jp910774z
We report herein a comprehensive theoretical study of the oxidation of propane by FeO+ on both the sextet and quartet potential energy surfaces (PESs) using density functional theory. The geometries and energies of all the stationary points involved are located. Interaction of FeO+ with propane could account for four types of encounters (i.e., α,β,γ-, 2α,β-, 3α-η3, and 2α,2γ-η4) complexes. Various mechanisms leading to the loss of CH3, H2O, C3H7OH (H2O + C3H6), and C3H6 are analyzed in terms of the topology of the PES. The reaction of FeO+ with propane involves initial C−H activation, while initial C−C activation is indeed unlikely to be important. The loss of CH3 takes place adiabatically on the sextet PES via the simple Cα-to-O H shift from η4-OFe+(C3H8) followed by CH3 shift. The C3H7OH elimination proceeds via direct Cα-to-O H shift followed by C−O coupling, while the loss of H2O, C3H6, and (H2O + C3H6) proceeds via the α,β-H and β,α-H abstraction mechanisms from all the η3 complexes. The most favorable channel is the α,β-H abstraction mechanism for the H2O loss because it not only is energetically and dynamically favorable but also has a high crossing probability between the sextet and quartet PESs. The computational results are in concert with the available experimental information and add new insight into the details of the individual elementary steps.
Co-reporter:Ming Li ; Wenyue Guo ; Ruibin Jiang ; Lianming Zhao ; Xiaoqing Lu ; Houyu Zhu ; Dianling Fu ;Honghong Shan
The Journal of Physical Chemistry C 2010 Volume 114(Issue 49) pp:21493-21503
Publication Date(Web):November 10, 2010
DOI:10.1021/jp106856n
Ethanol decomposition on Rh(111) is systematically investigated using periodic density functional theory (DFT) calculations. The various adsorption modes of the intermediates involved are located. It is determined that ethanol adsorbs weakly on the Rh(111) surface. CH3CH2O, CH, and H prefer 3-fold sites with adsorption energies of 49.9, 146.6, and 64.3 kcal/mol, respectively. CO binds stably at the top site with a binding energy of 42.5 kcal/mol. CH2CH2O (3-fold-η1(Cβ)-η1(O)) and CHCO (3-fold-η2(Cβ)−η1(Cα)) are inclined to adsorb on the surface to make the C and O atoms saturated. For the other intermediates, adsorption configurations are bridge-η1(Cβ)−η1(O) for CH2CHO, 3-fold-η1(Cβ)−η1(Cα)−η1(O) for CH2CO, and 3-fold-η2(Cβ)−η1(O) for CHCHO. For intermediates going along the decomposition pathway, energy barriers for the Cβ−H and C−C bond scission are gradually decreased; however, for the Cα−H or C−O bond cleavage, the energy barrier decreases first and then rises, presenting a V-shaped curve. The most favorable decomposition route for ethanol on Rh(111) is CH3CH2OH → CH3CH2O → CH2CH2O → CH2CHO → CH2CO → CHCO → CH + CO → C + CO, in which the dehydrogenation of CH3CH2O is the rate-determining step.
Co-reporter:Ruibin Jiang, Wenyue Guo, Ming Li, Houyu Zhu, Jing Li, Lianming Zhao, Dianling Fu and Honghong Shan
The Journal of Physical Chemistry C 2009 Volume 113(Issue 42) pp:18223-18232
Publication Date(Web):September 24, 2009
DOI:10.1021/jp9054999
The reaction of SO2 on Ir(111) has been systematically studied using self-consistent periodic density functional theory. Adsorptions of the reactants, intermediates, and products involved are calculated. In all possible adsorption modes of SO2, the parallel adsorption (η1(S)-η1(O)-η1(O)) is found to be the most favorable. Intermediate SO is also preferable for parallel adsorption. S and O are exclusively adsorbed at 3-fold sites. The oxidation product SO3 has two stable adsorption modes without any preference, i.e., η1(O)-η1(O)-η1(O) (crown) and η1(S)-η1(O)-η1(O) (chair). In the absence of oxygen, the disproportionated reaction, 3SO2 → S + 2SO3, is initiated by SO2 dissociation into SO and atomic O; then SO quickly decomposes into S and O; and lastly, the O atoms formed in the SO2 and SO dissociation are rapidly exhausted by other SO2. The fast reductions of SO and O accord well with the fact that they were not detected in the experiment [Fujitani, Nakamura, Kobayashi, Takahashi, Haneda, Hamada Surf. Sci. 2007, 601, 1615]. Further reaction of SO3 is expected to take place at high temperatures because of the high energy barriers involved (at least 2.06 eV), also in agreement with the experimental finding.
