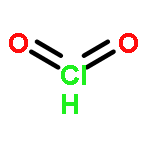
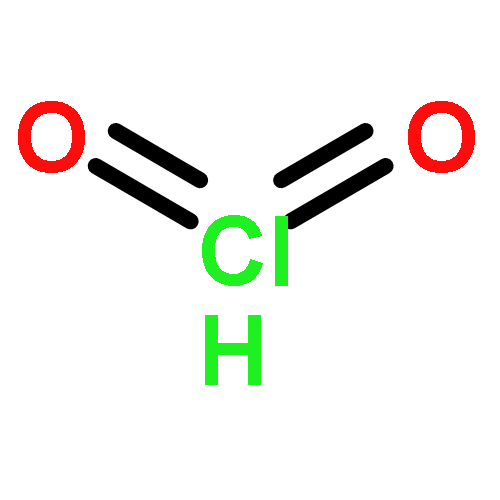
Co-reporter:Michael K. M. Ward;David M. Rowley
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 34) pp:23345-23356
Publication Date(Web):2017/08/30
DOI:10.1039/C7CP03854B
The kinetics of the reaction between gas phase BrO and HO2 radicals, BrO + HO2 → HOBr + O2 (1), have been studied over the atmospherically relevant temperature range T = 246–314 K and at ambient pressure, p = 760 ± 20 Torr, using laser flash photolysis coupled with ultraviolet absorption spectroscopy. The reaction was initiated by the generation of bromine monoxide radicals following laser photolytic generation of Br atoms from Br2/Cl2 containing mixtures and their reaction with ozone. Subsequently, the addition of methanol vapour to the reaction mixture, in the presence of excess oxygen, afforded the efficient simultaneous post-photolysis formation of HO2 radicals using well-defined chemistry. The decay of BrO radicals, in the presence and absence of HO2, was interrogated to determine the rate coefficients for the BrO + BrO and the BrO + HO2 reactions. A detailed sensitivity analysis was performed to ensure that the BrO + HO2 reaction was unequivocally monitored. The rate coefficient for reaction (1) is described by the Arrhenius expression: where statistical errors are 1σ. The negative temperature dependence of this reaction is in general accord with those reported by previous studies of this reaction. However, the present work reports greater absolute values for k1 than those of several previous studies. An assessment of previous laboratory studies of k1 is presented. This work confirms that reaction (1) plays a significant role in HOBr formation throughout the atmosphere following both anthropogenic, biogenic and volcanic emissions of brominated species. Reaction (1) therefore contributes to an efficient ozone depleting process in the atmosphere, and further confirms the significance of interactions between two different families of reactive atmospheric trace species.
Co-reporter:Michael K. M. Ward and David M. Rowley
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 8) pp:6301-6315
Publication Date(Web):02 Feb 2016
DOI:10.1039/C5CP07329D
The rate coefficient for the atmospherically important radical reaction: ClO + HO2 → Productswhich leads to ozone depletion, has been studied over the temperature range T = 210–298 K and at ambient pressure p = 760 ± 20 Torr. The reaction was studied using laser flash photolysis radical generation coupled with broadband charge coupled device absorption spectroscopy employing a two-dimensional charge-coupled-device (CCD) detection system. ClO radicals were generated following the photolysis of Cl2 and Cl2O gas mixtures diluted in nitrogen and oxygen. ClO radicals were monitored using broadband fingerprinting of their characteristic vibronic (A2Π ← X2Π) spectral structure, representing a definitive monitoring of this radical. Addition of hydroperoxy radical precursors to the gas mixture (methanol and oxygen) subsequently led to a competition for photolytically generated Cl atoms and a simultaneous prompt formation of both ClO and HO2 radicals. Detailed analysis and modelling of the radical production routes provided a degree of constraint into numerical integration simulations which were then used to interrogate and fit to ClO temporal profiles to extract the rate coefficient k1. The ambient temperature (T = 298 K) rate coefficient reported is k1 = (8.5 ± 1.5) × 10−12 cm3 molecule−1 s−1. The rate coefficient, k1, is described by the Arrhenius expression:where errors are 1σ statistical only. This significant rate coefficient is greater than previously reported, with a stronger negative temperature dependence than previously observed. Consequently this suggests that the contribution of reaction (1) to ozone loss, in particular at mid-latitudes might be currently underestimated in models. This work reports atmospheric pressure kinetic parameters for this reaction which are greater than those reported from low pressure studies, perhaps supporting ClO and HO2 association as predicted by previous theoretical studies of this process and highlighting the need for further pressure dependent experimental studies of the title reaction, which has been demonstrated here to be effective as an ozone loss process over a wide temperature range.
