Co-reporter:Darren W. Engers, Anna L. Blobaum, Rocco D. Gogliotti, Yiu-Yin Cheung, James M. Salovich, Pedro M. Garcia-Barrantes, J. Scott Daniels, Ryan Morrison, Carrie K. Jones, Matthew G. Soars, Xiaoliang Zhuo, Jeremy Hurley, John E. Macor, Joanne J. Bronson, P. Jeffrey Conn, Craig W. Lindsley, Colleen M. Niswender, and Corey R. Hopkins
ACS Chemical Neuroscience 2016 Volume 7(Issue 9) pp:1192
Publication Date(Web):April 13, 2016
DOI:10.1021/acschemneuro.6b00035
The efficacy of positive allosteric modulators (PAMs) of the metabotropic glutamate receptor 4 (mGlu4) in preclinical rodent models of Parkinson’s disease has been established by a number of groups. Here, we report an advanced preclinically characterized mGlu4 PAM, N-(3-chloro-4-fluorophenyl)-1H-pyrazolo[4,3-b]pyridin-3-amine (VU0418506). We detail the discovery of VU0418506 starting from a common picolinamide core scaffold and evaluation of a number of amide bioisosteres leading to the novel pyrazolo[4,3-b]pyridine head group. VU0418506 has been characterized as a potent and selective mGlu4 PAM with suitable in vivo pharmacokinetic properties in three preclinical safety species.Keywords: CYP induction; Metabotropic glutamate receptor 4; mGlu4; Parkinson’s disease; pyrazolo[4,3-b]pyridine
Co-reporter:Daniel R. Swale, Haruto Kurata, Sujay V. Kharade, Jonathan Sheehan, Rene Raphemot, Karl R. Voigtritter, Eric E. Figueroa, Jens Meiler, Anna L. Blobaum, Craig W. Lindsley, Corey R. Hopkins, and Jerod S. Denton
ACS Chemical Neuroscience 2016 Volume 7(Issue 7) pp:1013
Publication Date(Web):May 16, 2016
DOI:10.1021/acschemneuro.6b00111
The inward rectifier potassium (Kir) channel Kir7.1 (KCNJ13) has recently emerged as a key regulator of melanocortin signaling in the brain, electrolyte homeostasis in the eye, and uterine muscle contractility during pregnancy. The pharmacological tools available for exploring the physiology and therapeutic potential of Kir7.1 have been limited to relatively weak and nonselective small-molecule inhibitors. Here, we report the discovery in a fluorescence-based high-throughput screen of a novel Kir7.1 channel inhibitor, VU714. Site-directed mutagenesis of pore-lining amino acid residues identified glutamate 149 and alanine 150 as essential determinants of VU714 activity. Lead optimization with medicinal chemistry generated ML418, which exhibits sub-micromolar activity (IC50 = 310 nM) and superior selectivity over other Kir channels (at least 17-fold selective over Kir1.1, Kir2.1, Kir2.2, Kir2.3, Kir3.1/3.2, and Kir4.1) except for Kir6.2/SUR1 (equally potent). Evaluation in the EuroFins Lead Profiling panel of 64 GPCRs, ion-channels, and transporters for off-target activity of ML418 revealed a relatively clean ancillary pharmacology. While ML418 exhibited low CLHEP in human microsomes which could be modulated with lipophilicity adjustments, it showed high CLHEP in rat microsomes regardless of lipophilicity. A subsequent in vivo PK study of ML418 by intraperitoneal (IP) administration (30 mg/kg dosage) revealed a suitable PK profile (Cmax = 0.20 μM and Tmax = 3 h) and favorable CNS distribution (mouse brain/plasma Kp of 10.9 to support in vivo studies. ML418, which represents the current state-of-the-art in Kir7.1 inhibitors, should be useful for exploring the physiology of Kir7.1 in vitro and in vivo.Keywords: comparative modeling; electrophysiology; KCNJ13; melanocortin signaling; myometrium; thallium flux
Co-reporter:Jonathan O. Witt, Andrea L. McCollum, Miguel A. Hurtado, Eric D. Huseman, Daniel E. Jeffries, Kayla J. Temple, Hyekyung C. Plumley, Anna L. Blobaum, Craig W. Lindsley, Corey R. Hopkins
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 10) pp:2481-2488
Publication Date(Web):15 May 2016
DOI:10.1016/j.bmcl.2016.03.102
Herein, we report the synthesis and structure–activity relationship of a series of chiral alkoxymethyl morpholine analogs. Our efforts have culminated in the identification of (S)-2-(((6-chloropyridin-2-yl)oxy)methyl)-4-((6-fluoro-1H-indol-3-yl)methyl)morpholine as a novel potent and selective dopamine D4 receptor antagonist with selectivity against the other dopamine receptors tested (<10% inhibition at 1 μM against D1, D2L, D2S, D3, and D5).
