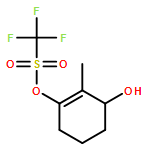
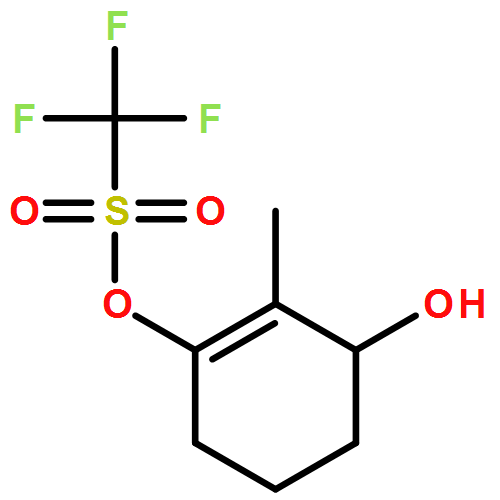
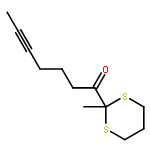
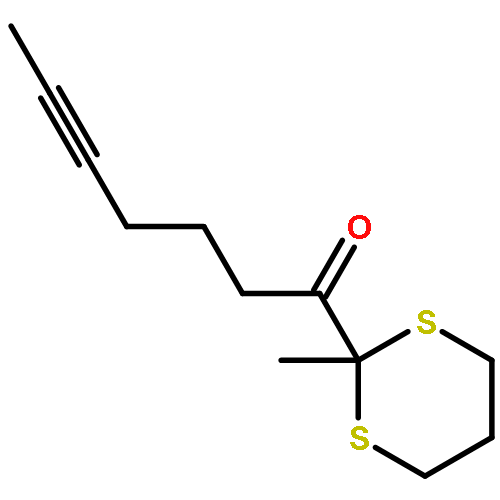
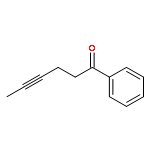
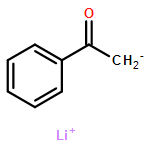
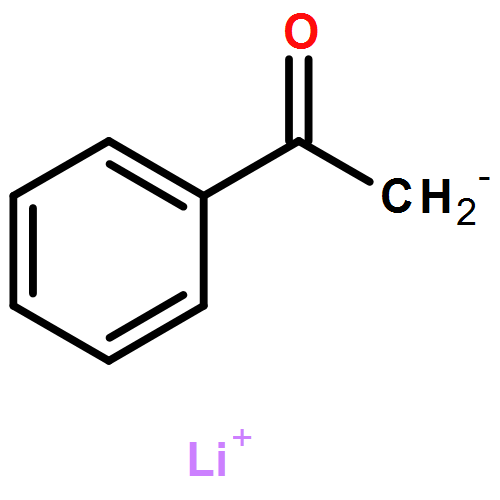
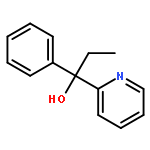
Co-reporter:Tung T. Hoang, Mélodie Birepinte, Nicholas J. Kramer, and Gregory B. Dudley
Organic Letters 2016 Volume 18(Issue 14) pp:3470-3473
Publication Date(Web):June 30, 2016
DOI:10.1021/acs.orglett.6b01665
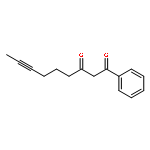
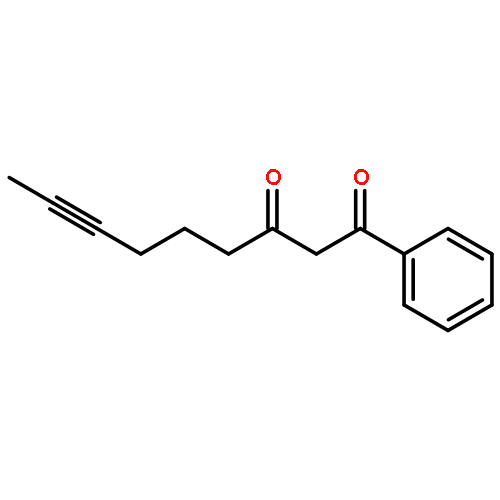
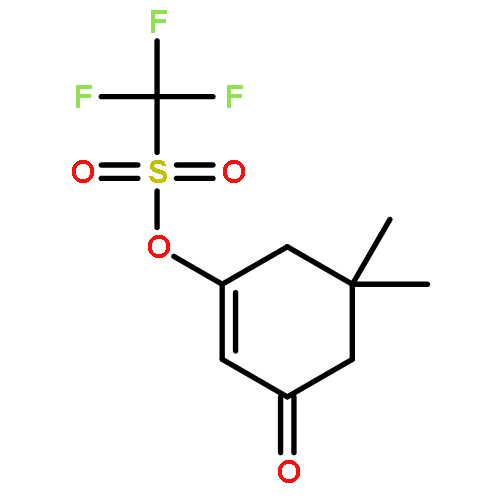
Strategic pairing of ring openings and cycloisomerization provides rapid and efficient “open and shut” entry into sparsely functionalized illudalanes, as exemplified here in the context of a six-step synthesis of alcyopterosin A. Key steps include a tandem ring-opening fragmentation/olefination process for preparing a neopentyl-tethered 1,6-enyne, ring-opening olefination telescoped with alkyne homologation, and Rh-catalyzed oxidative cycloisomerization.
Co-reporter:Ron R. Ramsubhag, Chelsea L. Massaro, Christina M. Dadich, Andrew J. Janeczek, Tung T. Hoang, Elizabeth A. Mazzio, Suresh Eyunni, Karam F. A. Soliman and Gregory B. Dudley
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 33) pp:7855-7858
Publication Date(Web):01 Aug 2016
DOI:10.1039/C6OB01351A
3,3-Dimethylcyclopentanes (neopentylenes) are ubiquitous in Nature but largely absent from synthetic pharmaceutical libraries. Neopentylenes define a hydrophobic and rigid 3-D topology with distinct molecular pharmacology, as exemplified here with two neopentylene-fused analogues of the synthetic anti-inflammatory drug, ibuprofen.
Co-reporter:Ron R. Ramsubhag and Gregory B. Dudley
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 22) pp:5028-5031
Publication Date(Web):13 May 2016
DOI:10.1039/C6OB00795C
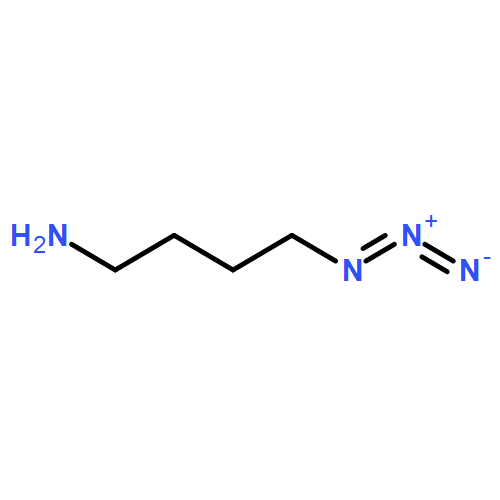
Carbamate-tethered propargyl and benzocyclononyne moieties within a single molecular unit undergo cycloaddition with azides under complementary CuAAC and/or SPAAC coupling conditions. The carbamate linker can be cleaved by analogy to the –CBz protecting group for click capture-and-release applications.
Co-reporter:Paratchata Batsomboon, Gregory B. Dudley
Tetrahedron Letters 2016 Volume 57(Issue 33) pp:3757-3759
Publication Date(Web):17 August 2016
DOI:10.1016/j.tetlet.2016.07.014
•Improved synthesis of C1–C15 of palmerolide A.•Observations on optimal protecting group strategies for polyketide synthesis.•Unique utility of DIP chloride for asymmetric reduction of linear alkyl vinyl ketones.•Use of barium hydroxide for mild Horner–Wadsworth–Emmons olefination.A streamlined synthesis of a C1–C15 fragment of palmerolide A is described. Key tactical advances (vis-à-vis previous reports from the Dudley laboratory) include diastereoselective reduction (>20:1 dr) of a C7 ketone using DIP chloride and a modified protecting group strategy that enables both improved step count and end-game efficiency. Asymmetric reduction of linear alkyl vinyl ketone derivatives is a largely unsolved problem in organic synthesis. DIP chloride has been uniquely effective for asymmetric reduction of the alkyl vinyl ketone derivatives in the present study.
Co-reporter:Rimantas Slegeris, Gregory B. Dudley
Tetrahedron 2016 Volume 72(Issue 26) pp:3666-3672
Publication Date(Web):30 June 2016
DOI:10.1016/j.tet.2016.03.041
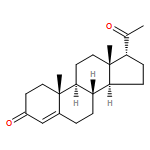
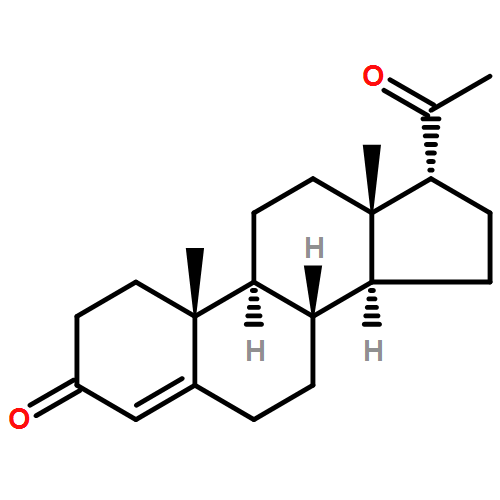
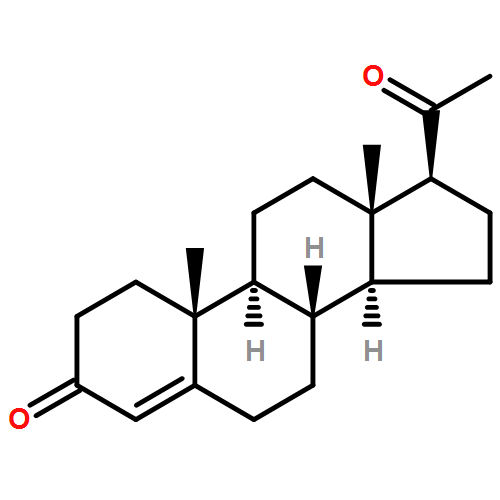
Three alternative synthetic entries into Johnson's classic synthesis of rac-progesterone are presented in this manuscript. ent-Progesterone, the non-natural enantiomer of progesterone, has recently been identified as a potential alternative to progesterone for investigations into possible prevention and treatment of traumatic brain injury (TBI). Difficulties in accessing ent-progesterone in large quantities prevent it from being studied more thoroughly. Strategies for producing synthetic rac-progesterone are described and discussed herein.
Co-reporter:Brian Gold, Paratchata Batsomboon, Gregory B. Dudley, and Igor V. Alabugin
The Journal of Organic Chemistry 2014 Volume 79(Issue 13) pp:6221-6232
Publication Date(Web):June 13, 2014
DOI:10.1021/jo500958n
Our recent work has provided an alternative strategy for acceleration of azide/alkyne cycloadditions via selective transition state (TS) stabilization. Optimization of hyperconjugative assistance, provided by the antiperiplanar arrangement of propargylic σ-acceptors relative to the forming bonds, is predicted to relieve strain in cyclooctynes while providing large acceleration to the cycloaddition. The present work investigates this strategy in alkynyl crown ethers, where propargylic C–O bonds contained within the macrocycle are constrained close to proper alignment for hyperconjugative assistance. Preorganization of σ-acceptors into the optimal arrangement for hyperconjugative interactions may alleviate a portion of the entropic penalty for reaching the TS. Optimal alignment can be reinforced, and transition-state stabilization can be further amplified by binding positively charged ions to the crown ether core, highlighting the potential for applications in ion sensing.
Co-reporter:Michael R. Rosana, Jacob Hunt, Anthony Ferrari, Taylor A. Southworth, Yuchuan Tao, Albert E. Stiegman, and Gregory B. Dudley
The Journal of Organic Chemistry 2014 Volume 79(Issue 16) pp:7437-7450
Publication Date(Web):July 22, 2014
DOI:10.1021/jo501153r
Thermally promoted Friedel–Crafts benzylation of arene solvents has been examined under both conventional convective heating with an oil bath and heating using microwave (MW) energy. Bulk solution temperatures—as measured by internal and external temperature probes and as defined by solvent reflux—were comparable in both sets of experiments. MW-specific rate enhancements were documented under certain conditions and not others. The observed rate enhancements at a given temperature are proposed to arise from selective MW heating of polar solutes, perturbing thermal equilibrium between the solute and bulk solution. Central to MW-specific thermal phenomena is the difference between heat and temperature. Temperature is a measure of the ensemble average kinetic molecular energy of all solution components, but temperature does not provide information about solute-specific energy differences that may arise as a consequence of selective MW heating. Enhanced chemical reactivity of the MW-absorbing solute can be described as a MW-specific “extra-temperature thermal effect”, because the measurable solution temperature only captures a portion of the solute kinetic molecular energy. Experimental factors that favor MW-specific rate enhancements are discussed with an eye toward future development of MW-actuated organic reactions, in which the observed thermal reactivity exceeds what is predicted from temperature-based Arrhenius calculations.
Co-reporter:Marilda P. Lisboa, David M. Jones, and Gregory B. Dudley
Organic Letters 2013 Volume 15(Issue 4) pp:886-889
Publication Date(Web):February 1, 2013
DOI:10.1021/ol400014e
