Co-reporter:Man-Rong Li, Jason P. Hodges, Maria Retuerto, Zheng Deng, Peter W. Stephens, Mark C. Croft, Xiaoyu Deng, Gabriel Kotliar, Javier Sánchez-Benítez, David Walker, and Martha Greenblatt
Chemistry of Materials 2016 Volume 28(Issue 9) pp:3148
Publication Date(Web):April 13, 2016
DOI:10.1021/acs.chemmater.6b00755
Transition-metal-only double perovskite oxides (A2BB′O6) are of great interest due to their strong and unusual magnetic interactions; only one compound, Mn2FeReO6, was reported in this category to date. Herein, we report the second transition-metal-only double perovskite, Mn2MnReO6, prepared at high pressure and temperature. Mn2MnReO6 crystallizes in a monoclinic P21/n structure, as established by synchrotron X-ray and powder neutron diffraction (PND) methods, with eight-coordinated A sites and rock-salt arrangement of the B and B′-site MnO6 and ReO6. Both the structural analysis and the X-ray absorption near edge spectroscopy results indicate mixed valence states of the B/B′-site in Mn2+2Mn2+/3+Re5+/6+O6. The magnetic and PND studies evidence an antiferromagnetic (AFM) transition at ∼110 K and a transition from a simple AFM to canted AFM with net ferromagnetic component at ∼50 K. The observed Efros–Shklovskii variable-range-hopping semiconducting behavior is attributed to the three (A-site Mn2+, B-site Mn2+/3+, and B′-site Re5+/6+) interpenetrating canted AFM lattices. Theoretical calculations demonstrate that the almost fully polarized Mn states in Mn2MnReO6 are driven away from the Fermi level by static on-site interactions and open a small gap, which is responsible for the insulating state in such a d-electron-rich system. These results provide insight of the electronic origin of the physical properties of Mn2MnReO6 with local electronic structure similar to that of Mn2FeReO6.
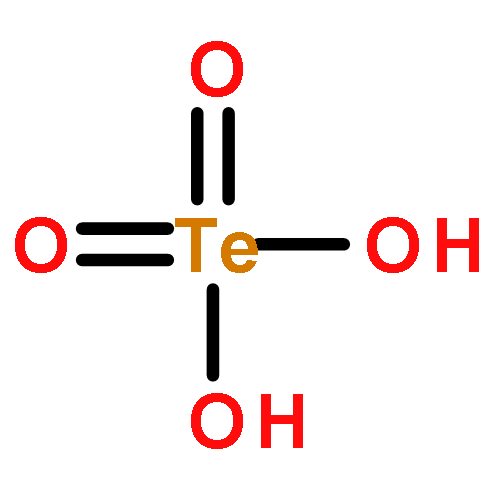
Co-reporter:Man-Rong Li, Zheng Deng, Saul H. Lapidus, Peter W. Stephens, Carlo U. Segre, Mark Croft, Robert Paria Sena, Joke Hadermann, David Walker, and Martha Greenblatt
Inorganic Chemistry 2016 Volume 55(Issue 20) pp:10135-10142
Publication Date(Web):September 28, 2016
DOI:10.1021/acs.inorgchem.6b01047
A novel 6H-type hexagonal perovskite Ba3(Cr0.97(1)Te0.03(1))2TeO9 was prepared at high pressure (6 GPa) and temperature (1773 K). Both transmission electron microscopy and synchrotron powder X-ray diffraction data demonstrate that Ba3(Cr0.97(1)Te0.03(1))2TeO9 crystallizes in P63/mmc with face-shared (Cr0.97(1)Te0.03(1))O6 octahedral pairs interconnected with TeO6 octahedra via corner-sharing. Structure analysis shows a mixed Cr2+/Cr3+ valence state with ∼10% Cr2+. The existence of Cr2+ in Ba3(Cr2+0.10(1)Cr3+0.87(1)Te6+0.03)2TeO9 is further evidenced by X-ray absorption near-edge spectroscopy. Magnetic properties measurements show a paramagnetic response down to 4 K and a small glassy-state curvature at low temperature. In this work, the octahedral Cr2+O6 component is stabilized in an oxide material for the first time; the expected Jahn–Teller distortion of high-spin (d4) Cr2+ is not found, which is attributed to the small proportion of Cr2+ (∼10%) and the face-sharing arrangement of CrO6 octahedral pairs, which structurally disfavor axial distortion.
Co-reporter:Dr. Man-Rong Li;Maria Retuerto; Peter W. Stephens; Mark Croft;Dr. Denis Sheptyakov;Vladimir Pomjakushin;Zheng Deng;Dr. Hirofumi Akamatsu;Venkatraman Gopalan;Dr. Javier Sánchez-Benítez;Felix O. Saouma; Joon I. Jang; David Walker; Martha Greenblatt
Angewandte Chemie International Edition 2016 Volume 55( Issue 34) pp:9862-9867
Publication Date(Web):
DOI:10.1002/anie.201511360
Abstract
Cationic rearrangement is a compelling strategy for producing desirable physical properties by atomic-scale manipulation. However, activating ionic diffusion typically requires high temperature, and in some cases also high pressure in bulk oxide materials. Herein, we present the cationic rearrangement in bulk Mn2FeMoO6 at unparalleled low temperatures of 150–300 oC. The irreversible ionic motion at ambient pressure, as evidenced by real-time powder synchrotron X-ray and neutron diffraction, and second harmonic generation, leads to a transition from a Ni3TeO6–type to an ordered-ilmenite structure, and dramatic changes of the electrical and magnetic properties. This work demonstrates a remarkable cationic rearrangement, with corresponding large changes in the physical properties in a bulk oxide at unprecedented low temperatures.
Co-reporter:Dr. Man-Rong Li;Maria Retuerto; Peter W. Stephens; Mark Croft;Dr. Denis Sheptyakov;Vladimir Pomjakushin;Zheng Deng;Dr. Hirofumi Akamatsu;Venkatraman Gopalan;Dr. Javier Sánchez-Benítez;Felix O. Saouma; Joon I. Jang; David Walker; Martha Greenblatt
Angewandte Chemie International Edition 2016 Volume 55( Issue 34) pp:
Publication Date(Web):
DOI:10.1002/anie.201683461
Co-reporter:Dr. Man-Rong Li;Maria Retuerto; Peter W. Stephens; Mark Croft;Dr. Denis Sheptyakov;Vladimir Pomjakushin;Zheng Deng;Dr. Hirofumi Akamatsu;Venkatraman Gopalan;Dr. Javier Sánchez-Benítez;Felix O. Saouma; Joon I. Jang; David Walker; Martha Greenblatt
Angewandte Chemie 2016 Volume 128( Issue 34) pp:10016-10021
Publication Date(Web):
DOI:10.1002/ange.201511360
Abstract
Cationic rearrangement is a compelling strategy for producing desirable physical properties by atomic-scale manipulation. However, activating ionic diffusion typically requires high temperature, and in some cases also high pressure in bulk oxide materials. Herein, we present the cationic rearrangement in bulk Mn2FeMoO6 at unparalleled low temperatures of 150–300 oC. The irreversible ionic motion at ambient pressure, as evidenced by real-time powder synchrotron X-ray and neutron diffraction, and second harmonic generation, leads to a transition from a Ni3TeO6–type to an ordered-ilmenite structure, and dramatic changes of the electrical and magnetic properties. This work demonstrates a remarkable cationic rearrangement, with corresponding large changes in the physical properties in a bulk oxide at unprecedented low temperatures.
