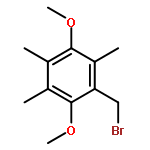
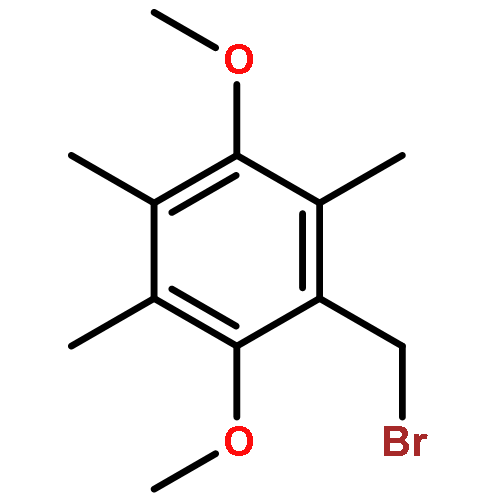
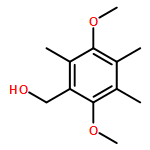
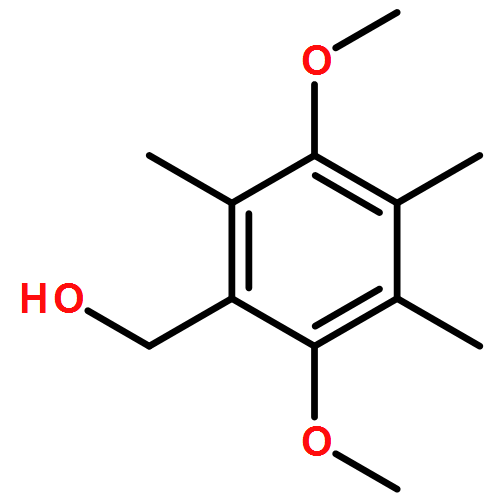
Co-reporter:Ellinor Haglund, Anna Pilko, Roy Wollman, Patricia Ann JenningsJosé Nelson Onuchic
The Journal of Physical Chemistry B 2017 Volume 121(Issue 4) pp:
Publication Date(Web):December 30, 2016
DOI:10.1021/acs.jpcb.6b11506
Protein engineering is a powerful tool in drug design and therapeutics, where disulphide bridges are commonly introduced to stabilize proteins. However, these bonds also introduce covalent loops, which are often neglected. These loops may entrap the protein backbone on opposite sides, leading to a “knotted” topology, forming a so-called Pierced Lasso (PL). In this elegant system, the “knot” is held together with a single disulphide bridge where part of the polypeptide chain is threaded through. The size and position of these covalent loops can be manipulated through protein design in vitro, whereas nature uses polymorphism to switch the PL topology. The PL protein leptin shows genetic modification of an N-terminal residue, adding a third cysteine to the same sequence. In an effort to understand the mechanism of threading of these diverse topologies, we designed three loop variants to mimic the polymorphic sequence. This adds elegance to the system under study, as it allows the generation of three possible covalent loops; they are the original wild-type C-terminal loop protein, the fully circularized unthreaded protein, and the N-terminal loop protein, responsible for different lasso topologies. The size of the loop changes the threading mechanism from a slipknotting to a plugging mechanism, with increasing loop size. Interestingly, the ground state of the native protein structure is largely unaffected, but biological assays show that the activity is maximized by properly controlled dynamics in the threaded state. A threaded topology with proper conformational dynamics is important for receptor interaction and activation of the signaling pathways in vivo.