Co-reporter:Rocco D. Gogliotti, Anna L. Blobaum, Ryan M. Morrison, J. Scott Daniels, James M. Salovich, Yiu-Yin Cheung, Alice L. Rodriguez, Matthew T. Loch, P. Jeffrey Conn, Craig W. Lindsley, Colleen M. Niswender, Corey R. Hopkins
Bioorganic & Medicinal Chemistry Letters 2016 26(13) pp: 2984-2987
Publication Date(Web):1 July 2016
DOI:10.1016/j.bmcl.2016.05.029
Herein we report the synthesis and characterization of a novel series of N-phenylsulfonyl-1H-pyrrole picolinamides as novel positive allosteric modulators of mGlu4. We detail our work towards finding phenyl replacements for the core scaffold of previously reported phenyl sulfonamides and phenyl sulfone compounds. Our efforts culminated in the identification of N-(1-((3,4-dimethylphenyl)sulfonyl)-1H-pyrrol-3-yl)picolinamide as a potent PAM of mGlu4.
Co-reporter:Sean R. Bollinger, Darren W. Engers, Elizabeth A. Ennis, Jane Wright, Charles W. Locuson, Craig W. Lindsley, Randy D. Blakely, Corey R. Hopkins
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 8) pp:1757-1760
Publication Date(Web):15 April 2015
DOI:10.1016/j.bmcl.2015.02.058
The synthesis and SAR of 4-methoxy-3-(piperidin-4-yl) benzamides identified after a high-throughput screen of the MLPCN library is reported. SAR was explored around the 3-piperidine substituent as well as the amide functionality of the reported compounds. Starting from the initial lead compounds, 1–7, iterative medicinal chemistry efforts led to the identification of ML352 (10m). ML352 represents a potent and selective inhibitor of CHT based on a drug-like scaffold.
Co-reporter:Wong Wen;Dr. Yan Wang;Zhe Li;Dr. Pang-Yen Tseng;Dr. Owen B. McManus;Dr. Meng Wu;Dr. Min Li; Craig W. Lindsley; Xinzhong Dong; Corey R. Hopkins
ChemMedChem 2015 Volume 10( Issue 1) pp:57-61
Publication Date(Web):
DOI:10.1002/cmdc.201402277
Abstract
Previous studies have shown that the activation of mouse MrgC11, a G-protein-coupled receptor, by its peptide ligand BAM8-22 can inhibit chronic pain. A large-scale screen has been carried out to isolate small-molecule allosteric agonists of MrgX1, the human homologue of MrgC11. The goal of this study is to improve the efficacy and potency of positive allosteric modulators (PAMs) with therapeutic implications in combating chronic pain. Herein we report an iterative parallel synthesis effort and a structure–activity relationship study of a series of arylsulfonamides which led to the discovery of the first PAM of MrgX1, ML382.
