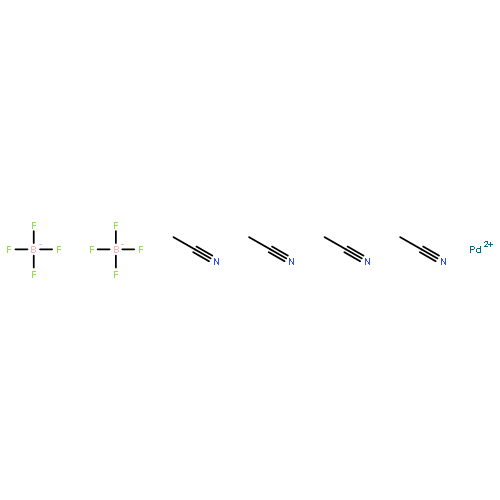Co-reporter:Evgueni G. Mednikov;Lawrence F. Dahl
Inorganic Chemistry 2015 Volume 54(Issue 3) pp:1145-1151
Publication Date(Web):November 26, 2014
DOI:10.1021/ic502470m
The monogold [(μ14-Au)Pd22(CO)20(PEt3)8]+ nanocation (2, with a [(CF3CO2)2H]− counterion) is shown to be a versatile precursor for the generation of three different neutral Au–Pd nanoclusters with double gold content in their distinctly dissimilar bimetallic architectures. These carbon monoxide (CO)-induced conversions are based on the reduction of AuI to Au0 that is controlled by the reaction medium. Under basic and acidic conditions, the known Au2Pd21(CO)20(PEt3)10 (3; >90% yield) and Au2Pd28(CO)26(PEt3)10 (4; ∼40% yield), respectively, were obtained, whereas neutral conditions gave rise to the new (μ12-Au)2Pd42(CO)30(PEt3)12 (1; ∼10–20% yield; all yields based on gold). The molecular structure of 1, established from a 100 K CCD X-ray diffraction study, consists of a five-layer hexoganol close-packed (hcp) Au2Pd42 framework of pseudo-D3h symmetry (crystallographic D3 site symmetry) of the Pd6/AuPd9/Pd12/AuPd9/Pd6 layer sequence, with the Au atoms centering two identical hcp (μ12-Au)Pd12 face-fused anti-cuboctahedral fragments. The 12 Et3-attached P atoms are coordinated to the triangular vertex Pd atoms in the four outer layers (except the middle Pd12); all five layers are stapled by interlayer bridging COs. The radial Aucent–Pd mean distance of 2.79 Å within the two symmetry-equivalent (μ12-Au)Pd12 anti-cuboctahedral fragments of 1 is identical with the radial Pdcent–Pd mean distances within hcp (μ12-Pd)Pd12 anti-cuboctahedral fragments of the two geometrically related nondistorted layered structures of Pd52(CO)36(PEt3)14 and [Ni9Pd33(CO)41(PPh3)6]4– ([PPh4]+ counterion), indicating a strain-free structural effect upon the substitution of Au for Pd in their analogous hcp layer-stacked arrangements. It provides prime evidence for an extension to 1 of our previous self-consistent experimental/theoretical-based hypothesis for delocalization of the 6s valence Au electrons in Au2Pd21 (3) and Au2Pd28 (4) toward a formal closed-shell Au+ configuration that is electronically equivalent to that of zerovalent Pd.