Co-reporter:Fang Bai;Yang-Sung Sohn;Faruck Morcos;Merav Darash-Yahana;Celso O. Rezende;Colin H. Lipper;Mark L. Paddock;Patricia A. Jennings;Yuting Luo;Emmanuel A. Theodorakis;Sarah H. Holt;Ron Mittler;José N. Onuchic;Luhua Song;Sagi Tamir;Rachel Nechushtai
PNAS 2015 Volume 112 (Issue 12 ) pp:3698-3703
Publication Date(Web):2015-03-24
DOI:10.1073/pnas.1502960112
Identification of novel drug targets and chemotherapeutic agents is a high priority in the fight against cancer. Here, we
report that MAD-28, a designed cluvenone (CLV) derivative, binds to and destabilizes two members of a unique class of mitochondrial
and endoplasmic reticulum (ER) 2Fe-2S proteins, mitoNEET (mNT) and nutrient-deprivation autophagy factor-1 (NAF-1), recently
implicated in cancer cell proliferation. Docking analysis of MAD-28 to mNT/NAF-1 revealed that in contrast to CLV, which formed
a hydrogen bond network that stabilized the 2Fe-2S clusters of these proteins, MAD-28 broke the coordinative bond between
the His ligand and the cluster’s Fe of mNT/NAF-1. Analysis of MAD-28 performed with control (Michigan Cancer Foundation; MCF-10A)
and malignant (M.D. Anderson–metastatic breast; MDA-MB-231 or MCF-7) human epithelial breast cells revealed that MAD-28 had
a high specificity in the selective killing of cancer cells, without any apparent effects on normal breast cells. MAD-28 was
found to target the mitochondria of cancer cells and displayed a surprising similarity in its effects to the effects of mNT/NAF-1
shRNA suppression in cancer cells, causing a decrease in respiration and mitochondrial membrane potential, as well as an increase
in mitochondrial iron content and glycolysis. As expected, if the NEET proteins are targets of MAD-28, cancer cells with suppressed
levels of NAF-1 or mNT were less susceptible to the drug. Taken together, our results suggest that NEET proteins are a novel
class of drug targets in the chemotherapeutic treatment of breast cancer, and that MAD-28 can now be used as a template for
rational drug design for NEET Fe-S cluster-destabilizing anticancer drugs.
Co-reporter:José N. Onuchic;Kendra L. Hailey;Shahar Rotem-Bamberger;Faruck Morcos;Andrea R. Conlan;Rachel Nechushtai;Sagi Tamir;Ron Mittler;Mark L. Paddock;Patricia A. Jennings;Assaf Friedler;John A. Zuris;Colin H. Lipper;Charles Wang;Chen Katz
PNAS 2014 Volume 111 (Issue 14 ) pp:5177-5182
Publication Date(Web):2014-04-08
DOI:10.1073/pnas.1403770111
Life requires orchestrated control of cell proliferation, cell maintenance, and cell death. Involved in these decisions are
protein complexes that assimilate a variety of inputs that report on the status of the cell and lead to an output response.
Among the proteins involved in this response are nutrient-deprivation autophagy factor-1 (NAF-1)- and Bcl-2. NAF-1 is a homodimeric
member of the novel Fe-S protein NEET family, which binds two 2Fe-2S clusters. NAF-1 is an important partner for Bcl-2 at
the endoplasmic reticulum to functionally antagonize Beclin 1-dependent autophagy [Chang NC, Nguyen M, Germain M, Shore GC
(2010) EMBO J 29(3):606–618]. We used an integrated approach involving peptide array, deuterium exchange mass spectrometry (DXMS), and
functional studies aided by the power of sufficient constraints from direct coupling analysis (DCA) to determine the dominant
docked conformation of the NAF-1–Bcl-2 complex. NAF-1 binds to both the pro- and antiapoptotic regions (BH3 and BH4) of Bcl-2,
as demonstrated by a nested protein fragment analysis in a peptide array and DXMS analysis. A combination of the solution
studies together with a new application of DCA to the eukaryotic proteins NAF-1 and Bcl-2 provided sufficient constraints
at amino acid resolution to predict the interaction surfaces and orientation of the protein–protein interactions involved
in the docked structure. The specific integrated approach described in this paper provides the first structural information,
to our knowledge, for future targeting of the NAF-1–Bcl-2 complex in the regulation of apoptosis/autophagy in cancer biology.