Applied Surface Science (31 May 2017) Volume 405() pp:
Publication Date(Web):31 May 2017
DOI:10.1016/j.apsusc.2017.02.054
•Three graphdiyne-like membranes were designed and their stabilities were confirmed.•The DFT and MD results claimed a tunable gas separation property of the membranes.•Graphdiyne modified with F or O can effectively separate CO2/N2/CH4 mixtures.Three graphdiyne-like monolayers were designed by substituting one-third diacetylenic linkages with heteroatoms hydrogen, fluorine, and oxygen (GDY_X, X = H, F, and O), respectively. The CO2/N2/CH4 separation performance of the designed graphdiyne-like monolayers was investigated by using both first-principle density functional theory (DFT) and molecular dynamic (MD) simulations. The stabilities of GDY_X monolayers were confirmed by the calculated cohesive energies and phonon dispersion spectra. Both the DFT and MD calculations demonstrated that although the GDY_H membrane has poor selectivity for CO2/N2/CH4 gases, the GDY_F and GDY_O membranes can excellently separate CO2 and N2 from CH4 in a wide temperature range. Moreover, the CO2/N2 mixture can be effectively separated by GDY_O at temperatures lower than 300 K. Based on the kinetic theory, extremely high permeances were found for CO2 and N2 passing through the GDY_X membranes (10−4–10−2 mol/m2 s Pa at 298 K). In addition, the influence of relative concentration on selectivity was also investigated for gases in the binary mixtures. This work provides an effective way to modify graphdiyne for the separation of large molecular gases, which is quite crucial in the gas separation industry.Graphdiyne monolayer membrane modified by fluorine or oxygen can effectively separate CO2/N2/CH4 mixtures.Figure optionsDownload full-size imageDownload high-quality image (245 K)Download as PowerPoint slide
International Journal of Hydrogen Energy (23 February 2017) Volume 42(Issue 8) pp:
Publication Date(Web):23 February 2017
DOI:10.1016/j.ijhydene.2016.11.158
•Two dumbbell-shaped porous γ-graphyne membranes were designed for H2 purification.•The H2 separation performance of the membranes were studied using both DFT and MD simulations.•Both the graphyne-like membranes can separate H2 with excellent selectivity and acceptable permeance.Two dumbbell-shaped porous γ-graphynes were designed by substituting one-third acetylenic linkages with heteroatoms nitrogen and hydrogen named γ-GYN and γ-GYH, respectively. The calculated cohesive energies and phonon dispersion spectra indicate the possibility to realize the new membranes in experiments. The separation performance of the designed monolayers for H2 from H2O, CO2, N2, CO, and CH4 was investigated using both first-principle density functional theory (DFT) and molecular dynamic (MD) simulations. The DFT calculations suggest the designed membranes are excellent in H2 separation because of the super high selectivities of H2 over other gases H2O, CO2, N2, CO, and CH4 (>1010, 1013, 1021, 1018, and 1046, respectively, at room temperature) together with the high/extremely low permeances for H2/other gases, e.g., reaching the industrial standard at 400 K (γ-GYN) and 425 K (γ-GYH)/lower than the industrial limit by 3–22 orders even at 600 K. The MD simulations indicates that only H2 in the gas mixture containing H2, H2O, CO2, N2, CO, and CH4 can penetrate across the membranes even at the temperature of 600 K and γ-GYN is more favorable for H2 penetration. All these results indicate both the designed membranes, especially γ-GYN, are excellent candidates for H2 purification from gas mixtures.Dumbbell-shaped porous γ-graphynes γ-GYN and γ-GYH can effectively separate H2 from H2O, CO2, N2, CO, and CH4.
