Co-reporter:
Chinese Journal of Chemistry 2017 Volume 35(Issue 3) pp:354-362
Publication Date(Web):2017/03/01
DOI:10.1002/cjoc.201600605
AbstractQM combined with ABEEMσπ (atom-bond electronegativity equalization method) fluctuating charge polarizable force field [QM/MM(ABEEM)] has been employed to investigate the hydrolysis mechanism of ruthenium(III) complex NAMI-A. Eleven possible hydrolysis paths of NAMI-A and its hydrolysates have been unveiled. Structures obtained by QM/MM (ABEEM) method were used to calculate the activation free energy by free energy perturbation theory. Based on analysis of structures and activation free energies, it is found that the rate of DMSO dissociation is faster than that of Cl− dissociation in the first step, which is in agreement with experimental results. Hydrolysates of the first step continue to hydrolyze with lower activation free energies than that of the first hydrolysis step, but the trans-position Cl− hydrolysis in the second step needs more time to perform. This can account for experimental phenomenon reasonably. We have simulated further hydrolysis paths of the hydrolysates, then found that both Cl− dissociation and DMSO dissociation take place in the third hydrolysis step and [RuCl(H2O)4(Im)]2+ is the main solute in aqueous solution after the fourth hydrolysis step.
Co-reporter:Yu Guo;Hui Li;Lan-Lan He;Dong-Xia Zhao;Li-Dong Gong;Zhong-Zhi Yang
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 21) pp:13909-13923
Publication Date(Web):2017/05/31
DOI:10.1039/C7CP01617D
The dioxygen formation mechanism of biological water oxidation in nature has long been the focus of argument; many diverse mechanistic hypotheses have been proposed. Based on a recent breakthrough in the resolution of the electronic and structural properties of the oxygen-evolving complex in the S3 state, our density functional theory (DFT) calculations reveal that the open-cubane oxo–oxyl coupling mechanism, whose substrates preferably originate from W2 and O5 in the S2 state, emerges as the best candidate for O–O bond formation in the S4 state. This is justified by the overwhelming energetic superiority of this mechanism over alternative mechanisms in both the isomeric open and closed-cubane forms of the Mn4CaO5 cluster; spin-dependent reactivity rooted in variable magnetic couplings was found to play an essential role. Importantly, this oxygen evolution mechanism is supported by the recent discovery of femtosecond X-ray free electron lasers (XFEL), and the origin of the observed structural changes from the S1 to S3 state has been analyzed. In this view, we corroborate the proposed water binding mechanism during S2–S3 transition and correlate the theoretical models with experimental findings from aspects of substrate selectivity according to water exchange kinetics. This theoretical consequence for native metalloenzymes may serve as a significant guide for improving the design and synthesis of biomimetic materials in the field of photocatalytic water splitting.
Co-reporter:Hui Li;Huiyuan Zou;Linlin Liu;Dongxia Zhao
Chemical Research in Chinese Universities 2017 Volume 33( Issue 2) pp:239-247
Publication Date(Web):2017 April
DOI:10.1007/s40242-017-6401-x
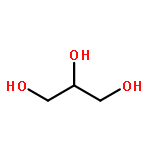
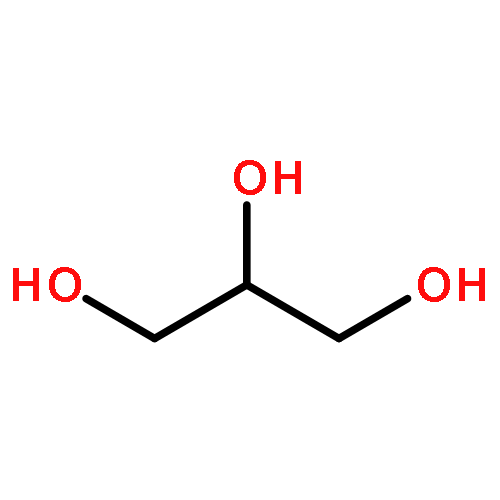
We used ABEEMσπ(atom-bond electronegativity equalization method) polarizable force field(ABEEMσπ PFF) method combined with QM and molecular dynamics-free energy perturbation(MD-FEP) methods to investigate the function of water molecules in hydrolysis process of ImH[trans-Ru(Im)2Cl4](ICR). The activation free energies obtained via MD-FEP calculation are in fair agreement with the experimental data. In addition, QM/MM(ABEEM) rationally describes the charge distributions and the electrostatic interaction between molecules. ABEEMσπ fluctuating charge model has the following good characteristics: (1) not only atomic charge regions but also σ, π bond and lone pair charge regions are explicitly represented for a molecule; (2) the region charges are geometry dependent and calculated from time to time in the dynamic simulation without any iterative procedure so that its performance is time-saving compared with the Drude model and induced dipole model.