Co-reporter:Yang-Sung Sohn;Sagi Tamir;Dorit Michaeli;Luhua Song;Andrea R. Conlan;Imad Matouk;Yael Harir;Sarah H. Holt;Vladimir Shulaev;Mark L. Paddock;Abraham Hochberg;Ioav Z. Cabanchick;José N. Onuchic;Patricia A. Jennings;Rachel Nechushtai;Ron Mittler
PNAS 2013 Volume 110 (Issue 36 ) pp:14676-14681
Publication Date(Web):2013-09-03
DOI:10.1073/pnas.1313198110
Mitochondria are emerging as important players in the transformation process of cells, maintaining the biosynthetic and energetic
capacities of cancer cells and serving as one of the primary sites of apoptosis and autophagy regulation. Although several
avenues of cancer therapy have focused on mitochondria, progress in developing mitochondria-targeting anticancer drugs nonetheless
has been slow, owing to the limited number of known mitochondrial target proteins that link metabolism with autophagy or cell
death. Recent studies have demonstrated that two members of the newly discovered family of NEET proteins, NAF-1 (CISD2) and
mitoNEET (mNT; CISD1), could play such a role in cancer cells. NAF-1 was shown to be a key player in regulating autophagy,
and mNT was proposed to mediate iron and reactive oxygen homeostasis in mitochondria. Here we show that the protein levels
of NAF-1 and mNT are elevated in human epithelial breast cancer cells, and that suppressing the level of these proteins using
shRNA results in significantly reduced cell proliferation and tumor growth, decreased mitochondrial performance, uncontrolled
accumulation of iron and reactive oxygen in mitochondria, and activation of autophagy. Our findings highlight NEET proteins
as promising mitochondrial targets for cancer therapy.
Co-reporter:Elizabeth Leigh Baxter;Herbert L. Axelrod;Mark L. Paddock;Rachel Nechushtai;Aina E. Cohen;Patricia A. Jennings;Charles Wang;Phu Luong T. Vo;John A. Zuris;Jose N. Onuchic
PNAS 2013 Volume 110 (Issue 3 ) pp:948-953
Publication Date(Web):2013-01-15
DOI:10.1073/pnas.1208286110
Metalloproteins (MPs) comprise one-third of all known protein structures. This diverse set of proteins contain a plethora
of unique inorganic moieties capable of performing chemistry that would otherwise be impossible using only the amino acids
found in nature. Most of the well-studied MPs are generally viewed as being very rigid in structure, and it is widely thought
that the properties of the metal centers are primarily determined by the small fraction of amino acids that make up the local
environment. Here we examine both theoretically and experimentally whether distal regions can influence the metal center in
the diabetes drug target mitoNEET. We demonstrate that a loop (L2) 20 Å away from the metal center exerts allosteric control
over the cluster binding domain and regulates multiple properties of the metal center. Mutagenesis of L2 results in significant
shifts in the redox potential of the [2Fe-2S] cluster and orders of magnitude effects on the rate of [2Fe-2S] cluster transfer
to an apo-acceptor protein. These surprising effects occur in the absence of any structural changes. An examination of the
native basin dynamics of the protein using all-atom simulations shows that twisting in L2 controls scissoring in the cluster
binding domain and results in perturbations to one of the cluster-coordinating histidines. These allosteric effects are in
agreement with previous folding simulations that predicted L2 could communicate with residues surrounding the metal center.
Our findings suggest that long-range dynamical changes in the protein backbone can have a significant effect on the functional
properties of MPs.
Co-reporter:Benjamin T. Andrews, Dominique T. Capraro, Joanna I. Sulkowska, José N. Onuchic, and Patricia A. Jennings
The Journal of Physical Chemistry Letters 2013 Volume 4(Issue 1) pp:180-188
Publication Date(Web):December 18, 2012
DOI:10.1021/jz301893w
Topologically complex proteins fold by multiple routes as a result of hard-to-fold regions of the proteins. Oftentimes, these regions are introduced into the protein scaffold for function and increase frustration in the otherwise smooth-funneled landscape. Interestingly, while functional regions add complexity to folding landscapes, they may also contribute to a unique behavior referred to as hysteresis. While hysteresis is predicted to be rare, it is observed in various proteins, including proteins containing a unique peptide cyclization to form a fluorescent chromophore as well as proteins containing a knotted topology in their native fold. Here, hysteresis is demonstrated to be a consequence of the decoupling of unfolding events from the isomerization or hula-twist of a chromophore in one protein and the untying of the knot in a second protein system. The question now is can hysteresis be a marker for the interplay of landscapes where complex folding and functional regions overlap?