An enantioselective route to palmerolide A is described. The approach features original syntheses of three subunits, which are then assembled to produce a known late-stage intermediate and formally provide the highest overall yield of the natural product reported to date. Recent innovations in alkynogenic fragmentation and vinylogous aldol methodology figure prominently in the synthesis of the C1–C15 and C16–C25 subunits, respectively.
Co-reporter:Tung T. Hoang and Gregory B. Dudley
Organic Letters 2013 Volume 15(Issue 15) pp:4026-4029
Publication Date(Web):July 25, 2013
DOI:10.1021/ol401839e
A tandem process provides high-value 1,6-enynes that are otherwise difficult to prepare. Two base-mediated reactions—fragmentation and olefination—are executed in a coordinated manner that is overall more efficient than either reaction on its own. The 1,6-enynes can be strategically employed in conjunction with carbocyclization to deliver important targets, as noted for reported syntheses of hirsutene and illudol.
Co-reporter:Jumreang Tummatorn, Gaspar Diaz Muñoz, Gregory B. Dudley
Tetrahedron Letters 2013 Volume 54(Issue 10) pp:1312-1314
Publication Date(Web):6 March 2013
DOI:10.1016/j.tetlet.2012.12.122
A new synthesis of (−)-angustureine is described, featuring an annulation and fragmentation sequence that formally results in alkynylation of a β-amino ester. The six-step sequence from a readily available chiral β-amino ester to the natural product was achieved in 46% yield, with no protecting groups and three purifications.
Co-reporter:Dr. Gregory B. Dudley;Dr. Albert E. Stiegman ;Michael R. Rosana
Angewandte Chemie International Edition 2013 Volume 52( Issue 31) pp:7918-7923
Publication Date(Web):
DOI:10.1002/anie.201301539
Co-reporter:Dr. Marilda P. Lisboa ;Dr. Gregory B. Dudley
Chemistry - A European Journal 2013 Volume 19( Issue 48) pp:16146-16168
Publication Date(Web):
DOI:10.1002/chem.201302167
Abstract
The chemical synthesis of the palmerolides is the subject of this review. The palmerolides are a family of Antarctic marine natural products, many of which display potent and selective cytotoxicity against melanoma cells. The confluence of promising bioactivities, limited natural supplies, and complex structures makes the palmerolides exciting targets for chemical synthesis. To date, several approaches have been reported, and a consensus strategy based on convergent fragment assembly has emerged. Collective wisdom from myriad approaches reviewed here may enable hybrid strategies capable of delivering larger amounts of synthetic palmerolides to support continued biological studies. Considering the relative lack of options for melanoma chemotherapy and the intriguing activity profile of the palmerolides, efforts aimed at developing an efficient, gram-scale synthesis of palmerolide A and congeneric structures should be given a high priority.
Co-reporter:Dr. Gregory B. Dudley;Dr. Albert E. Stiegman ;Michael R. Rosana
Angewandte Chemie 2013 Volume 125( Issue 31) pp:8074-8079
Publication Date(Web):
DOI:10.1002/ange.201301539
Co-reporter:Michael R. Rosana, Yuchuan Tao, Albert E. Stiegman and Gregory B. Dudley
Chemical Science 2012 vol. 3(Issue 4) pp:1240-1244
Publication Date(Web):17 Jan 2012
DOI:10.1039/C2SC01003H
Microwave-actuated organic reactions are defined herein as chemical reaction systems for which microwave irradiation provides a clear benefit over conventional heating to the same temperature. This study is focused on a rationally designed, microwave-actuated reaction (thermal Friedel–Crafts benzylation), in which a microwave-absorbing ionic solute reacts in a non-polar and largely microwave-transparent solvent. Steady-state microwave irradiation (ssMWi) induces observed levels of reactivity from the solute that cannot be duplicated by conventional heating of the homogeneous solution to similar temperatures. This observation is qualitatively consistent with the Arrhenius [k = Ae(−Ea/RT)] relationship between rate and molecular collisions (k ∝ A at constant T). A new paradigm for designing microwave-actuated organic reactions for microwave-assisted organic synthesis emerges from this study.
Co-reporter:Teng-wei Wang, Tanit Intaranukulkit, Michael R. Rosana, Rimantas Slegeris, Janet Simon and Gregory B. Dudley
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 2) pp:248-250
Publication Date(Web):13 Oct 2011
DOI:10.1039/C1OB06504A
Benzylation of alcohols and other substrates under thermal conditions translates smoothly from conventional heating into MW-assisted organic synthesis (MAOS). Reactions times are decreased from hours to minutes while good to excellent yields are maintained. MW heating should be considered for benzylation of high-value substrates using the title reagent.
Co-reporter:Jumreang Tummatorn, Paratchata Batsomboon, Ronald J. Clark, Igor V. Alabugin, and Gregory B. Dudley
The Journal of Organic Chemistry 2012 Volume 77(Issue 5) pp:2093-2097
Publication Date(Web):February 8, 2012
DOI:10.1021/jo300188y
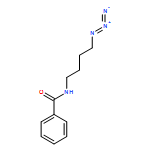
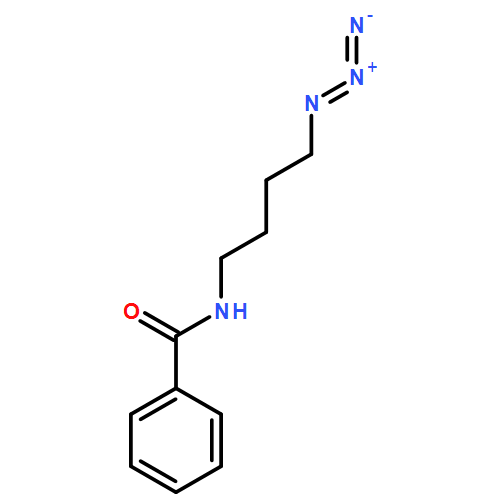
Preliminary studies related to the design and development of new cycloalkyne reagents for metal-free click coupling are reported. Cyclononynes are more stable than cyclooctynes, and the robust benzocyclononyne platform offers spontaneous reactivity toward azides at rates competitive with other azidophiles that have been employed for metal-free click coupling. Benzocyclononynes (e.g., 1) provide valuable insight into the design of new cycloalkynes for strain-promoted azide–alkyne cycloaddition (SPAAC) couplings for applications in which side reactions and decomposition of the reagent must be kept to a minimum.
Co-reporter:Jumreang Tummatorn and Gregory B. Dudley
Organic Letters 2011 Volume 13(Issue 1) pp:158-160
Publication Date(Web):November 24, 2010
DOI:10.1021/ol102760q
Dihydropyridone (DHPD) triflates undergo nucleophile-triggered fragmentation to provide homopropargyl amine derivatives, the stereochemistry of which is defined by starting from readily available β-amino esters.
Co-reporter:Sami F. Tlais and Gregory B. Dudley
Organic Letters 2010 Volume 12(Issue 20) pp:4698-4701
Publication Date(Web):September 22, 2010
DOI:10.1021/ol102201z
A blueprint for controlling the stereochemistry of oxygenated 5,5-spiroketals using chelation effects is provided. Chelation specifically of zinc salts (other protic and Lewis acids were less effective) between the spiroketal oxygen and an appropriately positioned alcohol group overrides normal biases in the preparation of 5,5-spiroketals, as illustrated by the stereocontrolled synthesis of epimeric cephalosporolide H isomers. This study provides new and valuable information for prescribing the chirality of the stereogenic core of 5,5-spiroketals.
Co-reporter:Jingyue Yang
Advanced Synthesis & Catalysis 2010 Volume 352( Issue 18) pp:3438-3442
Publication Date(Web):
DOI:10.1002/adsc.201000495
Abstract
A previously reported anionic rearrangement of benzyl 2-pyridyl ethers can now be accessed by a distinct and unusual mechanism: addition of alkyllithium reagents to α-(2-pyridyloxy)-styrene triggers an anionic rearrangement to afford tertiary pyridyl carbinols. The process is explained by invoking a contra-electronic, pyridine-directed carbolithiation of the enol ether π-system.
Co-reporter:David M. Jones, Gregory B. Dudley
Tetrahedron 2010 66(26) pp: 4860-4866
Publication Date(Web):
DOI:10.1016/j.tet.2010.03.014
Co-reporter:David M. Jones, Marilda P. Lisboa, Shin Kamijo and Gregory B. Dudley
The Journal of Organic Chemistry 2010 Volume 75(Issue 10) pp:3260-3267