Co-reporter:Valerio Ferracci and David M. Rowley
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 3) pp:1182-1196
Publication Date(Web):04 Oct 2013
DOI:10.1039/C3CP53440E
The kinetics of the atmospherically important gas phase radical reaction between BrO and ClO have been studied over the temperature range T = 246–314 K by means of laser flash photolysis coupled with UV absorption spectroscopy. Charge-coupled-device (CCD) detection allowed simultaneous monitoring of both free radicals and the OClO product using ‘differential’ spectroscopy, which minimised interference from underlying UV absorbing species. In this way, the total rate coefficient for BrO + ClO → products (1) was measured, along with that for the OClO producing channel of this process BrO + ClO → OClO + Br (1c). These reaction rate coefficients are described by the Arrhenius expressions: k1/cm3 molecule−1 s−1 = (2.5 ± 2.2) × 10−12 exp[(630 ± 240)/T] and k1c/cm3 molecule−1 s−1 = (4.6 ± 3.0) × 10−12 exp[(280 ± 180)/T], where errors are 2σ, statistical only. An extensive sensitivity analysis was performed to quantify the potential additional systematic uncertainties in this work arising from uncertainties in secondary chemistry, absorption cross-sections and precursor concentrations. This analysis identified the reactions of initial and secondarily generated bromine atoms (specifically Br + O3 and Br + Cl2O) as particularly important, along with the reversible combination of ClO with OClO forming Cl2O3. Potential uncertainty in this latter process was used to define the lowest temperature of the present study. Results from this work indicate larger absolute values for k1 and k1c than those reported in previous studies, but a weaker negative temperature dependence for k1c than previously observed, resulting in a branching ratio for channel (1c) with a positive temperature dependence, in disagreement with previous studies. Reaction (1c) is the principal source of OClO in the polar stratosphere and is commonly used in atmospheric models as an indicator of stratospheric bromine chemistry. Thus these measurements might lead to a reinterpretation of modelled stratospheric OClO, which has also been suggested by previous comparisons of observations with atmospheric model studies.
Co-reporter:Valerio Ferracci, Kaori Hino and David M. Rowley
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 17) pp:7997-8007
Publication Date(Web):25 Mar 2011
DOI:10.1039/C0CP02847A
The BrO self-reaction, BrO + BrO → products (1), has been studied using laser flash photolysis coupled with UV absorption spectroscopy over the temperature range T = 266.5–321.6 K, under atmospheric pressure. BrO radicals were generated via laser photolysis of Br2 in the presence of excess ozone. Both BrO and O3 were monitored via UV absorption spectroscopy using charge-coupled device (CCD) detection. Simultaneous fitting to both temporal concentration traces allowed determination of the rate constant of the two channels of reaction (1), BrO + BrO → 2Br + O2 (1a); BrO + BrO → Br2 + O2 (1b), hence the calculation of the overall rate of reaction (1) and the branching ratio, α: k1a/cm3 molecule−1 s−1 = (1.92 ± 1.54) × 10−12 exp[(126 ± 214)/T], k1b/cm3 molecule−1 s−1 = (3.4 ± 0.8) × 10−13 exp[(181 ± 70)/T], k1/cm3 molecule−1 s−1 = (2.3 ± 1.5) × 10−12 exp(134 ± 185 /T) and α = k1a/k1 = (0.84 ± 0.09) exp[(−7 ± 32)/T]. Errors are 1σ, statistical only. Results from this work show a weaker temperature dependence of the branching ratio for channel (1a) than that found in previous work, leading to values of α at temperatures typical of the Polar Boundary Layer higher than those reported by previous studies. This implies a shift of the partitioning between the two channels of the BrO self-reaction towards the bromine atom and hence directly ozone-depleting channel (1a).