Co-reporter:Evgueni G. Mednikov, Sergei A. Ivanov, and Lawrence F. Dahl
Inorganic Chemistry 2015 Volume 54(Issue 13) pp:6157-6168
Publication Date(Web):May 6, 2015
DOI:10.1021/acs.inorgchem.5b00208
This first homopalladium carbido cluster, {Pd4(μ4-C)}Pd32(CO)28(PMe3)14 (1), was isolated (3–7% yields) from an ultimately simplified procedure—the reaction of CHCl3 under N2 with either Pd8(CO)8(PMe3)7 or Pd10(CO)12(PMe3)6 at room temperature. Charge-coupled device (CCD) X-ray diffraction data at 100 K for 1·2.5 C6H14 (1a) and 1·3 CHCl3 (1b) produced closely related molecular parameters for 1. This {Pd4C}Pd32 cluster (1) possesses a highly unusual tetracoordinated carbide atom that causes a major distortion of a central regular Pd4 tetrahedron into a new symmetry type of encapsulated Pd4 cage of pseudo-D2 (222) symmetry. Mean Pd–Pd distances for the three pairs of opposite twofold-equivalent Pd–Pd tetrahedral-like edges for 1a are 2.71, 2.96, and 3.59 Å; the mean of the four Pd–C distances [range, 1.87(2)–1.94(2) Å] is 1.91 Å. An astonishing molecular feature is that this {Pd4C}Pd32 cluster (1) is an isostructural and electronically equivalent analogue of the nanosized Au4Pd32(CO)28(PMe3)14 (2). Cluster 2, likewise a pseudo-D2 molecule, contains a geometrically analogous tetrahedrally deformed interior Au4 entity encapsulated within an identical Pd32(CO)28(PMe3)14 shell; mean distances for the three corresponding symmetry-equivalent pairs of slightly smaller opposite tetrahedral-distorted Au–Au edges are 2.64, 2.90, and 3.51 Å. A computational study by both a natural population analysis (NPA) and an atoms-in-molecules (AIM) method performed on model analogues {Pd4C}Pd32(CO)28(PH3)14 (1-mod) and Au4Pd32(CO)28(PH3)14 (2-mod) suggested that the negatively charged Au4 entity in 2-mod may be described as two weakly interacting electron-pair Au2 intradimers. In contrast, an NPA of the {Pd4C} entity in 1-mod revealed that two similarly oriented identical Pd2 intradimers of 2.71 Å are primarily stabilized by Pd–C bonding with a negatively charged carbide atom. The isostructural stabilizations of 1 and 2 are then attributed to the similar sizes, shapes, and overall negative charge distributions of the electronically equivalent interior {Pd4C} and Au4 entities. This resulting remarkable structural/electronic equivalency between 1 and 2 is consistent with the greatly improved performances of commercial palladium catalysts for vinyl acetate synthesis by gold-atom incorporation to suppress carbonization of the Pd atoms, namely, that the extra Au 6s1 valence electron of each added Au atom provides an effective “negative charge protection” against electron-donating carbon atoms forming Pd carbido species such as {Pd4C}.
Co-reporter:Dr. Evgueni G. Mednikov; Lawrence F. Dahl
Angewandte Chemie 2013 Volume 125( Issue 30) pp:7967-7971
Publication Date(Web):
DOI:10.1002/ange.201301982
Co-reporter:Dr. Evgueni G. Mednikov; Lawrence F. Dahl
Angewandte Chemie International Edition 2013 Volume 52( Issue 30) pp:7813-7817
Publication Date(Web):
DOI:10.1002/anie.201301982
Co-reporter:Evgueni G. Mednikov, Nicky Vo, Charles G. Fry, and Lawrence F. Dahl
Organometallics 2012 Volume 31(Issue 7) pp:2878-2886
Publication Date(Web):March 5, 2012
DOI:10.1021/om201150x