Publication Date(Web):April 29, 2010
DOI:10.1021/jo100249g
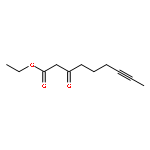
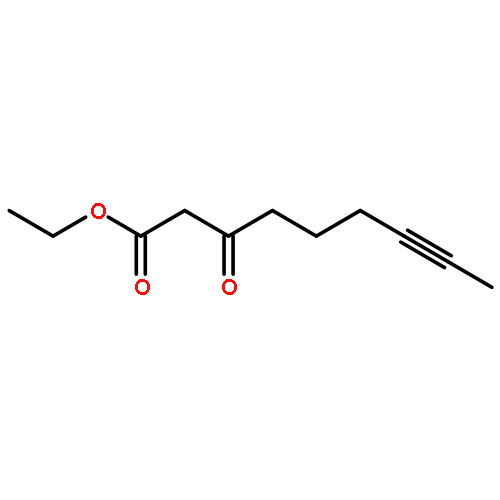
Addition of stabilized carbanionic nucleophiles to cyclic vinylogous acyl triflates (VATs) triggers a ring-opening fragmentation to give acyclic β-keto ester and related products, much like those observed traditionally in the Claisen condensation. Unlike in the classical Claisen condensation, however, the VAT-Claisen reaction described herein is rendered irreversible by C−C bond cleavage, not by deprotonation of the activated methylene product. Full details of this original reaction methodology are disclosed herein, including how subtle differences between the various nucleophiles impact the proper choice of reaction conditions for making 1,3-diketones, β-keto esters, and β-keto phosphonates.
Co-reporter:Douglas A. Engel and Gregory B. Dudley
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 20) pp:4149-4158
Publication Date(Web):14 Sep 2009
DOI:10.1039/B912099H
The Meyer–Schuster rearrangement is the conversion of propargyl alcohols into α,β-unsaturated carbonyl compounds via a formal 1,3-hydroxyl shift and tautomerization. The major challenge associated with the Meyer–Schuster reaction is that of selectively promoting the desired rearrangement over the myriad other reaction pathways available to propargyl alcohols. This Perspective Article features recent advances in the Meyer–Schuster reaction, including several demonstrated techniques for improving the scope. Strengths and weaknesses of each technique are discussed, and outstanding problems that warrant further study are highlighted. The primary motivation for research and development of the Meyer–Schuster rearrangement is as a means of preparing α,β-unsaturated carbonyl compounds as part of a two-stage olefination strategy.
Co-reporter:Jingyue Yang and Gregory B. Dudley
The Journal of Organic Chemistry 2009 Volume 74(Issue 20) pp:7998-8000
Publication Date(Web):September 17, 2009
DOI:10.1021/jo901707x
An anionic rearrangement of 2-benzyloxypyridine is described. Pyridine-directed metalation of the benzylic carbon leads to 1,2-migration of pyridine via a postulated associative mechanism (addition/elimination). Several aryl pyridyl carbinols were obtained in high yields. A formal synthesis of carbinoxamine, an antihistamine drug used for the treatment of seasonal allergies and hay fever, emerges from this methodology.
Co-reporter:Ernest O. Nwoye and Gregory B. Dudley
Chemical Communications 2007 (Issue 14) pp:1436-1437
Publication Date(Web):26 Feb 2007
DOI:10.1039/B617926F
2-(4-Methoxybenzyloxy)-4-methylquinoline reacts with methyl triflate in the presence of alcohols to generate a neutral organic salt that transfers the para-methoxybenzyl (PMB) protecting group onto alcohols in high yield and under mild conditions.
Co-reporter:Mariya V. Kozytska and Gregory B. Dudley
Chemical Communications 2005 (Issue 24) pp:3047-3049
Publication Date(Web):06 May 2005
DOI:10.1039/B503110A
The adjacent centres of electrophilicity and nucleophilicity lead to interesting chemical reactivity in the title reagent.
Co-reporter:Ernest O. Nwoye and Gregory B. Dudley
Chemical Communications 2007(Issue 14) pp:NaN1437-1437
Publication Date(Web):2007/02/26
DOI:10.1039/B617926F
2-(4-Methoxybenzyloxy)-4-methylquinoline reacts with methyl triflate in the presence of alcohols to generate a neutral organic salt that transfers the para-methoxybenzyl (PMB) protecting group onto alcohols in high yield and under mild conditions.
Co-reporter:Douglas A. Engel and Gregory B. Dudley
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 20) pp:NaN4158-4158
Publication Date(Web):2009/09/14
DOI:10.1039/B912099H
The Meyer–Schuster rearrangement is the conversion of propargyl alcohols into α,β-unsaturated carbonyl compounds via a formal 1,3-hydroxyl shift and tautomerization. The major challenge associated with the Meyer–Schuster reaction is that of selectively promoting the desired rearrangement over the myriad other reaction pathways available to propargyl alcohols. This Perspective Article features recent advances in the Meyer–Schuster reaction, including several demonstrated techniques for improving the scope. Strengths and weaknesses of each technique are discussed, and outstanding problems that warrant further study are highlighted. The primary motivation for research and development of the Meyer–Schuster rearrangement is as a means of preparing α,β-unsaturated carbonyl compounds as part of a two-stage olefination strategy.
Co-reporter:Ron R. Ramsubhag and Gregory B. Dudley
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 22) pp:NaN5031-5031
Publication Date(Web):2016/05/13
DOI:10.1039/C6OB00795C
Carbamate-tethered propargyl and benzocyclononyne moieties within a single molecular unit undergo cycloaddition with azides under complementary CuAAC and/or SPAAC coupling conditions. The carbamate linker can be cleaved by analogy to the –CBz protecting group for click capture-and-release applications.
Co-reporter:Ron R. Ramsubhag, Chelsea L. Massaro, Christina M. Dadich, Andrew J. Janeczek, Tung T. Hoang, Elizabeth A. Mazzio, Suresh Eyunni, Karam F. A. Soliman and Gregory B. Dudley
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 33) pp:NaN7858-7858
Publication Date(Web):2016/08/01
DOI:10.1039/C6OB01351A
3,3-Dimethylcyclopentanes (neopentylenes) are ubiquitous in Nature but largely absent from synthetic pharmaceutical libraries. Neopentylenes define a hydrophobic and rigid 3-D topology with distinct molecular pharmacology, as exemplified here with two neopentylene-fused analogues of the synthetic anti-inflammatory drug, ibuprofen.
Co-reporter:Teng-wei Wang, Tanit Intaranukulkit, Michael R. Rosana, Rimantas Slegeris, Janet Simon and Gregory B. Dudley
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 2) pp:NaN250-250
Publication Date(Web):2011/10/13
DOI:10.1039/C1OB06504A
Benzylation of alcohols and other substrates under thermal conditions translates smoothly from conventional heating into MW-assisted organic synthesis (MAOS). Reactions times are decreased from hours to minutes while good to excellent yields are maintained. MW heating should be considered for benzylation of high-value substrates using the title reagent.
Co-reporter:Michael R. Rosana, Yuchuan Tao, Albert E. Stiegman and Gregory B. Dudley
Chemical Science (2010-Present) 2012 - vol. 3(Issue 4) pp:NaN1244-1244
Publication Date(Web):2012/01/17
DOI:10.1039/C2SC01003H
Microwave-actuated organic reactions are defined herein as chemical reaction systems for which microwave irradiation provides a clear benefit over conventional heating to the same temperature. This study is focused on a rationally designed, microwave-actuated reaction (thermal Friedel–Crafts benzylation), in which a microwave-absorbing ionic solute reacts in a non-polar and largely microwave-transparent solvent. Steady-state microwave irradiation (ssMWi) induces observed levels of reactivity from the solute that cannot be duplicated by conventional heating of the homogeneous solution to similar temperatures. This observation is qualitatively consistent with the Arrhenius [k = Ae(−Ea/RT)] relationship between rate and molecular collisions (k ∝ A at constant T). A new paradigm for designing microwave-actuated organic reactions for microwave-assisted organic synthesis emerges from this study.
.jpg)


