Co-reporter:Daisuke Koyama, Matthew J. Milner, and Andrew J. Orr-Ewing
The Journal of Physical Chemistry B October 5, 2017 Volume 121(Issue 39) pp:9274-9274
Publication Date(Web):September 12, 2017
DOI:10.1021/acs.jpcb.7b06917
The computationally predicted presence of two structurally distinct minima in the first triplet excited (T1) state of 2-thiouracil (2TU) is substantiated by sub-picosecond transient vibrational absorption spectroscopy (TVAS) in deuterated acetonitrile solution. Following 300 nm ultraviolet excitation to the second singlet excited state of 2TU, a transient infrared absorption band centered at 1643 cm–1 is observed within our minimum time resolution of 0.3 ps. It is assigned either to 2TU molecules in the S1 state or to vibrationally hot T1-state molecules, with the latter assignment more consistent with recent computational and experimental studies. The 1643 cm–1 band decays with a time constant of 7.2 ± 0.8 ps, and there is corresponding growth of several further bands centered at 1234, 1410, 1424, 1443, 1511, 1626, and 1660 cm–1 which show no decline in intensity over the 1 ns time limit of our measurements. These spectral features are assigned to two different conformations of 2TU, corresponding to separate energy minima on the T1-state potential energy surface, on the basis of their extended lifetimes, computed infrared frequencies, and the observed quenching of the bands by addition of styrene. Corresponding measurements for the 4-thiouracil (4TU) isomer show sub-picosecond population of the T1 state, which vibrationally cools with a time constant of 5.2 ± 0.6 ps. However, TVAS measurements in the carbonyl stretching region do not distinguish the two computed T1-state conformers of 4TU because of the similarity of their vibrational frequencies.
Co-reporter:Katharina Röttger, Hugo J. B. Marroux, Arsène F. M. Chemin, Emma Elsdon, Thomas A. A. Oliver, Steven T. G. Street, Alexander S. Henderson, M. Carmen Galan, Andrew J. Orr-Ewing, and Gareth M. Roberts
The Journal of Physical Chemistry B May 4, 2017 Volume 121(Issue 17) pp:4448-4448
Publication Date(Web):April 10, 2017
DOI:10.1021/acs.jpcb.7b02679
Transient electronic and vibrational absorption spectroscopies have been used to investigate whether UV-induced electron-driven proton transfer (EDPT) mechanisms are active in a chemically modified adenine–thymine (A·T) DNA base pair. To enhance the fraction of biologically relevant Watson–Crick (WC) hydrogen-bonding motifs and eliminate undesired Hoogsteen structures, a chemically modified derivative of A was synthesized, 8-(tert-butyl)-9-ethyladenine (8tBA). Equimolar solutions of 8tBA and silyl-protected T nucleosides in chloroform yield a mixture of WC pairs, reverse WC pairs, and residual monomers. Unlike previous transient absorption studies of WC guanine–cytosine (G·C) pairs, no clear spectroscopic or kinetic evidence was identified for the participation of EDPT in the excited-state relaxation dynamics of 8tBA·T pairs, although ultrafast (sub-100 fs) EDPT cannot be discounted. Monomer-like dynamics are proposed to dominate in 8tBA·T.
Co-reporter:Daisuke Koyama;Paul M. Donaldson
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 20) pp:12981-12991
Publication Date(Web):2017/05/24
DOI:10.1039/C7CP01784G
The mechanism of the thiol–ene reaction induced by 330 nm ultraviolet excitation of 1,2-di(quinolin-2-yl)disulfide (QSSQ) in the presence of methyl methacrylate (MMA) is investigated by sub-picosecond to microsecond transient absorption spectroscopy. The measurements, spanning more than seven orders of magnitude of time, directly reveal multiple radical reaction steps. The ground state quinoliene-2-thiyl radical (QS) is formed with a time constant of ∼200 fs by photolysis of QSSQ, followed by (64 ± 1)% decay of the initially formed QS radical because of solvent cage induced geminate recombination and QS dimer formation with a rate coefficient of (3.4 ± 0.2) × 1010 M−1 s−1 in methanol solution. In MMA solution, the carbon centered radical QS-MMA forms with a bimolecular reaction rate coefficient of (2.8 ± 0.2) × 107 M−1 s−1. The distinct infrared band at 1653 cm−1 assigned to the CO stretch mode of the QS-MMA radical decays rapidly in aerated solution, in contrast to observations in a solution purged of O2 by N2 bubbling. This decay is attributed to reaction of the QS-MMA radicals with molecular oxygen, producing peroxy radicals. Kinetic analysis of the intensity of the band at 1653 cm−1 reveals a bimolecular reaction rate coefficient of (3.3 ± 0.3) × 109 M−1 s−1 for the reaction of the QS-MMA radicals with molecular oxygen, and indicates that this reaction step is reversible.
Co-reporter:Michael P. Grubb;Philip M. Coulter;Hugo J. B. Marroux;Michael N. R. Ashfold
Chemical Science (2010-Present) 2017 vol. 8(Issue 4) pp:3062-3069
Publication Date(Web):2017/03/28
DOI:10.1039/C6SC05234G
We present a systematic study of the mode-specific vibrational relaxation of NO2 in six weakly-interacting solvents (perfluorohexane, perfluoromethylcyclohexane, perfluorodecalin, carbon tetrachloride, chloroform, and d-chloroform), chosen to elucidate the dominant energy transfer mechanisms in the solution phase. Broadband transient vibrational absorption spectroscopy has allowed us to extract quantum state-resolved relaxation dynamics of the two distinct NO2 fragments produced from the 340 nm photolysis of N2O4 → NO2(X) + NO2(A) and their separate paths to thermal equilibrium. Distinct relaxation pathways are observed for the NO2 bending and stretching modes, even at energies as high as 7000 cm−1 above the potential minimum. Vibrational energy transfer is governed by different interaction mechanisms in the various solvent environments, and proceeds with timescales ranging from 20–1100 ps. NO2 relaxation rates in the perfluorocarbon solvents are identical despite differences in acceptor mode state densities, infrared absorption cross sections, and local solvent structure. Vibrational energy is shown to be transferred to non-vibrational solvent degrees of freedom (V-T) through impulsive collisions with the perfluorocarbon molecules. Conversely, NO2 relaxation in chlorinated solvents is reliant on vibrational resonances (V-V) while V-T energy transfer is inefficient and thermal excitation of the surrounding solvent molecules inhibits faster vibrational relaxation through direct complexation. Intramolecular vibrational redistribution allows the symmetric stretch of NO2 to act as a gateway for antisymmetric stretch energy to exit the molecule. This study establishes an unprecedented level of detail for the cooling dynamics of a solvated small molecule, and provides a benchmark system for future theoretical studies of vibrational relaxation processes in solution.
Co-reporter:Dr. Hugo J. B. Marroux;Dr. Basile F. E. Curchod;Charly A. Faradji;Dr. Timothy A. Shuttleworth;Dr. Hazel A. Sparkes; Paul G. Pringle; Andrew J. Orr-Ewing
Angewandte Chemie 2017 Volume 129(Issue 44) pp:13901-13904
Publication Date(Web):2017/10/23
DOI:10.1002/ange.201707508
AbstractUltrafast, reversible intersystem crossing (ISC) is reported under ambient conditions for the electronic ground state of the pentacoordinate cobalt nitrosyl complexes, [CoX2(NO)(PMePh2)2] (X=Cl, Br), in solution. ISCs on such short timescales are more typically observed in electronically excited states reached by absorption of ultraviolet or visible light. Singlet and triplet electron spin states of the complex, corresponding to two different isomers, are populated at room temperature, and the two isomers exchange on a timescale of a few picoseconds. Ultrafast two-dimensional infrared spectroscopy observes the change in wavenumber of the NO ligand band accompanying the isomerization and associated ISC on the (spin) adiabatic ground potential energy surface. Comparison of the dynamics of the chloro- and bromo-complexes shows that inertial effects of the ligand motion have a greater effect than spin–orbit coupling on determining the forward and reverse isomerization and ISC rates.
