Co-reporter:Pierrik Lassalas; Bryant Gay; Caroline Lasfargeas; Michael J. James; Van Tran; Krishna G. Vijayendran; Kurt R. Brunden; Marisa C. Kozlowski; Craig J. Thomas; Amos B. SmithIII; Donna M. Huryn;Carlo Ballatore
Journal of Medicinal Chemistry 2016 Volume 59(Issue 7) pp:3183-3203
Publication Date(Web):March 11, 2016
DOI:10.1021/acs.jmedchem.5b01963
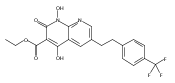
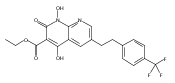
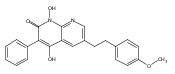
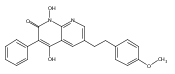
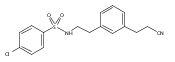
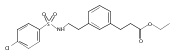
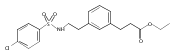
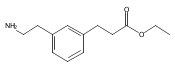
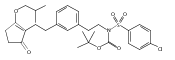
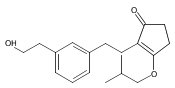
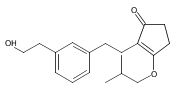
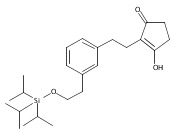
The replacement of a carboxylic acid with a surrogate structure, or (bio)-isostere, is a classical strategy in medicinal chemistry. The general underlying principle is that by maintaining the features of the carboxylic acid critical for biological activity, but appropriately modifying the physicochemical properties, improved analogs may result. In this context, a systematic assessment of the physicochemical properties of carboxylic acid isosteres would be desirable to enable more informed decisions of potential replacements to be used for analog design. Herein we report the structure–property relationships (SPR) of 35 phenylpropionic acid derivatives, in which the carboxylic acid moiety is replaced with a series of known isosteres. The data set generated provides an assessment of the relative impact on the physicochemical properties that these replacements may have compared to the carboxylic acid analog. As such, this study presents a framework for how to rationally apply isosteric replacements of the carboxylic acid functional group.
Co-reporter:Michael C. Myers, Parag P. Shah, Mary Pat Beavers, Andrew D. Napper, Scott L. Diamond, Amos B. Smith III, Donna M. Huryn
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 12) pp:3646-3651
Publication Date(Web):15 June 2008
DOI:10.1016/j.bmcl.2008.04.065
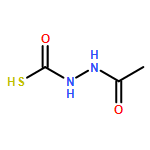
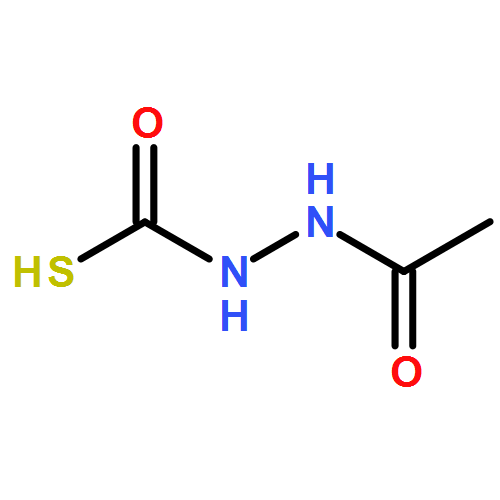
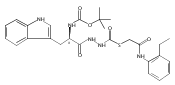
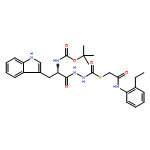
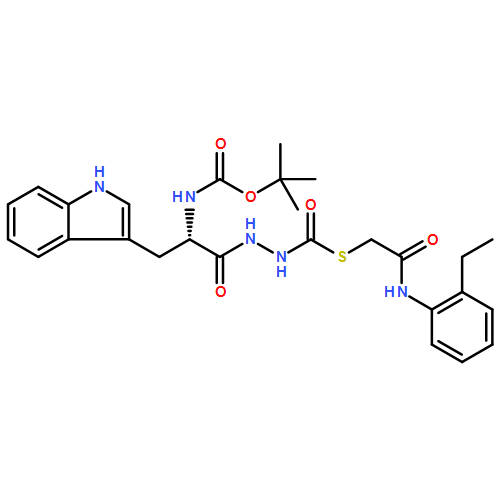
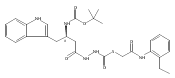
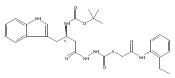
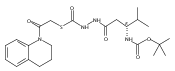
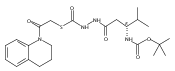
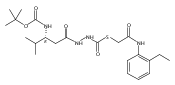
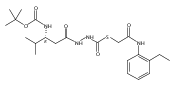
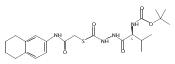
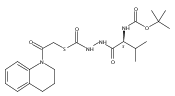