![PHOSPHONIC ACID, [1-(PHENYLTHIO)ETHYL]-, DIETHYL ESTER](http://img.cochemist.com/ccimg/55300/55264-89-6.png)
![PHOSPHONIC ACID, [1-(PHENYLTHIO)ETHYL]-, DIETHYL ESTER](http://img.cochemist.com/ccimg/55300/55264-89-6_b.png)
![Oxirane, 3-[(3E)-5-bromo-3-methyl-3-pentenyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/54400/54355-78-1.png)
![Oxirane, 3-[(3E)-5-bromo-3-methyl-3-pentenyl]-2,2-dimethyl-](http://img.cochemist.com/ccimg/54400/54355-78-1_b.png)
![OXIRANE, 3-[(3E)-5-CHLORO-3-METHYL-3-PENTENYL]-2,2-DIMETHYL-](http://img.cochemist.com/ccimg/43200/43119-82-0.png)
![OXIRANE, 3-[(3E)-5-CHLORO-3-METHYL-3-PENTENYL]-2,2-DIMETHYL-](http://img.cochemist.com/ccimg/43200/43119-82-0_b.png)
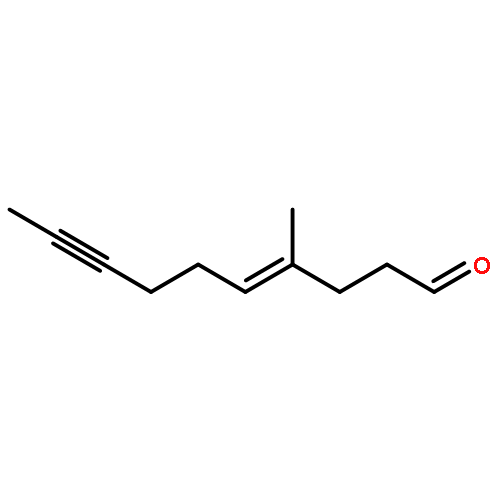
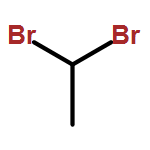
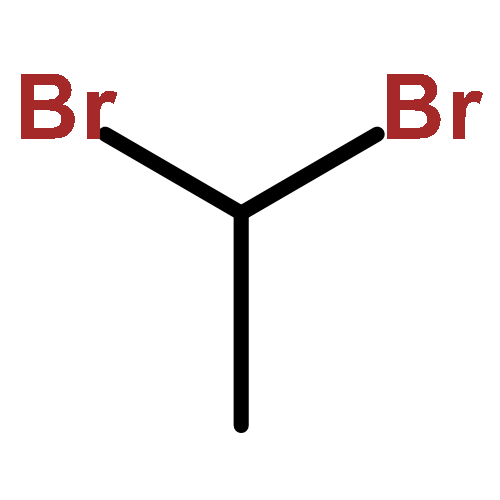
![9-methoxy-7,7-dimethyl-7,8-dihydro-6H-cyclopenta[g]isoquinoline-5-carboxylic acid](http://img.cochemist.com/ccimg/18600/18500-63-5.png)
![9-methoxy-7,7-dimethyl-7,8-dihydro-6H-cyclopenta[g]isoquinoline-5-carboxylic acid](http://img.cochemist.com/ccimg/18600/18500-63-5_b.png)
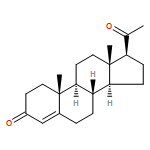