Co-reporter:Dr. Rabi Chhantyal-Pun;Dr. Max R. McGillen;Dr. Joseph M. Beames;Dr. M. Anwar H. Khan;Dr. Carl J. Percival; Dudley E. Shallcross; Andrew J. Orr-Ewing
Angewandte Chemie 2017 Volume 129(Issue 31) pp:9172-9175
Publication Date(Web):2017/07/24
DOI:10.1002/ange.201703700
AbstractThe rate coefficients for gas-phase reaction of trifluoroacetic acid (TFA) with two Criegee intermediates, formaldehyde oxide and acetone oxide, decrease with increasing temperature in the range 240–340 K. The rate coefficients k(CH2OO + CF3COOH)=(3.4±0.3)×10−10 cm3 s−1 and k((CH3)2COO + CF3COOH)=(6.1±0.2)×10−10 cm3 s−1 at 294 K exceed estimates for collision-limited values, suggesting rate enhancement by capture mechanisms because of the large permanent dipole moments of the two reactants. The observed temperature dependence is attributed to competitive stabilization of a pre-reactive complex. Fits to a model incorporating this complex formation give k [cm3 s−1]=(3.8±2.6)×10−18 T2 exp((1620±180)/T) + 2.5×10−10 and k [cm3 s−1]=(4.9±4.1)×10−18 T2 exp((1620±230)/T) + 5.2×10−10 for the CH2OO + CF3COOH and (CH3)2COO + CF3COOH reactions, respectively. The consequences are explored for removal of TFA from the atmosphere by reaction with biogenic Criegee intermediates.
Co-reporter:Dr. Rabi Chhantyal-Pun;Dr. Max R. McGillen;Dr. Joseph M. Beames;Dr. M. Anwar H. Khan;Dr. Carl J. Percival; Dudley E. Shallcross; Andrew J. Orr-Ewing
Angewandte Chemie International Edition 2017 Volume 56(Issue 31) pp:9044-9047
Publication Date(Web):2017/07/24
DOI:10.1002/anie.201703700
AbstractThe rate coefficients for gas-phase reaction of trifluoroacetic acid (TFA) with two Criegee intermediates, formaldehyde oxide and acetone oxide, decrease with increasing temperature in the range 240–340 K. The rate coefficients k(CH2OO + CF3COOH)=(3.4±0.3)×10−10 cm3 s−1 and k((CH3)2COO + CF3COOH)=(6.1±0.2)×10−10 cm3 s−1 at 294 K exceed estimates for collision-limited values, suggesting rate enhancement by capture mechanisms because of the large permanent dipole moments of the two reactants. The observed temperature dependence is attributed to competitive stabilization of a pre-reactive complex. Fits to a model incorporating this complex formation give k [cm3 s−1]=(3.8±2.6)×10−18 T2 exp((1620±180)/T) + 2.5×10−10 and k [cm3 s−1]=(4.9±4.1)×10−18 T2 exp((1620±230)/T) + 5.2×10−10 for the CH2OO + CF3COOH and (CH3)2COO + CF3COOH reactions, respectively. The consequences are explored for removal of TFA from the atmosphere by reaction with biogenic Criegee intermediates.
Co-reporter:Philip M. Coulter, Michael P. Grubb, Andrew J. Orr-Ewing
Chemical Physics Letters 2017 Volume 683(Volume 683) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.cplett.2017.01.068
•We observe geminate recombination of methyl nitrate after UV photolysis in solution.•There is a preference for relaxation to the higher-energy anti conformer.•The preference for the anti over the syn conformer is entropically driven.•We support Pfab’s assignment of the structured bands in the UV absorption spectrum.The dynamics of the ultraviolet (UV) photoexcitation of methyl nitrite in weakly interacting perfluoromethylcyclohexane solution are investigated using transient absorption spectroscopy. UV excitation in the structured S1 ← S0 absorption band induces dissociation, with geminate recombination and vibrational cooling of syn and anti-conformers of methyl nitrate on a timescale of ∼56 ps. Solvent-induced vibrational cooling favours relaxation to the higher-energy anti-conformer on entropic grounds, and subsequent inter-conversion to the lower-energy syn-conformer is prevented by a 3500 cm–1 barrier. UV excitation to the S2 state produces a transient electronic absorption band resembling the absorption spectrum of NO2.Download high-res image (99KB)Download full-size image
Co-reporter:Shubhrangshu Pandit;Balázs Hornung;Greg T. Dunning;Thomas J. Preston;Kristian Brazener
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 2) pp:1614-1626
Publication Date(Web):2017/01/04
DOI:10.1039/C6CP07164C
Velocity map imaging (VMI) measurements and quasi-classical trajectory (QCT) calculations on a newly developed, global potential energy surface (PES) combine to reveal the detailed mechanisms of reaction of Cl atoms with n-pentane. Images of the HCl (v = 0, J = 1, 2 and 3) products of reaction at a mean collision energy of 33.5 kJ mol−1 determine the centre-of-mass frame angular scattering and kinetic energy release distributions. The HCl products form with relative populations of J = 0–5 levels that fit to a rotational temperature of 138 ± 13 K. Product kinetic energy release distributions agree well with those derived from a previous VMI study of the pentyl radical co-product [Estillore et al., J. Chem. Phys. 2010, 132, 164313], but the angular distributions show more pronounced forward scattering. The QCT calculations reproduce many of the experimental observations, and allow comparison of the site-specific dynamics of abstraction of primary and secondary H-atoms. They also quantify the relative reactivity towards Cl atoms of the three different H-atom environments in n-pentane.
Co-reporter:Daisuke Koyama and Andrew J. Orr-Ewing
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 17) pp:12115-12127
Publication Date(Web):08 Apr 2016
DOI:10.1039/C6CP01290F
The photochemical reaction dynamics of the benzothiazole-2-thiyl (BS) radical, produced by 330 nm ultraviolet photolysis of 2,2′-dithiobis(benzothiazole) (BSSB), are examined on the picosecond time scale. The initial addition product of a thiol–ene reaction between the BS radical and styrene is directly observed by transient vibrational absorption spectroscopy (TVAS). Transient electronic absorption spectroscopy (TEAS) in the ultraviolet and visible spectral regions reveals rapid formation of the ground state BS radical with a time constant of ∼200 fs. The photolytically generated BS radical decays through geminate recombination to the parent molecule BSSB and competitive formation of a BS radical dimer with a rate coefficient of (3.7 ± 0.2) × 1010 M−1 s−1 in methanol, and thereafter (36 ± 1)% of the initially formed BS radicals survive at the longest time delay (1.3 ns). In styrene solution, in contrast to methanol and toluene solutions, kinetic traces of the BS radical show an additional decay with a time constant of 305 ± 13 ps, and a broad band at 345–500 nm grows with the same time constant, suggesting a bimolecular reaction of the BS radical with styrene. The TVAS measurements reveal an absorption band of the ground state BS radical at 1301 cm−1 in toluene solution, and the band decays with a time constant of 294 ± 32 ps in styrene solution. Two product bands grow at 1239 cm−1 and 1429 cm−1 with respective time constants of 312 ± 68 ps and 325 ± 33 ps, and are attributed to the addition product BS–St radical formed from the BS radical and styrene. A bimolecular reaction rate coefficient of kreact = (3.8 ± 0.2) × 108 M−1 s−1 is deduced and 22 ± 1% of the initially formed BS radicals are converted to the BS–St radical in neat styrene solution.
Co-reporter:Hugo J. B. Marroux and Andrew J. Orr-Ewing
The Journal of Physical Chemistry B 2016 Volume 120(Issue 17) pp:4125-4130
Publication Date(Web):April 12, 2016
DOI:10.1021/acs.jpcb.6b02979
Collection of two-dimensional infrared (2DIR) spectra using two ultrafast, broadband infrared pump pulses followed by an ultrafast probe pulse optimizes the experimental time and frequency resolution, but can also introduce quantum beat and coherence transfer pathways. The associated coherent dynamics create intensity oscillations and add extra features to 2DIR spectra. We describe a method to suppress these pathways using pump–pulse shaping, without significantly degrading the time and spectral resolution. We illustrate the method for a rhodium dicarbonyl complex, acetylacetonato dicarbonyl rhodium (RDC), to establish the relative importance of coherence and population transfer between carbonyl symmetric and asymmetric stretching modes. Our technique effectively suppresses the quantum beats. Comparison of peak intensities obtained with shaped and unshaped pump pulses demonstrates that coherence transfer does not play a significant role in the 2DIR spectrum of RDC in this spectral region.