Recently, we identified a thiocarbazate that exhibits potent inhibitory activity against human cathepsin L. Since this structure represents a novel chemotype with potential for activity against the entire cysteine protease family, we designed, synthesized, and assayed a series of analogs to probe the mechanism of action, as well as the structural requirements for cathepsin L activity. Molecular docking studies using coordinates of a papain–inhibitor complex as a model for cathepsin L provided useful insights.A novel series of thiocarbazates was designed and synthesized to probe the structural requirements for cathepsin L inhibitory activity. These studies were guided by molecular docking studies using coordinates of a papain–inhibitor complex as a model for cathepsin L, and led to an understanding of the specific binding interactions as well as appropriate carbonyl reactivity required for potent activity. Furthermore, a highly potent inhibitor of cathepsin L (IC50 7 nM) was identified a result of these studies.
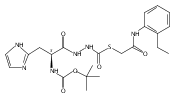
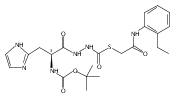
Co-reporter:Michael C. Myers, Parag P. Shah, Scott L. Diamond, Donna M. Huryn, Amos B. Smith III
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 1) pp:210-214
Publication Date(Web):1 January 2008
DOI:10.1016/j.bmcl.2007.10.107
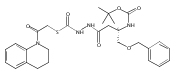
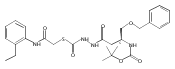
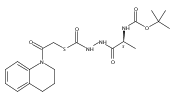
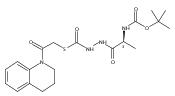
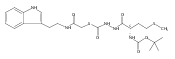
Library samples containing 2,5-disubstituted oxadiazoles were identified as potent hits in a high throughput screen (HTS) of the NIH Molecular Libraries Small Molecule Repository (MLSMR) directed at discovering inhibitors of cathepsin L. However, when synthesized in pure form, the putative actives were found to be devoid of biological activity. Analyses by LC–MS of original library samples indicated the presence of a number of impurities, in addition to the oxadiazoles. Synthesis and bioassay of the probable impurities led to the identification of a thiocarbazate that likely originated via ring opening of the oxadiazole. Previously unknown, thiocarbazates (−)-11 and (−)-12 were independently synthesized as single enantiomers and found to inhibit cathepsin L in the low nanomolar range.Library samples containing 2,5-disubstituted oxadiazoles were identified as potent hits in a high throughput screen (HTS) of the NIH Molecular Libraries Small Molecule Repository (MLSMR) directed at discovering inhibitors of cathepsin L. However, when synthesized in pure form, the putative actives were found to be devoid of biological activity. Analyses by LC–MS of original library samples indicated the presence of a number of impurities, in addition to the oxadiazoles. Synthesis and bioassay of the probable impurities led to the identification of a thiocarbazate that likely originated via ring opening of the oxadiazole. Previously unknown, thiocarbazates (−)-11 and (−)-12 were independently synthesized as single enantiomers and found to inhibit cathepsin L in the low nanomolar range.
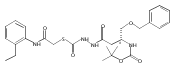
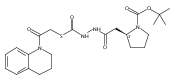
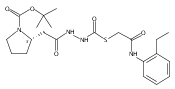