Co-reporter:Cynthia B. Berry, Michael Bubser, Carrie K. Jones, John P. Hayes, James A. Wepy, Charles W. Locuson, J. Scott Daniels, Craig W. Lindsley, and Corey R. Hopkins
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 9) pp:1060
Publication Date(Web):July 9, 2014
DOI:10.1021/ml500267c
Herein, we report the structure–activity relationship of a chiral morpholine-based scaffold, which led to the identification of a potent and selective dopamine 4 (D4) receptor antagonist. The 4-chlorobenzyl moiety was identified, and the compound was designated an MLPCN probe molecule, ML398. ML398 is potent against the D4 receptor with IC50 = 130 nM and Ki = 36 nM and shows no activity against the other dopamine receptors tested (>20 μM against D1, D2S, D2L, D3, and D5). Further in vivo studies showed that ML398 reversed cocaine-induced hyperlocomotion at 10 mg/kg.Keywords: addiction; Dopamine 4 receptor antagonist; ML398; MLPCN
Co-reporter:Darren W. Engers, Audrey Y. Frist, Craig W. Lindsley, Charles C. Hong, Corey R. Hopkins
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 11) pp:3248-3252
Publication Date(Web):1 June 2013
DOI:10.1016/j.bmcl.2013.03.113
A structure–activity relationship of the 3- and 6-positions of the pyrazolo[1,5-a]pyrimidine scaffold of the known BMP inhibitors dorsomorphin, 1, LDN-193189, 2, and DMH1, 3, led to the identification of a potent and selective compound for ALK2 versus ALK3. The potency contributions of several 3-position substituents were evaluated with subtle structural changes leading to significant changes in potency. From these studies, a novel 5-quinoline molecule was identified and designated an MLPCN probe molecule, ML347, which shows >300-fold selectivity for ALK2 and presents the community with a selective molecular probe for further biological evaluation.
Co-reporter:Uyen Le, Bruce J. Melancon, Thomas M. Bridges, Paige N. Vinson, Thomas J. Utley, Atin Lamsal, Alice L. Rodriguez, Daryl Venable, Douglas J. Sheffler, Carrie K. Jones, Anna L. Blobaum, Michael R. Wood, J. Scott Daniels, P. Jeffrey Conn, Colleen M. Niswender, Craig W. Lindsley, Corey R. Hopkins
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 1) pp:346-350
Publication Date(Web):1 January 2013
DOI:10.1016/j.bmcl.2012.10.073
Herein we report a next generation muscarinic receptor 4 (M4) positive allosteric modulator (PAM), ML253 which exhibits nanomolar activity at both the human (EC50 = 56 nM) and rat (EC50 = 176 nM) receptors and excellent efficacy by the left-ward shift of the ACh concentration response curve (fold shift, human = 106; rat = 50). In addition, ML253 is selective against the four other muscarinic subtypes, displays excellent CNS exposure and is active in an amphetamine-induced hyperlocomotion assay.
Co-reporter:Haibo Yu;Beiyan Zou;Cecile Terrenoire;Robert S. Kass;Corey R. Hopkins;Meng Wu;Min Li;Zhihong Lin;Owen B. McManus;Hongkang Zhang;Craig W. Lindsley;Margrith E. Mattmann
PNAS 2013 Volume 110 (Issue 21 ) pp:8732-8737
Publication Date(Web):2013-05-21
DOI:10.1073/pnas.1300684110
Voltage-gated KCNQ1 (Kv7.1) potassium channels are expressed abundantly in heart but they are also found in multiple other
tissues. Differential coassembly with single transmembrane KCNE beta subunits in different cell types gives rise to a variety
of biophysical properties, hence endowing distinct physiological roles for KCNQ1–KCNEx complexes. Mutations in either KCNQ1 or KCNE1 genes result in diseases in brain, heart, and the respiratory system. In addition to complexities arising from existence
of five KCNE subunits, KCNE1 to KCNE5, recent studies in heterologous systems suggest unorthodox stoichiometric dynamics in
subunit assembly is dependent on KCNE expression levels. The resultant KCNQ1–KCNE channel complexes may have a range of zero
to two or even up to four KCNE subunits coassembling per KCNQ1 tetramer. These findings underscore the need to assess the
selectivity of small-molecule KCNQ1 modulators on these different assemblies. Here we report a unique small-molecule gating
modulator, ML277, that potentiates both homomultimeric KCNQ1 channels and unsaturated heteromultimeric (KCNQ1)4(KCNE1)n (n < 4) channels. Progressive increase of KCNE1 or KCNE3 expression reduces efficacy of ML277 and eventually abolishes ML277-mediated
augmentation. In cardiomyocytes, the slowly activating delayed rectifier potassium current, or IKs, is believed to be a heteromultimeric combination of KCNQ1 and KCNE1, but it is not entirely clear whether IKs is mediated by KCNE-saturated KCNQ1 channels or by channels with intermediate stoichiometries. We found ML277 effectively
augments IKs current of cultured human cardiomyocytes and shortens action potential duration. These data indicate that unsaturated heteromultimeric
(KCNQ1)4(KCNE1)n channels are present as components of IKs and are pharmacologically distinct from KCNE-saturated KCNQ1–KCNE1 channels.