Co-reporter:Thomas J. Preston, Balázs Hornung, Shubhrangshu Pandit, Jeremy N. Harvey, and Andrew J. Orr-Ewing
The Journal of Physical Chemistry A 2016 Volume 120(Issue 27) pp:4672-4682
Publication Date(Web):January 26, 2016
DOI:10.1021/acs.jpca.5b09487
Dynamics of collisions between structured molecular species quickly become complex as molecules become large. Reactions of methane with halogen and oxygen atoms serve as model systems for polyatomic molecule chemical dynamics, and replacing the atomic reagent with a diatomic radical affords further insights. A new, full-dimensional potential energy surface for collisions between CN + CH4 to form HCN + CH3 is developed and then used to perform quasi-classical simulations of the reaction. Coupled-cluster energies serve as input to an empirical valence bonding (EVB) model, which provides an analytical function for the surface. Efficient sampling permits simulation of velocity-map ion images and exploration of dynamics over a range of collision energies. Reaction populates HCN vibration, and energy partitioning changes with collision energy. The reaction cross-section depends on the orientation of the diatomic CN radical. A two-dimensional extension of the cone of acceptance for an atom in the line-of-centers model appropriately describes its reactivity. The simulation results foster future experiments and diatomic extensions to existing atomic models of chemical collisions and reaction dynamics.
Co-reporter:Rabi Chhantyal-Pun, Anthony Davey, Dudley E. Shallcross, Carl J. Percival and Andrew J. Orr-Ewing
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 5) pp:3617-3626
Publication Date(Web):22 Dec 2014
DOI:10.1039/C4CP04198D
Criegee intermediates are important species formed during the ozonolysis of alkenes. Reaction of stabilized Criegee intermediates with various species like SO2 and NO2 may contribute significantly to tropospheric chemistry. In the laboratory, self-reaction can be an important loss pathway for Criegee intermediates and thus needs to be characterized to obtain accurate bimolecular reaction rate coefficients. Cavity ring-down spectroscopy was used to perform kinetic measurements for various reactions of CH2OO at 293 K and under low pressure (7 to 30 Torr) conditions. For the reaction CH2OO + CH2OO (8), a rate coefficient k8 = (7.35 ± 0.63) × 10−11 cm3 molecule−1 s−1 was derived from the measured CH2OO decay rates, using an absorption cross section value reported previously. A rate coefficient of k4 = (3.80 ± 0.04) × 10−11 cm3 molecule−1 s−1 was obtained for the CH2OO + SO2 (4) reaction. An upper limit for the unimolecular CH2OO loss rate coefficient of 11.6 ± 8.0 s−1 was deduced from studies of reaction (4). SO2 catalysed CH2OO isomerization or intersystem crossing is proposed to occur with a rate coefficient of (3.53 ± 0.32) × 10−11 cm3 molecule−1 s−1.
Co-reporter:Greg T. Dunning, Thomas J. Preston, Stuart J. Greaves, Gregory M. Greetham, Ian P. Clark, and Andrew J. Orr-Ewing
The Journal of Physical Chemistry A 2015 Volume 119(Issue 50) pp:12090-12101
Publication Date(Web):July 20, 2015
DOI:10.1021/acs.jpca.5b05624
Transient electronic and vibrational absorption spectroscopy unravel the mechanisms and dynamics of bimolecular reactions of CN radicals with acetone in deuterated chloroform solutions. The CN radicals are produced by ultrafast ultraviolet photolysis of dissolved ICN. Two reactive forms of CN radicals are distinguished by their electronic absorption bands: “free” (uncomplexed) CN radicals, and “solvated” CN radicals that are complexed with solvent molecules. The lifetimes of the free CN radicals are limited to a few picoseconds following their photolytic production because of geminate recombination to ICN and INC, complexation with CDCl3 molecules, and reaction with acetone. The acetone reaction occurs with a rate coefficient of (8.0 ± 0.5) × 1010 M–1 s–1 and transient vibrational spectra in the C═N and C═O stretching regions reveal that both the nascent HCN and 2-oxopropyl (CH3C(O)CH2) radical products are vibrationally excited. The rate coefficient for the reaction of solvated CN with acetone is 40 times slower than for free CN, with a rate coefficient of (2.0 ± 0.9) × 109 M–1 s–1 obtained from the rise in the HCN product v1(C═N stretch) IR absorption band. Evidence is also presented for CN complexes with acetone that are more strongly bound than the CN–CDCl3 complexes because of CN interactions with the carbonyl group. The rates of reactions of these more strongly associated radicals are slower still.
Co-reporter:Daisuke Koyama, Philip Coulter, Michael P. Grubb, Gregory M. Greetham, Ian P. Clark, and Andrew J. Orr-Ewing
The Journal of Physical Chemistry A 2015 Volume 119(Issue 52) pp:12924-12934
Publication Date(Web):December 4, 2015
DOI:10.1021/acs.jpca.5b10720
The bimolecular reactions that follow 267 nm ultraviolet photolysis of ICN in acetonitrile solution have been studied using transient absorption spectroscopy on the picosecond time scale. Time-resolved electronic absorption spectroscopy (TEAS) in the ultraviolet and visible spectral regions observes rapid production and loss (with a decay time constant of 0.6 ± 0.1 ps) of the photolytically generated free CN radicals. Some of these radicals convert to a solvated form which decays with a lifetime of 8.5 ± 2.1 ps. Time-resolved vibrational absorption spectroscopy (TVAS) reveals that the free and solvated CN-radicals undergo geminate recombination with I atoms to make ICN and INC, H atom abstraction reactions, and addition reactions to solvent molecules to make C3H3N2 radical species. These radical products have a characteristic absorption band at 2036 cm–1 that shifts to 2010 cm–1 when ICN is photolyzed in CD3CN. The HCN yield is low, suggesting the addition pathway competes effectively with H atom abstraction from CH3CN, but the delayed growth of the C3H3N2 radical band is best described by reaction of solvated CN radicals through an unobserved intermediate species. Addition of methanol or tetrahydrofuran as a cosolute promotes H atom abstraction reactions that produce vibrationally hot HCN. The combination of TEAS and TVAS measurements shows that the rate-limiting process for production of ground-state HCN is vibrational cooling, the rate of which is accelerated by the presence of methanol or tetrahydrofuran.
Co-reporter:Philip Coulter, Michael P. Grubb, Daisuke Koyama, Igor V. Sazanovich, Gregory M. Greetham, and Andrew J. Orr-Ewing
The Journal of Physical Chemistry A 2015 Volume 119(Issue 52) pp:12911-12923
Publication Date(Web):December 4, 2015
DOI:10.1021/acs.jpca.5b10716
The fates of CN radicals produced by ultraviolet (UV) photolysis of ICN in various organic solvents have been examined by transient electronic and vibrational absorption spectroscopy (TEAS and TVAS). Near-UV and visible bands in the TEAS measurement enable direct observation of the CN radicals and their complexes with the solvent molecules. Complementary TVAS measurements probe the products of CN–radical reactions. Geminate recombination to form ICN and INC is a minor pathway on the 150 fs −1300 ps time scales of our experiments in the chosen organic solvents; nonetheless, large infrared transition dipole moments permit direct observation of INC that is vibrationally excited in the C≡N stretching mode. The time constants for INC vibrational cooling range from 30 ps in tetrahydrofuran (THF) to 1400 ps in more weakly interacting solvents such as chloroform. The major channel for CN removal in the organic solvents is reaction with solvent molecules, as revealed by depletion of solvent absorption bands and growth of product bands in the TVA spectra. HCN is a reaction product of hydrogen atom abstraction in most of the photoexcited solutions, and forms with vibrational excitation in both the C–H and C≡N stretching modes. The vibrational cooling rate of the C≡N stretch in HCN depends on the solvent, and follows the same trend as the cooling rate of the C≡N stretch in INC. However, in acetonitrile solution an additional reaction pathway produces C3H3N2• radicals, which release HCN on a much longer time scale.
Co-reporter:Balázs Hornung, Jeremy N. Harvey, Thomas J. Preston, Greg T. Dunning, and Andrew J. Orr-Ewing
The Journal of Physical Chemistry A 2015 Volume 119(Issue 37) pp:9590-9598
Publication Date(Web):August 25, 2015
DOI:10.1021/acs.jpca.5b06418
We report a theoretical investigation of the CH4 + Cl hydrogen abstraction reaction in the framework of empirical valence bond (EVB) theory. The main purpose of this study is to benchmark the EVB method against previous experimental and theoretical work. Analytical potential energy surfaces for the reaction have been developed on which quasi-classical trajectory calculations were carried out. The surfaces agree well with ab initio calculations at stationary points along the reaction path and dynamically relevant regions outside the reaction path. The analysis of dynamical data obtained using the EVB method, such as vibrational, rotational, and angular distribution functions, shows that this method compares well to both experimental measurements and higher-level theoretical calculations, with the additional benefit of low computational cost.