Co-reporter:Dr. Man-Rong Li;Maria Retuerto; Peter W. Stephens; Mark Croft;Dr. Denis Sheptyakov;Vladimir Pomjakushin;Zheng Deng;Dr. Hirofumi Akamatsu;Venkatraman Gopalan;Dr. Javier Sánchez-Benítez;Felix O. Saouma; Joon I. Jang; David Walker; Martha Greenblatt
Angewandte Chemie 2016 Volume 128( Issue 34) pp:
Publication Date(Web):
DOI:10.1002/ange.201683461
Co-reporter:Maria Retuerto; Zhiping Yin; Thomas J. Emge; Peter W. Stephens; Man-Rong Li; Tapati Sarkar; Mark C. Croft; Alexander Ignatov; Z. Yuan; S. J. Zhang; Changqing Jin; Robert Paria Sena▽; Joke Hadermann▽; Gabriel Kotliar
Inorganic Chemistry 2015 Volume 54(Issue 3) pp:1066-1075
Publication Date(Web):December 9, 2014
DOI:10.1021/ic502400d
CsTlCl3 and CsTlF3 perovskites have been theoretically predicted to be superconductors when properly hole-doped. Both compounds have been previously prepared as pure compounds: CsTlCl3 in a tetragonal (I4/m) and a cubic (Fm3̅m) perovskite polymorph and CsTlF3 as a cubic perovskite (Fm3̅m). In this work, substitution of Tl in CsTlCl3 with Hg is reported, in an attempt to hole-dope the system and induce superconductivity. The whole series CsTl1–xHgxCl3 (x = 0.0, 0.1, 0.2, 0.4, 0.6, and 0.8) was prepared. CsTl0.9Hg0.1Cl3 is tetragonal as the more stable phase of CsTlCl3. However, CsTl0.8Hg0.2Cl3 is already cubic with the space group Fm3̅m and with two different positions for Tl+ and Tl3+. For x = 0.4 and 0.5, solid solutions could not be formed. For x ≥ 0.6, the samples are primitive cubic perovskites with one crystallographic position for Tl+, Tl3+, and Hg2+. All of the samples formed are insulating, and there is no signature of superconductivity. X-ray absorption spectroscopy indicates that all of the samples have a mixed-valence state of Tl+ and Tl3+. Raman spectroscopy shows the presence of the active Tl–Cl–Tl stretching mode over the whole series and the intensity of the Tl–Cl–Hg mode increases with increasing Hg content. First-principle calculations confirmed that the phases are insulators in their ground state and that Hg is not a good dopant in the search for superconductivity in this system.
Co-reporter:Dr. Man-Rong Li;Dr. Maria Retuerto;Zheng Deng;Dr. Peter W. Stephens;Dr. Mark Croft;Dr. Qingzhen Huang;Hui Wu;Xiaoyu Deng;Gabriel Kotliar;Dr. Javier Sánchez-Benítez;Dr. Joke Hadermann;Dr. David Walker; Martha Greenblatt
Angewandte Chemie 2015 Volume 127( Issue 41) pp:12237-12241
Publication Date(Web):
DOI:10.1002/ange.201506456
Abstract
The first transition-metal-only double perovskite compound, Mn2+2Fe3+Re5+O6, with 17 unpaired d electrons displays ferrimagnetic ordering up to 520 K and a giant positive magnetoresistance of up to 220 % at 5 K and 8 T. These properties result from the ferrimagnetically coupled Fe and Re sublattice and are affected by a two-to-one magnetic-structure transition of the Mn sublattice when a magnetic field is applied. Theoretical calculations indicate that the half-metallic state can be mainly attributed to the spin polarization of the Fe and Re sites.
Co-reporter:Dr. Man-Rong Li;Dr. Maria Retuerto;Zheng Deng;Dr. Peter W. Stephens;Dr. Mark Croft;Dr. Qingzhen Huang;Hui Wu;Xiaoyu Deng;Gabriel Kotliar;Dr. Javier Sánchez-Benítez;Dr. Joke Hadermann;Dr. David Walker; Martha Greenblatt
Angewandte Chemie International Edition 2015 Volume 54( Issue 41) pp:12069-12073
Publication Date(Web):
DOI:10.1002/anie.201506456
Abstract
The first transition-metal-only double perovskite compound, Mn2+2Fe3+Re5+O6, with 17 unpaired d electrons displays ferrimagnetic ordering up to 520 K and a giant positive magnetoresistance of up to 220 % at 5 K and 8 T. These properties result from the ferrimagnetically coupled Fe and Re sublattice and are affected by a two-to-one magnetic-structure transition of the Mn sublattice when a magnetic field is applied. Theoretical calculations indicate that the half-metallic state can be mainly attributed to the spin polarization of the Fe and Re sites.
Co-reporter:Man-Rong Li ; Peter W. Stephens ; Maria Retuerto ; Tapati Sarkar ; Christoph P. Grams ; Joachim Hemberger ; Mark C. Croft ; David Walker
Journal of the American Chemical Society 2014 Volume 136(Issue 24) pp:8508-8511
Publication Date(Web):May 19, 2014
DOI:10.1021/ja502774v
Polar oxides are technically of great interest but difficult to prepare. Our recent discoveries predicted that polar oxides can be synthesized in the corundum-derivative A2BB′O6 family with unusually small cations at the A-site and a d0 electron configuration ion at B′-site. When magnetic transition-metal ions are incorporated more interesting polar magnetic oxides can form. In this work we experimentally verified this prediction and prepared LiNbO3 (LN)-type polar magnetic Zn2FeTaO6 via high pressure and temperature synthesis. The crystal structure analysis indicates highly distorted ZnO6 and (Fe/Ta)O6 octahedra, and an estimated spontaneous polarization (PS) of ∼50 μC/cm2 along the c-axis was obtained from point charge model calculations. Zn2Fe3+Ta5+O6 has a lower magnetic transition temperature (TN ∼ 22 K) than the Mn2FeTaO6 analogue but is less conductive. The dielectric and polarization measurements indicate a potentially switchable component.
Co-reporter:Dr. Man-Rong Li;Maria Retuerto;Dr. David Walker;Tapati Sarkar;Dr. Peter W. Stephens;Dr. Swarnakamal Mukherjee;Tanusri Saha Dasgupta;Dr. Jason P. Hodges;Dr. Mark Croft;Dr. Christoph P. Grams;Joachim Hemberger;Dr. Javier Sánchez-Benítez;Ashfia Huq;Dr. Felix O. Saouma;Joon I. Jang
Angewandte Chemie 2014 Volume 126( Issue 40) pp:10950-10954
Publication Date(Web):
DOI:10.1002/ange.201406180
Abstract
Above-room-temperature polar magnets are of interest due to their practical applications in spintronics. Here we present a strategy to design high-temperature polar magnetic oxides in the corundum-derived A2BB′O6 family, exemplified by the non-centrosymmetric (R3) Ni3TeO6-type Mn2+2Fe3+Mo5+O6, which shows strong ferrimagnetic ordering with TC=337 K and demonstrates structural polarization without any ions with (n−1)d10ns0, d0, or stereoactive lone-pair electrons. Density functional theory calculations confirm the experimental results and suggest that the energy of the magnetically ordered structure, based on the Ni3TeO6 prototype, is significantly lower than that of any related structure, and accounts for the spontaneous polarization (68 μC cm−2) and non-centrosymmetry confirmed directly by second harmonic generation. These results motivate new directions in the search for practical magnetoelectric/multiferroic materials.
Co-reporter:Dr. Man-Rong Li;Maria Retuerto;Dr. David Walker;Tapati Sarkar;Dr. Peter W. Stephens;Dr. Swarnakamal Mukherjee;Tanusri Saha Dasgupta;Dr. Jason P. Hodges;Dr. Mark Croft;Dr. Christoph P. Grams;Joachim Hemberger;Dr. Javier Sánchez-Benítez;Ashfia Huq;Dr. Felix O. Saouma;Joon I. Jang
Angewandte Chemie International Edition 2014 Volume 53( Issue 40) pp:10774-10778
Publication Date(Web):
DOI:10.1002/anie.201406180
Abstract
Above-room-temperature polar magnets are of interest due to their practical applications in spintronics. Here we present a strategy to design high-temperature polar magnetic oxides in the corundum-derived A2BB′O6 family, exemplified by the non-centrosymmetric (R3) Ni3TeO6-type Mn2+2Fe3+Mo5+O6, which shows strong ferrimagnetic ordering with TC=337 K and demonstrates structural polarization without any ions with (n−1)d10ns0, d0, or stereoactive lone-pair electrons. Density functional theory calculations confirm the experimental results and suggest that the energy of the magnetically ordered structure, based on the Ni3TeO6 prototype, is significantly lower than that of any related structure, and accounts for the spontaneous polarization (68 μC cm−2) and non-centrosymmetry confirmed directly by second harmonic generation. These results motivate new directions in the search for practical magnetoelectric/multiferroic materials.
