Co-reporter:Cory M. Ayres, Timothy P. Riley, Steven A. Corcelli, and Brian M. Baker
Journal of Chemical Information and Modeling August 28, 2017 Volume 57(Issue 8) pp:1990-1990
Publication Date(Web):July 11, 2017
DOI:10.1021/acs.jcim.7b00118
In cellular immunity, T cells recognize peptide antigens bound and presented by major histocompatibility complex (MHC) proteins. The motions of peptides bound to MHC proteins play a significant role in determining immunogenicity. However, existing approaches for investigating peptide/MHC motional dynamics are challenging or of low throughput, hindering the development of algorithms for predicting immunogenicity from large databases, such as those of tumor or genetically unstable viral genomes. We addressed this by performing extensive molecular dynamics simulations on a large structural database of peptides bound to the most commonly expressed human class-I MHC protein, HLA-A*0201. The simulations reproduced experimental indicators of motion and were used to generate simple models for predicting site-specific, rapid motions of bound peptides through differences in their sequence and chemical composition alone. The models can easily be applied on their own or incorporated into immunogenicity prediction algorithms. Beyond their predictive power, the models provide insight into how amino acid substitutions can influence peptide and protein motions and how dynamic information is communicated across peptides. They also indicate a link between peptide rigidity and hydrophobicity, two features known to be important in influencing cellular immune responses.
Co-reporter:Céline Douat-Casassus ; Oleg Borbulevych ; Marion Tarbe ; Nadine Gervois ; Francine Jotereau ; Brian M. Baker ;Stéphane Quideau
Journal of Medicinal Chemistry 2010 Volume 53(Issue 19) pp:7061-7066
Publication Date(Web):August 31, 2010
DOI:10.1021/jm100683p
There is growing interest in using tumor associated antigens presented by class I major histocompatibility complex (MHC-I) proteins as cancer vaccines. As native peptides are poorly stable in biological fluids, researchers have sought to engineer synthetic peptidomimetics with greater biostability. Here, we demonstrate that antigenic peptidomimetics of the Melan-A/MART-126(27L)-35 melanoma antigen adopt strikingly different conformations when bound to MHC-I, highlighting the degeneracy of T cell recognition and revealing the challenges associated with mimicking native peptide conformation.
Co-reporter:Francis K. Insaidoo, Jaroslav Zajicek and Brian M. Baker
Biochemistry 2009 Volume 48(Issue 41) pp:
Publication Date(Web):September 17, 2009
DOI:10.1021/bi9008787
T-Cell receptor recognition of peptides bound by major histocompatibility complex (MHC) proteins initiates a cellular immune response. Dynamics of peptides within MHC binding grooves can influence TCR recognition, yet NMR studies which could address this rigorously have been hindered by the expense of isotopically labeled peptides and the large size of peptide−MHC complexes. Here we describe a methodology for characterizing peptide dynamics within MHC binding grooves via NMR, using a biosynthetic approach for producing labeled peptide. With the Tax11−19 peptide bound to the human class I MHC HLA-A*0201, we demonstrate that peptide generated in this manner can be well characterized in MHC binding grooves by NMR, providing opportunities to more precisely study the role of peptide dynamics in TCR recognition. Demonstrating the utility of such studies, the data with the Tax11−19 peptide indicate the presence of slow conformational exchange in the peptide, supporting an “induced-fit” style TCR binding mechanism.
Co-reporter:Kathryn M. Armstrong;Francis K. Insaidoo
Journal of Molecular Recognition 2008 Volume 21( Issue 4) pp:275-287
Publication Date(Web):
DOI:10.1002/jmr.896
Abstract
αβ T-cell receptors (TCRs) recognize peptide antigens presented by class I or class II major histocompatibility complex molecules (pMHC). Here we review the use of thermodynamic measurements in the study of TCR–pMHC interactions, with attention to the diversity in binding thermodynamics and how this is related to the variation in TCR–pMHC interfaces. We show that there is no enthalpic or entropic signature for TCR binding; rather, enthalpy and entropy changes vary in a compensatory manner that reflects a narrow free energy window for the interactions that have been characterized. Binding enthalpy and entropy changes do not correlate with structural features such as buried surface area or the number of hydrogen bonds within TCR–pMHC interfaces, possibly reflecting the myriad of contributors to binding thermodynamics, but likely also reflecting a reliance on van't Hoff over calorimetric measurements and the unaccounted influence of equilibria linked to binding. TCR–pMHC binding heat capacity changes likewise vary considerably. In some cases, the heat capacity changes are consistent with conformational differences between bound and free receptors, but there is little data indicating these conformational differences represent the need to organize disordered CDR loops. In this regard, we discuss how thermodynamics may provide additional insight into conformational changes occurring upon TCR binding. Finally, we highlight opportunities for the further use of thermodynamic measurements in the study of TCR–pMHC interactions, not only for understanding TCR binding in general, but also for understanding specifics of individual interactions and the engineering of TCRs with desired molecular recognition properties. Copyright © 2008 John Wiley & Sons, Ltd.
