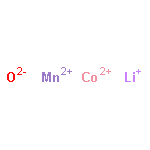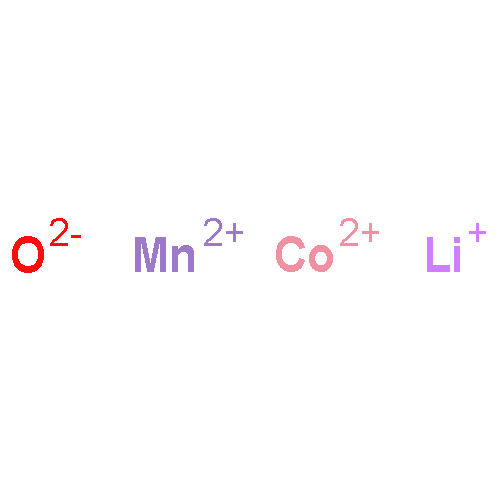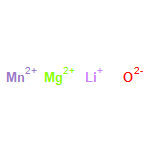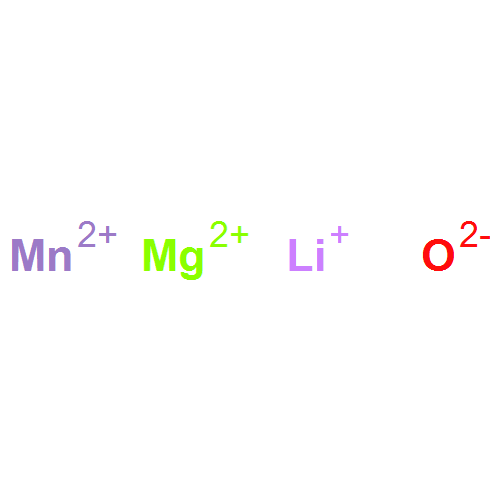Co-reporter:Julius Koettgen;Tobias Zacherle;Steffen Grieshammer
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 15) pp:9957-9973
Publication Date(Web):2017/04/12
DOI:10.1039/C6CP04802A
The rate of oxygen ion jumps in a solid oxide depends not only on the activation energy but also on the pre-exponential factor of diffusion. In order to allow a fully ab initio prediction of the oxygen ion conductivity in pure and samarium doped ceria, we calculated the attempt frequency for an oxygen ion jump from first principles combining DFT+U, the NEB method, phonon calculations and the transition state theory. Different definitions of the jump attempt frequency are presented. The equivalence of the Eyring and the Vineyard method is shown without restriction to the Gamma point. Convergence checks of the phonon mesh reveal that the common reduction to the Gamma point is not sufficient to calculate the attempt frequency. Calculations of Sm doped ceria revealed an increase of the prefactor. The attempt frequency for the constant pressure case in quasi-harmonic approximation is larger than the attempt frequency at constant volume in harmonic approximation. The calculated electronic energies, enthalpies and entropies of migration are in agreement with the experimental diffusion coefficients and activation energies.
Co-reporter:Markus Teusner
The Journal of Physical Chemistry C 2015 Volume 119(Issue 18) pp:9721-9727
Publication Date(Web):April 17, 2015
DOI:10.1021/jp512863u
Oxygen transport in mayenite single crystals was studied by means of 18O/16O isotope exchange experiments and time-of-flight secondary ion mass spectrometry (ToF-SIMS). Oxygen tracer diffusion coefficients D* and oxygen surface exchange coefficients k* were determined as a function of temperature, 773 ≤ T/K ≤ 1273, at an oxygen activity of aO2 = 0.1, and as a function of oxygen activity, 0.01 ≤ aO2 ≤ 0.9, at a temperature of T = 1123 K. Two diffusion processes were observed: a fast diffusion process that is attributed to the interstitialcy diffusion of free oxygen ions (O2–) and a slow diffusion process that is attributed to the interstitialcy diffusion of superoxide ions (O2–).