Co-reporter:M. Retuerto, T. Emge, J. Hadermann, P. W. Stephens, M. R. Li, Z. P. Yin, M. Croft, A. Ignatov, S. J. Zhang, Z. Yuan, C. Jin, J. W. Simonson, M. C. Aronson, A. Pan, D. N. Basov, G. Kotliar, and M. Greenblatt
Chemistry of Materials 2013 Volume 25(Issue 20) pp:4071
Publication Date(Web):September 18, 2013
DOI:10.1021/cm402423x
Recently, CsTlCl3 and CsTlF3 perovskites were theoretically predicted to be potential superconductors if they were optimally doped. The syntheses of these two compounds together with a complete characterization of the samples are reported. CsTlCl3 was obtained as orange crystals in two different polymorphs: a tetragonal phase (I4/m) and a cubic phase (Fm3̅m). CsTlF3 was formed as a light brown powder, and also as a double cubic perovskite (Fm3̅m). In all three CsTlX3 phases, Tl+ and Tl3+ were located in two different crystallographic positions that accommodate their different bond lengths. In CsTlCl3, some Tl vacancies were found in the Tl+ position. The charge ordering between Tl+ and Tl3+ was confirmed by X-ray absorption and Raman spectroscopy. The Raman spectroscopy of CsTlCl3 at high pressure (58 GPa) did not indicate any phase transition to a possible single Tl2+ state. However, the highly insulating material became less resistive with an increasing high pressure, while it underwent a change in its optical properties, from transparent to deeply opaque red, indicative of a decrease in the magnitude of the band gap. The theoretical design and experimental validation of the existence of CsTlF3 and CsTlCl3 cubic perovskites are the necessary first steps in confirming the theoretical prediction of superconductivity in these materials.Keywords: BaBiO3; charge order; CsAuCl3; CsTlCl3; CsTlF3; mixed valence; superconductivity;
Co-reporter:M. Retuerto ; M. R. Li ; A. Ignatov ; M. Croft ; K. V. Ramanujachary ; S. Chi ; J. P. Hodges ; W. Dachraoui ; J. Hadermann ; T. Thao Tran ; P. Shiv Halasyamani ; C. P. Grams ▽; J. Hemberger ▽;M. Greenblatt
Inorganic Chemistry 2013 Volume 52(Issue 21) pp:12482-12491
Publication Date(Web):October 18, 2013
DOI:10.1021/ic401491y
We have expanded the double perovskite family of materials with the unusual combination of layered order in the A sublattice and rock salt order over the B sublattice to compounds NaLaFeWO6 and NaNdFeWO6. The materials have been synthesized and studied by powder X-ray diffraction, neutron diffraction, electron diffraction, magnetic measurements, X-ray absorption spectroscopy, dielectric measurements, and second harmonic generation. At room temperature, the crystal structures of both compounds can be defined in the noncentrosymmetric monoclinic P21 space group resulting from the combination of ordering both in the A and B sublattices, the distortion of the cell due to tilting of the octahedra, and the displacement of certain cations. The magnetic studies show that both compounds are ordered antiferromagnetically below TN ≈ 25 K for NaLaFeWO6 and at ∼21 K for NaNdFeWO6. The magnetic structure of NaNdFeWO6 has been solved with a propagation vector k = (1/2 0 1/2) as an antiferromagnetic arrangement of Fe and Nd moments. Although the samples are potential multiferroics, the dielectric measurements do not show a ferroelectric response.
Co-reporter:Man-Rong Li, Maria Retuerto, Yong Bok Go, Thomas J. Emge, Mark Croft, Alex Ignatov, Kandalam V. Ramanujachary, Walid Dachraoui, Joke Hadermann, Mei-Bo Tang, Jing-Tai Zhao, Martha Greenblatt
Journal of Solid State Chemistry 2013 Volume 197() pp:543-549
Publication Date(Web):January 2013
DOI:10.1016/j.jssc.2012.07.038
Single crystals of Bi3Mn1.9Te1.1O11 were prepared from NaCl+KCl flux. This compound adopts KSbO3-type crystal structure as evidenced by electron and single crystal X-ray diffraction analysis. The three-dimensional channel structure is formed by corner-sharing octahedral (Mn0.63Te0.37)2O10 dimers and two identical (Bi1)4(Bi2)2 interpenetrating lattices. The intra-dimer Mn/Te–Mn/Te distances in Bi3Mn1.9Te1.1O11 are short and are consistent with weak metal–metal interactions. The mixed oxidation state of manganese and the edge-sharing octahedral features are confirmed by X-ray near edge absorption spectroscopy measurements, which indicate Bi3(MnIII1.1MnIV0.8)TeVI1.1O11 with 57.7% Mn3+ and 42.3% Mn4+. The partial substitution of Te for Mn perturbs long-range magnetic interactions, thereby destroying the ferromagnetic ordering found in Bi3Mn3O11 (TC=150 K).Single crystal of Bi3Mn1.9Te1.1O11 was grown from NaCl+KCl binary flux, suggesting that the high pressure Bi3Mn3O11 phase can be stabilized by partial substitution of Mn by Te at ambient pressure. Bi3Mn1.9Te1.1O11 adopts a typical three dimensional KSbO3-type crystal structure with three interpenetrating lattices and weak intra-dimmer metal–metal interaction caused by the d electrons of Mn. The edge-shared (Mn0.63Te0.37)2O10 octahedral dimer and mixed oxidation state of manganese (Bi3(MnIII1.1MnIV0.8)TeVI1.1O11 with 57.7% Mn3+ and 42.3% Mn4+) features were evidenced by X-ray absorption near edge spectroscopy. Compared with Bi3Mn3O11, the Te substituted Bi3Mn1.9Te1.1O11 relaxes the crystal structure, but destroys the long-range magnetic ordering and gives short-range magnetic ordering below 5 K.Highlights► High pressure Bi3Mn3O11 is stabilized by partial Te substitution at ambient pressure. ► New KSbO3-type Bi3Mn1.9Te1.1O11 single crystal was grown from binary flux. ► The presence of mixed oxidation state of manganese is evidenced by XANES study. ► The Te-substitution destroys the long-range magnetic ordering and relaxes the structure.
Co-reporter:M. Retuerto, M.-R. Li, Y.B. Go, A. Ignatov, M. Croft, K.V. Ramanujachary, R.H. Herber, I. Nowik, J.P. Hodges, W. Dachraoui, J. Hadermann, M. Greenblatt
Journal of Solid State Chemistry 2013 Volume 198() pp:246
Publication Date(Web):February 2013
DOI:10.1016/j.jssc.2012.09.038
Co-reporter:Dr. Man-Rong Li;Dr. David Walker;Maria Retuerto;Tapati Sarkar;Dr. Joke Hadermann;Dr. Peter W. Stephens;Dr. Mark Croft;Alexer Ignatov;Dr. Christoph P. Grams;Joachim Hemberger;Dr. Israel Nowik;Dr. P. Shiv Halasyamani;T. Thao Tran;Dr. Swarnakamal Mukherjee;Dr. Tanusri Saha Dasgupta
Angewandte Chemie International Edition 2013 Volume 52( Issue 32) pp:8406-8410
Publication Date(Web):
DOI:10.1002/anie.201302775
Co-reporter:Dr. Man-Rong Li;Dr. David Walker;Maria Retuerto;Tapati Sarkar;Dr. Joke Hadermann;Dr. Peter W. Stephens;Dr. Mark Croft;Alexer Ignatov;Dr. Christoph P. Grams;Joachim Hemberger;Dr. Israel Nowik;Dr. P. Shiv Halasyamani;T. Thao Tran;Dr. Swarnakamal Mukherjee;Dr. Tanusri Saha Dasgupta
Angewandte Chemie 2013 Volume 125( Issue 32) pp:8564-8568
Publication Date(Web):
DOI:10.1002/ange.201302775
Co-reporter:M. Retuerto, M.-R. Li, Y. B. Go, A. Ignatov, M. Croft, K. V. Ramanujachary, J. Hadermann, J. P. Hodges, R. H. Herber, I. Nowik, and M. Greenblatt
Inorganic Chemistry 2012 Volume 51(Issue 22) pp:12273-12280
Publication Date(Web):October 26, 2012
DOI:10.1021/ic301550m
SrFe0.75Mo0.25O3−δ has been recently discovered as an extremely efficient electrode for intermediate temperature solid oxide fuel cells (IT-SOFCs). We have performed structural and magnetic studies to fully characterize this multifunctional material. We have observed by powder neutron diffraction (PND) and transmission electron microscopy (TEM) that its crystal symmetry is better explained with a tetragonal symmetry (I4/mcm space group) than with the previously reported orthorhombic symmetry (Pnma space group). The temperature dependent magnetic properties indicate an exceptionally high magnetic ordering temperature (TN ∼ 750 K), well above room temperature. The ordered magnetic structure at low temperature was determined by PND to be an antiferromagnetic coupling of the Fe cations. Mössbauer spectroscopy corroborated the PND results. A detailed study, with X-ray absorption spectroscopy (XAS), in agreement with the Mössbauer results, confirmed the formal oxidation states of the cations to be mixed valence Fe3+/4+ and Mo6+.