Co-reporter:Oleg Y. Borbulevych, Priscilla Do, Brian M. Baker
Molecular Immunology (September 2010) Volume 47(Issue 15) pp:2519-2524
Publication Date(Web):1 September 2010
DOI:10.1016/j.molimm.2010.06.005
Presentation of peptides by class I or class II major histocompatibility complex (MHC) molecules is required for the initiation and propagation of a T cell-mediated immune response. Peptides from the Wilms Tumor 1 transcription factor (WT1), upregulated in many hematopoetic and solid tumors, can be recognized by T cells and numerous efforts are underway to engineer WT1-based cancer vaccines. Here we determined the structures of the class I MHC molecule HLA-A*0201 bound to the native 126–134 epitope of the WT1 peptide and a recently described variant (R1Y) with improved MHC binding. The R1Y variant, a potential vaccine candidate, alters the positions of MHC charged side chains near the peptide N-terminus and significantly reduces the peptide/MHC electrostatic surface potential. These alterations indicate that the R1Y variant is an imperfect mimic of the native WT1 peptide, and suggest caution in its use as a therapeutic vaccine. Stability measurements revealed how the R1Y substitution enhances MHC binding affinity, and together with the structures suggest a strategy for engineering WT1 variants with improved MHC binding that retain the structural features of the native peptide/MHC complex.
Co-reporter:Lance M. Hellman, Liusong Yin, Yuan Wang, Sydney J. Blevins, Timothy P. Riley, Orrin S. Belden, Timothy T. Spear, Michael I. Nishimura, Lawrence J. Stern, Brian M. Baker
Journal of Immunological Methods (May 2016) Volume 432() pp:95-101
Publication Date(Web):1 May 2016
DOI:10.1016/j.jim.2016.02.016
•The thermal stability of peptide/MHC complexes correlates with peptide affinity.•Differential scanning fluorimetry permits rapid assessment of peptide/MHC stability.•Substantially lower volume requirements of DSF provides for high throughput.•DSF also allows for measurements of peptide/MHC kinetic stability/dissociation rates.•Complexities not detectable with other binding assays can be observed with DSF.Measurements of thermal stability by circular dichroism (CD) spectroscopy have been widely used to assess the binding of peptides to MHC proteins, particularly within the structural immunology community. Although thermal stability assays offer advantages over other approaches such as IC50 measurements, CD-based stability measurements are hindered by large sample requirements and low throughput. Here we demonstrate that an alternative approach based on differential scanning fluorimetry (DSF) yields results comparable to those based on CD for both class I and class II complexes. As they require much less sample, DSF-based measurements reduce demands on protein production strategies and are amenable for high throughput studies. DSF can thus not only replace CD as a means to assess peptide/MHC thermal stability, but can complement other peptide-MHC binding assays used in screening, epitope discovery, and vaccine design. Due to the physical process probed, DSF can also uncover complexities not observed with other techniques. Lastly, we show that DSF can also be used to assess peptide/MHC kinetic stability, allowing for a single experimental setup to probe both binding equilibria and kinetics.
Co-reporter:Orrin S. Belden, Sarah Catherine Baker, Brian M. Baker
Trends in Immunology (July 2015) Volume 36(Issue 7) pp:385-387
Publication Date(Web):1 July 2015
DOI:10.1016/j.it.2015.05.004
Recruiting volunteers who can provide computational time, programming expertise, or puzzle-solving talent has emerged as a powerful tool for biomedical research. Recent projects demonstrate the potential for such ‘crowdsourcing’ efforts in immunology. Tools for developing applications, new funding opportunities, and an eager public make crowdsourcing a serious option for creative solutions for computationally-challenging problems. Expanded uses of crowdsourcing in immunology will allow for more efficient large-scale data collection and analysis. It will also involve, inspire, educate, and engage the public in a variety of meaningful ways. The benefits are real – it is time to jump in!
