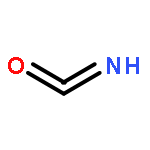
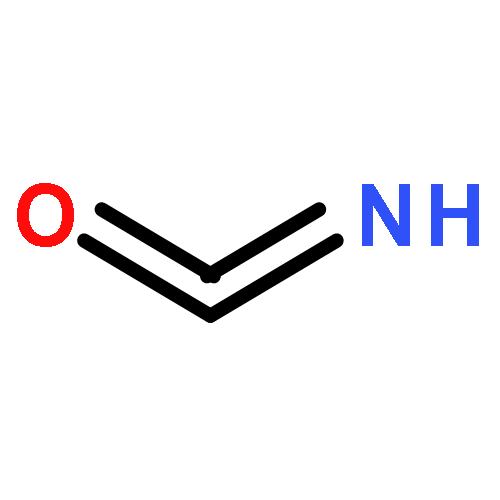
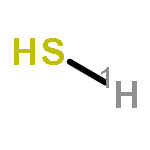
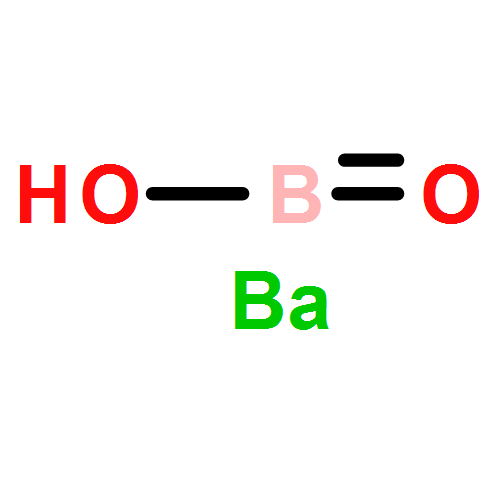Co-reporter:Colin M. Western;Brant E. Billinghurst
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 16) pp:10222-10226
Publication Date(Web):2017/04/19
DOI:10.1039/C7CP00266A
An initial implementation of a tool for automatic assignment of spectra within the PGOPHER program is presented, together with its application to rotational analysis of the ν11 band of cis-1,2-dichloroethene. It is based on the AUTOFIT algorithm presented by N. A. Seifert et al. (J. Mol. Spectrosc., 2015, 312, 13) but implemented in a more efficient and general way, allowing it to be applied to a much wider variety of spectra.
Co-reporter:Colin M. Western
Journal of Quantitative Spectroscopy and Radiative Transfer 2017 Volume 186() pp:221-242
Publication Date(Web):January 2017
DOI:10.1016/j.jqsrt.2016.04.010
•Easy-to-use graphical interface for assigning and understanding molecular spectra.•Simulates rotational and vibrational structure of many types of molecular spectra.•Fits molecular properties to line positions or spectral contours.•Handles linear molecules and symmetric and asymmetric tops.•Handles perturbations, nuclear and electron spin, and electric and magnetic fields.The pgopher program is a general purpose program for simulating and fitting molecular spectra, particularly the rotational structure. The current version can handle linear molecules, symmetric tops and asymmetric tops and many possible transitions, both allowed and forbidden, including multiphoton and Raman spectra in addition to the common electric dipole absorptions. Many different interactions can be included in the calculation, including those arising from electron and nuclear spin, and external electric and magnetic fields. Multiple states and interactions between them can also be accounted for, limited only by available memory. Fitting of experimental data can be to line positions (in many common formats), intensities or band contours and the parameters determined can be level populations as well as rotational constants. pgopher is provided with a powerful and flexible graphical user interface to simplify many of the tasks required in simulating, understanding and fitting molecular spectra, including Fortrat diagrams and energy level plots in addition to overlaying experimental and simulated spectra. The program is open source, and can be compiled with open source tools. This paper provides a formal description of the operation of version 9.1.
Co-reporter:Ashim Kumar Saha, Gautam Sarma, Chung-Hsin Yang, Sebastiaan Y. T. van de Meerakker, David H. Parker and Colin M. Western
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 21) pp:14145-14158
Publication Date(Web):30 Apr 2015
DOI:10.1039/C5CP01299F
Rotationally resolved resonance enhanced multiphoton ionisation spectra of the 1E′′ state of NH2D are presented and analysed. The analysis indicates a small (34.9 cm−1) lifting of the vibronic degeneracy of the zero point level, approximately equal in sign but opposite in magnitude to the splitting observed in NHD2 in previous work. This observation is consistent with previous measurements on systems with partial isotopic substitution subject to a mild Jahn–Teller effect. A model is developed to calculate the splitting induced by asymmetric isotopic substitution of a degenerate electronic state, based on a harmonic force field with linear and quadratic Jahn–Teller terms added. The force field is developed in internal co-ordinates to allow the same parameters to be used to calculate the pattern of vibronic levels for all four isotopologues. The lifting of the degeneracy of the zero point level on asymmetric substitution comes from the quadratic Jahn–Teller effect; the linear term does not lift the degeneracy.