Co-reporter:M. Retuerto, M.-R. Li, Y.B. Go, A. Ignatov, M. Croft, K.V. Ramanujachary, R.H. Herber, I. Nowik, J.P. Hodges, W. Dachraoui, J. Hadermann, M. Greenblatt
Journal of Solid State Chemistry 2012 Volume 194() pp:48-58
Publication Date(Web):October 2012
DOI:10.1016/j.jssc.2012.06.031
A series of perovskites Sr4−xLaxFe3ReO12 (x=0.0, 1.0, 2.0) has been prepared by wet chemistry methods. The structure analyses by powder X-ray and neutron diffraction and electron microscopy show that these compounds adopt simple perovskite structures without cation ordering over the B sites: tetragonal (I4/mcm) for x=0.0 and 1.0 and orthorhombic (Pbmn) for x=2.0. The oxidation states of the cations in the compound with x=0.0 appear to be Fe3+/4+ and Re7+ and decrease for both with La substitution as evidenced by X-ray absorption spectroscopy. All the compounds are antiferromagnetically ordered above room temperature, as demonstrated by Mössbauer spectroscopy and the magnetic structures, which were determined by powder neutron diffraction. The substitution of Sr by La strongly affects the magnetic properties with an increase of TN up to ∼750 K.Graphical abstractHigh resolution transmission electron microscopy image of Sr4−xLaxFe3ReO12 (x=2.0), showing twin domains. Fourier transforms are given of the areas indicated by the circles.Highlights► Sr4−xLaxFe3ReO12 (x=0.0, 1.0, 2.0) perovskites prepared by wet chemistry. ► PXD, PND, ED, indicate no cation ordering, I4/mcm) for x=0.0, 1.0, Pbmn for x=2. ► XAS show oxidation states Fe3+/4+ and Re7+; both decrease with increasing x. ► All order antiferromagnetically above RT, with highest TN ∼750 K.
Co-reporter:Graeme P. Gardner;Dr. Yong Bok Go;David M. Robinson;Paul F. Smith; Joke Hadermann; Artem Abakumov; Martha Greenblatt; G. Charles Dismukes
Angewandte Chemie International Edition 2012 Volume 51( Issue 7) pp:1616-1619
Publication Date(Web):
DOI:10.1002/anie.201107625
Co-reporter:Graeme P. Gardner;Dr. Yong Bok Go;David M. Robinson;Paul F. Smith; Joke Hadermann; Artem Abakumov; Martha Greenblatt; G. Charles Dismukes
Angewandte Chemie 2012 Volume 124( Issue 7) pp:1648-1651
Publication Date(Web):
DOI:10.1002/ange.201107625
Co-reporter:Walid Dachraoui, Tao Yang, Chang Liu, Graham King, Joke Hadermann, Gustaaf Van Tendeloo, Anna Llobet, and Martha Greenblatt
Chemistry of Materials 2011 Volume 23(Issue 9) pp:2398
Publication Date(Web):April 14, 2011
DOI:10.1021/cm200226u
The new compounds NaLaFeTaO6, NaLaFeNbO6, NaLaMnTaO6, and NaLaMnNbO6 have been synthesized and characterized with a combination of transmission electron microscopy, X-ray powder diffraction (XRPD), neutron powder diffraction (NPD), and magnetization measurements. Through electron microscopy study, a local layered order of the A-cations has been detected without the typical occurrence of rock salt order at the B-cation site. Satellite reflections in the electron diffraction related to the local layered order are not visible on the XRPD or NPD patterns. The occurrence of local layered order is supported by pair distribution function analysis, which also reveals the presence of uncorrelated displacements of the Nb and Ta cations. The octahedra are tilted according to the system a−b+a−, and the coordinates were refined from XRPD and NPD with a disordered cation distribution in the space group Pnma. The magnetic exchange interactions in NaLaFeTaO6 and NaLaFeNbO6 are antiferromagnetic, while they are ferromagnetic in NaLaMnTaO6 and NaLaMnNbO6. Long-range magnetic ordering is not observed down to 4 K for any of the compositions.Keywords: double perovskite; electron microscopy; layered ordering; magnetic;
Co-reporter:Tao Yang, Tyché Perkisas, Joke Hadermann, Mark Croft, Alexander Ignatov, Gustaaf Van Tendeloo and Martha Greenblatt
Journal of Materials Chemistry A 2011 vol. 21(Issue 1) pp:199-205
Publication Date(Web):26 Oct 2010
DOI:10.1039/C0JM02614J
LaAMnSnO6 (A = Sr, Ba) have been synthesized by high temperature solid-state reactions under dynamic 1% H2/Ar flow. Rietveld refinements on room temperature powder X-ray diffraction data indicate that LaSrMnSnO6 crystallizes in the GdFeO3-structure, with space groupPnma and, combined with transmission electron microscopy, LaBaMnSnO6 in Imma. Both space groups are common in disordered double-perovskites. The Mn3+ and Sn4+ ions whose valence states were confirmed by X-ray absorption spectroscopy, are completely disordered over the B-sites and the BO6 octahedra are slightly distorted. LaAMnSnO6 are ferromagnetic semiconductors with a TC = 83 K for the Sr- and 66 K for the Ba-compound. The title compounds, together with the previously reported LaCaMnSnO6 provide an interesting example of progression from Pnma to Imma as the tolerance factor increases. An analysis of the relationship between space group and tolerance factor for the series LaAMnMO6 (A = Ca, Sr, Ba; M = Sn, Ru) provides a better understanding of the symmetry determination for double perovskites.
Co-reporter:David M. Robinson ; Yong Bok Go ; Martha Greenblatt ;G. Charles Dismukes
Journal of the American Chemical Society 2010 Volume 132(Issue 33) pp:11467-11469
Publication Date(Web):July 30, 2010
DOI:10.1021/ja1055615
Nanocrystalline spinel LiMn2O4 has been prepared and treatment of LiMn2O4 with dilute nitric acid solution resulted in the delithiation of the framework, while maintaining the spinel structure, λ-MnO2. LiMn2O4 is not a catalyst for water oxidation. Upon removal of the lithium, the cubical Mn4O4 cores become active sites for oxidizing water to molecular oxygen, which was investigated with the photochemical [Ru2+(2,2′-bpy)3]/persulfate system at pH 5.8. The nanosize λ-MnO2 obtained from the nanocrystalline LiMn2O4, which was synthesized by the citrate route, shows a significantly higher water oxidation catalytic activity (Turnover Frequency: 3 × 10−5 mol O2/s/mol Mn) than that obtained via solid state reaction with micrometer and irregular particle sizes (Turnover Frequency: 5 × 10−6 mol O2/s/mol Mn).