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 38) pp:NaN16182-16182
Publication Date(Web):2013/08/07
DOI:10.1039/C3CP51948A
Periodic density functional theory (DFT) calculations have been performed to systematically investigate the effect of reaction intermediate on catalytic activity for hydrazine (N2H4) decomposition on Rh(111). Reaction mechanisms via intramolecular and NH2-assisted N2H4 decompositions are comparatively analyzed, including adsorption configuration, reaction energy and barrier of elementary step, and reaction network. Our results show that the most favorable N2H4 decomposition pathway starts with the initial N–N bond scission to the NH2 intermediate, followed by stepwise H stripping from adsorbed N2Hx (x = 1–4) species, and finally forms the N2 and NH3 products. Comparatively, the stepwise intramolecular dehydrogenation via N2H4 → N2H3 → N2H2 → N2H → N2, and N2H4 → NH2 → NH → N with or without NH2 promotion effect, are unfavorable due to higher energy barriers encountered. Energy barrier analysis, reaction rate constants, and electronic structures are used to identify the crucial competitive route. The promotion effect of the NH2 intermediate is structurally reflected in the weakening of the N–H bond and strengthening of the N–N bond in N2Hx in the coadsorption system; it results intrinsically from the less structural deformation of the adsorbate, and weakening of the interaction between dehydrogenated fragment and departing H in transition state. Our results highlight the crucial effect of reaction intermediate on catalytic activity and provide a theoretical approach to analyze the effect.
Co-reporter:Fuling Liu, Yuwen Xu, Lianming Zhao, Liangliang Zhang, Wenyue Guo, Rongming Wang and Daofeng Sun
Journal of Materials Chemistry A 2015 - vol. 3(Issue 43) pp:NaN21552-21552
Publication Date(Web):2015/08/04
DOI:10.1039/C5TA03680A
The current study describes the first barium–organic framework with permanent porosity, efficient catalytic capacity, and highly selective luminescence sensing of DMSO molecules and metal ions. Single-crystal-to-single-crystal transformations (from complex 1 to complexes 2 and 3) were used to thermally generate coordinatively unsaturated metal sites (CUSs) as catalytically active sites (CASs). Complex 3 exhibits efficient catalytic capacity for the cyanosilylation of aldehydes and ketones, and the cycloaddition of CO2 and epoxides. To the best of our knowledge, complex 3 keeps a record among the MOF-based catalysts for the cyanosilylation of aldehydes and ketones. The generation of Ba2+ CUSs with high catalytic activity in a SCSC fashion is responsible for the excellent properties of 3, which is further confirmed by the theoretical calculation. Besides, complex 2 can highly sense DMSO molecules through fluorescence enhancement.
Co-reporter:Dongliang Jin, Xiaoqing Lu, Mingmin Zhang, Shuxian Wei, Qing Zhu, Xiaofan Shi, Yang Shao, Weili Wang and Wenyue Guo
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 22) pp:NaN11046-11046
Publication Date(Web):2014/04/03
DOI:10.1039/C3CP55107E
The effects of chemical and structural surface heterogeneity on the CH4 adsorption behaviour on microporous carbons have been investigated using a hybrid theoretical approach, including the use of density functional theory (DFT), molecular dynamics (MD), and grand canonical Monte Carlo (GCMC) simulations. Bader charge analysis is first performed to analyze the surface atomic partial charges. The CH4 adsorption densities in defective and functionalized graphite slit pores are lower than that in the perfect pore according to the MD simulations. Finally, the CH4 adsorption isotherms for the perfect, defective and functionalized slit pores are analyzed using the GCMC simulations in combination with the DFT and MD results. For pores with a defective surface, the adsorption capacities decrease; the embedded functional groups decrease the adsorption capacity at low pressure and enhance it at high pressure. Our results demonstrate the significant effects of chemical and structural surface heterogeneity on the CH4 adsorption and provide a systematic approach to understand the gas adsorption behaviour.