Co-reporter:Yu Guo, Hui Li, Lan-Lan He, Dong-Xia Zhao, Li-Dong Gong, Zhong-Zhi Yang
Biochimica et Biophysica Acta (BBA) - Bioenergetics 2017 Volume 1858, Issue 10(Issue 10) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.bbabio.2017.08.001
•Multi-state reactivity is proposed for interconversion by O5 transfer in S2.•O5 is demonstrated to be deprotonated in both S1 and S2 states.•Structural polymorphism via Mn3 ligand exchange correlates to water exchange.•Structural variability translocates W2/O5 and facilitates their coupling in S4.•Alternative scenarios for OO bonding and water binding during S4-S0 are presented.The structural polymorphism of the oxygen-evolving complex is of great significance to photosynthetic water oxidation. Employing density functional theory calculations, we have made further advisement on the interconversion mechanism of O5 transfer in the S2 state, mainly focusing on the potentiality of multi-state reactivity and spin transitions. Then, O5 protonation is proven impossible in S2 for irreversibility of the interconversion, which serves as an auxiliary judgment for the protonation state of O5 in S1. Besides, the structural polymorphism could also be archived by alternative mechanisms involving Mn3 ligand exchange, one of which with Mn3(III) makes sense to substrate water exchange in S2, although being irresponsible for the derivations of the observed EPR signals. During the water exchange, high-spin states would prevail to facilitate electron transfer between the ferromagnetically coupled Mn centers. In addition, water exchange in S1 could account for the closed-cubane structure as the initial form entering S2 at cryogenic temperatures. With regard to water oxidation, the structural flexibility and variability in both S2 and S3 guarantee smooth W2-O5 coupling in S4, according to the substrate assignments from water exchange kinetics. Within this theoretical framework, the new XFEL findings on S1-S3 can be readily rationalized. Finally, an alternative mechanistic scenario for OO bond formation with ·OH radical near O4 is presented, followed by water binding to the pivot Mn4(III) from O4 side during S4-S0. This may diversify the substrate sources combined with the Ca channel in water delivery for the forthcoming S-cycle.Download high-res image (187KB)Download full-size image
Co-reporter:Lin-Lin Liu, Zhong-Zhi Yang, Dong-Xia Zhao, Li-Dong Gong and Cui Liu
RSC Advances 2014 vol. 4(Issue 94) pp:52083-52087
Publication Date(Web):09 Oct 2014
DOI:10.1039/C4RA09631B
This study describes theoretical simulations of morphological transitions for linear and cyclic block copolymers with polymer blending. Mesoscopic dissipative particle dynamics simulations with reliable interaction parameters from all-atom molecular dynamics simulations based on the ABEEM polarizable force field, can reproduce the self-assembly behavior well and morphological transitions observed by experiments.
Co-reporter:Dong-Xia Zhao and Zhong-Zhi Yang
The Journal of Physical Chemistry A 2014 Volume 118(Issue 39) pp:9045-9057
Publication Date(Web):August 11, 2014
DOI:10.1021/jp5020466
The potential and force acting on one electron within a molecule (PAEM and FAEM) have been investigated and analyzed. The PAEM, defined as the interaction energy on one electron provided by all the nuclei and the remaining electrons in a molecule, can be precisely expressed and calculated by ab initio method and our in-house program. Although the analysis of the scalar function PAEM is similar to that of the molecular electron density in the Bader’s AIM theory, the former is distinct from the latter mainly in three points: (a) The minus gradient of the PAEM is the force acting on one electron within a molecule (FAEM). (b) The bond center is defined in terms of the features of FAEM and PAEM between two bonded atoms, and it is a two-dimensional attractive center whereas a nucleus is a three-dimensional attractive source for electrons. We have calculated the physical quantities of one electron at the bond center, such as Dpb, the Hessian matrix, and its eigenvalues. Interestingly, it is found that the force constant and frequency of the electron interflow around the bond center are well correlated with those corresponding quantities for the nuclear vibration which relate to the bond strength, for some series of diatomic molecules. (c) The bond center locates at a different point from that of the critical point of the electron density in the Bader’s AIM theory, which will lead to different partitioning of the molecular space into the atomic regions.