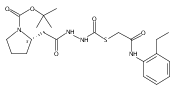
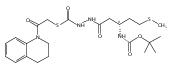
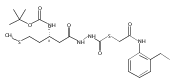
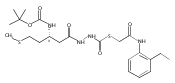
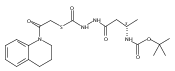
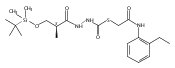
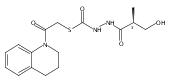
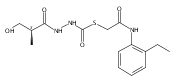
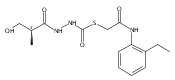
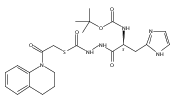
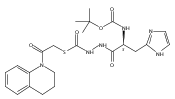
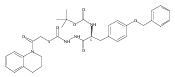
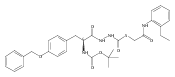
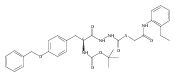
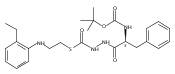
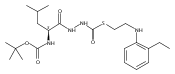
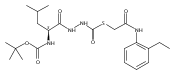
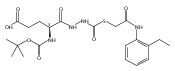
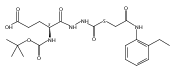
Co-reporter:Michael C. Myers, Andrew D. Napper, Nuzhat Motlekar, Parag P. Shah, Chun-Hao Chiu, Mary Pat Beavers, Scott L. Diamond, Donna M. Huryn, Amos B. Smith III
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 17) pp:4761-4766
Publication Date(Web):1 September 2007
DOI:10.1016/j.bmcl.2007.06.091
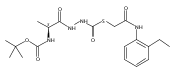
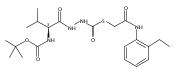
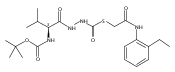
Substituted pyrazole esters were identified as hits in a high throughput screen (HTS) of the NIH Molecular Libraries Small Molecule Repository (MLSMR) to identify inhibitors of the enzyme cathepsin B. Members of this class, along with functional group analogs, were synthesized in an effort to define the structural requirements for activity. Analog characterization was hampered by the need to include a reducing agent such as dithiothreitol (DTT) or cysteine in the assay, highlighting the caution required in interpreting biological data gathered in the presence of such nucleophiles. Despite the confounding effects of DTT and cysteine, our studies demonstrate that the pyrazole 1 acts as alternate substrate for cathepsin B, rather than as an inhibitor.Substituted pyrazole esters were identified as hits in a high throughput screen (HTS) of the NIH Molecular Libraries Small Molecule Repository (MLSMR) to identify inhibitors of the enzyme cathepsin B. Members of this class, along with functional group analogs, were synthesized in an effort to define the structural requirements for activity. Analog characterization was hampered by the need to include a reducing agent such as dithiothreitol (DTT) or cysteine in the assay, highlighting the caution required in interpreting biological data gathered in the presence of such nucleophiles. Despite the confounding effects of DTT and cysteine, our studies demonstrate that the pyrazole 1 acts as alternative substrate for cathepsin B, rather than as an inhibitor.
.jpg)
![3-CYCLOBUTENE-1,2-DIONE, 3-HYDROXY-4-[(PHENYLMETHYL)AMINO]-](http://img.cochemist.com/ccimg/636700/636601-21-3.png)
![3-CYCLOBUTENE-1,2-DIONE, 3-HYDROXY-4-[(PHENYLMETHYL)AMINO]-](http://img.cochemist.com/ccimg/636700/636601-21-3_b.png)