Co-reporter:Tao Yang, Artem M. Abakumov, Joke Hadermann, Gustaaf Van Tendeloo, Israel Nowik, Peter W. Stephens, Joachim Hemberger, Alexander A. Tsirlin, Kandalam V. Ramanujachary, Samuel Lofland, Mark Croft, Alexander Ignatov, Junliang Sun and Martha Greenblatt
Chemical Science 2010 vol. 1(Issue 6) pp:751-762
Publication Date(Web):11 Oct 2010
DOI:10.1039/C0SC00348D
The most efficient use of spatial volume and the lowest potential energies in the metal oxide structures are based on cubic close packing (ccp) or hexagonal close packing (hcp) of anions with cations occupying the interstices. A promising way to tune the composition of close packed oxides and design new compounds is related to fragmenting the parent structure into modules by periodically spaced planar interfaces, such as twin planes at the unit cell scale. The unique crystal chemistry properties of cations with a lone electron pair, such as Bi3+ or Pb2+, when located at interfaces, enables them to act as “chemical scissors”, to help relieve configurational strain. With this approach, we synthesized a new oxide, BiMnFe2O6, where fragments of the hypothetical hcp oxygen-based MO structure (the NiAs structure type), for the first time, serve as the building modules in a complex transition metal oxide. Mn3+ and Fe3+ ions are randomly distributed in two crystallographically independent sites (M1 and M2). The structure consists of quasi two-dimensional blocks of the 2H hexagonal close packed MO structure cut along the (114) crystal plane of the hcp lattice and stacked along the c axis. The blocks are related by a mirror operation that allows BiMnFe2O6 to be considered as a polysynthetically twinned 2H hcp MO structure. The transition to an AFM state with an incommensurate spin configuration at ∼ 212 K is established by 57Fe Mössbauer spectroscopy, magnetic susceptibility, specific heat and low temperature powder neutron diffraction.
Co-reporter:Tao Yang, Junliang Sun, Jeongho Yeon, P. Shiv Halasyamani, Shiliang Huang, Joachim Hemberger and Martha Greenblatt
Chemistry of Materials 2010 Volume 22(Issue 16) pp:4814
Publication Date(Web):July 27, 2010
DOI:10.1021/cm101582j
The structure of the new polar oxide, Cd2InVO6, was determined by powder X-ray diffraction. Cd2InVO6 crystallizes in space group P31 with the unit cell parameters: a = 12.218 96(4), c = 9.256 75(4) Å. The Cd2+ and In3+ ions are statistically disordered in nine independent positions (M1−M9) with a certain level of site preference. M1−M3 form highly asymmetric oxygen-coordination polyhedra, which are similar to those formed by alkali or alkali-earth cations. M4−M9 are in distorted octahedral cavities. It is shown that M1−M3 are likely occupied mostly by Cd2+ while M4−M9 are extensively mixed by Cd2+ and In3+. The structure is best described as a framework of interconnected M4−M9 distorted octahedra with the M1−M3 polyhedra off-framework and the three independent VO4 tetrahedra filling the channels of the framework structure by corner-sharing with the MO6 octahedra. The polar framework is composed of M4O6∼M9O6 octahedra in a five-connected net with the topology nomination (33, 63, 94). The Bi3+-substituted compounds were also investigated with the rationale that the lone pair electrons of Bi3+ might enhance ferroelectricity. Single phase Cd1−xBix(Cd1+xIn1−x)VO6 forms limited solid solutions (0.02 ≤ x ≤ 0.14). Ferroelectricity was observed for neither the parent nor the Bi3+-substituted compounds, which suggest that the dipole moments are not switchable or too insignificant in magnitude. The powder second-harmonic generation measurements with 1064 nm radiation established that Cd1−xBix(Cd1+xIn1−x)VO6 are type-1 phase-matchable materials for x = 0, 0.14 with 70 and 90 times the efficiency of α-SiO2, respectively.
Co-reporter:Tao Yang, Mark Croft, Alexander Ignatov, Israel Nowik, Rihong Cong, and Martha Greenblatt
Chemistry of Materials 2010 Volume 22(Issue 21) pp:5876
Publication Date(Web):October 6, 2010
DOI:10.1021/cm1018053
Solid solutions of Ca1−δFe2−xMnxO4 (0.45 ≤ x ≤ 2) were synthesized from CaCl2 as flux at 850 °C in air. The entire series, even with x = 2, crystallizes in the CaFe2O4-type structure (Pnma), rather than in the CaMn2O4-type structure (Pbcm). Rietveld refinements confirmed mixed-valency Mn3+/Mn4+ and a substantial level of Ca2+ deficiency (δ ≈ 0.25) at high x. With increasing x, the unit-cell dimensions a and b decrease, while that of c increases. Detailed structural analyses, together with Mn K-edge X-ray absorption and 57Fe Mössbauer spectroscopy studies, revealed that the stabilization of CaFe2O4-type structure, even at high values of x, is due to the existence of non-Jahn−Teller active Mn4+ (and Fe3+), which is compensated by the formation of the Ca2+ deficiencies in the one-dimensional (1D) channels of Ca1−δFe2−xMnxO4 during the flux synthesis. Antiferromagnetic (AFM) long-range ordering is achieved for all compounds at low temperature, because of strong AFM interactions between Mn3+/Mn4+ and Fe3+. In addition, a spin (or cluster) glass component was also observed, as expected, because of the extensive Mn/Fe structural and Mn3+/Mn4+ charge disordering.
Co-reporter:Tao Yang, Tyché Perkisas, Joke Hadermann, Mark Croft, Alexander Ignatov, Martha Greenblatt
Journal of Solid State Chemistry 2010 Volume 183(Issue 11) pp:2689-2694
Publication Date(Web):November 2010
DOI:10.1016/j.jssc.2010.08.041
LaSrMnNbO6 has been synthesized by high temperature solid state reaction under 1% H2/Ar dynamic flow. The structure is determined by Rietveld refinement of the powder X-ray diffraction data. It crystallizes in the monoclinic space group P21/n with the unit cell parameters: a=5.69187(12), b=5.74732(10), c=8.07018(15) Å and β=90.0504(29)°, which were also confirmed by electron diffraction. The Mn2+ and Nb5+ ions, whose valence states are confirmed by X-ray absorption near-edge spectroscopy, are almost completely ordered over the B-site (<1% inversion) of the perovskite structure due to the large differences of both cationic size (0.19 Å) and charge. The octahedral framework displays significant tilting distortion according to Glazer’s tilt system a−b−c+. Upon heating, LaSrMnNbO6 decomposes at 690 °C under O2 flow or at 775 °C in air. The magnetic susceptibility data indicate the presence of long-range antiferromagnetic ordering at TN=8 K; the experimentally observed effective paramagnetic moment, μeff=5.76 μB for high spin Mn2+ (3d5, S=5/2) is in good agreement with the calculated value (μcalcd=5.92 μB).An ordered double perovskite, LaSrMnNbO6 has been synthesized in the monoclinic space group P21/n. The Mn2+ and Nb5+ ions, whose valence states are confirmed by X-ray absorption near-edge spectroscopy, are ordered over the B-site. The magnetic susceptibility data indicate long-range antiferromagnetic ordering at TN=8 K.
Co-reporter:Tao Yang, Junliang Sun, Mark Croft, Israel Nowik, Alexander Ignatov, Rihong Cong, Martha Greenblatt
Journal of Solid State Chemistry 2010 Volume 183(Issue 6) pp:1215-1220
Publication Date(Web):June 2010
DOI:10.1016/j.jssc.2010.03.019
Solid state solutions of Ca4Fe3−xMnxO8−δCl2 (0.92≤x≤1.79 (δ∼0.1) single crystals were synthesized in CaCl2-flux in air. The structure, determined by single-crystal X-ray diffraction, is related to the n=3 Ruddlesden–Popper phase in space group I4/mmm with strong deviations from the ideal structure. Mn and Fe are disordered over two transition metal sites. Due to the positional disordering of the equatorial oxygen atoms in the MO6 octahedra in Ca4Fe3−xMnxO8−δCl2 both tilting (∼9°) along the c-axis and rotation (∼10.5°) within the ab-plane are observed. All the Fe ions are trivalent, as confirmed by 57Fe Mössbauer spectroscopy and X-ray absorption near edge spectroscopy (XAS), while the formal valence state of Mn varies from very close to 4+ in the x=0.92 to mix-valent 3+/4+ in the x=1.79 member, as indicated by XAS. Magnetic investigations evidence short-range antiferromagnetic ordering already at room temperature and spin-glass behavior at low temperature due to the structural disordering of Mn/Fe.Structural analysis by the X-ray diffraction, the 57Fe Mössbauer and the X-ray absorption spectroscopy shows that all Fe ions are trivalent, the valence of Mn varies from ∼4+ to 3+/4+ with increasing x.