Co-reporter:Xia Du, Dong-Xia Zhao, Zhong-Zhi Yang
Chemical Physics 2013 Volume 412() pp:84-91
Publication Date(Web):1 February 2013
DOI:10.1016/j.chemphys.2012.12.016
Abstract
A new approach to characterize and measure bond strength has been developed. First, we propose a method to accurately calculate the potential acting on an electron in a molecule (PAEM) at the saddle point along a chemical bond in situ, denoted by Dpb. Then, a direct method to quickly evaluate bond strength is established. We choose some familiar molecules as models for benchmarking this method. As a practical application, the Dpb of base pairs in DNA along C–H and N–H bonds are obtained for the first time. All results show that C7–H of A–T and C8–H of G–C are the relatively weak bonds that are the injured positions in DNA damage. The significance of this work is twofold: (i) A method is developed to calculate Dpb of various sizable molecules in situ quickly and accurately; (ii) This work demonstrates the feasibility to quickly predict the bond strength in macromolecules.
Co-reporter:Hui-Yuan Zou, Dong-Xia Zhao, Zhong-Zhi Yang
Journal of Molecular Graphics and Modelling 2013 Volume 43() pp:58-65
Publication Date(Web):June 2013
DOI:10.1016/j.jmgm.2013.03.004
•The mechanism of CPT's E-ring-opening has been studied by theoretical methods.•E-ring-opening conforms to the addition coupled elimination reaction pathway.•The activation free energies increase as the number of participated water rises.•The solvent effect plays a key role in affecting the reaction.A reaction mechanism of the anticancer agent camptothecin (CPT)’s E-ring-opening has been studied by DFT method and IEF-PCM solvation model. Our results indicate that under the physiological PH, CPT's E-ring-opening is a spontaneous process, and it conforms to the addition coupled elimination reaction pathway with a proton translocation. The obtained activation free energies in the explicit water model are in agreement with the available experimental values. More than ten reactions have been studied to provide exhaustive analyses of the relationship between structure and reactivity. On the whole, our results accord with the experimental findings and the mechanism we proposed is reasonable.A reaction mechanism of the anticancer agent camptothecin (CPT)’s E-ring-opening studied by theoretical methods, which describes the conversion of CPT from an active lactone form to an inactive hydrolyzed carboxylate form.
Co-reporter:Chunyang Yu;Lidong Gong
Frontiers of Chemistry in China 2011 Volume 6( Issue 4) pp:287-299
Publication Date(Web):2011 December
DOI:10.1007/s11458-011-0259-0
In this paper, the interaction between hydrogen peroxide (HP) and water were systemically studied by atom-bond electronegativity equalization method fused into molecular mechanics (ABEEM/MM) and ab initio method. The results show that the optimized geometries, interaction energies and dipole moments of hydrated HP clusters HP(H2O)n (n = 1–6) calculated by ABEEM/MM model are fairly consistent with the MP2/aug-cc-pVTZ//MP2/aug-cc-pVDZ results. The ABEEM/MM results indicate that n = 4 is the transition state structure from 2D planar structure to 3D network structure. The variations of the average hydrogen bond length with the increasing number of water molecules given by ABEEM/MM model agree well with those of ab initio studies. Moreover, the radial distribution functions (RDFs) of water molecule around HP in HP aqueous solution have been analyzed in detail. It can be confirmed that HP is a good proton donor and poor proton acceptor in aqueous solution by analysis of the RDFs.