The new Tl(I)–Pd(0) cluster Pd9[μ3/3-Tl(acac)](μ2-CO)6(μ3-CO)3(PPh3)6 (1) was prepared in high yields (over 90%), both by reaction of Pd10(CO)12(PPh3)6 (4), PPh3, and TlPF6 in THF in the presence of acetylacetone (Hacac) and base (NEt3) and by direct reaction of Pd10(CO)12(PPh3)6 with PPh3 and Tl(acac). The composition and molecular structure of 1 were unambiguously established from 100 K CCD X-ray diffractometry studies of two solvated crystals, 1·1.5Hacac·0.5THF (1A) and 1·0.3THF (1B), which showed essentially identical geometries for the entire Pd9Tl(CO)9P6 fragment of pseudo-C3v symmetry; its composition is in agreement with X-ray Tl/Pd field-emission microanalysis with a scanning electron microscope for crystals of 1B. This cluster can be viewed as a markedly deformed Pd6 octahedron (oct) with the three Pd(oct) atoms of one of its eight triangular faces connected both by three edge-bridging wingtip (wt) Pd(μ2-CO)2PPh3 fragments and by a symmetrical capping Tl(I). Three triply bridging carbonyl ligands asymmetrically cap the lower alternate 3-fold-related triangular faces of the Pd6 octahedron, and the three other PPh3 ligands are each coordinated to Pd atoms in the geometrically opposite staggered Pd(oct)3 face. The 6s25d10 Tl(I) is also equivalently attached to both chelating O atoms of a bidentate acetylacetonate (acac) monoanion. Although the C2 axis of the pseudo-C2v planar Tl(acac) fragment is approximately parallel to the pseudo-C3 axis of the TlPd9 core, the orientation of the Tl(acac) plane relative to the octahedral-based Pd9 geometry is considerably different for each of the three independent nondisordered molecules of 1 in 1A and 1B; these different planar Tl(acac) orientations may be mainly attributed to anisotropic crystal-packing effects. Coordination of the Tl(I) atom to the three Pd(oct) atoms of the Pd9 core presumably occurs via its so-called “inert” 6s2 electron pair with resulting three short Tl–Pd(oct) connectivities of mean distance 2.83 Å; these connectivities together with three longer Tl–Pd(wt) ones of mean distance 3.15 Å give rise to a (crown-like)Pd6 sextuple (μ3/3-Tl) coordination mode. Of particular stereochemical interest is a comparison of solution behavior of 1 with that for the known structurally related analogue, Pd9[μ3-TlCo(CO)3L](μ2-CO)6(μ3-CO)3L6 (2) (with L = PEt3 instead of PPh3). In 2 the Tl(I) is alternatively attached to a trigonal-bipyramidal Co(CO)3L monoanion and primarily coordinated to the three inner Pd(oct) atoms of a similar PR3/CO-ligated octahedron; corresponding Tl–Pd(oct) and Tl–Pd(wt) mean distances for two independent molecules in 2 are 2.77 and 3.31 Å, respectively. Variable-temperature 31P{1H} NMR solution data of 1 indicate the occurrence of presumed fast wobbling-like motion of the [μ3/3-Tl(acac)] entity about the pseudo-C3 axis of the Pd9(μ2-CO)6(μ3-CO)3P6 fragment without Pd–Tl detachment (i.e., the entire cluster of 1 remains intact). In direct contrast, corresponding temperature-dependent 31P and 13C NMR data of 2 instead are consistent with rapid, reversible dissociation/association of the entire [μ3-TlCo(CO)3L] ligand from the analogous Pd9(μ2-CO)6(μ3-CO)3P6 fragment of 2. This highly dissimilar dynamic solution behavior that points to a stronger Tl(I) attachment to the Pd9 core in 1 than that in 2 may be attributed from the above crystallographic evidence to greater involvement of the outer three wingtip Pd(wt) atoms in bonding connectivities to the Tl(I) in 1 compared to predominant bonding connectivities of only the three inner Pd(oct) atoms to the Tl(I) in 2. 1H NMR solution spectra of 1 also suggest significant covalent character in the bidentate Tl–O(acac) bonding in 1 based upon the observation of H(acac)–Tl coupling; this premise is consistent with its Tl–O distances of 2.35 Å (av) being ca. 0.2 Å shorter than those of 2.52 Å (av) found in crystalline Tl(acac), which with no observed H–Tl NMR coupling in solution implies ionicity of its bidentate Tl–O bonding. Both 1 and 2 conform to an 86 CVE count expected for an octahedral metal polyhedron based upon the Tl(I) and each wingtip Pd(μ2-CO)2L fragment contributing 2 and 4 CVEs, respectively.