Co-reporter:Tapas Kumar Mandal, Mark Croft, Joke Hadermann, Gustaaf Van Tendeloo, Peter W. Stephens and Martha Greenblatt
Journal of Materials Chemistry A 2009 vol. 19(Issue 25) pp:4382-4390
Publication Date(Web):24 Apr 2009
DOI:10.1039/B823513A
The synthesis, electron diffraction (ED), synchrotron X-ray and neutron structure, X-ray absorption spectroscopy (XAS) and magnetic property studies of La2MnVO6 double perovskite are described. Analysis of the synchrotron powder X-ray diffraction data for La2MnVO6 indicates a disordered arrangement of Mn and V at the B-site of the perovskite structure. Absence of super-lattice reflections in the ED patterns for La2MnVO6 supports the disordered cation arrangement. Room temperature time-of-flight (TOF) neutron powder diffraction (NPD) data show no evidence of cation ordering, in corroboration with the ED and synchrotron studies (orthorhombic Pnma, a = 5.6097(3), b = 7.8837(5) and c = 5.5668(3) Å; 295 K, NPD). A comparison of XAS analyses of La2TVO6 with T = Ni and Co shows T2+ formal oxidation state while the T = Mn material evidences a Mn3+ admixture into a dominantly Mn2+ ground state. V-K edge measurements manifest a mirror image behavior with a V4+ state for T = Ni and Co with a V3+ admixture arising in the T = Mn material. The magnetic susceptibility data for La2MnVO6 show ferromagnetic correlations; the observed effective moment, µeff (5.72 µB) is much smaller than the calculated moment (6.16 µB) based on the spin-only formula for Mn2+ (d5, HS) /V4+ (d1), supportive of the partly oxidized Mn and reduced V scenario (Mn3+/V3+).
Co-reporter:Tapas Kumar Mandal, Artem M. Abakumov, Maxim V. Lobanov, Mark Croft, Viktor V. Poltavets and Martha Greenblatt
Chemistry of Materials 2008 Volume 20(Issue 14) pp:4653
Publication Date(Web):June 25, 2008
DOI:10.1021/cm800583e
The synthesis, structural characterization, and magnetic property studies of SrLaMnSbO6 double perovskite oxide are reported. The crystal structure of SrLaMnSbO6 has been solved by powder X-ray (PXD) and neutron diffraction (NPD) data in the monoclinic space group P21/n (a = 5.6878(3) Å, b = 5.6990(2) Å, c = 8.0499(4) Å and β = 89.98(2)°; 295 K, NPD data). The Mn and Sb atoms are nearly completely ordered over the B-site of the perovskite structure. The octahedral framework displays significant tilting distortion according to the Glazer’s tilt system a−a−c+. X-ray absorption near-edge spectroscopic (XAS) studies show the presence of Mn2+ and Sb5+ formal oxidation states. The magnetic susceptibility data of SrLaMnSbO6 indicate the presence of ferromagnetic correlations; the calculated effective paramagnetic moment, μcalcd = 5.92 μB (for HS Mn2+(3d5), S = 5/2; as evidenced by XAS data) is in good agreement with the value obtained experimentally (μexp = 5.70 μB). Variable temperature neutron diffraction data show no evidence of structural transition down to 3.7 K. A long-range antiferromagnetic ordering is established at TN = 8 K as evidenced by the magnetic susceptibility and specific heat measurements. The magnetic structure at 3.7 K is characterized by k = 0 propagation vector and m1x = −m2x, m1y = m2y = 0, m1z = −m2z (mx = 1.26(7) μB, mz = 1.82(6) μB) coupling of magnetic moments on the Mn1 (1/2,0,0) and Mn2 (0,1/2,1/2) atoms with the ordered magnetic moment of 2.21(4) μB.
Co-reporter:Tapas Kumar Mandal, Viktor V. Poltavets, Mark Croft, Martha Greenblatt
Journal of Solid State Chemistry 2008 Volume 181(Issue 9) pp:2325-2331
Publication Date(Web):September 2008
DOI:10.1016/j.jssc.2008.04.038
A2MnB′O6 (A=Ca, Sr; B=Sb, Ta) double perovskites have been synthesized and their structural and magnetic properties have been investigated. Rietveld refinement of the powder X-ray diffraction data for Sr2MnSbO6 indicated significant ordering of Mn and Sb at the B-site while all other phases showed mostly a random distribution of the B-site cations. X-ray absorption spectroscopic data established the presence of Mn in the 3+ and Sb/Ta in the 5+ oxidation states in all the phases. Magnetic susceptibility data indicated ferromagnetic correlations for all the A2MnB′O6 phases with Weiss temperatures varying from 64 to 107 K.The M vs. H plots for the ordered Sr2MnSbO6. The magnetization data at 5 K shows hysteresis loop (inset) with a Brillouin-like curvature indicating significant ferromagnetic correlations in the system.
Co-reporter:Tapas Kumar Mandal, Artem M. Abakumov, Joke Hadermann, Gustaaf Van Tendeloo, Mark Croft and Martha Greenblatt
Chemistry of Materials 2007 Volume 19(Issue 25) pp:6158
Publication Date(Web):November 13, 2007
DOI:10.1021/cm071840g
We report the synthesis, structural investigation, and magnetic property studies of Sr1.31Co0.63Mn0.37O3 that adopts an incommensurate composite hexagonal perovskite-related structure. The crystal structure has been solved using a (3 + 1)-dimensional superspace approach from powder X-ray and neutron diffraction data (SSG R3̅m(00γ)0s, a = 9.5548(1) Å, c = 2.5599(1) Å, q = 0.65581(4)c*, RB = 0.041, RP = 0.059). The structure consists of face-sharing chains of octahedra and trigonal prisms, wherein the trigonal prismatic sites are preferentially occupied by Co with some cation disorder. A combination of electron diffraction and high-resolution electron microscopic analysis has demonstrated that the compound possesses a complicated microstructure related to the formation of domains with slightly different lengths of the modulation vector. X-ray absorption near-edge spectroscopic (XAS) studies clearly indicate the presence of Mn in the 4+ and Co in the 3+ oxidation state. While the magnetic susceptibility data indicates the presence of antiferromagnetic correlations in the system, the calculation of effective paramagnetic moment (μcal = 3.561 μB), assuming the metal oxidation states as obtained from XAS and the cation distribution as obtained from neutron refinement, is in agreement with the value obtained experimentally (μexp = 3.676 μB).
Co-reporter:Qisheng Lin, Martha Greenblatt, El’ad N. Caspi, Maxim Avdeev
Journal of Solid State Chemistry 2006 Volume 179(Issue 7) pp:2086-2092
Publication Date(Web):July 2006
DOI:10.1016/j.jssc.2006.03.049
Powder neutron diffraction studies show that CaLaMnMoO6 double perovskite crystallizes in monoclinic P21/n , with a=5.56961(9)a=5.56961(9), b=5.71514(9)b=5.71514(9), c=7.9358(1)Å and β=90.043(1)°β=90.043(1)°. Mn and Mo occupy the 2c and 2d positions, respectively, with 6.0(4)% Mn/Mo anti-site mixing. Temperature-dependent magnetic susceptibility measurements reveal that CaLaMnMoO6 is ferrimagnetic, with TN=92(3) K, below which large magnetic frustration is detected. The zero-field magnetic moment measured at 5 K is about 1.2 μB, comparable to that of ALaMnMoO6 (A=Ba and Sr), but much lower than expected for antiparallel ordering of formally Mn2+ (d5) and Mo5+ (d1). Moreover, no long-range magnetic ordering is observed in neutron diffraction data down to 4 K. The magnetic frustration is discussed in the framework of nearest-neighbors next-nearest-neighbors magnetic frustration.Temperature-dependent magnetic susceptibility, χ and inverse susceptibility, 1/χ for the double perovskite, CaLaMnMoO6 at H=10kOe. Solid line is a fit of the ferrimagnetic modified Curie–Weiss model, 1/χ=T/C+1/χ0-σ/T-θ1/χ=T/C+1/χ0-σ/T-θ to the inverse susceptibility data.