Co-reporter:Steffen Grieshammer, Benjamin O. H. Grope, Julius Koettgen and Manfred Martin
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 21) pp:9974-9986
Publication Date(Web):20 Jan 2014
DOI:10.1039/C3CP54811B
We investigate the dopant distribution and its influence on the oxygen ion conductivity of ceria doped with rare earth oxides by combining density functional theory and Monte Carlo simulations. We calculate the association energies of dopant pairs, oxygen vacancy pairs and between dopant ions and oxygen vacancies by means of DFT + U including finite size corrections. The cation coordination numbers from ensuing Metropolis Monte Carlo simulations show remarkable agreement with experimental data. Combining Metropolis and Kinetic Monte Carlo simulations we find a distinct dependence of the ionic conductivity on the dopant distribution and predict long term degradation of electrolytes based on doped ceria.
Co-reporter:Judith Hinterberg, Alina Adams, Bernhard Blümich, Paul Heitjans, Sangtae Kim, Zuhair A. Munir and Manfred Martin
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 45) pp:19825-19830
Publication Date(Web):09 Oct 2013
DOI:10.1039/C3CP53039F
We report nuclear magnetic resonance (NMR) results on water saturated, dense, nano-crystalline YSZ samples (9.5 mol% yttria doped zirconia) which exhibit proton conductivity at temperatures as low as room temperature. 1H-NMR spectra recorded under static and magic angle spinning conditions show two distinct signals. Their temperature-dependent behavior and their linewidths suggest that one can be attributed to (free) water adsorbed on the surface of the sample and the other one to mobile protons within the sample. This interpretation is supported by comparison with measurements on a single-crystalline sample. For the nano-crystalline samples motional narrowing is observed for the signal originating from protons in the sample interior. For these protons, the analysis of temperature and field dependent spin-lattice relaxation time T1 points towards diffusion in a confined two-dimensional geometry. We attribute this quasi two-dimensional motion to protons that are mobile along internal interfaces or nanopores of nano-crystalline YSZ.
Co-reporter:B.O.H. Grope, T. Zacherle, M. Nakayama, M. Martin
Solid State Ionics 2012 Volume 225() pp:476-483
Publication Date(Web):4 October 2012
DOI:10.1016/j.ssi.2012.01.028
Co-reporter:Han-Ill Yoo and Manfred Martin
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 44) pp:14699-14705
Publication Date(Web):14 Oct 2010
DOI:10.1039/C0CP00977F
It is well known that the open-cell voltage (U) of a galvanic cell involving a binary compound, or a multinary compound with a single kind of mobile ionic species, is a state property under a gradient of chemical potential of the mobile component. It is not so transparent, however, whether U is still a state property when involving a ternary or multinary compound with two or more kinds of mobile ions under multiple chemical potential gradients of those mobile components. We clarify this issue with a multinary oxide that conducts oxide ions, protons and electron holes and is exposed to the chemical potential gradients of both water and oxygen. We show that U is path- and history-dependent, and manifests itself along the diffusion paths of the two mobile components H and O under given boundary conditions.
Co-reporter:D. Roehrens, J. Brendt, D. Samuelis, M. Martin
Journal of Solid State Chemistry 2010 Volume 183(Issue 3) pp:532-541
Publication Date(Web):March 2010
DOI:10.1016/j.jssc.2009.12.024
We investigated the ammonolysis of β-Ga2O3 at elevated temperatures by means of ex situ X-ray diffraction, ex situ neutron diffraction and in situ X-ray absorption spectroscopy. Within the detection limits of these methods, we can rule out the existence of a crystalline or amorphous oxynitride phase that is not derived from wurtzite-type GaN. No evidence for a β-Ga2O3 related oxynitride phase was found, and the nitrogen solubility in β-Ga2O3 was found to be below the detection limit of about 2–3 at% in the anionic sublattice. These findings were obtained by monitoring the anionic occupancy factors and the lattice parameters of the β-Ga2O3 phase obtained from total diffraction pattern refinement with the Rietveld method and by linear combination fitting of the X-ray absorption spectra that were recorded during the ammonolysis.The ammonolysis of β-Ga2O3 powders forming GaN at temperatures of 600–780 °C was monitored by means of XRD, neutron diffraction and X-ray absorption spectroscopy in order to identify the possible intermediates and the solubility limit of nitrogen in the oxide lattice.