Co-reporter:Daniel T. Harris, Nishant K. Singh, Qi Cai, Sheena N. Smith, ... Brian M. Baker
Structure (6 July 2016) Volume 24(Issue 7) pp:1142-1154
Publication Date(Web):6 July 2016
DOI:10.1016/j.str.2016.04.011
•Switching TCR specificity via in vitro evolution leads to an unusual binding mode•The engineered TCR still signals in an antigen-specific manner•Disruption of canonical TCR-MHC interactions catalyzes the specificity switch•Further contributions to the switch are distributed throughout the interfaceUtilizing a diverse binding site, T cell receptors (TCRs) specifically recognize a composite ligand comprised of a foreign peptide and a major histocompatibility complex protein (MHC). To help understand the determinants of TCR specificity, we studied a parental and engineered receptor whose peptide specificity had been switched via molecular evolution. Altered specificity was associated with a significant change in TCR-binding geometry, but this did not impact the ability of the TCR to signal in an antigen-specific manner. The determinants of binding and specificity were distributed among contact and non-contact residues in germline and hypervariable loops, and included disruption of key TCR-MHC interactions that bias αβ TCRs toward particular binding modes. Sequence-fitness landscapes identified additional mutations that further enhanced specificity. Our results demonstrate that TCR specificity arises from the distributed action of numerous sites throughout the interface, with significant implications for engineering therapeutic TCRs with novel and functional recognition properties.Download high-res image (240KB)Download full-size image
Co-reporter:Nishant K. Singh, Brian M. Baker
Structure (4 October 2016) Volume 24(Issue 10) pp:1623-1624
Publication Date(Web):4 October 2016
DOI:10.1016/j.str.2016.09.004
Recognition of antigens by T cell receptors (TCRs) underlies cellular immunity. By comparing how different TCRs recognize the key antigens associated with celiac disease, Petersen et al. (2016), in this issue of Structure, show how celiac antigen properties select immunologically distinct yet structurally and physically compatible TCRs, ultimately driving autoimmunity.
Co-reporter:Daniel R. Scott, Oleg Y. Borbulevych, Kurt H. Piepenbrink, Steven A. Corcelli, Brian M. Baker
Journal of Molecular Biology (2 December 2011) Volume 414(Issue 3) pp:385-400
Publication Date(Web):2 December 2011
DOI:10.1016/j.jmb.2011.10.006
αβ T-cell receptors (TCRs) recognize multiple antigenic peptides bound and presented by major histocompatibility complex molecules. TCR cross-reactivity has been attributed in part to the flexibility of TCR complementarity-determining region (CDR) loops, yet there have been limited direct studies of loop dynamics to determine the extent of its role. Here we studied the flexibility of the binding loops of the αβ TCR A6 using crystallographic, spectroscopic, and computational methods. A significant role for flexibility in binding and cross-reactivity was indicated only for the CDR3α and CDR3β hypervariable loops. Examination of the energy landscapes of these two loops indicated that CDR3β possesses a broad, smooth energy landscape, leading to rapid sampling in the free TCR of a range of conformations compatible with different ligands. The landscape for CDR3α is more rugged, resulting in more limited conformational sampling that leads to specificity for a reduced set of peptides as well as the major histocompatibility complex protein. In addition to informing on the mechanisms of cross-reactivity and specificity, the energy landscapes of the two loops indicate a complex mechanism for TCR binding, incorporating elements of both conformational selection and induced fit in a manner that blends features of popular models for TCR recognition.Download high-res image (133KB)Download full-size imageResearch Highlights► The CDR3α and CDR3β loops of the A6 TCR adjust their conformations upon binding. ► CDR3α interconverts slowly between bound and free conformations in the free TCR. ► Unlike CDR3α, CDR3β rapidly samples a spectrum of conformations in the free TCR. ► Both conformational selection and induced fit are implicated in binding. ► Cross-reactivity and specificity emerge from the dynamics of the free CDR3 loops.