Co-reporter:G. T. Dunning;D. R. Glowacki;T. J. Preston;S. J. Greaves;G. M. Greetham;I. P. Clark;J. N. Harvey;M. Towrie;A. J. Orr-Ewing
Science 2015 Volume 347(Issue 6221) pp:530-533
Publication Date(Web):30 Jan 2015
DOI:10.1126/science.aaa0103
Deuterium fluoride gets born shivering
Modern spectroscopic techniques can analyze collisions between gas phase molecules in exquisite detail, highlighting exactly which vibrations and rotations come into play. However, much chemistry of interest takes place in solution, where it's harder to tease out what happens. Dunning et al. applied infrared spectroscopy to study solution-phase formation of deuterium fluoride (DF) from F atoms, a longstanding test bed of gas phase dynamics. The DF product vibrated for a surprisingly long time before dissipating its energy to the surrounding solvent molecules.
Science, this issue p. 530
Co-reporter:Greg T. Dunning, Thomas J. Preston, Andrew J. Orr-Ewing, Stuart J. Greaves, Gregory M. Greetham, Ian P. Clark and Michael Towrie
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 30) pp:16095-16102
Publication Date(Web):23 Jun 2014
DOI:10.1039/C4CP01854K
Transient electronic absorption measurements with 1 ps time resolution follow XeF2 photoproducts in acetonitrile and chlorinated solvents. Ultraviolet light near 266 nm promptly breaks one Xe–F bond, and probe light covering 320–700 nm monitors the products. Some of the cleaved F atoms remain in close proximity to an XeF fragment and perturb the electronic states of XeF. The time evolution of a perturbed spectral feature is used to monitor the FXe–F complex population, which decays in less than 5 ps. Decay can occur through geminate recombination, diffusive separation or reaction of the complex with the solvent.
Co-reporter:Bernard J. Mason, Jim S. Walker, Jonathan P. Reid, and Andrew J. Orr-Ewing
The Journal of Physical Chemistry A 2014 Volume 118(Issue 11) pp:2083-2088
Publication Date(Web):February 28, 2014
DOI:10.1021/jp5014863
The extinction cross-sections of individual, optically confined aerosol particles with radii of a micrometer or less can, in principle, be measured using cavity ring-down spectroscopy (CRDS). However, when the particle radius is comparable in magnitude to the wavelength of light stored in a high-finesse cavity, the phenomenological cross-section retrieved from a CRDS experiment depends on the location of the particle in the intracavity standing wave and differs from the Mie scattering cross-section for plane-wave irradiation. Using an evaporating 1,2,6-hexanetriol particle of initial radius ∼1.75 μm confined within the 4.5 μm diameter core of a Bessel beam, we demonstrate that the scatter in the retrieved extinction efficiency of a single particle is determined by its lateral motion, which spans a few wavelengths of the intracavity standing wave used for CRDS measurements. Fits of experimental measurements to Mie calculations, modified to account for the intracavity standing wave, allow precise retrieval of the refractive index of 1,2,6-hexanetriol particles (with relative humidity, RH < 10%) of 1.47824 ± 0.00072.
Co-reporter:Christina M. Higgins, Louise A. Evans, Guy C. Lloyd-Jones, Dudley E. Shallcross, David P. Tew, and Andrew J. Orr-Ewing
The Journal of Physical Chemistry A 2014 Volume 118(Issue 15) pp:2756-2764
Publication Date(Web):March 31, 2014
DOI:10.1021/jp501517t
Absorption cross-sections and quantum yields for NO2 production (ΦNO2) are reported for gaseous methyl, ethyl, n-propyl, and isopropyl nitrate at 294 K. Absorption cross-sections in the wavelength range of 240–320 nm agree well with prior determinations. NO2 quantum yields at photoexcitation wavelengths of 290, 295, and 315 nm are unity within experimental uncertainties for all of the alkyl nitrates studied and are independent of bath gas (N2) pressure for total sample pressures in the range of 250–700 Torr. When averaged over all wavelengths and sample pressures, values of ΦNO2 are 1.03 ± 0.05 (methyl nitrate), 0.98 ± 0.09 (ethyl nitrate), 1.01 ± 0.04 (n-propyl nitrate), and 1.00 ± 0.05 (isopropyl nitrate), with uncertainties corresponding to 1 standard deviation. Absorption cross-sections for ethyl nitrate, isopropyl nitrate, and two unsaturated dinitrate compounds, but-3-ene-1,2-diyl dinitrate and (Z)-but-2-ene-1,4-diyl dinitrate in acetonitrile solution, are compared to gas-phase values, and over the wavelength range of 260–315 nm, the gas-phase values are well-reproduced by dividing the liquid-phase cross-sections by 2.0, 1.6, 1.7, and 2.2, respectively. Reasonable estimates of the gas-phase absorption cross-sections for low-volatility organic nitrates can therefore be obtained by halving the values for acetonitrile solutions. The quantum yield for NO2 formation from photoexcitation of but-3-ene-1,2-diyl dinitrate at 290 nm is significantly lower than those for the alkyl (mono) nitrates: a best estimate of ΦNO2 ≤ 0.25 is obtained from the experimental measurements.
Co-reporter:Thomas J. Preston, Greg T. Dunning, and Andrew J. Orr-Ewing and Saulo A. Vázquez
The Journal of Physical Chemistry A 2014 Volume 118(Issue 30) pp:5595-5607
Publication Date(Web):June 10, 2014
DOI:10.1021/jp5042734
Reactions between Cl atoms and propene can lead to HCl formation either by direct H abstraction or through a chloropropyl addition complex. Barring stabilizing collisions, the chloropropyl radical will either decompose to reactants or form HCl and allyl products. Using velocity-map imaging to measure the quantum state and velocity of the HCl products provides a view into the reaction dynamics, which show signs of both direct and indirect reaction mechanisms. Simulated trajectories of the reaction highlight the role of the direct H-abstraction pathways, and the resultant simulated scattering images show reasonable agreement with measurement. The simulations also show the importance of large excursions of the Cl atom far from equilibrium geometries within the chloropropyl complex, and these large-amplitude motions are the ultimate drivers toward HCl + allyl fragmentation. Gas-phase measurements of larger alkenes, 2-methylpropene and 2,3-dimethylbut-2-ene, show slightly different product distributions but still feature similar reaction dynamics. The current suite of experiments offers ready extensions to liquid-phase bimolecular reactions.
Co-reporter:Gareth M. Roberts, Hugo J. B. Marroux, Michael P. Grubb, Michael N. R. Ashfold, and Andrew J. Orr-Ewing
The Journal of Physical Chemistry A 2014 Volume 118(Issue 47) pp:11211-11225
Publication Date(Web):October 8, 2014
DOI:10.1021/jp508501w
A combination of ultrafast transient electronic absorption spectroscopy (TEAS) and transient vibrational absorption spectroscopy (TVAS) is used to investigate whether photoinduced N–H bond fission, mediated by a dissociative 1πσ* state, is active in aqueous adenine (Ade) at 266 and 220 nm. In order to isolate UV/visible and IR spectral signatures of the adeninyl radical (Ade[-H]), formed as a result of N–H bond fission, TEAS and TVAS are performed on Ade in D2O under basic conditions (pD = 12.5), which forms Ade[-H]− anions via deprotonation at the N7 or N9 sites of Ade’s 7H and 9H tautomers. At 220 nm we observe one-photon detachment of an electron from Ade[-H]−, which generates solvated electrons (eaq–) together with Ade[-H] radicals, with clear signatures in both TEAS and TVAS. Additional wavelength dependent TEAS measurements between 240–260 nm identify a threshold of 4.9 ± 0.1 eV (∼250 nm) for this photodetachment process in D2O. Analogous TEAS experiments on aqueous Ade at pD = 7.4 generate a similar photoproduct signal together with eaq– after excitation at 266 and 220 nm. These eaq– are born from ionization of Ade, together with Ade+ cations, which are indistinguishable from Ade[-H] radicals in TEAS. Ade+ and Ade[-H] are found to have different signatures in TVAS and we verify that the pD = 7.4 photoproduct signal observed in TEAS following 220 nm excitation is solely due to Ade+ cations. Based on these observations, we conclude that: (i) N–H bond fission in aqueous Ade is inactive at wavelengths ≥220 nm; and (ii) if such a channel exists in aqueous solution, its threshold is strongly blue-shifted relative to the onset of the same process in gas phase 9H-Ade (≤233 nm). In addition, we extract excited state lifetimes and vibrational cooling dynamics for 9H-Ade and Ade[-H]−. In both cases, excited state lifetimes of <500 fs are identified, while vibrational cooling occurs within a time frame of 4–5 ps. In contrast, 7H-Ade is confirmed to have a longer excited state lifetime of ∼5–10 ps through both TEAS and TVAS.