Co-reporter:Manfred Martin, Richard Dronskowski, Jürgen Janek, Klaus-Dieter Becker, Daniel Roehrens, Jochen Brendt, Marck W. Lumey, Lakshmi Nagarajan, Ilia Valov, Alexander Börger
Progress in Solid State Chemistry 2009 Volume 37(2–3) pp:132-152
Publication Date(Web):December 2009
DOI:10.1016/j.progsolidstchem.2009.11.005
Within the ternary system Ga–O–N we performed experimental and theoretical investigations on the thermodynamics, structure and kinetics of new stable and metastable compounds.We studied the ammonolysis of β-Ga2O3 at elevated temperatures by means of ex situ X-ray diffraction, ex situ neutron diffraction, and in situ X-ray absorption spectroscopy (XAS). From total diffraction pattern refinement with the Rietveld method we analyzed the anionic occupancy factors and the lattice parameters of β-Ga2O3 during the reaction. Within the detection limits of these methods, we can rule out the existence of a crystalline oxynitride phase that is not derived from wurtzite-type GaN. The nitrogen solubility in β-Ga2O3 was found to be below the detection limit of about 2–3 at.% in the anionic sublattice. The kinetics of the ammonolysis of β-Ga2O3 to α-GaN and of the oxidation of α-GaN to β-Ga2O3 was studied by means of in situ X-ray absorption spectroscopy. In both cases the reaction kinetics could be described well by fitting linear combinations of β-Ga2O3 and α-GaN spectra only, excluding that other crystalline or amorphous phases appear during these reactions. The kinetics of the ammonolysis can be described well by an extended Johnson–Mehl–Avrami–Kolmogorow model with nucleation and growth of GaN nuclei, while the oxidation kinetics can be modeled by a shrinking core model where Ga2O3 grows as a layer. Investigations by means of TEM and SEM support the assumptions in both models.To investigate the structure and energetics of spinel-type gallium oxynitrides (γ-galons) we performed first-principles calculations using density-functional theory. In addition to the ideal cubic γ-Ga3O3N we studied gallium deficient γ-galons within the Constant-Anion-Model.In highly non-stoichiometric, amorphous gallium oxide of approximate composition GaO1.2 we found at a temperature around 670 K an insulator–metal transition, with a conductivity jump of seven orders of magnitude. We demonstrate through experimental studies and density-functional theory calculations that the conductivity jump takes place at a critical gallium concentration and is induced by crystallization of stoichiometric β-Ga2O3 within the metastable oxide matrix. By doping with nitrogen the critical temperature and the conductivity in the highly conducting state can be tuned.
Co-reporter:J. Brendt, D. Samuelis, T. E. Weirich and M. Martin
Physical Chemistry Chemical Physics 2009 vol. 11(Issue 17) pp:3127-3137
Publication Date(Web):19 Mar 2009
DOI:10.1039/B901819K
The ammonolysis of β-Ga2O3 to α-GaN and the oxidation of α-GaN to β-Ga2O3 have been studied by means of in situX-ray absorption spectroscopy (XAS), transmission electron microscopy (TEM) and scanning electron microscopy (SEM). In situ X-ray absorption measurements on polycrystalline powder particles on the gallium K-edge during both reactions give detailed information about the reaction kinetics. We were able to extract this kinetics by fitting linear combinations of β-Ga2O3 and α-GaN spectra only. The kinetics of the ammonolysis can be described well by an extended Johnson–Mehl–Avrami–Kolmogorow model, while the oxidation kinetics can be modelled by a shrinking core model. Investigations by means of TEM and SEM support the assumptions in both models. Our experimental results and the models are discussed in terms of the reaction energetics and the reaction mechanisms.
Co-reporter:Sylvia Koerfer, Roger A. De Souza, Han-Ill Yoo, Manfred Martin
Solid State Sciences 2008 Volume 10(Issue 6) pp:725-734
Publication Date(Web):June 2008
DOI:10.1016/j.solidstatesciences.2007.06.017
The diffusion of strontium and zirconium in single crystal BaTiO3 was investigated in air at temperatures between 1000 °C and 1250 °C. Thin films of SrTiO3, deposited by spin coating a precursor solution and thin films of zirconium, deposited onto the sample surfaces by sputtering, were used as diffusion sources. The diffusion profiles were measured by SIMS depth profiling on a time-of-flight secondary ion mass spectrometer (ToF-SIMS). The diffusion coefficients of strontium and zirconium were given by DSr = 3.6 × 102.0±4.4 exp[−(543 ± 117) kJ mol−1/(RT)] cm2 s−1 and DZr = 1.1 × 101.0±2.1 exp[−(489 ± 56) kJ mol−1/(RT)] cm2 s−1. The results are discussed in terms of different diffusion mechanisms in the perovskite structure of BaTiO3.