Ethanol decomposition on Pd(110) is comprehensively investigated using self-consistent periodic density functional theory. Geometries and energies for all the intermediates involved are analyzed, and the decomposition network is mapped out to illustrate the reaction mechanism. On Pd(110), the most stable adsorption of the involved species tends to follow the gas-phase bond order rules, wherein C is tetravalent and O is divalent with the missing H atoms replaced by metal atoms. The most likely decomposition pathway of ethanol on Pd(110) is CH3CH2OH → CH3CH2O → CH3CHO → CH3CO → CH3 + CO → CO + H + CH4 + C, in which the initial dehydrogenation is the rate-limited step. No C–O scission pathway is identified. Comparing with ethanol decomposition on Pd(111) [Langmuir, 2010, 26, 1879–1888], Pd(110) characterizes relatively high activity and different selectivity. Two crucial factors controlling the variations of reactivity and selectivity from Pd(111) to Pd(110), i.e., the local electronic effect of the metals and the geometrical effect of the relevant transition states, are identified. Four distinct Brønsted-Evans-Polanyi (BEP) relations are identified for the three types of bond scission (C–H, C–O, and C–C) if we consider Pd(111) and Pd(110) as a whole, one for C–H bond scission, one for C–O bond scission, and two for C–C bond scission.
Co-reporter:Rongming Wang, Qingguo Meng, Liangliang Zhang, Haifeng Wang, Fangna Dai, Wenyue Guo, Lianming Zhao and Daofeng Sun
Chemical Communications 2014 - vol. 50(Issue 38) pp:NaN4914-4914
Publication Date(Web):2014/03/24
DOI:10.1039/C4CC00477A
Two porous metal–organic frameworks (1 and 2) with a fsc topology based on mixed ligands have been assembled and characterized. The different pillared ligands (pyrazine for 1 and 4,4′-bipyridine for 2) significantly influence the pore size of the frameworks. Gas uptake measurements reveal that complex 1 possesses higher H2, CO2, and CH4 uptake capacities than 2, although the surface area of 1 is lower than that of complex 2. These results further experimentally prove that the pore size plays an important role in gas uptake in porous MOFs, and the slit pore with a size of ∼6 Å exhibits stronger interactions with gas molecules.
The geometrical and electronic structures and photocatalytic performance of subnanometer Agn clusters (n = 2–6) deposited on AgBr(110) are studied under the framework of density functional theory (DFT) plus Hubbard U contributions. The most stable adsorption is facilitated by AgBr(110) interacting with the most stable structure of Agn and results in a new metal-induced gap band (MIGB) located between the valence (VB) and the conduction bands (CB). Both the MIGB and CB are mainly contributed by the sp hybridization states from the metal clusters, while the VB is composed primarily of the 4p states of the surface Br and the 4d states of Ag from both the adsorbate and the surface. The variety of the electronic structures favors visible and infrared light absorption, which strengthens substantially as the cluster size is enlarged. The dominant localization of the photo-excited electrons on the Agn clusters facilitates the oxidation–reduction reactions occurring on the surface and reduces effectively the photolysis of AgBr under sunlight irradiation. The overpotentials of the CB and VB edges indicate that photocatalytic conversion of CO2 with H2O to methanol is possible on AgBr(110) deposited with the Agn clusters which has been realized experimentally.
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 26) pp:NaN17460-17460
Publication Date(Web):2017/06/19
DOI:10.1039/C7CP01859B
The hydrodesulfurization (HDS) of thiophene on clean and S-modified MoP(010) is investigated to understand the HDS mechanism as well as the surface sulfur (S) atom effect using periodic density functional theory (DFT). The results show that thiophene prefers strongly flat adsorption on both the clean and S-modified surfaces, in either the molecular state or the dissociative state breaking simultaneously one C–S bond, and the adsorption of thiophene can be slightly weakened by the surface S atom. Thermodynamic and kinetic analysis indicates that the HDS of thiophene in both the molecular and dissociative adsorption states prefers to take place along the direct desulfurization (DDS) pathway rather than hydrogenation on both the clean and S-modified MoP(010) surfaces. Surface S shows a promotion effect on the HDS catalytic activity of MoP(010), because the energy barrier/rate constant of the rate-determining step on the DDS pathway is decreased/enlarged under the S modification. Compared with the situation of MoP(001), MoP(010) should have relatively low HDS activity, since a higher energy barrier as well as weaker exothermicity is involved in the reaction on the latter surface.