Co-reporter:Chung-Hsin Yang, Gautam Sarma, Ashim Kumar Saha, David H. Parker and Colin M. Western
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 17) pp:6390-6399
Publication Date(Web):15 Mar 2013
DOI:10.1039/C3CP00012E
Rotational analysis of the (2 + 1) resonance enhanced multiphoton ionization (REMPI) spectrum of the 1E′′ Rydberg state of the ammonia isotopologue NHD2 is reported. While the electronic degeneracy is lifted in NHD2 the splitting is small enough that interactions between the two states must be considered, particularly to model the intensity of the transitions. A simple model is developed to account for these interactions, relating them to terms present in the symmetric isotopologues. Spectroscopic parameters for the zero point and (ν2′ = 1–6) vibrational levels of the B 1E′′ state have been derived using this model and the spectra are accurately simulated for the first time using the PGOPHER program. The current work provides the basis for on-going velocity map imaging studies of rotational energy transfer in the mixed isotopologues of ammonia.
Co-reporter:C.-H. Yang, G. Sarma, J. J. ter Meulen, D. H. Parker and C. M. Western
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 42) pp:13983-13991
Publication Date(Web):22 Sep 2010
DOI:10.1039/C0CP00946F
Rotational analysis of the (2 + 1) resonance enhanced multiphoton ionization (REMPI) spectrum of the 1B1 Rydberg state of the water isotopomers H2O, HOD and D2O is reported. Spectroscopic parameters for the v = 0 vibrational level of the 1B1 state of the mixed isotopomer HOD are derived and its spectra are accurately simulated for the first time using the PGOPHER program. Simulation of two photon spectra of the 1B1–1A1 transition of HOD requires two transition moments, and the ratio of these is determined and explained by a simple geometrical model. Optimal transitions for state-selective detection of low energy rotational states are identified for all three molecules. Analysis of the linewidths in the present work, combined with previous work [H. H. Kuge and K. Kleinermanns, J. Chem. Phys., 1989, 90, 46–52; K. J. Yuan et al., Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 19148–19153; M. N. R. Ashfold et al., Chem. Phys., 1984, 84, 35–50; G. Meijer et al., J. Chem. Phys., 1986, 85, 6914–6922.], suggests that while a simple 〈Ja′2〉-dependent model for heterogeneous predissociation of the 1B1 Rydberg state accounts for much of the quantum number dependence, it is not sufficient for describing the predissociation in any of the three isotopomers. The component of the linewidth due to the homogeneous predissociation attributed to predissociation of the 1B1 by the Ã1B1 state was found to be significantly narrower than in previous work, indicating a longer lifetime of the 1B1 Rydberg state. The current work provides the basis for on-going studies of rotational energy transfer in the mixed isotopomers of water using the velocity map imaging technique.
Co-reporter:Eyad H. Al-Samra, Colin M. Western
Journal of Molecular Spectroscopy 2010 260(2) pp: 135-137
Publication Date(Web):
DOI:10.1016/j.jms.2010.02.001
Co-reporter:Ching-Ping Liu, Nicola L. Elliott, Colin M. Western, Yuan-Pern Lee, Reginald Colin
Journal of Molecular Spectroscopy 2006 Volume 238(Issue 2) pp:213-223
Publication Date(Web):August 2006
DOI:10.1016/j.jms.2006.05.005
Spectra of the B3Σ−– X3Σ− transition in SO above the first dissociation limit are recorded using degenerate four wave mixing. These spectra are combined with earlier work involving laser induced fluorescence, absorption spectra and Fourier transform emission spectra, to enable a rotational analysis and deperturbation of vibrational levels of the B state up to v′ = 16. Numerous perturbations were noted within the B3Σ− state, and the origin of these is discussed. In a number of cases, these perturbations can be attributed to interactions with specific other electronic states of SO, such as A3Π, C3Π, d1Π, and A″ 3Σ+.