Co-reporter:Valerio Ferracci and David M. Rowley
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 37) pp:11596-11608
Publication Date(Web):02 Aug 2010
DOI:10.1039/C0CP00308E
Recent work by von Hobe et al. [Atmos. Chem. Phys., 2007, 7, 3055] has highlighted significant inconsistencies between laboratory results, theoretical calculations and field observations concerning the ClO dimer ozone destruction cycle. This work investigates the temperature dependence of the equilibrium constant of one of the key reactions in this cycle, ClO + ClO + M ⇆ Cl2O2 + M (1, −1), by means of laser flash photolysis and time-resolved UV absorption spectroscopy. ClO radicals were generated via laser flash photolysis of Cl2/Cl2O mixtures in synthetic air. Radicals were monitored via UV absorption spectroscopy: the use of a charge coupled device (CCD) detector allowed time resolution over a broad range of wavelengths giving unequivocal concentrations of radicals. The equilibrium constant Keq was determined as the ratio of the rate constants of the forward and reverse reactions (1, −1) over the temperature range T = 256.55–312.65 K. Second Law and Third Law thermodynamic methods were employed to determine the standard enthalpy and entropy changes of reaction (1), ΔrH° and ΔrS°, from the measured equilibrium constants. The values obtained from Second Law analysis were ΔrH° = − 80.7 ± 2.2 kJ mol−1 and ΔrS° = −168.1 ± 7.8 J K−1 mol−1. Third Law analysis gave ΔrH° = −74.65 ± 0.4 kJ mol−1 and ΔrS° = −148.0 ± 0.4 J K−1 mol−1. These values are in good agreement with previous work by Nickolaisen et al. [J. Phys. Chem., 1994, 98, 155] but greater in (negative) magnitude than current JPL-NASA recommendations [Sander et al., Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, JPL Publication 06-2, NASA Jet Propulsion Laboratory, Pasadena, 2006 (interim update to this reference, 2009)]. The discrepancy between the Second and Third Law analyses also agrees with Nickolaisen et al., possibly indicating an aspect of the ClO recombination reaction not yet fully elucidated. The atmospheric implications of the results and their impact on the current understanding on polar ozone depletion are briefly discussed.
Co-reporter:William J. Bloss, David M. Rowley, R. Anthony Cox and Roderic L. Jones
Physical Chemistry Chemical Physics 2002 vol. 4(Issue 15) pp:3639-3647
Publication Date(Web):17 Jun 2002
DOI:10.1039/B201653B
The kinetics of the BrO+HO2reaction (1) have been studied using the technique of flash photolysis/time resolved UV absorption spectroscopy. BrO and HO2 radicals were generated via the Br+O3 and Cl+CH3OH/O2 methods respectively. The rate coefficient for the reaction was found to be k1=(2.35±0.82)×10−11 molecule−1 cm3 s−1 at 298 K and 760 Torr O2. Uncertainty limits correspond to combined 2 standard deviation statistical variation and systematic factors. This result is briefly compared with previous determinations of k1. A study of the enhancement of the HO2 self-reaction rate in the presence of methanol
was also performed, with the observed second-order loss rate coefficient given by kobs=(2.9×10−12+αβ[CH3OH])/(1+α[CH3OH])2, with α=(6.15±0.90)×10−19 molecule−1 cm3 and β=(3.2±0.5)×10−11 molecule−1 cm3 s−1 at 298 K and 760 Torr O2, for [CH3OH]⩽5.5×1017 molecule cm−3.
Co-reporter:Michael K. M. Ward and David M. Rowley
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 8) pp:NaN6315-6315
Publication Date(Web):2016/02/02
DOI:10.1039/C5CP07329D
The rate coefficient for the atmospherically important radical reaction: ClO + HO2 → Productswhich leads to ozone depletion, has been studied over the temperature range T = 210–298 K and at ambient pressure p = 760 ± 20 Torr. The reaction was studied using laser flash photolysis radical generation coupled with broadband charge coupled device absorption spectroscopy employing a two-dimensional charge-coupled-device (CCD) detection system. ClO radicals were generated following the photolysis of Cl2 and Cl2O gas mixtures diluted in nitrogen and oxygen. ClO radicals were monitored using broadband fingerprinting of their characteristic vibronic (A2Π ← X2Π) spectral structure, representing a definitive monitoring of this radical. Addition of hydroperoxy radical precursors to the gas mixture (methanol and oxygen) subsequently led to a competition for photolytically generated Cl atoms and a simultaneous prompt formation of both ClO and HO2 radicals. Detailed analysis and modelling of the radical production routes provided a degree of constraint into numerical integration simulations which were then used to interrogate and fit to ClO temporal profiles to extract the rate coefficient k1. The ambient temperature (T = 298 K) rate coefficient reported is k1 = (8.5 ± 1.5) × 10−12 cm3 molecule−1 s−1. The rate coefficient, k1, is described by the Arrhenius expression:where errors are 1σ statistical only. This significant rate coefficient is greater than previously reported, with a stronger negative temperature dependence than previously observed. Consequently this suggests that the contribution of reaction (1) to ozone loss, in particular at mid-latitudes might be currently underestimated in models. This work reports atmospheric pressure kinetic parameters for this reaction which are greater than those reported from low pressure studies, perhaps supporting ClO and HO2 association as predicted by previous theoretical studies of this process and highlighting the need for further pressure dependent experimental studies of the title reaction, which has been demonstrated here to be effective as an ozone loss process over a wide temperature range.