Co-reporter:Chun-Yang Yu and Zhong-Zhi Yang
The Journal of Physical Chemistry A 2011 Volume 115(Issue 12) pp:2615-2626
Publication Date(Web):March 9, 2011
DOI:10.1021/jp111284t
Hydrogen peroxide (HP) clusters (H2O2)n (n = 1−6) and liquid-state HP have been systemically investigated by the newly constructed ABEEM/MM fluctuating charge model. Because of the explicit description of charge distribution and special treatment of the hydrogen-bond interaction region, the ABEEM/MM potential model gives reasonable properties of HP clusters, including geometries, interaction energies, and dipole moments, when comparing with the present ab initio results. Meanwhile, the average dipole moment, static dielectric constant, heats of vaporization, radial distribution function, and diffusion constant for the dynamic properties of liquid HP at 273 K and 1 atm are fairly consistent with the available experimental data. To the best of our knowledge, this is the first theoretical investigation of condensed HP. The properties of HP monomer are studied in detail involving the structure, torsion potentials, molecular orbital analysis, charge distribution, dipole moment, and vibrational frequency.
Co-reporter:Dong-Xia Zhao, Cui Liu, Fang-Fang Wang, Chun-Yang Yu, Li-Dong Gong, Shu-Bin Liu and Zhong-Zhi Yang
Journal of Chemical Theory and Computation 2010 Volume 6(Issue 3) pp:795-804
Publication Date(Web):February 4, 2010
DOI:10.1021/ct9006647
A polarizable force field (PFF) using multiple fluctuating charges per atom, ABEEMσπ PFF, is presented in this work. The fluctuating partial charges are obtained from the electronegativity equalization principle applied to the decomposition scheme of atom-bond regions into multiple charge sites: atomic, lone-pair electron, and σ and π bond regions. These multiple partial charges per atom should better account for the polarization effect than single atomic charge in other PFFs. To evaluate the PFF, structural and energetic properties for some organic and biochemical systems, including rotational barriers; binding energies of base pairs; a base−base interaction in a B-DNA decamer; and interaction energies of ten stationary conformers of a water dimer, peptides, and bases with water molecules, have been calculated and compared with the experimental data or ab initio MP2 results. Molecular dynamics simulations using the PFF have been performed for crambin and BPTI protein systems. Better performances in modeling root-mean-square deviations of backbone bond lengths, bond angles, key dihedral angles, the coordinate root-mean-square shift of atoms, and the distribution of hydrogen bonds have been observed in comparison with other PFFs. These results indicate that the fluctuating charge force field, ABEEMσπ/MM, is accurate and reliable and can be applied to wide ranges of organic and biomolecular systems.
Co-reporter:Shuling Chen ; Dr. Zhongzhi Yang
Chinese Journal of Chemistry 2010 Volume 28( Issue 11) pp:2109-2118
Publication Date(Web):
DOI:10.1002/cjoc.201090350
Abstract
There are some controversial opinions about the origin of folding β-hairpin stability in aqueous solution. In this study, the structural and dynamic behavior of a 16-residue β-hairpin from B1 domain of protein G has been investigated at 280, 300, 350 and 450 K using molecular dynamics (MD) simulations by means of Atom-Bond Electronegativity Equalization Method Fused into Molecular Mechanics i.e., ABEEMδπ/MM and the explicit ABEEM-7P water solvent model. In addition, a 300 K simulation of one mutant having the aromatic residues substituted with alanines has been performed. The hydrophobic surface area, hydrophilic surface area and some structural properties have been used to measure the role of the hydrophobic interactions. It is found that the aromatic residues substituted with alanines have shown an evident destabilization of the structure and unfolding started after 1.5 ns. It is also found that the number of the main chain hydrogen bonds have different distributions through three different simulations. All above demonstrate that the hydrophobic interactions and the main chain hydrogen bonds play an important role in the stability of the folding structure of β-hairpin in solution. Furthermore, through the structural analyses of the β-hairpin structures from four temperature simulations and the comparison with other MD simulations of β-hairpin peptides, the new ABEEMδπ force field can reproduce the structural data in good agreement with the experimental data.