![bis[(2,3,5,6-η)-bicyclo[2.2.1]hepta-2,5-diene]di-μ-chlorodirhodium](http://img.cochemist.com/ccimg/12300/12257-42-0.png)
![bis[(2,3,5,6-η)-bicyclo[2.2.1]hepta-2,5-diene]di-μ-chlorodirhodium](http://img.cochemist.com/ccimg/12300/12257-42-0_b.png)




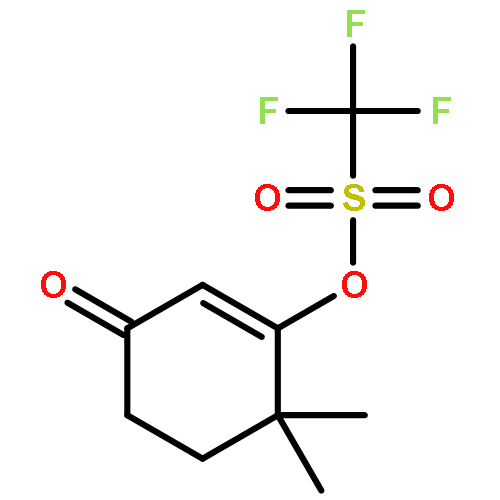
![3-Cyclohexene-1-acetic acid,3-methyl-2-oxo-4-[[(trifluoromethyl)sulfonyl]oxy]-, ethyl ester](http://img.cochemist.com/ccimg/892900/892875-46-6.png)
![3-Cyclohexene-1-acetic acid,3-methyl-2-oxo-4-[[(trifluoromethyl)sulfonyl]oxy]-, ethyl ester](http://img.cochemist.com/ccimg/892900/892875-46-6_b.png)
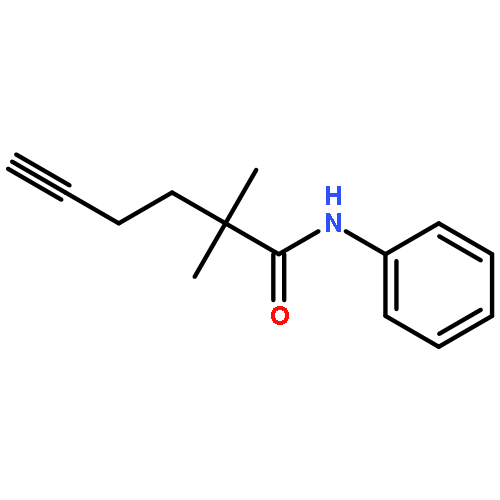
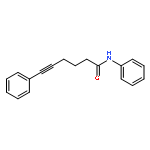
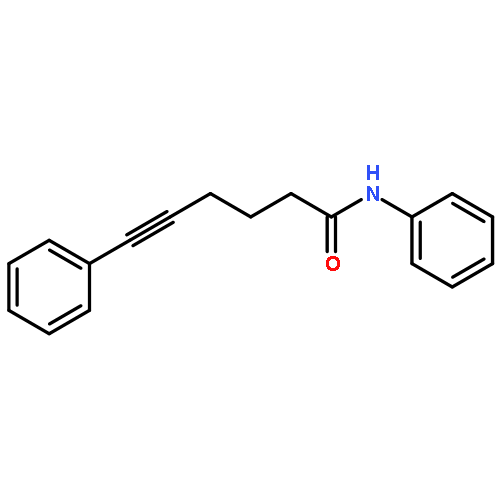
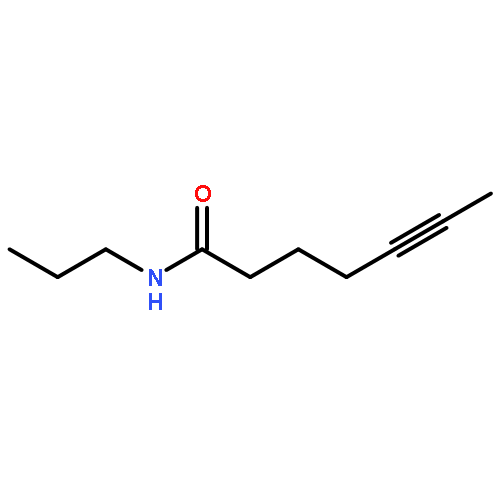
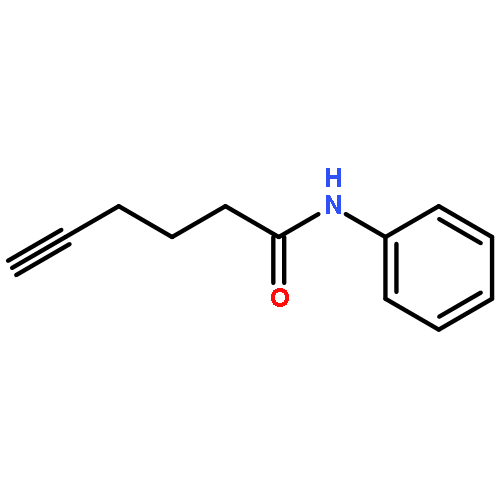
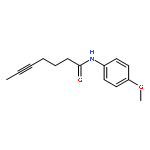
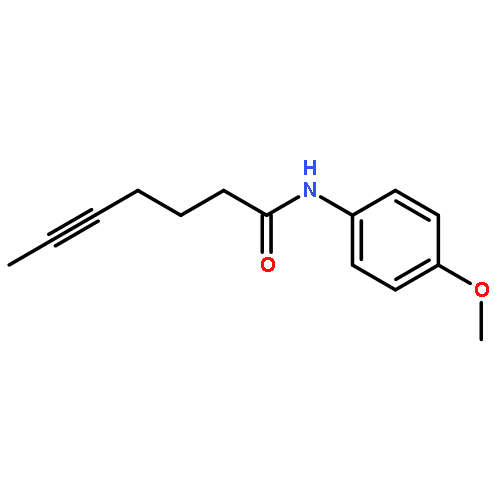
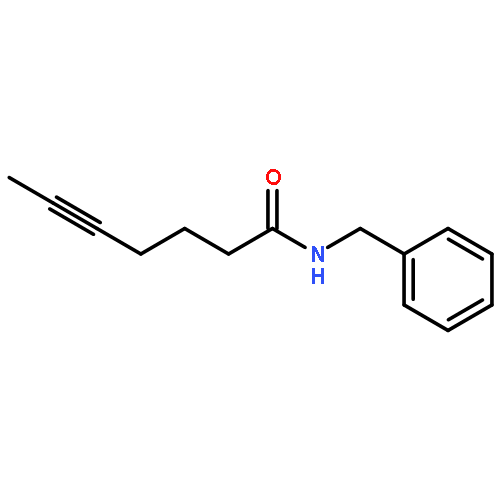
![5-Heptynamide, N-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/892900/892875-32-0.png)
![5-Heptynamide, N-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/892900/892875-32-0_b.png)
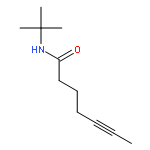
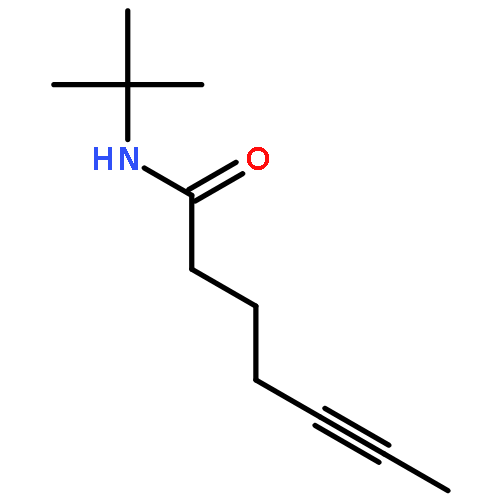
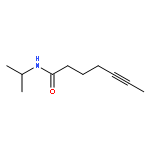
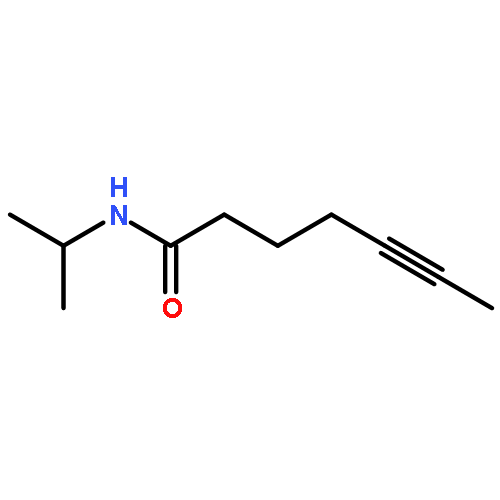
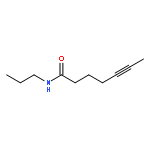