Co-reporter:Jim S. Walker, Antonia E. Carruthers, Andrew J. Orr-Ewing, and Jonathan P. Reid
The Journal of Physical Chemistry Letters 2013 Volume 4(Issue 10) pp:1748-1752
Publication Date(Web):May 8, 2013
DOI:10.1021/jz4008068
A Bessel beam optical trap is combined with continuous wave cavity ringdown spectroscopy to measure the extinction cross section of individual aerosol particles. Particles, ∼1 μm in size, can be captured indefinitely and processes that transform size or refractive index studied. The measured light extinction induced by the particle is shown to depend on the position of the particle in the cavity, allowing accurate measurements of the mode structure of a high finesse optical cavity without significant perturbation. The variation in extinction efficiency of a sodium chloride droplet with relative humidity is shown to agree well with predictions from Mie scattering theory.Keywords: aerosols; cavity ringdown spectroscopy; light scattering and extinction; optical manipulation;
Co-reporter:Fawzi Abou-Chahine, Thomas J. Preston, Greg T. Dunning, and Andrew J. Orr-Ewing, Gregory M. Greetham, Ian P. Clark, and Mike Towrie , Scott A. Reid
The Journal of Physical Chemistry A 2013 Volume 117(Issue 50) pp:13388-13398
Publication Date(Web):August 21, 2013
DOI:10.1021/jp406687x
Transient absorption spectroscopy is used to follow the reactive intermediates involved in the first steps in the photochemistry initiated by ultraviolet (266-nm wavelength) excitation of solutions of 1,5-hexadiene, isoprene, and 2,3-dimethylbut-2-ene in carbon tetrachloride or chloroform. Ultraviolet and visible bands centered close to 330 and 500 nm in both solvents are assigned respectively to a charge transfer band of Cl-solvent complexes and the strong absorption band of a higher energy isomeric form of the solvent molecules (iso-CCl3–Cl or iso-CHCl2–Cl). These assignments are supported by calculations of electronic excitation energies. The isomeric forms have significant contributions to their structures from charge-separated resonance forms and offer a reinterpretation of previous assignments of the carriers of the visible bands that were based on pulsed radiolysis experiments. Kinetic analysis demonstrates that the isomeric forms are produced via the Cl–solvent complexes. Addition of the unsaturated hydrocarbons provides a reactive loss channel for the Cl–solvent complexes, and reaction radii and bimolecular rate coefficients are derived from analysis using a Smoluchowski theory model. For reactions of Cl with 1,5-hexadiene, isoprene, and 2,3-dimethylbut-2-ene in CCl4, rate coefficients at 294 K are, respectively, (8.6 ± 0.8) × 109, (9.5 ± 1.6) × 109, and (1.7 ± 0.1) × 1010 M–1 s–1. The larger reaction radius and rate coefficient for 2,3-dimethylbut-2-ene are interpreted as evidence for an H-atom abstraction channel that competes effectively with the channel involving addition of a Cl-atom to a C═C bond. However, the addition mechanism appears to dominate the reactions of 1,5-hexadiene and isoprene. Two-photon excited CCl4 or CHCl3 can also ionize the diene or alkene solute.
Co-reporter:M. S. I. Aziz and Andrew J. Orr-Ewing
Environmental Science: Nano 2012 vol. 14(Issue 12) pp:3094-3100
Publication Date(Web):06 Nov 2012
DOI:10.1039/C2EM30801K
An automated near infra-red (IR) continuous wave cavity ring down spectrometer with sample preconcentration has been developed for the measurement of ethene (C2H4) in air. The spectrometer incorporated a distributed feedback diode laser operating at wavelengths λ ∼ 1.6 μm and a pre-concentration system containing an adsorbent, molecular sieve 4A (MS4A). An absorption line located at 6148.58 cm−1, and free from spectral overlap with other atmospheric molecules, was used for ethene detection. The spectrometer has a capacity for determination of atmospheric ethene mixing ratios at half hour time intervals, with a detection limit (2 SD above baseline noise) of 280 ppt. Both weekday and weekend measurements were performed in ambient air for periods of up to 30 hours. Average daytime mixing ratios of ethene were observed to be 2 ppbv and 1 ppbv during weekdays and weekends respectively. The mixing ratios of ethene varied from 0.6 ppbv to 1.2 ppbv in Bristol air during the weekend, with influence of meteorological conditions. The observed variations are discussed with consideration of probable sources and various meteorological parameters. A correlation is observed in the mixing ratio of ethene and nitrogen dioxide.
Co-reporter:Claudio Roscini;KaraL. Cubbage;Malcolm Berry Dr.;AndrewJ. Orr-Ewing Dr.;KevinI. Booker-Milburn Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 46) pp:8716-8720
Publication Date(Web):
DOI:10.1002/anie.200904059
Co-reporter:Manik Pradhan, M. S. I. Aziz, Roberto Grilli and Andrew J. Orr-Ewing
Environmental Science & Technology 2008 Volume 42(Issue 19) pp:7354-7359
Publication Date(Web):August 19, 2008
DOI:10.1021/es801378r
A fully automated instrument combining a continuous wave cavity ring-down spectrometer and dual-trap sample preconcentration has been implemented for monitoring C2H2 mixing ratios in ambient air. A distributed feedback diode laser operating in the near-infrared region (λ ∼ 1534.973 nm in air) detects C2H2 in absorption via the P(17) rotational line of the (ν1 + ν3) vibrational combination band. The instrument is shown to be capable of fast, quantitative, and precise monitoring of C2H2 mixing ratios, with a detection limit of ∼8 pptv (parts per trillion by volume). It thus has potential to be deployed for analysis of air samples in many rural and urban environments. In situ measurements were carried out at 30 min intervals over periods of up to 15 h on several days for indoor and outdoor air samples. For indoor air monitored on a Sunday, the C2H2 mixing ratio was stable at 1.45 ± 0.04 ppbv (parts per billion by volume). On weekdays, both indoor and outside air analyses showed peaks in the range 2−4 ppbv in the early morning and late afternoon that coincided with periods of busy road traffic.
Co-reporter:Phyllis A.Y. Fiadzomor, Anthony M. Keen, Robert B. Grant, Andrew J. Orr-Ewing
Chemical Physics Letters 2008 Volume 462(4–6) pp:188-191
Publication Date(Web):10 September 2008
DOI:10.1016/j.cplett.2008.08.023
Pressure broadening coefficients have been determined for three spectroscopic transitions of water vapour at wavelengths near 1.393 μm in the presence of noble gases. The interaction energy of the water dimer at room temperature was estimated from these pressure broadening coefficients using the Parmenter–Seaver relation. For the transitions investigated, the estimated interaction energies for the water dimer are: 20.4 ± 3.7, 12.3 ± 3.8 and 13.7 ± 3.3 kJ mol−1. The variation of the interaction energies reflects the rotational level dependence of the pressure broadening of these lines by the noble gases. The average of these values is compared to other experimental determinations.The interaction energy of the water dimer is estimated from the pressure broadening coefficients of three ro-vibrational transitions of water vapour with noble gases using the Parmenter–Seaver relation.