Co-reporter:Dominique T. Capraro;José N. Onuchic;Melinda Roy;Shachi Gosavi;Patricia A. Jennings
PNAS 2012 Volume 109 (Issue 5 ) pp:
Publication Date(Web):2012-01-31
DOI:10.1073/pnas.1114430109
Proteins fold into three-dimensional structures in a funneled energy landscape. This landscape is also used for functional
activity. Frustration in this landscape can arise from the competing evolutionary pressures of biological function and reliable
folding. Thus, the ensemble of partially folded states can populate multiple routes on this journey to the native state. Although
protein folding kinetics experiments have shown the presence of such routes for several proteins, there has been sparse information
about the structural diversity of these routes. In addition, why a given protein populates a particular route more often than
another protein of similar structure and sequence is not clear. Whereas multiple routes are observed in theoretical studies
on the folding of interleukin-1β (IL-1β), experimental results indicate one dominant route where the central portion of the
protein folds first, and is then followed by closure of the barrel in this β-trefoil fold. Here we show, using a combination
of computation and experiment, that the presence of functionally important regions like the β-bulge in the signaling protein
IL-1β strongly influences the choice of folding routes. By deleting the β-bulge, we directly observe the presence of route-switching.
This route-switching provides a direct link between route selection and the folding and functional landscapes of a protein.
Co-reporter:Elizabeth Leigh Baxter;José N. Onuchic;Patricia A. Jennings
PNAS 2012 Volume 109 (Issue 6 ) pp:
Publication Date(Web):2012-02-07
DOI:10.1073/pnas.1116369109
MitoNEET is a recently identified diabetes drug target that coordinates a transferable 2Fe-2S cluster, and additionally contains
an unusual strand swap. In this manuscript, we use a dual basin structure-based model to predict and characterize the folding
and functionality of strand swapping in mitoNEET. We demonstrate that a strand unswapped conformation is kinetically accessible
and that multiple levels of control are employed to regulate the conformational dynamics of the system. Environmental factors
such as temperature can shift route preference toward the unswapped pathway. Additionally we see that a region recently identified
as contributing to frustration in folding acts as a regulatory hinge loop that modulates conformational balance. Interestingly,
strand unswapping transfers strain specifically to cluster-coordinating residues, opening the cluster-coordinating pocket.
Strengthening contacts within the cluster-coordinating pocket opens a new pathway between the swapped and unswapped conformation
that utilizes cracking to bypass the unfolded basin. These results suggest that local control within distinct regions affect
motions important in regulating mitoNEET’s 2Fe-2S clusters.
Co-reporter:Elizabeth L. Baxter;Patricia A. Jennings;José N. Onuchic
PNAS 2011 Volume 108 (Issue 13 ) pp:5266-5271
Publication Date(Web):2011-03-29
DOI:10.1073/pnas.1017604108
MitoNEET is a recently identified drug target for a commonly prescribed diabetes drug, Pioglitazone. It belongs to a previously
uncharacterized ancient family of proteins for which the hallmark is the presence of a unique 39 amino acid CDGSH domain.
In order to characterize the folding landscape of this novel fold, we performed thermodynamic simulations on MitoNEET using
a structure-based model. Additionally, we implement a method of contact map clustering to partition out alternate pathways
in folding. This cluster analysis reveals a detour late in folding and enables us to carefully examine the folding mechanism
of each pathway rather than the macroscopic average. We observe that tightness in a region distal to the iron–sulfur cluster
creates a constraint in folding and additionally appears to mediate communication in folding between the two domains of the
protein. We demonstrate that by making changes at this site we are able to tweak the order of folding events in the cluster
binding domain as well as decrease the barrier to folding.