Co-reporter:Shu-Ling Chen;Dong-Xia Zhao;Li-Dong Gong
Theoretical Chemistry Accounts 2010 Volume 127( Issue 5-6) pp:627-639
Publication Date(Web):2010 November
DOI:10.1007/s00214-010-0762-2
The interaction between formic acid (FA) and water was systemically investigated by atom-bond electronegativity equalization method fused into molecular mechanics (ABEEMσπ/MM) and ab initio methods. The geometries of 20 formic acid–water complexes (FA–water) were obtained using B3LYP/aug-cc-pVTZ level optimizations, and the energies were determined at the MP2/aug-cc-pVTZ level with basis set superposition error (BSSE) and zero-point vibrational energy (ZPVE) corrections. The ABEEMσπ potential model gives reasonable properties of these clusters when compared with the present ab initio data. For interaction energies, the root mean square deviation is 0.74 kcal/mol, and the linear coefficient reaches 0.993. Next, FA in aqueous solution was also studied. The hydrogen-bonding pattern due to the interactions with water has been analyzed in detail. Furthermore, the ABEEMσπ charges changed when H2O interacted with the FA molecule, especially at the sites where the hydrogen bonds form. These results show that the ABEEMσπ fluctuating charge model is fine giving the overall characteristic hydration properties of FA–water systems in good agreement with the high-level ab initio calculations.
Co-reporter:Fang-Fang Wang, Dong-Xia Zhao, Zhong-Zhi Yang
Chemical Physics 2009 360(1–3) pp: 141-149
Publication Date(Web):12 June 2009
DOI:10.1016/j.chemphys.2009.04.022
Uracil–(H2O)n (n = 1–7) clusters were systemically investigated by ab initio methods and the newly constructed ABEEMσπ/MM fluctuating charge model. Water molecules have been gradually placed in an average plane containing uracil. The geometries of 38 uracil–water complexes were obtained using B3LYP/6-311++G∗∗ level optimizations, and the energies were determined at the MP2/6-311++G∗∗ level with BSSE corrections. The ABEEMσπ/MM potential model gives reasonable properties of these clusters when comparing with the present ab initio data. For interaction energies, the root mean square deviation is 0.96 kcal/mol, and the linear coefficient reaches 0.997. Furthermore, the ABEEMσπ charges changed when H2O interacted with the uracil molecule, especially at the sites where the hydrogen bond form. These results show that the ABEEMσπ/MM model is fine giving the overall characteristic hydration properties of uracil–water systems in good agreement with the high-level ab initio calculations.
Co-reporter:Zhong-Zhi Yang, Shi-Fei Qi, Dong-Xia Zhao and Li-Dong Gong
The Journal of Physical Chemistry B 2009 Volume 113(Issue 1) pp:254-259
Publication Date(Web):December 15, 2008
DOI:10.1021/jp804128s
For the most important arylnitrenium ion−guanosine C8 adducts in the reactions involving arylamine-initiated carcinogenesis, a detailed mechanism of their formation still remains unclear. In this paper, we employ quantum chemistry methods to explore this issue. Our study indicates that formation of these C8 adducts proceeds directly by additions of arylnitrenium ions to C8 position of nucleoside bases in DNA. The good agreements of theoretical rate constants, pKa value, and NMR chemical shifts of C8 intermediate with experimental data support this theoretical finding. Excitingly, predictions of what adducts can be observed in reactions of arylnitrenium ions with guanine and hypoxanthine are in fair agreement with experimental observations. This study answers an important question, in carcinogenesis researches, of what is the mechanism for formation of C8 adducts.