Co-reporter:Yiu-Yin Cheung ; Haibo Yu ; Kaiping Xu ; Beiyan Zou ; Meng Wu ; Owen B. McManus ; Min Li ; Craig W. Lindsley ;Corey R. Hopkins
Journal of Medicinal Chemistry 2012 Volume 55(Issue 15) pp:6975-6979
Publication Date(Web):July 13, 2012
DOI:10.1021/jm300700v
A potent and selective inhibitor of KCNQ2, (S)-5 (ML252, IC50 = 69 nM), was discovered after a high-throughput screen of the MLPCN library was performed. SAR studies revealed a small structural change (ethyl group to hydrogen) caused a functional shift from antagonist to agonist activity (37, EC50 = 170 nM), suggesting an interaction at a critical site for controlling gating of KCNQ2 channels.
Co-reporter:James M. Salovich, Paige N. Vinson, Douglas J. Sheffler, Atin Lamsal, Thomas J. Utley, Anna L. Blobaum, Thomas M. Bridges, Uyen Le, Carrie K. Jones, Michael R. Wood, J. Scott Daniels, P. Jeffrey Conn, Colleen M. Niswender, Craig W. Lindsley, Corey R. Hopkins
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 15) pp:5084-5088
Publication Date(Web):1 August 2012
DOI:10.1016/j.bmcl.2012.05.109
Herein we describe the discovery and development of a novel class of M4 positive allosteric modulators, culminating in the discovery of ML293. ML293 exhibited modest potency at the human M4 receptor (EC50 = 1.3 μM) and excellent efficacy as noted by the 14.6-fold leftward shift of the agonist concentration–response curve. ML293 was also selective versus the other muscarinic subtypes and displayed excellent in vivo PK properties in rat with low IV clearance (11.6 mL/min/kg) and excellent brain exposure (PO PBL, 10 mg/kg at 1 h, [Brain] = 10.3 μM, B:P = 0.85).
Co-reporter:Darren W. Engers ▼; Julie R. Field ; Uyen Le ; Ya Zhou ; Julie D. Bolinger ; Rocio Zamorano ; Anna L. Blobaum ; Carrie K. Jones ; Satyawan Jadhav ; C. David Weaver ; P. Jeffrey Conn ; Craig W. Lindsley ▼; Colleen M. Niswender ;Corey R. Hopkins ▼
Journal of Medicinal Chemistry 2011 Volume 54(Issue 4) pp:1106-1110
Publication Date(Web):January 19, 2011
DOI:10.1021/jm101271s
Herein we report the discovery, synthesis, and evaluation of a series of N-(4-acetamido)-phenylpicolinamides as positive allosteric modulators of mGlu4. Compounds from the series show submicromolar potency at both human and rat mGlu4. In addition, pharmacokinetic studies utilizing subcutaneous dosing demonstrated good brain exposure in rats.