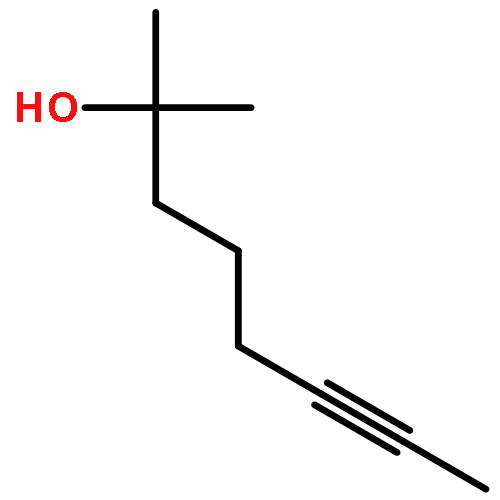
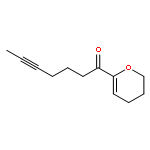
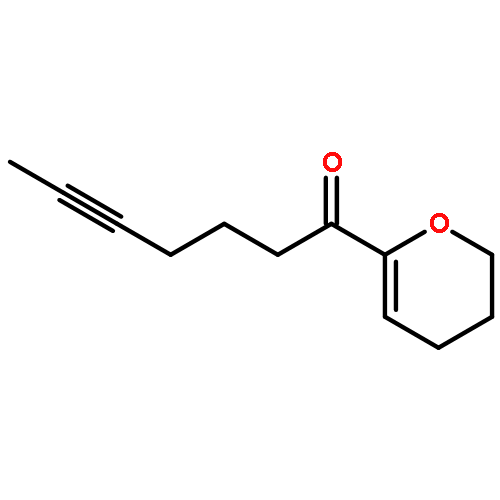
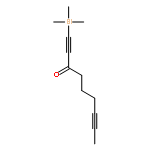
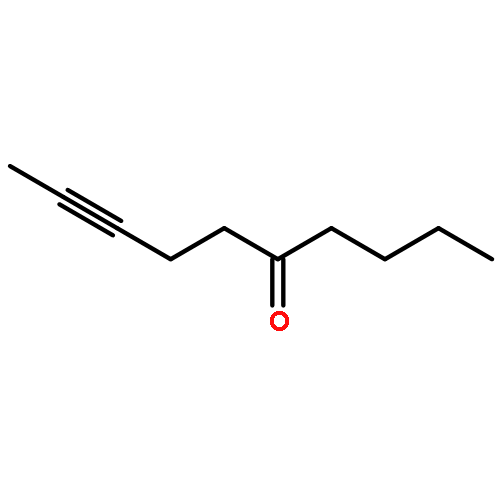
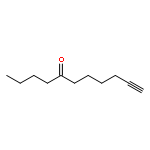
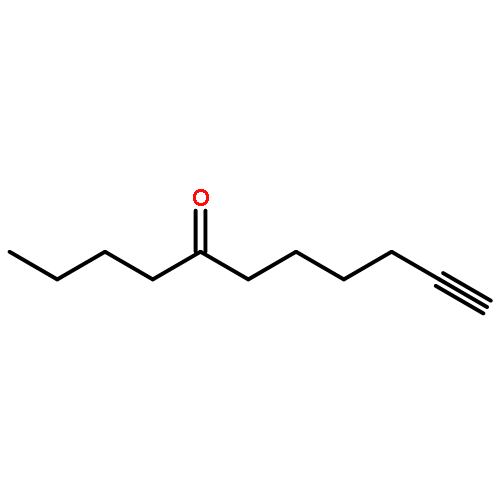
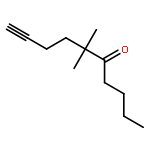
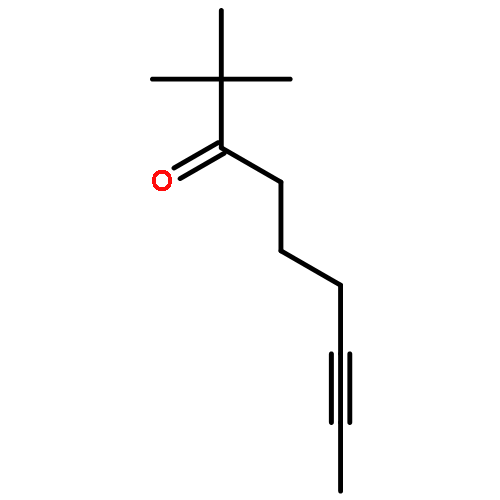
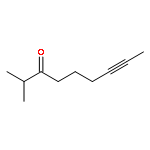
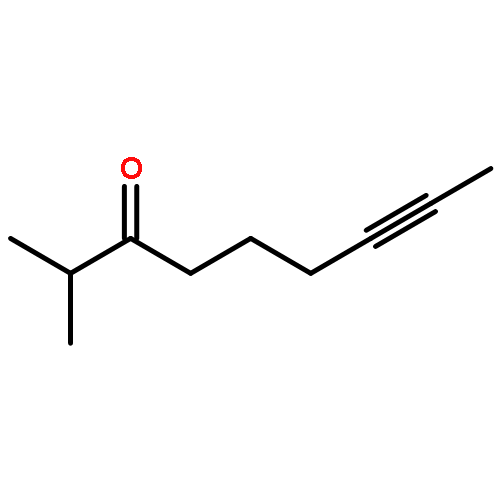