Co-reporter:Zhong-Zhi Yang, Yan-Li Ding and Dong-Xia Zhao
The Journal of Physical Chemistry A 2009 Volume 113(Issue 18) pp:5432-5445
Publication Date(Web):April 8, 2009
DOI:10.1021/jp804951w
Gas-phase front-side attack identity SN2(C) and SN2(Si) reactions, CH3X1 + X2− → CH3X2 + X1− and SiH3X1 + X2− → SiH3X2 + X1− (X = F, Cl), are investigated by the ab initio method and molecular face (MF) theory. The computations have been performed at the CCSD(T)/aug-cc-pVTZ//MP2/6-311++G(3df,3pd) and CISD/aug-cc-pVDZ levels. Front-side attack identity SN2 reactions for both SiH3X and CH3X have double-well potential energy surfaces (PESs), but their transition-state positions are different relative to the positions of reactants and products: it is lower for SiH3X, and it is higher for CH3X. The minimum energy path for an SN2(Si) reaction with retention of configuration proceeds from a stable pentacoordinated anion intermediate of Cs symmetry (TBP) via a Cs transition state (SP) to a complementary pentacoordinated intermediate (TBP) and finally up to separate products. Berry pseudorotation has been observed in the front-side attack identity SN2(Si) reactions with F− and Cl− along the intrinsic reaction coordinate (IRC) routes. In addition, the geometrical transformations of front-side attack identity SN2(C) and SN2(Si) reactions based on the IRC calculations at the MP2/6-311++G(3df, 3pd) level of theory are described compared with those of corresponding back-side attack reactions. The difference between front-side attack identity SN2(C) and SN2(Si) reactions has been demonstrated. In MF theory, the potential acting on an electron in a molecule (PAEM) is an important quantity; in particular, its Dpb can measure the strength of a chemical bond in a molecule. It is found that the difference between Dpb values of reactant and transition state may be related to the activation energy for front-side and back-side attack SN2(C) and SN2(Si) reactions, and the Dpb curves along the IRC routes have features similar to those of the potential energy profiles for all of the back-side attack SN2 reactions and front-side attack SN2(Si) reaction with F−. Furthermore, according to the MF theory, the spatial dynamic changing features of the molecular shapes and the face electron density are vividly depicted for the course of the reactions.
Co-reporter:Xin Li;LiDong Gong
Science China Chemistry 2008 Volume 51( Issue 12) pp:1221-1230
Publication Date(Web):2008 December
DOI:10.1007/s11426-008-0129-x
Constrained molecular dynamics simulations have been used to investigate the LiCl and NaCl ionic association in water in terms of atom-bond electronegativity equalization method fused into molecular mechanics (ABEEM/MM). The simulations make use of the seven-site fluctuating charge and flexible ABEEM-7P water model, based on which an ion-water interaction potential has been constructed. The mean force and the potential of mean force for LiCl and NaCl in water, the charge distributions, as well as the structural and dynamical properties of contact ion pair dissociation have been investigated. The results are reasonable and informative. For LiCl ion pair in water, the solvent-separated ion pair configurations are more stable than contact ion pair configurations. The calculated PMF for NaCl in water indicates that contact ion pair and solvent-separated ion pair configurations are of comparable stability.
Co-reporter:Zhong-Zhi Yang ;Yan-Li Ding ;Dong-Xia Zhao
ChemPhysChem 2008 Volume 9( Issue 16) pp:2379-2389
Publication Date(Web):
DOI:10.1002/cphc.200800364
Abstract
Electrophilic additions of hydrogen halides to alkenes in the gas phase are investigated with a high-level ab initio method, MP2/6-311+G(3df,2p). Based on this, the interesting features of these reactions along the IRC routes are characterized by the molecular face (MF) theory. For an alkene at the initial state, if the representative electron density (ED) encoded on the molecular face (MF) of the Markovnikov (M) carbon atom (the carbon with more hydrogen atoms) is larger than that of the anti-Markovnikov (AM) carbon atom (the carbon with fewer hydrogen atoms), the electrophilic addition reaction is predicted to proceed along the Markovnikov addition route; in the reverse situtation, the anti-Markovnikov addition route would be slightly preferred. It is then demonstrated that for a series of alkenes, the difference between activation energies of Markovnikov and anti-Markovnikov addition routes [ΔE#(M−AM)] has a good linear correlation with sign(KED)K2ED, where KED is characteristic of the electron density (ED) at the π region in the initial state of the alkenes. Interestingly, there is a good linear correlation between our sign (KED)K2ED and the absolute values of difference in the core ionization energy between M and AM carbon atoms obtained by others (L. J. Sæthre, T. D. Thomas, S. Svensson J. Chem. Soc. P21997, 2, 749.) in terms of the experimental study. In addition, the spatial dynamic changing features of the MF faces and interesting pictures of the electron transfer are clearly shown during the course of the electrophilic addition reactions. These results indicate that not only regioselectivity, but also activation energy and reactivity correlate with the π charge distribution in the initial state of the alkenes for electrophilic addition reactions.