Co-reporter:Chung-Hsin Yang, Gautam Sarma, Ashim Kumar Saha, David H. Parker and Colin M. Western
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 17) pp:NaN6399-6399
Publication Date(Web):2013/03/15
DOI:10.1039/C3CP00012E
Rotational analysis of the (2 + 1) resonance enhanced multiphoton ionization (REMPI) spectrum of the 1E′′ Rydberg state of the ammonia isotopologue NHD2 is reported. While the electronic degeneracy is lifted in NHD2 the splitting is small enough that interactions between the two states must be considered, particularly to model the intensity of the transitions. A simple model is developed to account for these interactions, relating them to terms present in the symmetric isotopologues. Spectroscopic parameters for the zero point and (ν2′ = 1–6) vibrational levels of the B 1E′′ state have been derived using this model and the spectra are accurately simulated for the first time using the PGOPHER program. The current work provides the basis for on-going velocity map imaging studies of rotational energy transfer in the mixed isotopologues of ammonia.
Co-reporter:Ashim Kumar Saha, Gautam Sarma, Chung-Hsin Yang, Sebastiaan Y. T. van de Meerakker, David H. Parker and Colin M. Western
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 21) pp:NaN14158-14158
Publication Date(Web):2015/04/30
DOI:10.1039/C5CP01299F
Rotationally resolved resonance enhanced multiphoton ionisation spectra of the 1E′′ state of NH2D are presented and analysed. The analysis indicates a small (34.9 cm−1) lifting of the vibronic degeneracy of the zero point level, approximately equal in sign but opposite in magnitude to the splitting observed in NHD2 in previous work. This observation is consistent with previous measurements on systems with partial isotopic substitution subject to a mild Jahn–Teller effect. A model is developed to calculate the splitting induced by asymmetric isotopic substitution of a degenerate electronic state, based on a harmonic force field with linear and quadratic Jahn–Teller terms added. The force field is developed in internal co-ordinates to allow the same parameters to be used to calculate the pattern of vibronic levels for all four isotopologues. The lifting of the degeneracy of the zero point level on asymmetric substitution comes from the quadratic Jahn–Teller effect; the linear term does not lift the degeneracy.
Co-reporter:C.-H. Yang, G. Sarma, J. J. ter Meulen, D. H. Parker and C. M. Western
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 42) pp:NaN13991-13991
Publication Date(Web):2010/09/22
DOI:10.1039/C0CP00946F
Rotational analysis of the (2 + 1) resonance enhanced multiphoton ionization (REMPI) spectrum of the 1B1 Rydberg state of the water isotopomers H2O, HOD and D2O is reported. Spectroscopic parameters for the v = 0 vibrational level of the 1B1 state of the mixed isotopomer HOD are derived and its spectra are accurately simulated for the first time using the PGOPHER program. Simulation of two photon spectra of the 1B1–1A1 transition of HOD requires two transition moments, and the ratio of these is determined and explained by a simple geometrical model. Optimal transitions for state-selective detection of low energy rotational states are identified for all three molecules. Analysis of the linewidths in the present work, combined with previous work [H. H. Kuge and K. Kleinermanns, J. Chem. Phys., 1989, 90, 46–52; K. J. Yuan et al., Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 19148–19153; M. N. R. Ashfold et al., Chem. Phys., 1984, 84, 35–50; G. Meijer et al., J. Chem. Phys., 1986, 85, 6914–6922.], suggests that while a simple 〈Ja′2〉-dependent model for heterogeneous predissociation of the 1B1 Rydberg state accounts for much of the quantum number dependence, it is not sufficient for describing the predissociation in any of the three isotopomers. The component of the linewidth due to the homogeneous predissociation attributed to predissociation of the 1B1 by the Ã1B1 state was found to be significantly narrower than in previous work, indicating a longer lifetime of the 1B1 Rydberg state. The current work provides the basis for on-going studies of rotational energy transfer in the mixed isotopomers of water using the velocity map imaging technique.
Co-reporter:Colin M. Western and Brant E. Billinghurst
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 16) pp:NaN10226-10226
Publication Date(Web):2017/04/04
DOI:10.1039/C7CP00266A
An initial implementation of a tool for automatic assignment of spectra within the PGOPHER program is presented, together with its application to rotational analysis of the ν11 band of cis-1,2-dichloroethene. It is based on the AUTOFIT algorithm presented by N. A. Seifert et al. (J. Mol. Spectrosc., 2015, 312, 13) but implemented in a more efficient and general way, allowing it to be applied to a much wider variety of spectra.

![Pyridine, 4,4'-([1,1'-biphenyl]-4,4'-diyldi-2,1-ethenediyl)bis-](http://img.cochemist.com/ccimg/178000/177987-12-1.png)
![Pyridine, 4,4'-([1,1'-biphenyl]-4,4'-diyldi-2,1-ethenediyl)bis-](http://img.cochemist.com/ccimg/178000/177987-12-1_b.png)