Co-reporter:Evgueni G. Mednikov ; Sergei A. Ivanov ;Lawrence F. Dahl
Inorganic Chemistry 2011 Volume 50(Issue 22) pp:11795-11806
Publication Date(Web):October 25, 2011
DOI:10.1021/ic201923y
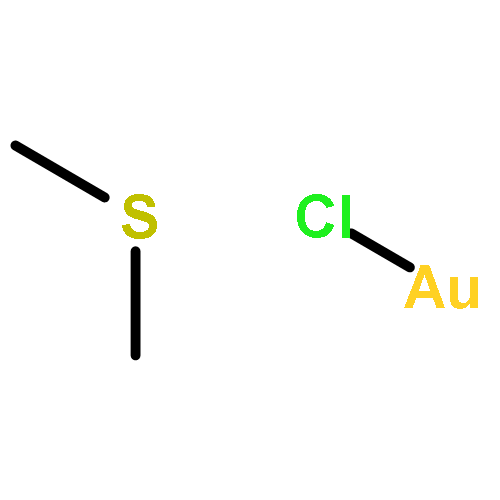
Initially isolated from Pd10(CO)12(PEt3)6 (5) and Au(SMe2)Cl precursors in a two-step carbon monoxide (CO)-involved procedure, the nanosized interpenetrating bicuboctahedral gold (Au)–palladium (Pd) Au2Pd28(CO)26(PEt3)10 (1) was then directly obtained in 25–30% yield from the CO-induced reaction of the CO-stable Au-centered cuboctahedral Au2Pd21(CO)20(PEt3)10 (3) with the structurally analogous CO-unstable Pd23(CO)20(PEt3)10 (4). Our hypothesis that this latter synthesis is initiated by the reaction of 3 with coordinatively unsaturated homopalladium species resulting from CO-induced fragmentation of 4 was subsequently substantiated by the alternatively designed synthesis of 1 (∼25% yield) from the CO-induced reaction of 3 with the structurally dissimilar CO-unstable Pd38(CO)28(PEt3)12 (6). The composition of 1, unambiguously established from a 100 K CCD X-ray diffractometry study, is in accordance with single-crystal X-ray Au–Pd field-emission microanalysis. The pseudo-C2h 30-atom Au2Pd28 geometry of 1 may be formally derived via substitution of the interior (μ12-Pd)2 moiety in the interpenetrating bicuboctahedral Pd20 kernel of the known isostructural Pd30(CO)26(PEt3)10 (2) with the corresponding interior (μ12-Au)2 moiety, in which the otherwise entire metal-core geometry and CO/PR3-ligated environment are essentially not altered. Of major significance is that this interior nonisovalent Pd-by-Au replacement in 2 produces CO-stable 1, whereas nanosized CO/PR3-ligated homopalladium Pdn clusters with n > 10 are generally unstable under CO. Because the two adjacent encapsulated Au atoms of 2.811(1) Å separation are not present on the metal surface, isolation of 1 under CO is ascribed to an electronic property. The virtually ideal geometrical site-occupancy fit between 1 and 2 provides definite crystallographic evidence for extensive delocalization in 1 of the two valence Au 6s electrons over the entire cluster (instead of a “localized” covalent Au–Au electron-pair interaction). Gradient-corrected (pseudo-scalar-relativistic) density functional theory (DFT) calculations were performed on the isostructural Au2Pd28(CO)26(PH3)10 (1-H) and Pd30(CO)26(PH3)10 (2-H) model clusters along with hypothetical [Au2Pd28(1-H)]2+ and [Pd30(2-H)]2– analogues (with phosphine ethyl substituents replaced by hydrogen ones). Natural population analysis of these four model clusters revealed similar highly positively charged metal surfaces of 28 Pd atoms relative to the two negatively charged interior metal atoms, which reflect a partially oxidized metal surface due to dominant CO back-bonding. The surprising observation that each less electronegative interior Pd atom in 2-H is more negatively charged by 0.30e than each interior Au atom in 1-H points to a more cationic Au in 1 than interior Pd in 2; this unexpected (opposite) charge difference is consistent with delocalization of each Au 6s valence electron toward a Au+ configuration. This premise is in agreement with the calculated Wiberg bond index (WBI) value of 0.055 for the Au–Au bond order in 1-H versus the WBI single-bond value of 1.01 obtained from analogous DFT calculations for the bare, neutral Au2 dimer, which has a much shorter spectroscopically determined gas-phase distance of 2.472 Å (that corresponds to a “localized” electron-pair interaction). Isolation of 1 under CO is of prime importance in nanoscience/nanotechnology in establishing relative stabilizations toward CO in well-defined CO/PEt3-ligated nonisovalent Pd2-by-Au2-substituted Au2Pdn–2 clusters [namely, n = 30 (1) and 23 (3)]. These important stereochemical implications have a direct relevance to the recent report of the higher tolerance to CO poisoning of highly active Au–Pd nanoparticle catalysts used for the complete conversion of formic acid into high-purity hydrogen (and CO2) for chemical hydrogen storage.