Co-reporter:Carrie K. Jones ; Darren W. Engers ; Analisa D. Thompson ; Julie R. Field ; Anna L. Blobaum ; Stacey R. Lindsley ; Ya Zhou ; Rocco D. Gogliotti ; Satyawan Jadhav ; Rocio Zamorano ; Jim Bogenpohl ∞; Yoland Smith ∞; Ryan Morrison ; J. Scott Daniels ; C. David Weaver ; P. Jeffrey Conn ; Craig W. Lindsley ; Colleen M. Niswender ;Corey R. Hopkins
Journal of Medicinal Chemistry 2011 Volume 54(Issue 21) pp:7639-7647
Publication Date(Web):October 3, 2011
DOI:10.1021/jm200956q
There is an increasing amount of literature data showing the positive effects on preclinical antiparkinsonian rodent models with selective positive allosteric modulators of metabotropic glutamate receptor 4 (mGlu4). However, most of the data generated utilize compounds that have not been optimized for druglike properties, and as a consequence, they exhibit poor pharmacokinetic properties and thus do not cross the blood–brain barrier. Herein, we report on a series of N-4-(2,5-dioxopyrrolidin-1-yl)phenylpicolinamides with improved PK properties with excellent potency and selectivity as well as improved brain exposure in rodents. Finally, ML182 was shown to be orally active in the haloperidol induced catalepsy model, a well-established antiparkinsonian model.
Co-reporter:Albert J. Robichaud, Darren W. Engers, Craig W. Lindsley, and Corey R. Hopkins
ACS Chemical Neuroscience 2011 Volume 2(Issue 8) pp:433
Publication Date(Web):June 14, 2011
DOI:10.1021/cn200043e
This Review describes recent activity in the advancement of ligands for the metabotropic glutamate 4 receptor subtype and their potential utility as central nervous system (CNS) therapeutics. Until recently, there was a paucity of compounds with suitable selectivity and druglike properties to elucidate the value of this target. The search for selective entities has led several groups to the investigation of allosteric modulators as a path to optimization of potential ligands. Recent efforts, discussed here, have afforded a variety of derivatives with improvements in potency, solubility, and pharmacokinetic properties that garner support for continued investigation and optimization.Keywords: allosteric modulator; anxiety; Class C GPCR; cognitive disorders; Metabotropic glutamate receptor 4; neurodegenerative disorders; orthosteric ligand; pain; Parkinson’s disease; psychiatric disorders
Co-reporter:Haibo Yu, Meng Wu, Steven D. Townsend, Beiyan Zou, Shunyou Long, J. Scott Daniels, Owen B. McManus, Min Li, Craig W. Lindsley, and Corey R. Hopkins
ACS Chemical Neuroscience 2011 Volume 2(Issue 10) pp:572
Publication Date(Web):July 15, 2011
DOI:10.1021/cn200065b
Herein we report the discovery, synthesis, and evaluation of a series of N-aryl-bicyclo[2.2.1]heptane-2-carboxamides as selective KCNQ2 (Kv7.2) and KCNQ4 (Kv7.4) channel openers. The best compound, 1 (ML213), has an EC50 of 230 nM (KCNQ2) and 510 nM (KCNQ4) and is selective for KCNQ2 and KCNQ4 channels versus a large battery of related potassium channels, as well as affording modest brain levels. This represents the first report of unique selectivity profiles for KCNQ2 and KCNQ4 over the other channels (KCNQ1/3/5) and as such should prove to be a valuable tool compound for understanding these channels in regulating neuronal activity.Keywords: activator; ion channels; KCNQ2; KCNQ4; Kv7; ML218; MLPCN probe
Co-reporter:Yiu-Yin Cheung, Rocio Zamorano, Anna L. Blobaum, C. David Weaver, P. Jeffrey Conn, Craig W. Lindsley, Colleen M. Niswender, and Corey R. Hopkins
ACS Combinatorial Science 2011 Volume 13(Issue 2) pp:159
Publication Date(Web):February 21, 2011
DOI:10.1021/co1000508
Using a functional high-throughput screening (HTS) and subsequent solution-phase parallel synthesis approach, we have discovered a novel series of positive allosteric modulators for mGlu4, a G-protein coupled receptor. This series is comprised of a homopiperazine central core. The solution-phase parallel synthesis and SAR of analogs derived from this series will be presented. This series of positive allosteric modulators of mGlu4 provide critical research tools to further probe the mGlu4-mediated effects in Parkinson’s disease.Keywords (keywords): metabotropic glutamate receptor 4; mGlu4; parallel synthesis; structure−activity relationship