Co-reporter:Paula Gorrotxategi Carbajo, Shona C. Smith, Anne-Louise Holloway, Carina A. Smith, Francis D. Pope, Dudley E. Shallcross and Andrew J. Orr-Ewing
The Journal of Physical Chemistry A 2008 Volume 112(Issue 48) pp:12437-12448
Publication Date(Web):November 10, 2008
DOI:10.1021/jp8070508
Absolute quantum yields for the radical (H + HCO) channel of HCHO photolysis, ΦHCO, have been measured for the tropospherically relevant range of wavelengths (λ) between 300 and 330 nm. The HCO photoproduct was directly detected by using a custom-built, combined ultra-violet (UV) absorption and cavity ring down (CRD) detection spectrometer. This instrument was previously employed for high-resolution (spectral resolution ∼0.0035 nm) measurements of absorption cross-sections of HCHO, σHCHO(λ), and relative HCO quantum yields. Absolute ΦHCO values were measured at seven wavelengths, λ = 303.70, 305.13, 308.87, 314.31, 320.67, 325.59, and 329.51 nm, using an independent calibration technique based on the simultaneous UV photolysis of HCHO and Cl2. These ΦHCO measurements display greater variability as a function of wavelength than the current NASA-JPL recommendations for ΦHCO. The absolute ΦHCO(λ) determinations and previously measured σHCHO(λ) were used to scale an extensive set of relative HCO yield measurements. The outcome of this procedure is a full suite of data for the product of the absolute radical quantum yield and HCHO absorption cross-section, ΦHCO(λ)σHCHO(λ), at wavelengths from 302.6 to 331.0 nm with a wavelength resolution of 0.005 nm. This product of photochemical parameters is combined with high-resolution solar photon flux data to calculate the integrated photolysis rate of HCHO to the radical (H + HCO) channel, J(HCO). Comparison with the latest NASA-JPL recommendations, reported at 1 nm wavelength resolution, suggests an increased J(HCO) of 25% at 0° solar zenith angle (SZA) increasing to 33% at high SZA (80°). The differences in the calculated photolysis rate compared with the current HCHO data arise, in part, from the higher wavelength resolution of the current data set and highlight the importance of using high-resolution spectroscopic techniques to achieve a complete and accurate picture of HCHO photodissociation processes. All experimental ΦHCO(λ)σHCHO(λ) data are available for the wavelength range 302.6−331.0 nm (at 294 and 245 K and under 200 Torr of N2 bath gas) as Supporting Information with wavelength resolutions of 0.005, 0.1, and 1.0 nm. Equivalent data sets of ΦH2+CO(λ)σHCHO(λ) for the molecular (H2 + CO) photofragmentation channel, produced using the measured ΦHCO(λ) σHCHO(τ) values, are also provided at 0.1 and 1.0 nm resolution.
Co-reporter:Stuart J. Greaves and Andrew J. Orr-Ewing, Diego Troya
The Journal of Physical Chemistry A 2008 Volume 112(Issue 39) pp:9387-9395
Publication Date(Web):July 17, 2008
DOI:10.1021/jp802347v
We present an electronic-structure and dynamics study of the Cl + C2H6 → HCl + C2H5 reaction. The stationary points of the ground-state potential energy surface have been characterized using various electronic-structure methods and basis sets. Our best calculations, CCSD(T) extrapolated to the complete basis limit, using geometries and harmonic frequencies obtained at the MP2/aug-cc-pVTZ level, are in agreement with the experimental reaction energy. Ab initio information has been used to reparameterize a semiempirical Hamiltonian so that the predictions of the improved Hamiltonian agree with the higher-level calculations in key regions of the potential energy surface. The improved semiempirical Hamiltonian is then used to propagate quasiclassical trajectories. Computed kinetic energy release and scattering angle distributions at a collision energy of ∼5.5 kcal mol−1 are in reasonable agreement with experiments, but no evidence was found for the low translational energy HCl products scattered in the backward hemisphere reported in recent experiments.
Co-reporter:Claudio Roscini;DavidM.E. Davies;Malcolm Berry Dr.;AndrewJ. Orr-Ewing ;KevinI. Booker-Milburn
Angewandte Chemie International Edition 2008 Volume 47( Issue 12) pp:2283-2286
Publication Date(Web):
DOI:10.1002/anie.200704816
Co-reporter:Claudio Roscini;DavidM.E. Davies;Malcolm Berry Dr.;AndrewJ. Orr-Ewing ;KevinI. Booker-Milburn
Angewandte Chemie 2008 Volume 120( Issue 12) pp:2315-2318
Publication Date(Web):
DOI:10.1002/ange.200704816
Co-reporter:Stuart J. Greaves, Jin Kim, Andrew J. Orr-Ewing, Diego Troya
Chemical Physics Letters 2007 Volume 441(4–6) pp:171-175
Publication Date(Web):25 June 2007
DOI:10.1016/j.cplett.2007.05.010
Classical trajectories have been computed using highly oriented reagents for the reaction Cl + C2H6 → HCl + C2H5 to examine the validity of a mechanism proposed in a recent experimental study by Huang et al. The low translational energies of some HCl products scattered in the backwards hemisphere in these experiments were attributed to ‘chattering collisions’ that internally excite the ethyl co-product. While trajectories involving oscillations of the H atom between Cl and C are observed in the current calculations, the resultant distributions of product internal energies and angular scattering are nearly identical to trajectories displaying direct scattering dynamics.The dynamical consequences of chattering collisions are explored for the Cl + ethane reaction using fully dimensional trajectory calculations.
Co-reporter:Ruth E. Lindley, Manik Pradhan and Andrew J. Orr-Ewing
Analyst 2006 vol. 131(Issue 6) pp:731-738
Publication Date(Web):24 Apr 2006
DOI:10.1039/B600506C
Two frequency chirped continuous wave diode lasers operating in the near infrared (IR) at wavelengths of λ
∼ 1.535 µm and λ
∼ 1.520 µm have been used to measure acetylene concentrations using the P(17) and R(9) rotational lines of the (ν1
+
ν3) vibrational combination band. The diode lasers were frequency chirped by applying an electrical current pulse to the laser driver at a repetition rate of greater than 1 kHz. As the laser is operated at high repetition rates, more than 1000 spectra per second can, in principle, be acquired and summed, allowing fast accumulation of data, rapid averaging and consequent improvement of the signal to noise ratio and detection limit. Experiments were performed using a single-pass cell with a path length of 16.4 cm, and also an astigmatic multi-pass absorption cell aligned to give a path length of 56 m. Detection limits corresponding to minimum detectable absorption coefficients, αmin, of 5.6 × 10−5 and 7.8 × 10−8 cm−1, respectively, were obtained over a 4 s detection bandwidth. These detection limits would correspond to mixing ratios of 21 parts per million by volume (ppmv) and 59 parts per billion by volume (ppbv) of acetylene at 1 atm in air, with the deleterious effects of pressure broadening accounted for. The single-pass cell was used to perform breakthrough volume (BTV) experiments for the low volume adsorbent traps used to pre-concentrate organic compounds in air, taking advantage of the capability of the system to measure concentrations in real time.
Co-reporter:Greg T. Dunning, Thomas J. Preston, Andrew J. Orr-Ewing, Stuart J. Greaves, Gregory M. Greetham, Ian P. Clark and Michael Towrie
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 30) pp:NaN16102-16102
Publication Date(Web):2014/06/23
DOI:10.1039/C4CP01854K
Transient electronic absorption measurements with 1 ps time resolution follow XeF2 photoproducts in acetonitrile and chlorinated solvents. Ultraviolet light near 266 nm promptly breaks one Xe–F bond, and probe light covering 320–700 nm monitors the products. Some of the cleaved F atoms remain in close proximity to an XeF fragment and perturb the electronic states of XeF. The time evolution of a perturbed spectral feature is used to monitor the FXe–F complex population, which decays in less than 5 ps. Decay can occur through geminate recombination, diffusive separation or reaction of the complex with the solvent.
Co-reporter:Michael P. Grubb, Philip M. Coulter, Hugo J. B. Marroux, Andrew J. Orr-Ewing and Michael N. R. Ashfold
Chemical Science (2010-Present) 2017 - vol. 8(Issue 4) pp:NaN3069-3069
Publication Date(Web):2017/02/10
DOI:10.1039/C6SC05234G
We present a systematic study of the mode-specific vibrational relaxation of NO2 in six weakly-interacting solvents (perfluorohexane, perfluoromethylcyclohexane, perfluorodecalin, carbon tetrachloride, chloroform, and d-chloroform), chosen to elucidate the dominant energy transfer mechanisms in the solution phase. Broadband transient vibrational absorption spectroscopy has allowed us to extract quantum state-resolved relaxation dynamics of the two distinct NO2 fragments produced from the 340 nm photolysis of N2O4 → NO2(X) + NO2(A) and their separate paths to thermal equilibrium. Distinct relaxation pathways are observed for the NO2 bending and stretching modes, even at energies as high as 7000 cm−1 above the potential minimum. Vibrational energy transfer is governed by different interaction mechanisms in the various solvent environments, and proceeds with timescales ranging from 20–1100 ps. NO2 relaxation rates in the perfluorocarbon solvents are identical despite differences in acceptor mode state densities, infrared absorption cross sections, and local solvent structure. Vibrational energy is shown to be transferred to non-vibrational solvent degrees of freedom (V-T) through impulsive collisions with the perfluorocarbon molecules. Conversely, NO2 relaxation in chlorinated solvents is reliant on vibrational resonances (V-V) while V-T energy transfer is inefficient and thermal excitation of the surrounding solvent molecules inhibits faster vibrational relaxation through direct complexation. Intramolecular vibrational redistribution allows the symmetric stretch of NO2 to act as a gateway for antisymmetric stretch energy to exit the molecule. This study establishes an unprecedented level of detail for the cooling dynamics of a solvated small molecule, and provides a benchmark system for future theoretical studies of vibrational relaxation processes in solution.