Co-reporter:Qisheng Lin, Martha Greenblatt, Mark Croft
Journal of Solid State Chemistry 2005 Volume 178(Issue 5) pp:1356-1366
Publication Date(Web):May 2005
DOI:10.1016/j.jssc.2004.12.031
Powder neutron and X-ray diffraction studies show that the double perovskites in the region 0⩽x ⩽1 exhibit two crystallographic modifications at room temperature: monoclinic P21/nP21/n and tetragonal I4/mI4/m, with a boundary at 0.75
Co-reporter:Viktor V. Poltavets, Konstantin A. Lokshin, Martha Greenblatt
Solid State Sciences 2005 Volume 7(Issue 11) pp:1312-1316
Publication Date(Web):November 2005
DOI:10.1016/j.solidstatesciences.2005.05.001
An isothermal section of the Na0.3CoO2H2O system phase diagram at 22 °C from 11 to 100% relative humidity is presented. The superconducting Na0.3CoO2⋅1.2H2O phase is stable at a relative humidity (RH) higher than 30%; Na0.3CoO2⋅0.6H2O is the stable phase at RH below 30%. The unit cell parameters and temperature of superconducting transition of Na0.3CoO2⋅1.2H2O do not depend on relative humidity. The Na0.3CoO2⋅1.2H2O and Na0.3CoO2⋅0.6H2O hydrates are line phases and have a constant water content over the water vapor pressure range of their stability.
Co-reporter:Gabriel M. Veith, Maxim V. Lobanov, Thomas J. Emge, Martha Greenblatt, Mark Croft, Frank Stowasser, Joke Hadermann and Gustaaf Van Tendeloo
Journal of Materials Chemistry A 2004 vol. 14(Issue 10) pp:1623-1630
Publication Date(Web):15 Apr 2004
DOI:10.1039/B315028C
The new compounds Ln2FeMoO7
(Ln = Y, Dy, Ho) have been synthesized by solid-state reaction in evacuated silica tubes. The crystal structure of Dy2FeMoO7 and Ho2FeMoO7 was determined ab initio by simulated annealing in the R space group. Transmission electron microscopy study of Y2FeMoO7 revealed an unusual lamellar microstructure, composed of blocks with the structure of different zirconolite polymorphs. The dominant form corresponds to the zirconolite-2M type structure (space group C2/c) with a partially ordered array of Fe and Mo ions. X-Ray near edge spectroscopy data are consistent with formally Fe3+ and Mo5+ oxidation states for all compounds. Magnetic and transport measurements are reported.
Co-reporter:S. Dikmen, P. Shuk, M. Greenblatt, H. Gocmez
Solid State Sciences 2002 Volume 4(Issue 5) pp:585-590
Publication Date(Web):April 2002
DOI:10.1016/S1293-2558(02)01301-8
The structure, ionic and electronic conductivities of Ce1−xGdxO2−δ (x=0–0.30) solid solutions, prepared in a wide substitutional range for the first time hydrothermally, were investigated. The uniformly small particle size (41–68 nm) of the hydrothermally prepared materials allows sintering of the samples into highly dense ceramic pellets at 1300–1400 °C, a significantly lower temperature than 1600–1650 °C, which is required for ceria solid electrolytes prepared by solid state techniques. The solubility limit of Gd2O3 in CeO2 was determined to be over 30 mol %. The maximum conductivity, σ600 °C∼7.53×10−3 S cm−1 with Ea=0.58 eV, was found at x=0.25.Graphic
Co-reporter:Z. Zeng, I.D. Fawcett, M. Greenblatt, M. Croft
Materials Research Bulletin 2001 Volume 36(3–4) pp:705-715
Publication Date(Web):February–March 2001
DOI:10.1016/S0025-5408(01)00520-7
Sr2Cr1.2Mo0.8O6-δ (δ=0, 0.2) with a double perovskite structure was prepared by solid state reaction in evacuated quartz tubes. Cr and Mo are partially ordered on the B site. Oxygen vacancies decrease the ordering, but increase the lattice parameters. X-ray absorption spectroscopy results are consistent with Cr being 3+, and Mo being close to 5+ for δ = 0.2 and 5.5 for δ = 0. The spin of Cr3+ (d3) and Mo5+ (d1) order in an anti-parallel arrangement by superexchange interaction, and lead to ferrimagnetic ordering below 465 K. Both compounds are n-type narrow gap semiconductors. Large magnetoresistance (-43%) is observed in Sr2Cr1.2Mo0.8O6. The MR behavior is attributed to an intra-grain tunneling mechanism.
Co-reporter:G.M. Veith, M. Greenblatt, M. Croft, J.B. Goodenough
Materials Research Bulletin 2001 Volume 36(7–8) pp:1521-1530
Publication Date(Web):May–June 2001
DOI:10.1016/S0025-5408(01)00610-9
Sm2Mo2O3.83N3.17, the first molybdenum oxynitride pyrochlore, was synthesized by heating the Sm2Mo2O7 pyrochlore in flowing ammonia at 625°C for 24 hours. Sm2Mo2O3.83N3.17 forms with the cubic pyrochlore structure, space group Fd3m (a = 10.4975 Å). The sample is semiconducting and the temperature-dependent magnetic susceptibility follows Curie-Weiss behavior. X-ray absorption near-edge spectroscopy measurements indicate that in the oxynitride, molybdenum has a formal oxidation state significantly larger than 4+.
Co-reporter:G. Kakali, K.V. Ramanujachary, M. Greenblatt
Sensors and Actuators B: Chemical 2001 Volume 79(Issue 1) pp:58-62
Publication Date(Web):25 September 2001
DOI:10.1016/S0925-4005(01)00848-6
Single crystals of the alkali metal bronzes Na0.9Mo6O17, (Li0.5Na0.5)0.9Mo6O17, Li0.9Mo6O17 and Li0.33MoO3 were tested as Na+-ion sensors. It is shown that single crystals of Na0.9Mo6O17 fabricated into electrodes by direct solid-state contact can perform as Na+-selective electrodes at temperatures from ambient to 70°C. These sensors respond rapidly and linearly to changing Na+ concentration in the range 0.1–1 M and exhibit good stability versus time and temperature. The (Li0.5Na0.5)0.9Mo6O17 crystal was insensitive to changing Na+ concentration while the Li0.9Mo6O17 and Li0.33MoO3 showed nonlinear behavior.
Co-reporter:P Shuk, M Greenblatt, M Croft
Journal of Alloys and Compounds 2000 Volumes 303–304() pp:465-471
Publication Date(Web):24 May 2000
DOI:10.1016/S0925-8388(00)00627-7
The structure, thermal expansion coefficients and ionic and electronic conductivities of Ce1−xEuxO2−δ (x=0–0.50) solid solutions, prepared hydrothermally for the first time, were investigated. The uniformly small particle size (40–55 nm) of the hydrothermally prepared materials allows sintering of the samples into highly dense ceramic pellets at 1300–1400°C, a significantly lower temperature than 1600–1650°C, which is required for samples prepared by solid-state techniques. X-ray absorption spectroscopy (XAS) was used to establish the pure Eu3+ character of the europium ions; XAS also reflects local disorder effects. The highest conductivity was found for Ce0.85Eu0.15O1.925−δ (σ700°C=2.6×10−2 S/cm, Ea=0.66 eV) with predominantly oxide-ion mobility.