Co-reporter:Valerio Ferracci and David M. Rowley
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 3) pp:NaN1196-1196
Publication Date(Web):2013/10/04
DOI:10.1039/C3CP53440E
The kinetics of the atmospherically important gas phase radical reaction between BrO and ClO have been studied over the temperature range T = 246–314 K by means of laser flash photolysis coupled with UV absorption spectroscopy. Charge-coupled-device (CCD) detection allowed simultaneous monitoring of both free radicals and the OClO product using ‘differential’ spectroscopy, which minimised interference from underlying UV absorbing species. In this way, the total rate coefficient for BrO + ClO → products (1) was measured, along with that for the OClO producing channel of this process BrO + ClO → OClO + Br (1c). These reaction rate coefficients are described by the Arrhenius expressions: k1/cm3 molecule−1 s−1 = (2.5 ± 2.2) × 10−12 exp[(630 ± 240)/T] and k1c/cm3 molecule−1 s−1 = (4.6 ± 3.0) × 10−12 exp[(280 ± 180)/T], where errors are 2σ, statistical only. An extensive sensitivity analysis was performed to quantify the potential additional systematic uncertainties in this work arising from uncertainties in secondary chemistry, absorption cross-sections and precursor concentrations. This analysis identified the reactions of initial and secondarily generated bromine atoms (specifically Br + O3 and Br + Cl2O) as particularly important, along with the reversible combination of ClO with OClO forming Cl2O3. Potential uncertainty in this latter process was used to define the lowest temperature of the present study. Results from this work indicate larger absolute values for k1 and k1c than those reported in previous studies, but a weaker negative temperature dependence for k1c than previously observed, resulting in a branching ratio for channel (1c) with a positive temperature dependence, in disagreement with previous studies. Reaction (1c) is the principal source of OClO in the polar stratosphere and is commonly used in atmospheric models as an indicator of stratospheric bromine chemistry. Thus these measurements might lead to a reinterpretation of modelled stratospheric OClO, which has also been suggested by previous comparisons of observations with atmospheric model studies.
Co-reporter:Valerio Ferracci, Kaori Hino and David M. Rowley
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 17) pp:NaN8007-8007
Publication Date(Web):2011/03/25
DOI:10.1039/C0CP02847A
The BrO self-reaction, BrO + BrO → products (1), has been studied using laser flash photolysis coupled with UV absorption spectroscopy over the temperature range T = 266.5–321.6 K, under atmospheric pressure. BrO radicals were generated via laser photolysis of Br2 in the presence of excess ozone. Both BrO and O3 were monitored via UV absorption spectroscopy using charge-coupled device (CCD) detection. Simultaneous fitting to both temporal concentration traces allowed determination of the rate constant of the two channels of reaction (1), BrO + BrO → 2Br + O2 (1a); BrO + BrO → Br2 + O2 (1b), hence the calculation of the overall rate of reaction (1) and the branching ratio, α: k1a/cm3 molecule−1 s−1 = (1.92 ± 1.54) × 10−12 exp[(126 ± 214)/T], k1b/cm3 molecule−1 s−1 = (3.4 ± 0.8) × 10−13 exp[(181 ± 70)/T], k1/cm3 molecule−1 s−1 = (2.3 ± 1.5) × 10−12 exp(134 ± 185 /T) and α = k1a/k1 = (0.84 ± 0.09) exp[(−7 ± 32)/T]. Errors are 1σ, statistical only. Results from this work show a weaker temperature dependence of the branching ratio for channel (1a) than that found in previous work, leading to values of α at temperatures typical of the Polar Boundary Layer higher than those reported by previous studies. This implies a shift of the partitioning between the two channels of the BrO self-reaction towards the bromine atom and hence directly ozone-depleting channel (1a).