Co-reporter:Daisuke Koyama, Paul M. Donaldson and Andrew J. Orr-Ewing
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 20) pp:NaN12991-12991
Publication Date(Web):2017/05/03
DOI:10.1039/C7CP01784G
The mechanism of the thiol–ene reaction induced by 330 nm ultraviolet excitation of 1,2-di(quinolin-2-yl)disulfide (QSSQ) in the presence of methyl methacrylate (MMA) is investigated by sub-picosecond to microsecond transient absorption spectroscopy. The measurements, spanning more than seven orders of magnitude of time, directly reveal multiple radical reaction steps. The ground state quinoliene-2-thiyl radical (QS) is formed with a time constant of ∼200 fs by photolysis of QSSQ, followed by (64 ± 1)% decay of the initially formed QS radical because of solvent cage induced geminate recombination and QS dimer formation with a rate coefficient of (3.4 ± 0.2) × 1010 M−1 s−1 in methanol solution. In MMA solution, the carbon centered radical QS-MMA forms with a bimolecular reaction rate coefficient of (2.8 ± 0.2) × 107 M−1 s−1. The distinct infrared band at 1653 cm−1 assigned to the CO stretch mode of the QS-MMA radical decays rapidly in aerated solution, in contrast to observations in a solution purged of O2 by N2 bubbling. This decay is attributed to reaction of the QS-MMA radicals with molecular oxygen, producing peroxy radicals. Kinetic analysis of the intensity of the band at 1653 cm−1 reveals a bimolecular reaction rate coefficient of (3.3 ± 0.3) × 109 M−1 s−1 for the reaction of the QS-MMA radicals with molecular oxygen, and indicates that this reaction step is reversible.
Co-reporter:Daisuke Koyama and Andrew J. Orr-Ewing
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 17) pp:NaN12127-12127
Publication Date(Web):2016/04/08
DOI:10.1039/C6CP01290F
The photochemical reaction dynamics of the benzothiazole-2-thiyl (BS) radical, produced by 330 nm ultraviolet photolysis of 2,2′-dithiobis(benzothiazole) (BSSB), are examined on the picosecond time scale. The initial addition product of a thiol–ene reaction between the BS radical and styrene is directly observed by transient vibrational absorption spectroscopy (TVAS). Transient electronic absorption spectroscopy (TEAS) in the ultraviolet and visible spectral regions reveals rapid formation of the ground state BS radical with a time constant of ∼200 fs. The photolytically generated BS radical decays through geminate recombination to the parent molecule BSSB and competitive formation of a BS radical dimer with a rate coefficient of (3.7 ± 0.2) × 1010 M−1 s−1 in methanol, and thereafter (36 ± 1)% of the initially formed BS radicals survive at the longest time delay (1.3 ns). In styrene solution, in contrast to methanol and toluene solutions, kinetic traces of the BS radical show an additional decay with a time constant of 305 ± 13 ps, and a broad band at 345–500 nm grows with the same time constant, suggesting a bimolecular reaction of the BS radical with styrene. The TVAS measurements reveal an absorption band of the ground state BS radical at 1301 cm−1 in toluene solution, and the band decays with a time constant of 294 ± 32 ps in styrene solution. Two product bands grow at 1239 cm−1 and 1429 cm−1 with respective time constants of 312 ± 68 ps and 325 ± 33 ps, and are attributed to the addition product BS–St radical formed from the BS radical and styrene. A bimolecular reaction rate coefficient of kreact = (3.8 ± 0.2) × 108 M−1 s−1 is deduced and 22 ± 1% of the initially formed BS radicals are converted to the BS–St radical in neat styrene solution.
Co-reporter:M. S. I. Aziz and Andrew J. Orr-Ewing
Environmental Science: Nano 2012 - vol. 14(Issue 12) pp:NaN3100-3100
Publication Date(Web):2012/11/06
DOI:10.1039/C2EM30801K
An automated near infra-red (IR) continuous wave cavity ring down spectrometer with sample preconcentration has been developed for the measurement of ethene (C2H4) in air. The spectrometer incorporated a distributed feedback diode laser operating at wavelengths λ ∼ 1.6 μm and a pre-concentration system containing an adsorbent, molecular sieve 4A (MS4A). An absorption line located at 6148.58 cm−1, and free from spectral overlap with other atmospheric molecules, was used for ethene detection. The spectrometer has a capacity for determination of atmospheric ethene mixing ratios at half hour time intervals, with a detection limit (2 SD above baseline noise) of 280 ppt. Both weekday and weekend measurements were performed in ambient air for periods of up to 30 hours. Average daytime mixing ratios of ethene were observed to be 2 ppbv and 1 ppbv during weekdays and weekends respectively. The mixing ratios of ethene varied from 0.6 ppbv to 1.2 ppbv in Bristol air during the weekend, with influence of meteorological conditions. The observed variations are discussed with consideration of probable sources and various meteorological parameters. A correlation is observed in the mixing ratio of ethene and nitrogen dioxide.
Co-reporter:Rabi Chhantyal-Pun, Anthony Davey, Dudley E. Shallcross, Carl J. Percival and Andrew J. Orr-Ewing
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 5) pp:NaN3626-3626
Publication Date(Web):2014/12/22
DOI:10.1039/C4CP04198D
Criegee intermediates are important species formed during the ozonolysis of alkenes. Reaction of stabilized Criegee intermediates with various species like SO2 and NO2 may contribute significantly to tropospheric chemistry. In the laboratory, self-reaction can be an important loss pathway for Criegee intermediates and thus needs to be characterized to obtain accurate bimolecular reaction rate coefficients. Cavity ring-down spectroscopy was used to perform kinetic measurements for various reactions of CH2OO at 293 K and under low pressure (7 to 30 Torr) conditions. For the reaction CH2OO + CH2OO (8), a rate coefficient k8 = (7.35 ± 0.63) × 10−11 cm3 molecule−1 s−1 was derived from the measured CH2OO decay rates, using an absorption cross section value reported previously. A rate coefficient of k4 = (3.80 ± 0.04) × 10−11 cm3 molecule−1 s−1 was obtained for the CH2OO + SO2 (4) reaction. An upper limit for the unimolecular CH2OO loss rate coefficient of 11.6 ± 8.0 s−1 was deduced from studies of reaction (4). SO2 catalysed CH2OO isomerization or intersystem crossing is proposed to occur with a rate coefficient of (3.53 ± 0.32) × 10−11 cm3 molecule−1 s−1.
Co-reporter:Shubhrangshu Pandit, Balázs Hornung and Andrew J. Orr-Ewing
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 40) pp:NaN28364-28364
Publication Date(Web):2016/09/21
DOI:10.1039/C6CP05393A
Elimination of HBr from UV-photoexcited vinyl bromides can occur through both 3-centre and 4-centre transition states (TSs). The competition between these pathways is examined using velocity map imaging of HBr (v = 0–2, J) photofragments. The three vinyl bromides chosen for study have methyl substituents that block either the 3-centre or the 4-centre TS, or leave both pathways open. The kinetic energy distributions extracted from velocity map images of HBr from 193 nm photolysis of the three vinyl bromide compounds are approximately described by a statistical model of energy disposal among the degrees of freedom of the photoproducts, and are attributed to dissociation on the lowest electronic state of the molecule after internal conversion. Dissociation via the 4-centre TS gives greater average kinetic energy release than for the 3-centre TS pathway. The resonance enhanced multi-photon ionization (REMPI) schemes used to detect HBr restrict measurements to J ≤ 7 for v = 2 and J ≤ 15 for v = 0. Within this spectroscopic range, the HBr rotational temperature is colder for the 4-centre than for the 3-centre elimination pathway. Calculations of the intrinsic reaction coordinates and RRKM calculations of HBr elimination rate coefficients provide mechanistic insights into the competition between the pathways.