Co-reporter:Richard Williams, Ya Zhou, Colleen M. Niswender, Qingwei Luo, P. Jeffrey Conn, Craig W. Lindsley and Corey R. Hopkins
ACS Chemical Neuroscience 2010 Volume 1(Issue 6) pp:411
Publication Date(Web):March 8, 2010
DOI:10.1021/cn9000318
This paper describes a detailed structure−activity relationship (SAR) analysis of the metabotropic glutamate receptor 4 (mGluR4) positive allosteric modulator, (−)-N-phenyl-7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxamide (PHCCC). We have now developed compounds with improved potency and efficacy; in addition, compounds are presented that show selectivity for mGluR4 versus the other mGluR subtypes.Keywords (keywords): allosteric modulation; Metabotropic glutamate receptor 4; Parkinson’s disease; PHCCC; positive allosteric modulator; SAR; structure−activity relationship
Co-reporter:Darren W. Engers, Patrick R. Gentry, Richard Williams, Julie D. Bolinger, C. David Weaver, Usha N. Menon, P. Jeffrey Conn, Craig W. Lindsley, Colleen M. Niswender, Corey R. Hopkins
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 17) pp:5175-5178
Publication Date(Web):1 September 2010
DOI:10.1016/j.bmcl.2010.07.007
Herein we disclose the synthesis and SAR of a series of 4-(phenylsulfamoyl)phenylacetamide compounds as mGlu4 positive allosteric modulators (PAMs) that were identified via a functional HTS. An iterative parallel approach to these compounds culminated in the discovery of VU0364439 (11) which represents the most potent (19.8 nM) mGlu4 PAM reported to date.Herein we disclose the synthesis and SAR of a series of 4-(phenylsulfamoyl)phenylacetamide compounds as mGlu4 positive allosteric modulators (PAMs) that were identified via a functional HTS. An iterative parallel approach to these compounds culminated in the discovery of VU0364439 (11) which represents the most potent (19.8 nM) mGlu4 PAM reported to date.
Co-reporter:James M. Salovich, Craig W. Lindsley, Corey R. Hopkins
Tetrahedron Letters 2010 Volume 51(Issue 29) pp:3796-3799
Publication Date(Web):21 July 2010
DOI:10.1016/j.tetlet.2010.05.060
Herein we report a general synthesis of 1,3-diarylsubstituted indazoles utilizing a two-step Suzuki cross-coupling/deprotection/N-arylation sequence. This procedure proceeds in excellent overall yield starting from the 3-iodo-N-Boc indazole derivative allowing for rapid access to these compounds.
Co-reporter:Darren W. Engers ; Colleen M. Niswender ; C. David Weaver ; Satyawan Jadhav ; Usha N. Menon ; Rocio Zamorano ; P. Jeffrey Conn ; Craig W. Lindsley ;Corey R. Hopkins
Journal of Medicinal Chemistry 2009 Volume 52(Issue 14) pp:4115-4118
Publication Date(Web):May 27, 2009
DOI:10.1021/jm9005065
We report the synthesis and evaluation of a series of heterobiaryl amides as positive allosteric modulators of mGluR4. Compounds 9b and 9c showed submicromolar potency at both human and rat mGluR4. In addition, both 9b and 9c were shown to be centrally penetrant in rats using nontoxic vehicles, a major advance for the mGluR4 field.
Co-reporter:Richard Williams, Kari A. Johnson, Patrick R. Gentry, Colleen M. Niswender, Charles D. Weaver, P. Jeffrey Conn, Craig W. Lindsley, Corey R. Hopkins
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 17) pp:4967-4970
Publication Date(Web):1 September 2009
DOI:10.1016/j.bmcl.2009.07.072
This Letter describes the synthesis and SAR of the novel positive allosteric modulator, VU0155041, a compound that has shown in vivo efficacy in rodent models of Parkinson’s disease. The synthesis takes advantage of an iterative parallel synthesis approach to rapidly synthesize and evaluate a number of analogs of VU0155041.This Letter describes the synthesis and SAR of the novel positive allosteric modulator, VU0155041, a compound that has shown in vivo efficacy in rodent models of Parkinson’s disease. The synthesis takes advantage of an iterative parallel synthesis approach to rapidly synthesize and evaluate a number of analogs of VU0155041.