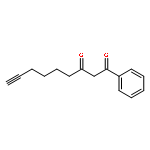
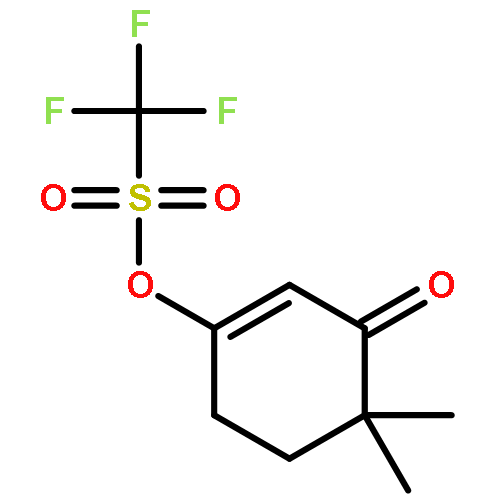
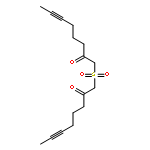
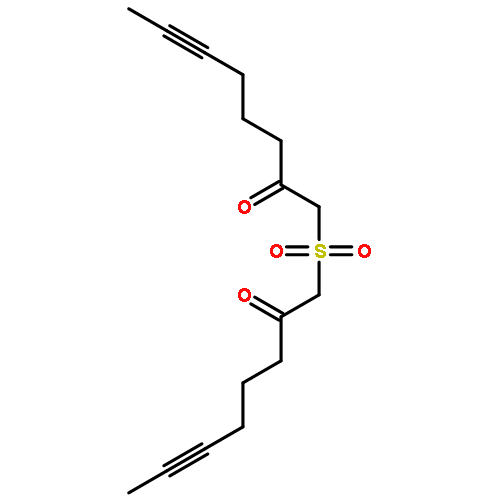
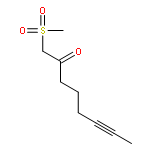
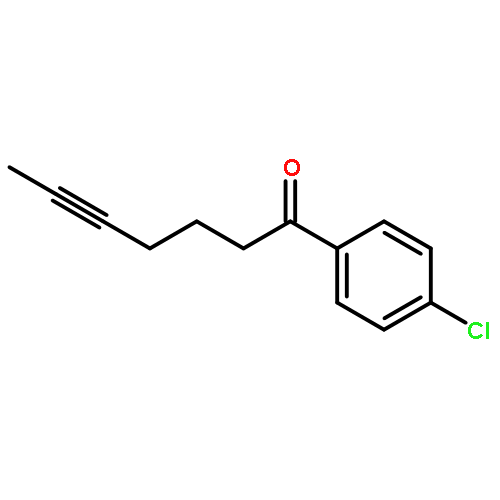
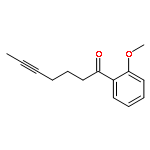
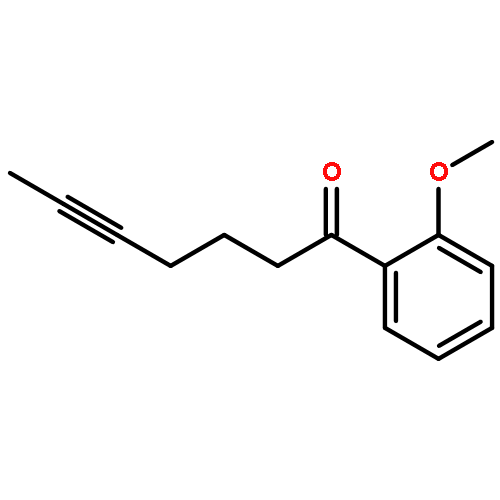
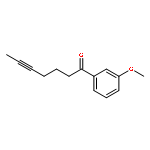