Co-reporter:Ian D. Fawcett, Gabriel M. Veith, Martha Greenblatt, Mark Croft, Israel Nowik
Solid State Sciences 2000 Volume 2(Issue 8) pp:821-831
Publication Date(Web):1 December 2000
DOI:10.1016/S1293-2558(00)01097-9
The SrMn1−xFexO3−δ (x=1/3, 1/2, 2/3) phases have been prepared and are shown by powder X-ray and neutron (for x=1/2) diffraction to adopt an ideal cubic perovskite structure with a disordered distribution of transition-metal cations over the six-coordinate B-site. Due to synthesis in air, the phases are oxygen deficient and formally contain both Fe3+ and Fe4+. Magnetic susceptibility data show an antiferromagnetic transition at 180 and 140 K for x=1/3 and 1/2, respectively and a spin-glass transition at 5, 25, 45 K for x=1/3, 1/2 and 2/3, respectively. The magnetic properties are explained in terms of super-exchange interactions between Mn4+, Fe(4+δ)+ and Fe(3+ε)+. The XAS results for the Mn-sites in these compounds indicate small Mn-valence changes, however, the Mn-pre-edge spectra indicate increased localization of the Mn-eg orbitals with Fe substitution. The Mössbauer results show the distinct two-site Fe(3+ε)+/Fe(4+δ)+ disproportionation in the Mn- substituted materials with strong covalency effects at both sites. This disproportionation is a very concrete reflection of a localization of the Fe-d states due to the Mn-substitution.
Co-reporter:K.V Ramanujachary, J.E Sunstrom IV, I Fawcett, P Shuk, M Greenblatt, M Croft, I Nowik, R.H Herber, S Khalid
Materials Research Bulletin 1999 Volume 34(Issue 5) pp:803-816
Publication Date(Web):15 March 1999
DOI:10.1016/S0025-5408(99)00063-X
Eu2VO4 with the K2NiF4-type structure was prepared by heating Eu2O3, V2O5, and V metal at 1473 K in an evacuated silica ampoule. Rietveld refinement of the powder X-ray diffraction (XRD) data confirmed the presence of layers of corner-shared VO6 octahedra with V–O bond distances of 1.928(1) Å (× 4) and 2.04(2) Å (× 2) perpendicular to the tetragonal c axis. X-ray absorption near-edge spectroscopy (XANES) indicated that the Eu is mixed-valent [Eu(II)/Eu(III)], while vanadium exists exclusively in the trivalent state. These valence assignments were supported by the results of temperature-dependent magnetic susceptibility and Mössbauer spectroscopy data. Variable temperature electrical resistivity measurements indicated that the sample was insulating (ρRT ≈ 104 Ω-cm). The compound undergoes a charge-ordering transition associated with the Eu sublattice at 445 K, as evidenced by Mössbauer spectroscopy, electrical resistivity, and in situ high temperature X-ray diffraction studies.
Co-reporter:Tapas Kumar Mandal, Mark Croft, Joke Hadermann, Gustaaf Van Tendeloo, Peter W. Stephens and Martha Greenblatt
Journal of Materials Chemistry A 2009 - vol. 19(Issue 25) pp:NaN4390-4390
Publication Date(Web):2009/04/24
DOI:10.1039/B823513A
The synthesis, electron diffraction (ED), synchrotron X-ray and neutron structure, X-ray absorption spectroscopy (XAS) and magnetic property studies of La2MnVO6 double perovskite are described. Analysis of the synchrotron powder X-ray diffraction data for La2MnVO6 indicates a disordered arrangement of Mn and V at the B-site of the perovskite structure. Absence of super-lattice reflections in the ED patterns for La2MnVO6 supports the disordered cation arrangement. Room temperature time-of-flight (TOF) neutron powder diffraction (NPD) data show no evidence of cation ordering, in corroboration with the ED and synchrotron studies (orthorhombic Pnma, a = 5.6097(3), b = 7.8837(5) and c = 5.5668(3) Å; 295 K, NPD). A comparison of XAS analyses of La2TVO6 with T = Ni and Co shows T2+ formal oxidation state while the T = Mn material evidences a Mn3+ admixture into a dominantly Mn2+ ground state. V-K edge measurements manifest a mirror image behavior with a V4+ state for T = Ni and Co with a V3+ admixture arising in the T = Mn material. The magnetic susceptibility data for La2MnVO6 show ferromagnetic correlations; the observed effective moment, µeff (5.72 µB) is much smaller than the calculated moment (6.16 µB) based on the spin-only formula for Mn2+ (d5, HS) /V4+ (d1), supportive of the partly oxidized Mn and reduced V scenario (Mn3+/V3+).
Co-reporter:Tao Yang, Artem M. Abakumov, Joke Hadermann, Gustaaf Van Tendeloo, Israel Nowik, Peter W. Stephens, Joachim Hemberger, Alexander A. Tsirlin, Kandalam V. Ramanujachary, Samuel Lofland, Mark Croft, Alexander Ignatov, Junliang Sun and Martha Greenblatt
Chemical Science (2010-Present) 2010 - vol. 1(Issue 6) pp:NaN762-762
Publication Date(Web):2010/10/11
DOI:10.1039/C0SC00348D
The most efficient use of spatial volume and the lowest potential energies in the metal oxide structures are based on cubic close packing (ccp) or hexagonal close packing (hcp) of anions with cations occupying the interstices. A promising way to tune the composition of close packed oxides and design new compounds is related to fragmenting the parent structure into modules by periodically spaced planar interfaces, such as twin planes at the unit cell scale. The unique crystal chemistry properties of cations with a lone electron pair, such as Bi3+ or Pb2+, when located at interfaces, enables them to act as “chemical scissors”, to help relieve configurational strain. With this approach, we synthesized a new oxide, BiMnFe2O6, where fragments of the hypothetical hcp oxygen-based MO structure (the NiAs structure type), for the first time, serve as the building modules in a complex transition metal oxide. Mn3+ and Fe3+ ions are randomly distributed in two crystallographically independent sites (M1 and M2). The structure consists of quasi two-dimensional blocks of the 2H hexagonal close packed MO structure cut along the (114) crystal plane of the hcp lattice and stacked along the c axis. The blocks are related by a mirror operation that allows BiMnFe2O6 to be considered as a polysynthetically twinned 2H hcp MO structure. The transition to an AFM state with an incommensurate spin configuration at ∼ 212 K is established by 57Fe Mössbauer spectroscopy, magnetic susceptibility, specific heat and low temperature powder neutron diffraction.
Co-reporter:Tao Yang, Tyché Perkisas, Joke Hadermann, Mark Croft, Alexander Ignatov, Gustaaf Van Tendeloo and Martha Greenblatt
Journal of Materials Chemistry A 2011 - vol. 21(Issue 1) pp:NaN205-205
Publication Date(Web):2010/10/26
DOI:10.1039/C0JM02614J
LaAMnSnO6 (A = Sr, Ba) have been synthesized by high temperature solid-state reactions under dynamic 1% H2/Ar flow. Rietveld refinements on room temperature powder X-ray diffraction data indicate that LaSrMnSnO6 crystallizes in the GdFeO3-structure, with space groupPnma and, combined with transmission electron microscopy, LaBaMnSnO6 in Imma. Both space groups are common in disordered double-perovskites. The Mn3+ and Sn4+ ions whose valence states were confirmed by X-ray absorption spectroscopy, are completely disordered over the B-sites and the BO6 octahedra are slightly distorted. LaAMnSnO6 are ferromagnetic semiconductors with a TC = 83 K for the Sr- and 66 K for the Ba-compound. The title compounds, together with the previously reported LaCaMnSnO6 provide an interesting example of progression from Pnma to Imma as the tolerance factor increases. An analysis of the relationship between space group and tolerance factor for the series LaAMnMO6 (A = Ca, Sr, Ba; M = Sn, Ru) provides a better understanding of the symmetry determination for double perovskites.
.png)


![[(sulfonatoperoxy)sulfonyl]oxidanide](http://img.cochemist.com/ccimg/15100/15092-81-6.png)
![[(sulfonatoperoxy)sulfonyl]oxidanide](http://img.cochemist.com/ccimg/15100/15092-81-6_b.png)