Co-reporter:Xiaoqing Lu, Li Liu, Yang Li, Wenyue Guo, Lianming Zhao and Honghong Shan
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 16) pp:NaN5650-5650
Publication Date(Web):2012/02/17
DOI:10.1039/C2CP40149E
The conversion of acetylene to ethylidyne on Pt(111) has been comprehensively investigated using self-consistent periodic density functional theory. Geometries and energies for all of the intermediates involved as well as the conversion mechanism were analyzed. On Pt(111), the carbon atoms in the majority of stable C2Hx (x = 1–4) intermediates prefer saturated sp3 configurations with the missing H atoms substituted by the adjacent metal atoms. The most favorable conversion pathway for acetylene to ethylidyne is via a three-step reaction mechanism, acetylene → vinyl → vinylidene → ethylidyne. The first step, acetylene → vinyl, depends on the availability of surface H atoms: without preadsorbed H the reaction occurs via the initial disproportionation of acetylene, which resulted in adsorbed vinyl; with an abundance of preadsorbed H, acetylene could transform to vinyl via both the disproportionation and hydrogenation reactions. Conversions through initial dehydrogenation of acetylene and isomerizations of acetylene and vinyl are unfavorable due to high energy barriers along the relevant pathways. The conversion rate involving vinylidene as an intermediate is at least 100 times larger than that involving ethylidene.
Owing to far-ranging industrial applications and theoretical researches, tailored synthesis of well-defined nanocrystals has attracted substantial research interest. Herein, β-AgI nanoplates have been synthesized through a facile polyvinylpyrrolidone (PVP)-assisted-aqueous-solution (PAAS) method under mild conditions. The parametric studies on the effect of ratio of reactants, solvents and surfactants were performed, revealing that a molar ratio of I− to Ag+ of 1.2 in deionized water and the presence of appropriate PVP as stabilizing agent can stimulate the preferred orientation growth of AgI nanoplates. The as-synthesized AgI nanoplates exhibit excellent photocatalytic activity and enhanced durability towards the degradation of organics, i.e., rhodamine B (RhB), under visible light illumination in comparison with corresponding bulk nanoparticles. A possible photocatalytic reaction mechanism was discussed, revealing O2˙− and h+ are main reactive species and free ˙OH radicals in solution also contribute to the degradation reaction. The superior photocatalytic performance renders the as-achieved AgI nanoplates promising candidates for applications in the fields of environmental purification or water disinfection. The present work opens an avenue to the synthesis of other shaped silver halide nanophotocatalysts.
Co-reporter:Xiaoqing Lu, Dongliang Jin, Shuxian Wei, Zhaojie Wang, Changhua An and Wenyue Guo
Journal of Materials Chemistry A 2015 - vol. 3(Issue 23) pp:NaN12132-12132
Publication Date(Web):2015/03/19
DOI:10.1039/C4TA06829G
Uncontrolled massive CO2 emission into the atmosphere is becoming a huge threat to our global climate and environment. Carbon capture and storage (CCS), starting with the crucial step of CO2 capture and separation, provides a promising approach to alleviate this issue. The major challenge for CO2 capture and separation is exploring efficient adsorbent materials with high storage capacity and selectivity. This review firstly summarized the significant advancement in a variety of state-of-the-art adsorbent materials. Then, particular attention was focused on the practical strategies to enhance CO2 capture and separation based on current adsorbent materials by topological structure design, chemical doping, chemical functionalization, open metal sites, and electric fields. These strategies paved constructive ways for the design and synthesis of novel adsorbent materials. Finally, we gave a perspective view on future directions in this rapidly growing field.