Co-reporter:Dominique T. Capraro, Patricia A. Jennings
Biophysical Journal (8 March 2016) Volume 110(Issue 5) pp:
Publication Date(Web):8 March 2016
DOI:10.1016/j.bpj.2016.01.017
Entanglement and knots occur across all aspects of the physical world. Despite the common belief that knots are too complicated for incorporation into proteins, knots have been identified in the native fold of a growing number of proteins. The discovery of proteins with this unique backbone characteristic has challenged the preconceptions about the complexity of biological structures, as well as current folding theories. Given the intricacies of the knotted geometry, the interplay between a protein’s fold, structure, and function is of particular interest. Interestingly, for most of these proteins, the knotted region appears critical both in folding and function, although full understanding of these contributions is still incomplete. Here, we experimentally reveal the impact of the knot on the landscape, the origin of the bistable nature of the knotted protein, and broaden the view of knot formation as uniquely decoupled from folding.
Co-reporter:Dominique T. Capraro, Heiko Lammert, David K. Heidary, Melinda Roy, Larry A. Gross, José N. Onuchic, Patricia A. Jennings
Biophysical Journal (20 August 2013) Volume 105(Issue 4) pp:
Publication Date(Web):20 August 2013
DOI:10.1016/j.bpj.2013.06.019
Deletion of the β-bulge trigger-loop results in both a switch in the preferred folding route, from the functional loop packing folding route to barrel closure, as well as conversion of the agonist activity of IL-1β into antagonist activity. Conversely, circular permutations of IL-1β conserve the functional folding route as well as the agonist activity. These two extremes in the folding-functional interplay beg the question of whether mutations in IL-1β would result in changes in the populations of heterogeneous folding routes and the signaling activity. A series of topologically equivalent water-mediated β-strand bridging interactions within the pseudosymmetric β-trefoil fold of IL-1β highlight the backbone water interactions that stabilize the secondary and tertiary structure of the protein. Additionally, conserved aromatic residues lining the central cavity appear to be essential for both stability and folding. Here, we probe these protein backbone-water molecule and side chain-side chain interactions and the role they play in the folding mechanism of this geometrically stressed molecule. We used folding simulations with structure-based models, as well as a series of folding kinetic experiments to examine the effects of the F42W core mutation on the folding landscape of IL-1β. This mutation alters water-mediated backbone interactions essential for maintaining the trefoil fold. Our results clearly indicate that this perturbation in the primary structure alters a structural water interaction and consequently modulates the population of folding routes accessed during folding and signaling activity.
Co-reporter:Andrea R. Conlan, Herbert L. Axelrod, Aina E. Cohen, Edward C. Abresch, ... Mark L. Paddock
Journal of Molecular Biology (11 September 2009) Volume 392(Issue 1) pp:143-153
Publication Date(Web):11 September 2009
DOI:10.1016/j.jmb.2009.06.079
The endoplasmic reticulum protein Miner1 is essential for health and longevity. Mis-splicing of CISD2, which codes for Miner1, is causative in Wolfram Syndrome 2 (WFS2) resulting in early onset optic atrophy, diabetes mellitus, deafness and decreased lifespan. In knock-out studies, disruption of CISD2 leads to accelerated aging, blindness and muscle atrophy. In this work, we characterized the soluble region of human Miner1 and solved its crystal structure to a resolution of 2.1 Å (R-factor = 17%). Although originally annotated as a zinc finger, we show that Miner1 is a homodimer harboring two redox-active 2Fe-2S clusters, indicating for the first time an association of a redox-active FeS protein with WFS2. Each 2Fe-2S cluster is bound by a rare Cys3-His motif within a 17 amino acid segment. Miner1 is the first functionally different protein that shares the NEET fold with its recently identified paralog mitoNEET, an outer mitochondrial membrane protein. We report the first measurement of the redox potentials (Em) of Miner1 and mitoNEET, showing that they are proton-coupled with Em ∼ 0 mV at pH 7.5. Changes in the pH sensitivity of their cluster stabilities are attributed to significant differences in the electrostatic distribution and surfaces between the two proteins. The structural and biophysical results are discussed in relation to possible roles of Miner1 in cellular Fe-S management and redox reactions.