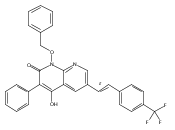
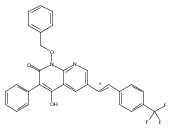
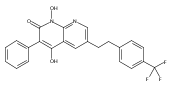
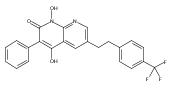


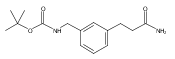
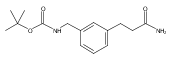

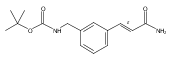
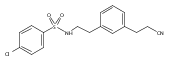
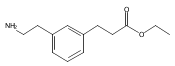
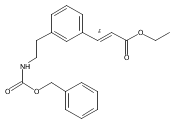
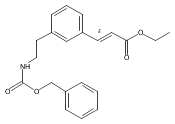

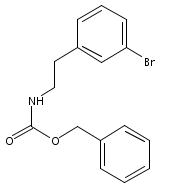

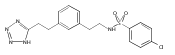
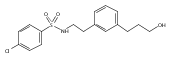
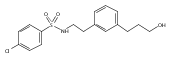
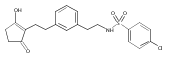
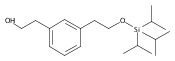
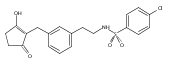
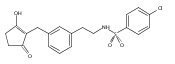
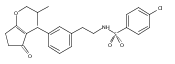
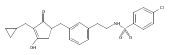
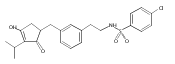
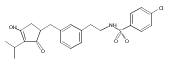
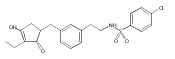
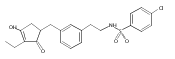
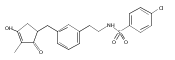
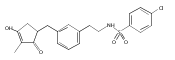

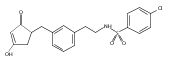



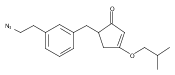
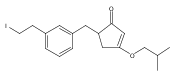
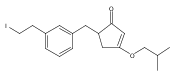
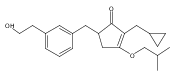
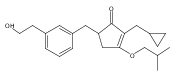
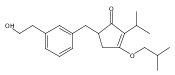
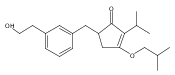
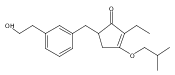






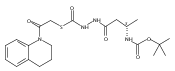
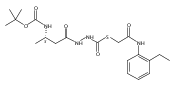
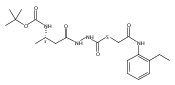
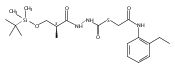

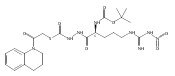
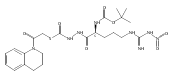
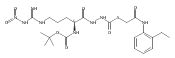
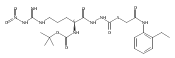
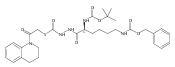
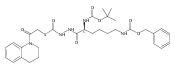
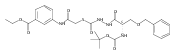
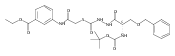

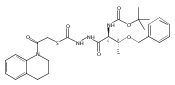
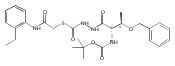
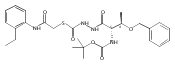
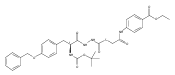


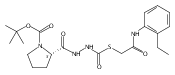
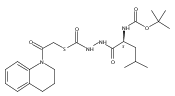
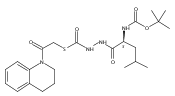
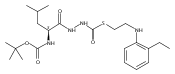




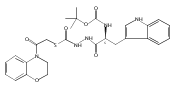
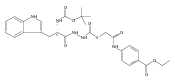
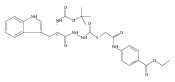

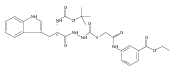
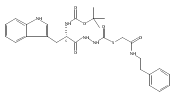
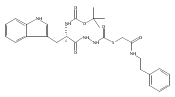
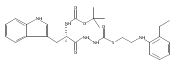
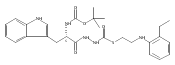





![4-[(3,5-dibromo-4-methoxyphenyl)methyl]-6-methyl-2-(methylamino)imidazo[4,5-d]azepin-5-one](http://img.cochemist.com/ccimg/634200/634151-15-8.png)
![4-[(3,5-dibromo-4-methoxyphenyl)methyl]-6-methyl-2-(methylamino)imidazo[4,5-d]azepin-5-one](http://img.cochemist.com/ccimg/634200/634151-15-8_b.png)










![2,3-Oxiranedicarboxamide,N-[(1S)-2-(dimethylamino)-2-oxo-1-(phenylmethyl)ethyl]-N'-[2-(2-pyridinyl)ethyl]-, (2R,3R)-](/data/chemimg/118000/215098-93-4.png)
![2,3-Oxiranedicarboxamide,N-[(1S)-2-(dimethylamino)-2-oxo-1-(phenylmethyl)ethyl]-N'-[2-(2-pyridinyl)ethyl]-, (2R,3R)-](/data/chemimg/118000/215098-93-4_b.png)