Co-reporter:Xiaoqing Lu, Yang Shao, Shuxian Wei, Zigang Zhao, Ke Li, Chen Guo, Weili Wang, Mingmin Zhang and Wenyue Guo
Journal of Materials Chemistry A 2015 - vol. 3(Issue 39) pp:NaN10139-10139
Publication Date(Web):2015/09/02
DOI:10.1039/C5TC02286J
A series of porphyrin sensitizers for dye-sensitized solar cells (DSSCs) have been systematically investigated by density functional theory (DFT) and time-dependent DFT (TD-DFT) in tetrahydrofuran (THF) solution. The effects of π-bridge length, heteroaromatic unit, longitudinal conjugation, and relative position of functionalized groups on the optical and electrical properties are elucidated by analyzing the geometry, electronic structure, electron excitation, spectrum, photo-induced intramolecular electron transfer (IET), and light-harvesting efficiency (LHE). Our results show that the increase in π-bridge length by the addition of phenyl groups distances the electron distribution of LUMO away from the anchoring group and sharply decreases the effective electron excitation at the long wavelength region. The introduction of heteroaromatics in the π-bridge, especially electron-deficient units, stabilizes LUMO levels and improves the light-harvesting capability and donor-to-acceptor IET characteristics significantly. The extension of longitudinal π-conjugation in the π-bridge broadens the B band and slightly strengthens and redshifts the Q band but results in undesired orbital overlap. Repositioning the phenyl/thiophene group away from carboxylic acid increases the energy gap but leads to more effective long-range IET processes with more electrons, longer distance, lower orbital overlap, and moderate transfer rate. Our results highlight the significant effect of the functionalized π-bridge on porphyrin sensitizers, and provide a new insight into the design and screening of sensitizers for DSSCs.
Co-reporter:Ruibin Jiang, Wenyue Guo, Ming Li, Xiaoqing Lu, Jianye Yuan and Honghong Shan
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 28) pp:NaN7803-7803
Publication Date(Web):2010/05/19
DOI:10.1039/B927050G
Dehydrogenation of methanol on Pd(100) is systematically investigated using self-consistent periodic density functional theory. The theoretical results are compared with those of the same reaction on Pd(111) published very recently [J. Phys. Chem. C, 2009, 113, 4188–4197]. Switching from (111) to (100), adsorptions are strengthened for most species except for CHO, CO and H at hollow sites. Moreover, Pd(100) affords relatively low energy barriers and higher rate constants for most elementary dehydrogenation steps as well as smaller desorption rates for the saturated adsorbates (methanol and formaldehyde), suggesting that the more open Pd surface indeed possesses the higher activity and selectivity for the complete dehydrogenation of methanol. At lower temperatures (e.g., 250 K), Pd(100) affords the same dehydrogenation path as Pd(111) for methanol, which is unchanged on the latter surface at both lower and higher temperatures; whereas at the typical steam re-forming (MSR) temperature (500 K), the path on Pd(100), i.e., CH3OH → CH3O and/or CH2OH → CH2O → CHO → CO, is different from the situation of Pd(111). In both cases, the initial bond scission process constitutes the rate-determining step.

![Zincate(1-), [4-[7-[2-[15-[bis[2',4'-bis(hexyloxy)[1,1'-biphenyl]-4-yl]amino]-10,20-bis[2,6-bis(octyloxy)phenyl]-21H,23H-porphin-5-yl-](/data/chemimg/101700/1565809-59-7.png)
![Zincate(1-), [4-[7-[2-[15-[bis[2',4'-bis(hexyloxy)[1,1'-biphenyl]-4-yl]amino]-10,20-bis[2,6-bis(octyloxy)phenyl]-21H,23H-porphin-5-yl-](/data/chemimg/101700/1565809-59-7_b.png)
![[1,1':4',1''-Terphenyl]-2',3,3'',5,5''-pentacarboxylic acid](/data/chemimg/3717600/1918145-07-9.png)
![[1,1':4',1''-Terphenyl]-2',3,3'',5,5''-pentacarboxylic acid](/data/chemimg/3717600/1918145-07-9_b.png)