Co-reporter:Daisuke Koyama and Andrew J. Orr-Ewing
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 37) pp:NaN26235-26235
Publication Date(Web):2016/09/07
DOI:10.1039/C6CP05110C
The photochemical dynamics of the thione 2-mercaptobenzothiazole (MBT) initiated by absorption of 330 nm ultraviolet light are investigated by ultrafast transient absorption spectroscopy. The lowest energy triplet state (T1) has mixed 3ππ*/3nπ* character and is populated with a quantum yield of 0.58 ± 0.01 from the photo-excited 1ππ* S2 state in methanol solution via rapid internal conversion to the 1nπ* S1 state (with time constant τ1 < 150 fs). The spectroscopic evidence points to a mechanism involving intersystem crossing from S1 to the 3nπ*/3ππ* T2 state (τ2 = 400 ± 100 fs) and internal conversion to T1 (with time constant for growth τ3 = 6.1 ± 0.4 ps). The remainder of the photoexcited molecules return to the ground state by S1 → S0 internal conversion. In methanol solution, the T1 state is long-lived but when the solvent is changed to styrene, triplet quenching is observed with a time constant of 107 ± 8 ps and assigned to the adduct-mediated energy transfer process MBT (T1) + styrene (S0) → 3[MBT–styrene] → MBT (S0) + Styrene (T1). Transient vibrational absorption spectroscopy observes the 3[MBT–styrene] biradical intermediate and determines its lifetime to be 700 ± 80 ps. Computational studies identify the mechanistic pathway for triplet quenching, which involves a curve crossing between two triplet states of the MBT–styrene adduct. The quenching process occurs with high efficiency, and no long-lived isomers of the initial adduct are observed.
Co-reporter:Shubhrangshu Pandit, Balázs Hornung, Greg T. Dunning, Thomas J. Preston, Kristian Brazener and Andrew J. Orr-Ewing
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 2) pp:NaN1626-1626
Publication Date(Web):2016/12/12
DOI:10.1039/C6CP07164C
Velocity map imaging (VMI) measurements and quasi-classical trajectory (QCT) calculations on a newly developed, global potential energy surface (PES) combine to reveal the detailed mechanisms of reaction of Cl atoms with n-pentane. Images of the HCl (v = 0, J = 1, 2 and 3) products of reaction at a mean collision energy of 33.5 kJ mol−1 determine the centre-of-mass frame angular scattering and kinetic energy release distributions. The HCl products form with relative populations of J = 0–5 levels that fit to a rotational temperature of 138 ± 13 K. Product kinetic energy release distributions agree well with those derived from a previous VMI study of the pentyl radical co-product [Estillore et al., J. Chem. Phys. 2010, 132, 164313], but the angular distributions show more pronounced forward scattering. The QCT calculations reproduce many of the experimental observations, and allow comparison of the site-specific dynamics of abstraction of primary and secondary H-atoms. They also quantify the relative reactivity towards Cl atoms of the three different H-atom environments in n-pentane.

![1H-Pyrrolo[1,2-a]azepine-5,8-dione, 2,3,9,9a-tetrahydro-6,7-dimethyl-](http://img.cochemist.com/ccimg/215000/214913-37-8.png)
![1H-Pyrrolo[1,2-a]azepine-5,8-dione, 2,3,9,9a-tetrahydro-6,7-dimethyl-](http://img.cochemist.com/ccimg/215000/214913-37-8_b.png)




![4-Pentenoic acid, 3-[(tetrahydro-2H-pyran-2-yl)oxy]-, ethyl ester](http://img.cochemist.com/ccimg/873300/873296-29-8.png)
![4-Pentenoic acid, 3-[(tetrahydro-2H-pyran-2-yl)oxy]-, ethyl ester](http://img.cochemist.com/ccimg/873300/873296-29-8_b.png)




















![7,8-dihydro-5H-benzo[7]annulene-5,9(6H)-dione](http://img.cochemist.com/ccimg/54100/54034-10-5.png)
![7,8-dihydro-5H-benzo[7]annulene-5,9(6H)-dione](http://img.cochemist.com/ccimg/54100/54034-10-5_b.png)

































![CALCIUM BIS[2-(4-CHLOROPHENOXY)-2-METHYLPROPANOATE]](http://img.cochemist.com/ccimg/7300/7272-58-4.png)
![CALCIUM BIS[2-(4-CHLOROPHENOXY)-2-METHYLPROPANOATE]](http://img.cochemist.com/ccimg/7300/7272-58-4_b.png)





























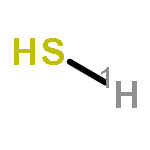
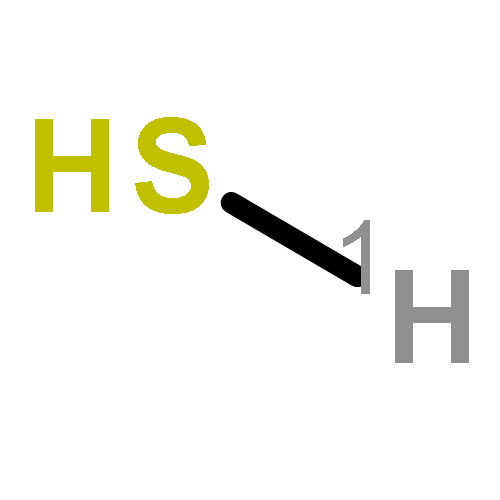
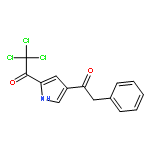
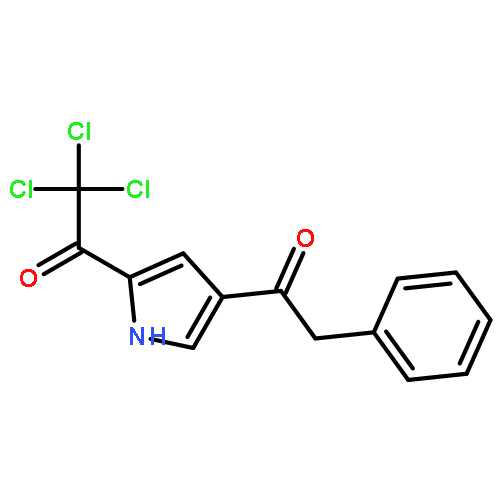
![6,7-DICHLORO-2,3,9,9A-TETRAHYDRO-1H-PYRROLO[1,2-A]AZEPINE-5,8-DIONE](http://img.cochemist.com/ccimg/346800/346701-48-2.png)
![6,7-DICHLORO-2,3,9,9A-TETRAHYDRO-1H-PYRROLO[1,2-A]AZEPINE-5,8-DIONE](http://img.cochemist.com/ccimg/346800/346701-48-2_b.png)






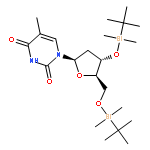
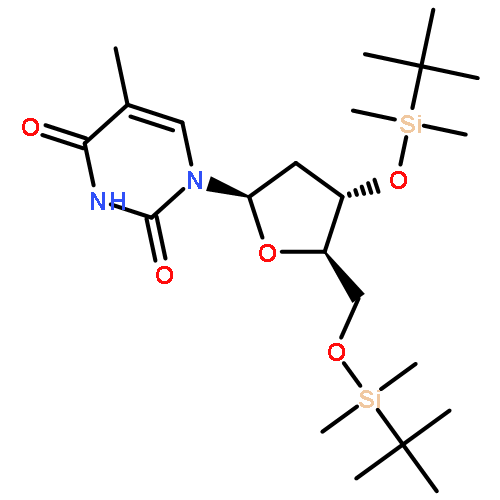
![Poly(oxy[1,1':3',1''-terphenyl]-2',5'-diyl)](http://img.cochemist.com/ccimg/25000/24938-68-9.png)
![Poly(oxy[1,1':3',1''-terphenyl]-2',5'-diyl)](http://img.cochemist.com/ccimg/25000/24938-68-9_b.png)