Co-reporter:Daniel R. Scott, Charles F. Vardeman II, Steven A. Corcelli, Brian M. Baker
Biophysical Journal (19 December 2012) Volume 103(Issue 12) pp:
Publication Date(Web):19 December 2012
DOI:10.1016/j.bpj.2012.10.037
Time-resolved fluorescence anisotropy (TRFA) has a rich history in evaluating protein dynamics. Yet as often employed, TRFA assumes that the motional properties of a covalently tethered fluorescent probe accurately portray the motional properties of the protein backbone at the probe attachment site. In an extensive survey using TRFA to study the dynamics of the binding loops of a αβ T cell receptor, we observed multiple discrepancies between the TRFA data and previously published results that led us to question this assumption. We thus simulated several of the experimentally probed systems using a protocol that permitted accurate determination of probe and protein time correlation functions. We found excellent agreement in the decays of the experimental and simulated correlation functions. However, the motional properties of the probe were poorly correlated with those of the backbone of both the labeled and unlabeled protein. Our results warrant caution in the interpretation of TRFA data and suggest further studies to ascertain the extent to which probe dynamics reflect those of the protein backbone. Meanwhile, the agreement between experiment and computation validates the use of molecular dynamics simulations as an accurate tool for exploring the molecular motion of T cell receptors and their binding loops.
Co-reporter:Oleg Y. Borbulevych, Kurt H. Piepenbrink, Brian E. Gloor, Daniel R. Scott, ... Brian M. Baker
Immunity (18 December 2009) Volume 31(Issue 6) pp:885-896
Publication Date(Web):18 December 2009
DOI:10.1016/j.immuni.2009.11.003
T cell-mediated immunity requires T cell receptor (TCR) cross-reactivity, the mechanisms behind which remain incompletely elucidated. The αβ TCR A6 recognizes both the Tax (LLFGYPVYV) and Tel1p (MLWGYLQYV) peptides presented by the human class I MHC molecule HLA-A2. Here we found that although the two ligands are ideal structural mimics, they form substantially different interfaces with A6, with conformational differences in the peptide, the TCR, and unexpectedly, the MHC molecule. The differences between the Tax and Tel1p ternary complexes could not be predicted from the free peptide-MHC structures and are inconsistent with a traditional induced-fit mechanism. Instead, the differences were attributable to peptide and MHC molecular motion present in Tel1p-HLA-A2 but absent in Tax-HLA-A2. Differential “tuning” of the dynamic properties of HLA-A2 by the Tax and Tel1p peptides thus facilitates cross-recognition and impacts how structural diversity can be presented to and accommodated by receptors of the immune system.

![Borate(1-),[5-[(3,5-dimethyl-2H-pyrrol-2-ylidene-kN)methyl]-1H-pyrrole-2-propanoato(2-)-kN1]difluoro-, hydrogen (1:1),(T-4)-](/data/chemimg/109100/165599-63-3.png)
![Borate(1-),[5-[(3,5-dimethyl-2H-pyrrol-2-ylidene-kN)methyl]-1H-pyrrole-2-propanoato(2-)-kN1]difluoro-, hydrogen (1:1),(T-4)-](/data/chemimg/109100/165599-63-3_b.png)

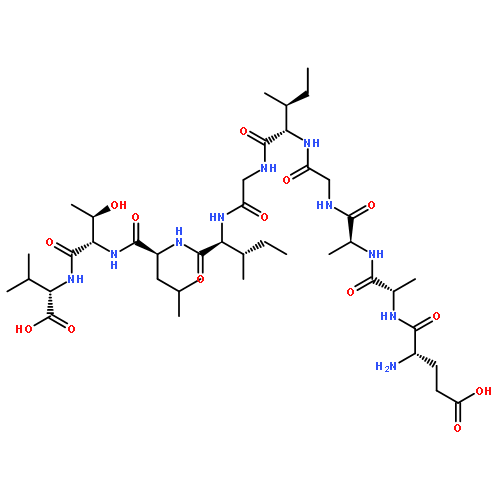








![4-Thia-1-azabicyclo[3.2.0]heptane-2-carboxylicacid, 6-[[(2'-carboxy[1,1'-biphenyl]-2-yl)carbonyl]amino]-3,3-dimethyl-7-oxo-,(2S,5R,6R)-](http://img.cochemist.com/ccimg/14800/14796-35-1.png)
![4-Thia-1-azabicyclo[3.2.0]heptane-2-carboxylicacid, 6-[[(2'-carboxy[1,1'-biphenyl]-2-yl)carbonyl]amino]-3,3-dimethyl-7-oxo-,(2S,5R,6R)-](http://img.cochemist.com/ccimg/14800/14796-35-1_b.png)