Co-reporter:Valerio Ferracci and David M. Rowley
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 37) pp:NaN11608-11608
Publication Date(Web):2010/08/02
DOI:10.1039/C0CP00308E
Recent work by von Hobe et al. [Atmos. Chem. Phys., 2007, 7, 3055] has highlighted significant inconsistencies between laboratory results, theoretical calculations and field observations concerning the ClO dimer ozone destruction cycle. This work investigates the temperature dependence of the equilibrium constant of one of the key reactions in this cycle, ClO + ClO + M ⇆ Cl2O2 + M (1, −1), by means of laser flash photolysis and time-resolved UV absorption spectroscopy. ClO radicals were generated via laser flash photolysis of Cl2/Cl2O mixtures in synthetic air. Radicals were monitored via UV absorption spectroscopy: the use of a charge coupled device (CCD) detector allowed time resolution over a broad range of wavelengths giving unequivocal concentrations of radicals. The equilibrium constant Keq was determined as the ratio of the rate constants of the forward and reverse reactions (1, −1) over the temperature range T = 256.55–312.65 K. Second Law and Third Law thermodynamic methods were employed to determine the standard enthalpy and entropy changes of reaction (1), ΔrH° and ΔrS°, from the measured equilibrium constants. The values obtained from Second Law analysis were ΔrH° = − 80.7 ± 2.2 kJ mol−1 and ΔrS° = −168.1 ± 7.8 J K−1 mol−1. Third Law analysis gave ΔrH° = −74.65 ± 0.4 kJ mol−1 and ΔrS° = −148.0 ± 0.4 J K−1 mol−1. These values are in good agreement with previous work by Nickolaisen et al. [J. Phys. Chem., 1994, 98, 155] but greater in (negative) magnitude than current JPL-NASA recommendations [Sander et al., Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, JPL Publication 06-2, NASA Jet Propulsion Laboratory, Pasadena, 2006 (interim update to this reference, 2009)]. The discrepancy between the Second and Third Law analyses also agrees with Nickolaisen et al., possibly indicating an aspect of the ClO recombination reaction not yet fully elucidated. The atmospheric implications of the results and their impact on the current understanding on polar ozone depletion are briefly discussed.

![2-[2-[BIS(2-OXIDO-2-OXOETHYL)AMINO]ETHYL-(CARBOXYMETHYL)AMINO]ACETATE; IRON(3+)](http://img.cochemist.com/ccimg/17100/17099-81-9.png)
![2-[2-[BIS(2-OXIDO-2-OXOETHYL)AMINO]ETHYL-(CARBOXYMETHYL)AMINO]ACETATE; IRON(3+)](http://img.cochemist.com/ccimg/17100/17099-81-9_b.png)








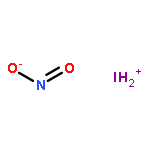
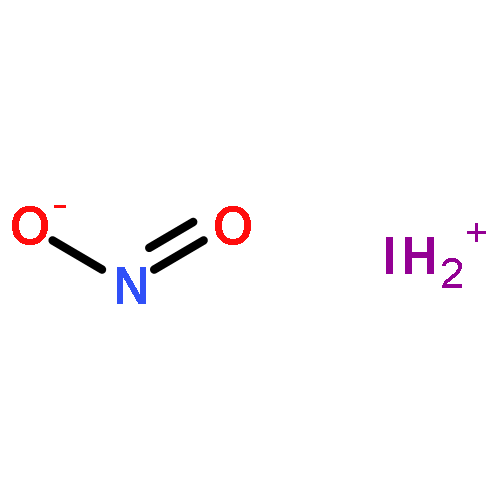


![2-{[(4-METHOXY-3,5-DIMETHYL-2-PYRIDINYL)METHYL]SULFINYL}-5-[(<SUP>2</SUP>H<SUB>3</SUB>)METHYLOXY]-1H-BENZIMIDAZOLE](http://img.cochemist.com/ccimg/7800/7791-21-1.png)
![2-{[(4-METHOXY-3,5-DIMETHYL-2-PYRIDINYL)METHYL]SULFINYL}-5-[(<SUP>2</SUP>H<SUB>3</SUB>)METHYLOXY]-1H-BENZIMIDAZOLE](http://img.cochemist.com/ccimg/7800/7791-21-1_b.png)