![Benzene, [(1Z)-3-bromo-1-iodo-2-methyl-1-propenyl]-](http://img.cochemist.com/ccimg/495500/495414-11-4.png)
![Benzene, [(1Z)-3-bromo-1-iodo-2-methyl-1-propenyl]-](http://img.cochemist.com/ccimg/495500/495414-11-4_b.png)


![Benzoic acid, 2-[methyl(phenylacetyl)amino]-, methyl ester](http://img.cochemist.com/ccimg/206400/206363-35-1.png)
![Benzoic acid, 2-[methyl(phenylacetyl)amino]-, methyl ester](http://img.cochemist.com/ccimg/206400/206363-35-1_b.png)
![Silane, [[(2E)-3-bromo-2-butenyl]oxy](1,1-dimethylethyl)diphenyl-](http://img.cochemist.com/ccimg/173100/173044-28-5.png)
![Silane, [[(2E)-3-bromo-2-butenyl]oxy](1,1-dimethylethyl)diphenyl-](http://img.cochemist.com/ccimg/173100/173044-28-5_b.png)














![Propanoic acid, 2-methyl-3-[(tetrahydro-2H-pyran-2-yl)oxy]-, methyl ester, (2S)-](/data/chemimg/2074700/96895-76-0.png)
![Propanoic acid, 2-methyl-3-[(tetrahydro-2H-pyran-2-yl)oxy]-, methyl ester, (2S)-](/data/chemimg/2074700/96895-76-0_b.png)
![1-Propanol, 2-methyl-3-[(tetrahydro-2H-pyran-2-yl)oxy]-, (2R)-](http://img.cochemist.com/ccimg/88800/88728-99-8.png)
![1-Propanol, 2-methyl-3-[(tetrahydro-2H-pyran-2-yl)oxy]-, (2R)-](http://img.cochemist.com/ccimg/88800/88728-99-8_b.png)









![Benzene, [[2-(2-methoxyethoxy)ethoxy]methyl]-](http://img.cochemist.com/ccimg/66300/66226-69-5.png)
![Benzene, [[2-(2-methoxyethoxy)ethoxy]methyl]-](http://img.cochemist.com/ccimg/66300/66226-69-5_b.png)


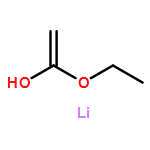
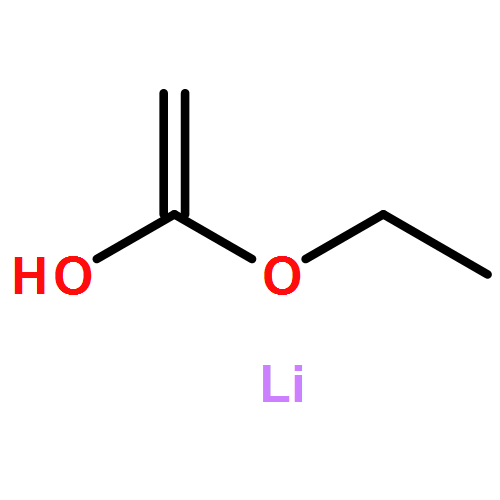

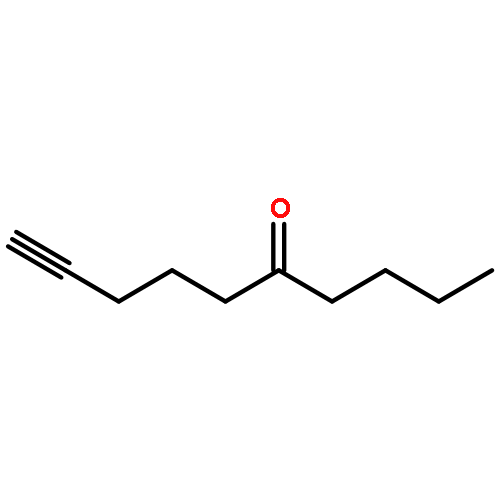
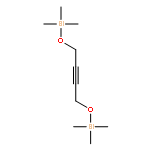


![Benzene, [[(1,1-dimethyl-2-propynyl)oxy]methyl]-](http://img.cochemist.com/ccimg/50500/50420-73-0.png)
![Benzene, [[(1,1-dimethyl-2-propynyl)oxy]methyl]-](http://img.cochemist.com/ccimg/50500/50420-73-0_b.png)
![Ethanol, 2,2'-[1,2-phenylenebis(oxy-2,1-ethanediyloxy)]bis-](/data/chemimg/2251300/41757-99-7.png)
![Ethanol, 2,2'-[1,2-phenylenebis(oxy-2,1-ethanediyloxy)]bis-](/data/chemimg/2251300/41757-99-7_b.png)




![1H-Benz[f]inden-4-ol, 2,3-dihydro-](http://img.cochemist.com/ccimg/27600/27532-59-8.png)
![1H-Benz[f]inden-4-ol, 2,3-dihydro-](http://img.cochemist.com/ccimg/27600/27532-59-8_b.png)




![Lithium, [(diphenylphosphinyl)methyl]-](http://img.cochemist.com/ccimg/23200/23182-99-2.png)
![Lithium, [(diphenylphosphinyl)methyl]-](http://img.cochemist.com/ccimg/23200/23182-99-2_b.png)
















![5,6-Dihydro[1,1'-biphenyl]-3(4H)-one](http://img.cochemist.com/ccimg/10400/10345-87-6.png)
![5,6-Dihydro[1,1'-biphenyl]-3(4H)-one](http://img.cochemist.com/ccimg/10400/10345-87-6_b.png)


![Benzenemethanol, a-[(2-methoxyethoxy)methyl]-](http://img.cochemist.com/ccimg/7000/6942-08-1.png)
![Benzenemethanol, a-[(2-methoxyethoxy)methyl]-](http://img.cochemist.com/ccimg/7000/6942-08-1_b.png)


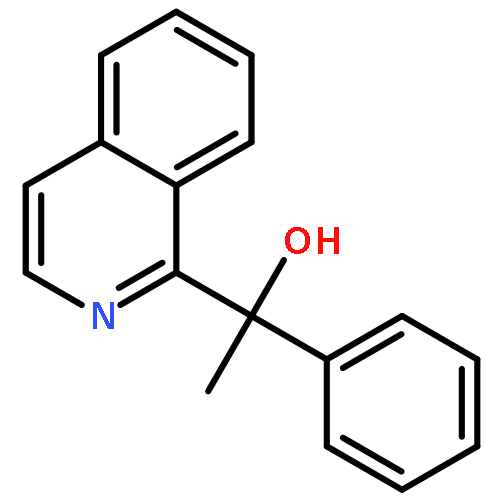


![Benzene, [(1-phenylethoxy)methyl]-](http://img.cochemist.com/ccimg/2100/2040-37-1.png)
![Benzene, [(1-phenylethoxy)methyl]-](http://img.cochemist.com/ccimg/2100/2040-37-1_b.png)