Co-reporter:Chunhui Luo
Journal of Materials Science 2007 Volume 42( Issue 6) pp:1955-1964
Publication Date(Web):2007 March
DOI:10.1007/s10853-006-0452-6
The stability field of the Li–Mn–O spinel, Li1 + xMn2 − xO4 − δ, was investigated as a function of temperature, T, cation composition, nLi/nMn, and oxygen partial pressure, pO2, by means of in situ X-ray diffraction (XRD) and thermogravimetry (TG). In a T-nLi/nMn phase diagram, the stability field is described by the upper and lower critical temperatures, Tc1 and TcL, respectively. Above Tc1, Li2MnO3 is formed as a second phase, and below TcL Mn2O3 is formed. Both Tc1 and TcL decrease continuously with increasing nLi/nMn and increase with increasing pO2. The single phase region contains lithium-deficient and lithium-excess spinels, and no discontinuous change of the critical temperature curves was found at nLi/nMn = 0.5, corresponding to LiMn2O4. With the experimental data obtained in this work, a three-dimensional stability field of the Li–Mn–O spinel phase diagram is put forward to describe the relationship between T, nLi/nMn and pO2. In addition, the upper critical temperature, Tc1, was investigated for spinels doped with Ni, Co and Mg. For all three dopants, Tc1 decreases with increasing dopant concentration.
Co-reporter:Manfred Martin
Journal of Materials Science 2007 Volume 42( Issue 6) pp:1865
Publication Date(Web):2007 March
DOI:10.1007/s10853-006-1324-9
Co-reporter:Olaf Schulz, Manfred Martin, Christos Argirusis and Günter Borchardt
Physical Chemistry Chemical Physics 2003 vol. 5(Issue 11) pp:2308-2313
Publication Date(Web):06 May 2003
DOI:10.1039/B301882M
Cation tracer diffusion of 138La, 84Sr and 25Mg in polycrystalline samples of doped lanthanum gallate, La0.9Sr0.1Ga0.9Mg0.1O2.9, was investigated by SIMS for temperatures between 900°C and 1400°C. It was found that diffusion takes place through the bulk and along the grain boundaries. The bulk diffusion coefficients are similar for all cations with activation energies which are strongly dependent on temperature. At high temperatures, the activation energies are about 4.5 eV, while at low temperatures values of about 1.5 eV are found. These results are explained by a frozen in defect structure at low temperatures. This means that the observed activation energy at low temperatures represents only the migration energy of the different cations while the observed activation energy at high temperatures is the sum of the defect formation energy and the migration energy. The migration energies of all cations are nearly identical, although 138La and 84Sr are occupying A-sites while 25Mg is occupying B-sites in the perovskite-structure. To explain these experimental findings we propose a defect cluster containing cation vacancies of both the A- and the B-sublattice and anion vacancies as well.
Co-reporter:Julius Koettgen, Tobias Zacherle, Steffen Grieshammer and Manfred Martin
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 15) pp:NaN9973-9973
Publication Date(Web):2017/03/24
DOI:10.1039/C6CP04802A
The rate of oxygen ion jumps in a solid oxide depends not only on the activation energy but also on the pre-exponential factor of diffusion. In order to allow a fully ab initio prediction of the oxygen ion conductivity in pure and samarium doped ceria, we calculated the attempt frequency for an oxygen ion jump from first principles combining DFT+U, the NEB method, phonon calculations and the transition state theory. Different definitions of the jump attempt frequency are presented. The equivalence of the Eyring and the Vineyard method is shown without restriction to the Gamma point. Convergence checks of the phonon mesh reveal that the common reduction to the Gamma point is not sufficient to calculate the attempt frequency. Calculations of Sm doped ceria revealed an increase of the prefactor. The attempt frequency for the constant pressure case in quasi-harmonic approximation is larger than the attempt frequency at constant volume in harmonic approximation. The calculated electronic energies, enthalpies and entropies of migration are in agreement with the experimental diffusion coefficients and activation energies.