Co-reporter:Kendra L. Hailey, Dominique T. Capraro, Sulyman Barkho, Patricia A. Jennings
Journal of Molecular Biology (10 July 2013) Volume 425(Issue 13) pp:2382-2392
Publication Date(Web):10 July 2013
DOI:10.1016/j.jmb.2013.03.016
The pleiotropic pro-inflammatory cytokine interleukin (IL)-1β has co-evolved with a competitive inhibitor, IL-1 receptor antagonist (IL-1Ra). IL-1β initiates cell signaling by binding the IL-1 receptor (IL-1R) whereas IL-1Ra acts as an antagonist, blocking receptor signaling. The current paradigm for agonist/antagonist functions for these two proteins is based on the receptor–ligand interaction observed in the crystal structures of the receptor–ligand complexes. While IL-1Ra and IL-1β are structurally homologous, IL-1Ra engages only two of the three extracellular domains of the receptor, whereas IL-1β engages all three. We find that an allosteric functional switch exists within a highly conserved pocket of residues, residues 111–120. This region is maintained across all IL-1 family members and serves as a hydrophobic mini-core for IL-1β folding. A key difference across species is a conserved aromatic residue at position 117 in IL-1β, versus a conserved cysteine in IL-1Ra at the analogous position, 116. We find that the replacement of C116 with a phenylalanine switches the protein from an antagonist to an agonist despite the distant location of C116 relative to receptor interaction sites. These results suggest new ways to develop designer cytokine activity into the β-trefoil fold and may be of general use in regulation of this large family of signaling proteins.Download high-res image (112KB)Download full-size imageHighlights► Is the structural plasticity of the β-trefoil fold elucidated by IL-1β studies consistent in IL-1Ra? ► C116F mutant shows similar overall structure and folding behavior and has agonist activity. ► A site in IL-1Ra diametrically opposite to that of receptor binding confirms the allosteric potential.
Co-reporter:Kendra L. Hailey, Dominique T. Capraro, Sulyman Barkho, Patricia A. Jennings
Journal of Molecular Biology (10 July 2013) Volume 425(Issue 13) pp:2382-2392
Publication Date(Web):10 July 2013
DOI:10.1016/j.jmb.2013.03.016
The pleiotropic pro-inflammatory cytokine interleukin (IL)-1β has co-evolved with a competitive inhibitor, IL-1 receptor antagonist (IL-1Ra). IL-1β initiates cell signaling by binding the IL-1 receptor (IL-1R) whereas IL-1Ra acts as an antagonist, blocking receptor signaling. The current paradigm for agonist/antagonist functions for these two proteins is based on the receptor–ligand interaction observed in the crystal structures of the receptor–ligand complexes. While IL-1Ra and IL-1β are structurally homologous, IL-1Ra engages only two of the three extracellular domains of the receptor, whereas IL-1β engages all three. We find that an allosteric functional switch exists within a highly conserved pocket of residues, residues 111–120. This region is maintained across all IL-1 family members and serves as a hydrophobic mini-core for IL-1β folding. A key difference across species is a conserved aromatic residue at position 117 in IL-1β, versus a conserved cysteine in IL-1Ra at the analogous position, 116. We find that the replacement of C116 with a phenylalanine switches the protein from an antagonist to an agonist despite the distant location of C116 relative to receptor interaction sites. These results suggest new ways to develop designer cytokine activity into the β-trefoil fold and may be of general use in regulation of this large family of signaling proteins.Download high-res image (112KB)Download full-size imageHighlights► Is the structural plasticity of the β-trefoil fold elucidated by IL-1β studies consistent in IL-1Ra? ► C116F mutant shows similar overall structure and folding behavior and has agonist activity. ► A site in IL-1Ra diametrically opposite to that of receptor binding confirms the allosteric potential.
.jpg)