![7-Bromothieno[2,3-b]pyrazine](http://img.cochemist.com/ccimg/1126900/1126824-72-3.png)
![7-Bromothieno[2,3-b]pyrazine](http://img.cochemist.com/ccimg/1126900/1126824-72-3_b.png)
![4-(6-(4-(piperazin-1-yl)phenyl)pyrazolo[1,5-a]pyrimidin-3-yl)quinoline](http://img.cochemist.com/ccimg/1062400/1062368-24-4.png)
![4-(6-(4-(piperazin-1-yl)phenyl)pyrazolo[1,5-a]pyrimidin-3-yl)quinoline](http://img.cochemist.com/ccimg/1062400/1062368-24-4_b.png)

![Benzenamine, 3-chloro-4-[(5-chloro-2-pyridinyl)oxy]-](http://img.cochemist.com/ccimg/875800/875774-83-7.png)
![Benzenamine, 3-chloro-4-[(5-chloro-2-pyridinyl)oxy]-](http://img.cochemist.com/ccimg/875800/875774-83-7_b.png)
![6-(4-(2-(Piperidin-1-yl)ethoxy)phenyl)-3-(pyridin-4-yl)pyrazolo[1,5-a]pyrimidine](http://img.cochemist.com/ccimg/866500/866405-64-3.png)
![6-(4-(2-(Piperidin-1-yl)ethoxy)phenyl)-3-(pyridin-4-yl)pyrazolo[1,5-a]pyrimidine](http://img.cochemist.com/ccimg/866500/866405-64-3_b.png)

![1-(5,6-Dimethyl-thieno[2,3-d]pyrimidin-4-yl)-piperidine-4-carboxylic acid](http://img.cochemist.com/ccimg/843000/842971-60-2.png)
![1-(5,6-Dimethyl-thieno[2,3-d]pyrimidin-4-yl)-piperidine-4-carboxylic acid](http://img.cochemist.com/ccimg/843000/842971-60-2_b.png)



![Pyridine, 2-[5-(3-chlorophenyl)-1,2,4-oxadiazol-3-yl]-](http://img.cochemist.com/ccimg/327100/327056-08-6.png)
![Pyridine, 2-[5-(3-chlorophenyl)-1,2,4-oxadiazol-3-yl]-](http://img.cochemist.com/ccimg/327100/327056-08-6_b.png)


![9H-Xanthene-9-propanoicacid, a-amino-a-[(1S,2S)-2-carboxycyclopropyl]-,(aS)-](http://img.cochemist.com/ccimg/202000/201943-63-7.png)
![9H-Xanthene-9-propanoicacid, a-amino-a-[(1S,2S)-2-carboxycyclopropyl]-,(aS)-](http://img.cochemist.com/ccimg/202000/201943-63-7_b.png)









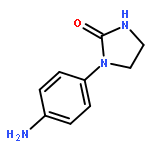
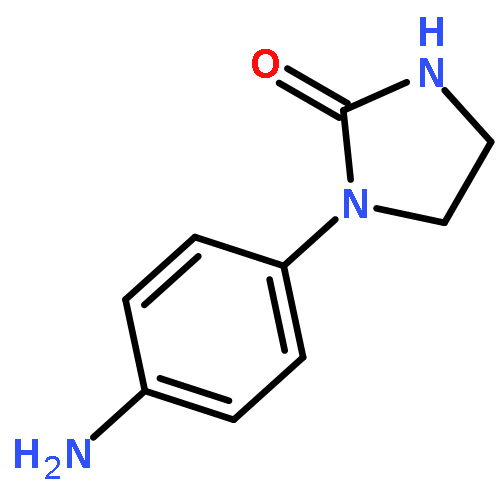
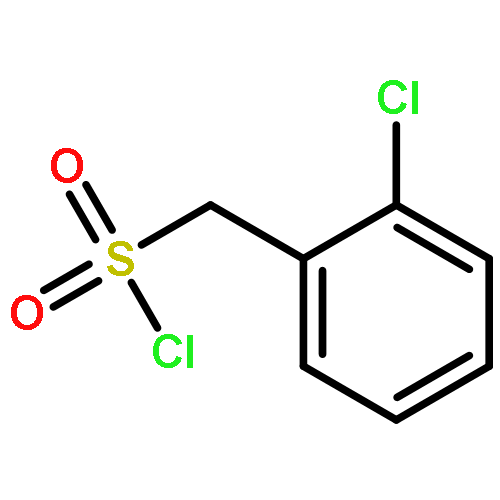
![Benzoic acid,2-[[2-[(methylsulfonyl)amino]benzoyl]amino]-, ethyl ester](http://img.cochemist.com/ccimg/89400/89369-45-9.png)