Co-reporter:Evgueni G. Mednikov;Nguyet T. Tran;Nicholas L. Aschbrenner
Journal of Cluster Science 2007 Volume 18( Issue 1) pp:253-269
Publication Date(Web):2007 March
DOI:10.1007/s10876-006-0103-8
Our synthetic exploratory efforts to obtain new nanosized Au–Pd carbonyl/phosphine clusters by use of the trimethylphosphine precursor Pd10(CO)12(PMe3)6 with smaller-sized PMe3 ligands (versus precursors with relatively larger PEt3 ligands) have previously produced via reaction with Au(SMe2)Cl an unusual Au4Pd32(CO)28(PMe3)14 (2) containing a pseudo-D2 36-atom Au4Pd32 core-geometry with a highly distorted encapsulated Au4 tetrahedron. Herein we report a striking illustration that analogous precursors under different reaction conditions have given rise to another new type of Au–Pd cluster, Au4Pd28(CO)22(PMe3)16 (1), that not only has a completely dissimilar Au4Pd28 core-geometry with a nearly regular encapsulated Au4 tetrahedron but also a totally different postulated multitwinned-composite growth-pattern. This extraordinary cluster, which was obtained from the reaction of Pd10(CO)12(PMe3)6 with Au(PPh3)Cl or Au(SMe2)Cl (estimated yield, ∼20–40%), has a 32-atom Au4Pd28 framework that roughly conforms to cubic T (23) symmetry that is maintained by inclusion of the 16 PMe3 P atoms but is completely reduced to general C1 (1) symmetry by inclusion of the 22 bridging CO ligands. 1 was isolated under different crystallization conditions to give two solvated crystal forms: namely, 1a as diisopropyl-solvated triclinic (\(\hbox{P}\bar 1 \) with Z = 2), and 1b as THF/hexane-solvated monoclinic (P2/c with Z = 4). A comparative analysis of resulting low-temperature CCD X-ray diffractometry determinations revealed an amazing molecular similarity between the actual shapes of the highly deformed Au4Pd28 architectures and ligand connectivities of 1 within the two dissimilar crystal structures. These results clearly indicate that the large observed localized geometrical distortions of 1 are primarily induced by intracluster strain-releasing effects and not by crystal-packing interactions. We propose a multitwinned growth-pattern of its Au4Pd28 core involving the formation of a Au4Pd24 composite-twinned framework formed from four markedly deformed interpenetrating three-layer Au-centered (Pd3)A(Au(n)Pd6)B(Au3)C cuboctahedra (n = 1–4) that are oriented along the four localized threefold axes of the Au4 tetrahedron. The other four outermost (external) Pd atoms that are tetrahedrally disposed about the Au4Pd24 composite presumably provide stabilization by face-condensations (i.e., three tetracapped, one tricapped). This new type of multitwinned bimetallic cluster has direct relevance to both ligated and non-ligated (bare) non-crystalline metal nanoparticles, of which many have been postulated to be multitwinned.
Co-reporter:Evgueni G. Mednikov Dr.;Sergei A. Ivanov Dr.;Irina V. Slovokhotova;Lawrence F. Dahl
Angewandte Chemie 2005 Volume 117(Issue 42) pp:
Publication Date(Web):25 OCT 2005
DOI:10.1002/ange.200502307
Einer geht noch: Die Strukturen eines Pd52- und eines Pd66-Nanoclusters haben als Gemeinsamkeit eine abgestumpfte ν-oktaedrische Pd38-Anordnung. Einer der 45 CO-Liganden von [Pd66(CO)45(PEt3)16] (Pd66-Kern abgebildet) ist überzählig. Um Platz für diesen 45. CO-Liganden zu schaffen, gehen zwei der 44 normalen Liganden eine Umlagerung ein, bei der die Anordnung der Metallzentren nicht grundlegend verändert wird.
Co-reporter:Evgueni G. Mednikov, Sergei A. Ivanov, Irina V. Slovokhotova,Lawrence F. Dahl
Angewandte Chemie International Edition 2005 44(42) pp:6848-6854
Publication Date(Web):
DOI:10.1002/anie.200502307