Co-reporter:Steffen Grieshammer, Benjamin O. H. Grope, Julius Koettgen and Manfred Martin
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 21) pp:NaN9986-9986
Publication Date(Web):2014/01/20
DOI:10.1039/C3CP54811B
We investigate the dopant distribution and its influence on the oxygen ion conductivity of ceria doped with rare earth oxides by combining density functional theory and Monte Carlo simulations. We calculate the association energies of dopant pairs, oxygen vacancy pairs and between dopant ions and oxygen vacancies by means of DFT + U including finite size corrections. The cation coordination numbers from ensuing Metropolis Monte Carlo simulations show remarkable agreement with experimental data. Combining Metropolis and Kinetic Monte Carlo simulations we find a distinct dependence of the ionic conductivity on the dopant distribution and predict long term degradation of electrolytes based on doped ceria.
Co-reporter:Judith Hinterberg, Alina Adams, Bernhard Blümich, Paul Heitjans, Sangtae Kim, Zuhair A. Munir and Manfred Martin
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 45) pp:NaN19830-19830
Publication Date(Web):2013/10/09
DOI:10.1039/C3CP53039F
We report nuclear magnetic resonance (NMR) results on water saturated, dense, nano-crystalline YSZ samples (9.5 mol% yttria doped zirconia) which exhibit proton conductivity at temperatures as low as room temperature. 1H-NMR spectra recorded under static and magic angle spinning conditions show two distinct signals. Their temperature-dependent behavior and their linewidths suggest that one can be attributed to (free) water adsorbed on the surface of the sample and the other one to mobile protons within the sample. This interpretation is supported by comparison with measurements on a single-crystalline sample. For the nano-crystalline samples motional narrowing is observed for the signal originating from protons in the sample interior. For these protons, the analysis of temperature and field dependent spin-lattice relaxation time T1 points towards diffusion in a confined two-dimensional geometry. We attribute this quasi two-dimensional motion to protons that are mobile along internal interfaces or nanopores of nano-crystalline YSZ.
Co-reporter:J. Brendt, D. Samuelis, T. E. Weirich and M. Martin
Physical Chemistry Chemical Physics 2009 - vol. 11(Issue 17) pp:NaN3137-3137
Publication Date(Web):2009/03/19
DOI:10.1039/B901819K
The ammonolysis of β-Ga2O3 to α-GaN and the oxidation of α-GaN to β-Ga2O3 have been studied by means of in situX-ray absorption spectroscopy (XAS), transmission electron microscopy (TEM) and scanning electron microscopy (SEM). In situ X-ray absorption measurements on polycrystalline powder particles on the gallium K-edge during both reactions give detailed information about the reaction kinetics. We were able to extract this kinetics by fitting linear combinations of β-Ga2O3 and α-GaN spectra only. The kinetics of the ammonolysis can be described well by an extended Johnson–Mehl–Avrami–Kolmogorow model, while the oxidation kinetics can be modelled by a shrinking core model. Investigations by means of TEM and SEM support the assumptions in both models. Our experimental results and the models are discussed in terms of the reaction energetics and the reaction mechanisms.
Co-reporter:Han-Ill Yoo and Manfred Martin
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 44) pp:NaN14705-14705
Publication Date(Web):2010/10/14
DOI:10.1039/C0CP00977F
It is well known that the open-cell voltage (U) of a galvanic cell involving a binary compound, or a multinary compound with a single kind of mobile ionic species, is a state property under a gradient of chemical potential of the mobile component. It is not so transparent, however, whether U is still a state property when involving a ternary or multinary compound with two or more kinds of mobile ions under multiple chemical potential gradients of those mobile components. We clarify this issue with a multinary oxide that conducts oxide ions, protons and electron holes and is exposed to the chemical potential gradients of both water and oxygen. We show that U is path- and history-dependent, and manifests itself along the diffusion paths of the two mobile components H and O under given boundary conditions.