![Benzoic acid,2-[[2-[(methylsulfonyl)amino]benzoyl]amino]-, ethyl ester](http://img.cochemist.com/ccimg/89400/89369-45-9_b.png)

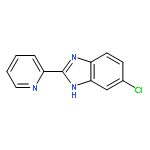
![2-[3-(3-CHLOROPHENYL)-1H-1,2,4-TRIAZOL-5-YL]PYRIDINE](http://img.cochemist.com/ccimg/76600/76591-82-7.png)
![2-[3-(3-CHLOROPHENYL)-1H-1,2,4-TRIAZOL-5-YL]PYRIDINE](http://img.cochemist.com/ccimg/76600/76591-82-7_b.png)

![2-[2-phenylethyl]-morpholine](http://img.cochemist.com/ccimg/58100/58039-64-8.png)
![2-[2-phenylethyl]-morpholine](http://img.cochemist.com/ccimg/58100/58039-64-8_b.png)
![Imidazo[1,5-a]pyridine-1-carbaldehyde](http://img.cochemist.com/ccimg/56700/56671-67-1.png)
![Imidazo[1,5-a]pyridine-1-carbaldehyde](http://img.cochemist.com/ccimg/56700/56671-67-1_b.png)
![Morpholine, 2-[(4-methoxyphenoxy)methyl]-, (S)- (9CI)](/data/chemimg/99700/56287-60-6.png)
![Morpholine, 2-[(4-methoxyphenoxy)methyl]-, (S)- (9CI)](/data/chemimg/99700/56287-60-6_b.png)
![(2s)-2-[(3-methoxyphenoxy)methyl]morpholine;hydrochloride](http://img.cochemist.com/ccimg/55300/55253-30-0.png)
![(2s)-2-[(3-methoxyphenoxy)methyl]morpholine;hydrochloride](http://img.cochemist.com/ccimg/55300/55253-30-0_b.png)

![2-BROMO-1-METHYL-1H-BENZO[D]IMIDAZOLE](http://img.cochemist.com/ccimg/49600/49572-60-3.png)
![2-BROMO-1-METHYL-1H-BENZO[D]IMIDAZOLE](http://img.cochemist.com/ccimg/49600/49572-60-3_b.png)








![Morpholinium,2,2'-[1,1'-biphenyl]-4,4'-diylbis[2-hydroxy-4,4-dimethyl-, bromide (1:2)](http://img.cochemist.com/ccimg/400/312-45-8.png)
![Morpholinium,2,2'-[1,1'-biphenyl]-4,4'-diylbis[2-hydroxy-4,4-dimethyl-, bromide (1:2)](http://img.cochemist.com/ccimg/400/312-45-8_b.png)
![3-Azaspiro[5.5]undecane](http://img.cochemist.com/ccimg/200/180-44-9.png)
![3-Azaspiro[5.5]undecane](http://img.cochemist.com/ccimg/200/180-44-9_b.png)
![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2.png)
![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2_b.png)