Co-reporter:Qing-Mei Guan;Zhong-Zhi Yang
Chinese Journal of Chemistry 2007 Volume 25(Issue 6) pp:
Publication Date(Web):14 JUN 2007
DOI:10.1002/cjoc.200790136
A detailed theoretical investigation on Co3+ hydration in aqueous solution has been carried out by means of molecular dynamics (MD) simulations based on the atom-bond electronegativity equalization method fused into molecular mechanics (ABEEM/MM). The effective Co3+ ion-water potential has been constructed by fitting to ab initio structures and binding energies for ionic clusters. And then the ion-water interaction potential was applied in combination with the ABEEM-7P water model to molecular dynamics simulations of single Co3+(aq.) solution, managing to reproduce many experimental structural and dynamical properties of the solution. Here, not only the common properties (radial distribution function, angular distribution function and solvation energy) obtained for Co3+ in ABEEM-7P water solution were in good agreement with those from the experimental methods and other molecular dynamics simulations but also very interesting properties of charge distributions, geometries of water molecules, hydrogen bond, diffusion coefficients, vibrational spectra are investigated by ABEEM/MM model.
Co-reporter:Zhongzhi Yang, Baoqiu Cui
Acta Physico-Chimica Sinica 2007 Volume 23(Issue 9) pp:1332-1336
Publication Date(Web):September 2007
DOI:10.1016/S1872-1508(07)60068-0
The structures of sperm whale myoglobin (Mb) and mutants were investigated in terms of the ABEEM/MM method. The molecular dynamic simulations showed that the bifurcated hydrogen-bondings in the proximal side of the heme in Mb were not stable. These simulations indicated that hydrogen-bondings could not determine the overall orientation of imidazole, which could be related to the histidine residue. The amide acids and the bulk of the imidazole can have effects on the flexibility of proximal ligands.
Co-reporter:Ping Qian
Science China Chemistry 2007 Volume 50( Issue 2) pp:190-204
Publication Date(Web):2007 April
DOI:10.1007/s11426-007-0003-2
ABEEM/MM model has been applied to compute the various properties characterizing water clusters (H2O)n (n = 7−10), such as optimized geometries, the hydrogen bonds number, cluster interaction energies, stabilities, ABEEM charge distributions, dipole moments, structural parameters, and so on, and to describe the transition reflected by the hexamer region from two-dimensional (from dimer to pentamer) to three-dimensional structures (for clusters larger than the hexamer).
Co-reporter:Yu Guo, Hui Li, Lan-Lan He, Dong-Xia Zhao, Li-Dong Gong and Zhong-Zhi Yang
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 21) pp:NaN13923-13923
Publication Date(Web):2017/05/03
DOI:10.1039/C7CP01617D
The dioxygen formation mechanism of biological water oxidation in nature has long been the focus of argument; many diverse mechanistic hypotheses have been proposed. Based on a recent breakthrough in the resolution of the electronic and structural properties of the oxygen-evolving complex in the S3 state, our density functional theory (DFT) calculations reveal that the open-cubane oxo–oxyl coupling mechanism, whose substrates preferably originate from W2 and O5 in the S2 state, emerges as the best candidate for O–O bond formation in the S4 state. This is justified by the overwhelming energetic superiority of this mechanism over alternative mechanisms in both the isomeric open and closed-cubane forms of the Mn4CaO5 cluster; spin-dependent reactivity rooted in variable magnetic couplings was found to play an essential role. Importantly, this oxygen evolution mechanism is supported by the recent discovery of femtosecond X-ray free electron lasers (XFEL), and the origin of the observed structural changes from the S1 to S3 state has been analyzed. In this view, we corroborate the proposed water binding mechanism during S2–S3 transition and correlate the theoretical models with experimental findings from aspects of substrate selectivity according to water exchange kinetics. This theoretical consequence for native metalloenzymes may serve as a significant guide for improving the design and synthesis of biomimetic materials in the field of photocatalytic water splitting.